Back to Archived Journals » Open Access Journal of Sports Medicine » Volume 13
Cross-Communication Between Knee Osteoarthritis and Fibrosis: Molecular Pathways and Key Molecules
Authors Bolia IK ![]() , Mertz K
, Mertz K ![]() , Faye E
, Faye E ![]() , Sheppard J, Telang S
, Sheppard J, Telang S ![]() , Bogdanov J
, Bogdanov J ![]() , Hasan LK
, Hasan LK ![]() , Haratian A
, Haratian A ![]() , Evseenko D, Weber AE, Petrigliano FA
, Evseenko D, Weber AE, Petrigliano FA ![]()
Received 10 November 2021
Accepted for publication 18 February 2022
Published 1 March 2022 Volume 2022:13 Pages 1—15
DOI https://doi.org/10.2147/OAJSM.S321139
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Andreas Imhoff
Ioanna K Bolia, Kevin Mertz, Ethan Faye, Justin Sheppard, Sagar Telang, Jacob Bogdanov, Laith K Hasan, Aryan Haratian, Denis Evseenko, Alexander E Weber, Frank A Petrigliano
USC Epstein Family Center for Sports Medicine at Keck Medicine of USC, Los Angeles, CA, USA
Correspondence: Ioanna K Bolia, 1520 San Pablo Street Suite 2000, Los Angeles, CA, 90033, USA, Tel +1 9703432813, Fax +1 818-658-5925, Email [email protected]
Abstract: Knee fibrosis is characterized by the presence of excessive connective tissue due to dysregulated fibroblast activation following local or systemic tissue damage. Knee fibrosis constitutes a major clinical problem in orthopaedics due to the severe limitation in the knee range of motion that leads to compromised function and patient disability. Knee osteoarthritis is an extremely common orthopedic condition that is associated with patient disability and major costs to the health-care systems worldwide. Although knee fibrosis and osteoarthritis (OA) have traditionally been perceived as two separate pathologic entities, recent research has shown common ground between the pathophysiologic processes that lead to the development of these two conditions. The purpose of this review was to identify the pathophysiologic pathways as well as key molecules that are implicated in the development of both knee OA and knee fibrosis in order to understand the relationship between the two diagnoses and potentially identify novel therapeutic targets.
Keywords: fibrosis, molecules, knee, osteoarthritis
Introductions
Historically, knee fibrosis and knee osteoarthritis (OA) have been perceived as distinct diseases with unique pathophysiologies. Knee fibrosis, or knee arthrofibrosis, classically arises following injury or surgery to the knee and is characterized by excessive scar formation and restricted range of motion of the joint.1 This development of knee fibrosis is thought to be caused by dysregulated fibroblasts that produce excessive extracellular matrix.2 Knee OA results of chronic mechanical stress of the knee joint and an age-related decreased in proteoglycans, proteins that function to absorb shock and mediate load distribution.3 Over time, the joint cartilage loses its elasticity and increased friability leads to degeneration of the joint and narrowing of the joint space with concurrent sclerosis.3
While fibrosis has not historically been considered a pre-requisite for osteoarthritis, recent studies have shown that it is a hallmark characteristic of OA that may play an essential role.2,4 The fact that fibrotic processes such as synovial fibrosis are triggered as a result of osteoarthritic cartilage damage shows that the inflammatory cascades that are involved in osteoarthritis include many molecules that are commonly seen in fibrosis pathways as well.2 When articular cartilage degrades in osteoarthritis, the damaged cartilage secretes a variety of molecules that initiates an inflammatory cascade.4 These molecules cause the release of cytokines that help trigger synovitis, the initiation of the de-differentiation of articular chondrocytes into chondrocytes that have a fibroblastic phenotype, and synovial fibrosis in over half of osteoarthritis patients.4–6 In osteoarthritis progression, inflammatory cells and the de-differentiated chondrocytes and their secretory products which include fibrocartilage then secrete other cytokines, thereby triggering further synovitis and cartilage damage.4
Although there are clear similarities between the inflammatory pathways of knee fibrosis and OA, these relationships have not been previously well documented. The purpose of this review was to identify the pathophysiologic pathways as well as key molecules that are implicated in the development of both knee OA and knee fibrosis in order to understand the relationship between the two diagnoses and potentially identify novel therapeutic targets.
Collagen Related Molecules
Normal articular cartilage contains organized layers of ECM surrounding mature chondrocytes that produce primarily type II collagen (>90%), as well as type IX collagen and type XI collagen linked together by proteoglycans such as aggrecan.7 In pathological states such as osteoarthritis, post-traumatic remodeling of the extracellular matrix (ECM) with abnormal subtypes of collagen is an established finding, contributing to pathophysiology by damaging the integrity and stability of the ECM.7,8 The breakdown of type II collagen is a key feature of OA and is thought to be vital pathway for the loss of cartilage integrity.7,9 Chondrocytes in OA display a hypertrophic, dedifferentiated phenotype involving the production of new collagens, the most commonly reported being type I collagen,7,10,11 type III collagen,7,12 and excessive type X collagen, all of which contrast with the normal phenotype seen in healthy articular tissue.7
Mechanisms Implicated in Collagen Pathways in Osteoarthritis and Fibrosis Development
Accumulation of excess collagen within articular cartilage is a hallmark of knee fibrosis.6 The collagen subtypes expressed in knee OA are also regular markers of fibrosis in other organs such as the liver or skin.13,14 In patients with arthrofibrosis following total knee arthroplasty, the genes controlling collagen types I, III, and VI (COL1A1, COL3A1, and COL6A1) have been reported to be particularly upregulated.15 Given these findings, the molecular pathways that lead to degradation of cartilaginous collagens (Type II, IX, XI) and the production of fibrous collagens (Type I, III, VI) are a valuable area of study. Furthermore, cartilage oligomeric matrix protein and Coll2-1 are also related to knee osteoarthritis and are often studied biomarkers of cartilage degeneration that are also involved in liver fibrosis and idiopathic pulmonary fibrosis.16,17
Medication development to alter this process is complicated as collagen subtypes implicated in OA and fibrosis also play a role in healthy cartilage. For example, Type VI Collagen is involved in mechanotransduction in the pericellular matrix of cartilage,18 and site-specific developmental roles throughout the body,19 but Li et al found that COL6A1 and COL6A3 play a pathogenic role in OA and Fibrosis through focal adhesion pathways.10
There are several pathways resulting in pathologic imbalance of collagen deposition and breakdown. As will be discussed later in this paper, TGF-β induces a well-studied pathway resulting in type 2 collagen degradation, type 1/3 collagen production, myofibroblast transition, and persistent fibrosis. While investigating OA-related fibrosis induced by TGF-β and collagenase, Remst et al found increased levels of lysyl hydroxylase 2 (LH2) and pyridinoline cross-linked type 1 collagen in murine OA fibrotic joints.20 They propose that TGF-β induces LH2, resulting in a higher amount of cross-linked type I collagen, which is resistant to degradation by proteinases.6,20 LH2 is universally upregulated in fibrosis and is encoded by procollagen-lysine, 2-oxoglutarate 5-dioxygenase 2b (PLOD2), which may be targeted in drug development to prevent persistent fibrosis by blocking cross-link formation, and resulting in reversible fibrosis.21 TGF-β is the top of the fibrotic cascade but has many functions and cannot be blocked without negative health effects. As such, LH2 is an attractive medication target as it is not shown to be involved in other healthy processes.21 Reduction of pyridinoline cross-linked type 1 collagen is also desirable as it is mechanically inferior in human joints.22
The NOTCH pathway has been identified as a regulator of cartilage homeostasis, cartilage development, and development of post-traumatic OA.23,24 Liu et al reported joint fibrosis and osteoarthritic pathology in a mouse model with genetically induced sustained NOTCH signaling. The mechanism of fibrosis induction was shown to be through the IL-6-STAT3 signaling pathway, which may be shared with the mechanism of NOTCH-induced OA.24
The Wnt/β-catenin pathway (Figure 1) is typically quiescent in adults but upregulated in response to injury and is associated with fibrotic repair. Wnt signaling has also been linked to osteoarthritis in previous studies.25 This pathway is activated following traumatic injury in mice and elevated levels of Wnt are seen in load-induced fibrosis in OA mice.26,27 Inhibition of this pathway in murine joints has been shown to reduce OA severity, reduce type 1 collagen levels, and reduce proliferation of synovial fibroblasts. In human chondrocytes, the addition of Wnt decreased COL2A1 mRNA expression levels, but was rescued via Wnt inhibition.26
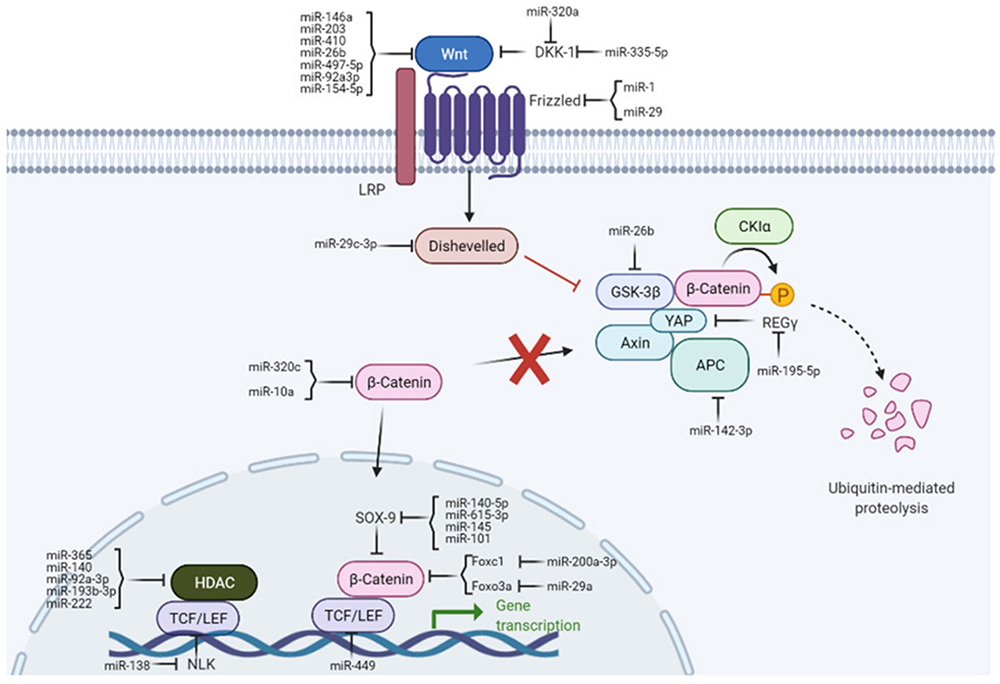 |
Figure 1 Wnt/B-catenin signaling pathway. Note: Figure reproduced from Shang X, Böker KO, Taheri S, Hawellek T, Lehmann W, Schilling AF. The Interaction between microRNAs and the Wnt/β-Catenin Signaling Pathway in Osteoarthritis. International Journal of Molecular Sciences. 2021; 22(18):9887. 10.3390/ijms22189887.101 |
The PI3K/AKT signaling pathway offers another opportunity for prevention of fibrosis and OA. Liu et al has shown that inhibition of the PI3K/AKT signaling pathway via phosphorylation of AMPK in OA-induced rat-joints results in preservation of type 2 collagen and histologically preserved cartilage.28 Additionally, markers of enhanced fibrosis (COL-1, -SMA) and chondrocyte hypertrophy (COL-X, MMP-13, and Runx2) were reduced in the treatment arm compared to control.28 The PI3K/AKT pathway is not fully understood, with some conflicting studies on its effect upon chondrocyte hypertrophy.29,30 However, in a recent study analyzing the gene expression of early-stage fibrosis development in OA-associated mice, the PI3K pathway was the most robustly expressed in the 2 weeks following fibrosis induction, indicating its importance as a potential therapeutic target.27
Zhang et al recently found that the HIF1-/NLRP3 inflammatory pathway aggravates synovitis and fibrosis in Murine osteoarthritic knees.31 Zhang et al later reported that inhibition of this pathway resulted in decreased type 1 collagen deposition and other pro-fibrotic factors.32
Nidogens and Laminins are macromolecules associated with the basement membrane and pericellular matrix of articular cartilage. Schminke et al have shown that an increase laminin expression in cultured cells results in increased collagen type II synthesis and decreased collagen type I synthesis.33 Nidogen-2 was shown to increase the expression of SOX9 (SRY-type high-mobility group box-9),33 which is commonly found to be decreased in OA and has been explored as a regenerative target of OA treatment.34,35 Interestingly, SOX9 expression was greatly increased in fibrosis compared to control in the previously mentioned study analyzing the gene expression of early-stage fibrosis development in OA-associated mice.27 These findings support the previously reported regulatory function of SOX9,29,36 in cartilage function and present an opportunity for targeted research.
Peroxisome proliferator activated receptor gamma (PPARg) plays a key role in a range of pathophysiological processes and has been identified a therapeutic target for OA because PPARg agonists can reduce production of inflammatory factors involved in OA.37 Vasheghani et al studied cartilage-specific PPARg knockout mice and showed increased synovial inflammation and fibrosis when compared to control, as well as increased type II collagen breakdown.37 Additionally, a recent study demonstrated a protective effect against synovial fibrosis using a PPARg agonist combined with a glucocorticoid.38
Endothelin-1 (ET-1) is an inflammatory cytokine mediator involved in bone formation and implicated in fibrosis of various organs.39 ET-1 is elevated in OA and cooperates with TGF-β to induce the fibrotic cascade.39 As such, it may play a role in the overproduction of collagen type 1 seen in OA fibrosis.
Denatured Collagen types I and II that are taken orally have been found to be anti-inflammatory and chondroprotective in OA, though the mechanism by which they act is not fully understood.40,41
Interleukins
The initiation and progression of both knee fibrosis and osteoarthritis involve pro-inflammatory cascades often involving multiple pathways regulated by various cytokines. Interleukins (IL) are one of the most prominent types of cytokines and can have either pro-inflammatory or anti-inflammatory effects, with both types being implicated in fibrosis and OA. Interleukins are often secreted by cells of the immune system in response to event prompting and acute inflammatory reaction. Among these cells, macrophages are responsible for a large proportion of interleukin release in knee osteoarthritis and synovial fibrosis, and are often found in large amounts in the intima of the synovium.42,43 M1 macrophages are responsible for secreting pro-inflammatory interleukins including IL-1, IL-6, IL-8, and IL-18, while M2 macrophages typically secrete anti-inflammatory interleukins IL-10 and IL-13.2,44
Mechanisms Implicated in Promoting Macrophage Interleukin Secretion in Osteoarthritis
Inflammasomes are protein complexes found in cells like macrophages that are activated by cellular stress that can initiate cascades that promote cell death and release IL-1β and IL-18.45 During osteoarthritis when there is stress produced by cartilage damage and the production of molecules like uric acid, these inflammasome complexes are triggered, particularly the inflammasome NLRP3 which is a NOD-like receptor.44 NLRP3 stimulation in macrophages leads to the activation of caspase proteins which triggers cell death in a process known as pyroptosis which is caspase-induced cell death in a pro-inflammatory environment.44
Pyroptosis of these macrophages leads to large releases of IL-1β and IL-18 which trigger not only large-scale cartilage degeneration through MMP and aggrecanase-related pathways, but also stimulation of pro-fibrotic pathways as well that can cause further synovial fibrosis.42 Synovial macrophage pyroptosis inhibition in knee osteoarthritis has been found to reduce not only fibrosis, but the effects of osteoarthritis as well.42 It can be deduced that this effect is largely due to the fact that preventing cell death of macrophages helps reduce the quantity of IL-1β and IL-18 that is released. These conclusions are further supported by the fact that inhibition of the NLRP3 inflammasome complex with a compound called vanillic acid and thus the release of IL-1β and IL-18 not only reduced the progression of osteoarthritis but fibrosis as well.46 IL-1β and IL-18 are both prominently involved in fibrosis and osteoarthritis which shows a significant overlap between these two processes.
A member of the IL-1 family, IL-18 is another interleukin that has been shown to play a role in knee osteoarthritis. IL-18 upregulates MMP’s and the molecule PGE2 which causes cartilage damage in the knee.47,48 PGE2 has been found to be a stimulator of both IL-6.49 IL-18 expression has been found to cause an increased release of IL-6 from chondrocytes through its impact on PGE2 in osteoarthritic knees.50 There seems to be a potential connection between IL-18 and knee fibrosis through its ability to impact PGE2 expression and thus IL-6 expression.
Pro-Inflammatory Cytokines Involved in Osteoarthritis and Fibrosis
Experimental studies of osteoarthritic knee joints have revealed increased expression of IL-6 and IL-1β.51 IL-1β, which is primarily produced by macrophages in inflammatory processes, is also prominently involved in fibrosis and is a key regulator of other pro-fibrotic factors such as PDGF and TGF-β.2 IL-1β also activates matrix metalloproteinases and promotes the migration of cells that are responsible for fibrotic processes.2,52 These matrix metalloproteinases (MMP) are also pivotal in the progression of osteoarthritis as they facilitate the breakdown of type II collagen and aggrecan in hyaline cartilage.4 In osteoarthritis IL-1β stimulates the activation and release of MMP-1, MMP-3, and MMP-13, of which MMP-13 is the most prominent.4,53 Additionally, in osteoarthritis, IL-1β release in the synovium stimulates synovial fibroblasts which contain IL-1β receptors IL-1RI and IL-1RII.52 The stimulation of the synovial fibroblasts leads to the production of fibrotic tissue, often resembling pannus tissue typically observed in rheumatoid arthritis.52,54 IL-1β’s dual role in both promoting knee cartilage degeneration and fibrotic tissue formation demonstrates a potential shared pathway for joint fibrosis and development of OA.
IL-6 is another interleukin with overlap in both knee fibrosis and osteoarthritis. Primarily secreted from macrophages, IL-6ʹs release is promoted by other molecules like PGE2, TGF-β, IL-1β and TNFα.53,55 In osteoarthritis of the knee, IL-6 induces MMP-1 and MMP-13 release which promotes cartilage degeneration.56 IL-6 also is involved in osteoclast activation, bone resorption, and thus changes in the subchondral bone.56 The NOTCH signaling pathway is significantly involved in IL-6 downstream expression.24 This pathway and family of cell receptors is prominently found both in adult hyaline cartilage including cartilage of developing growth plates.24 While occasional NOTCH signaling is necessary for cartilage and joint maintenance, continuous and abnormal activation of this pathway can result in both osteoarthritic and fibrotic processes.24 A significant proportion of the NOTCH pathway’s contribution to both knee fibrosis and cartilage destruction in osteoarthritis is through its upregulation of the IL-6-STAT3 pathway.24 IL-6 has downstream effects when expressed as a result of activated STAT3, a transcription factor.24 It has been found that IL-6 is significantly upregulated in knee joints with fibrosis and osteoarthritis compared to joints without fibrosis and osteoarthritis.24 IL-6 was found in the largest quantity in regions of cartilage with the most severe fibrosis, which shows IL-6 role in both osteoarthritic and fibrotic processes.24 The inflammation of the infrapatellar fat pad also contributes to increased IL-6 release in osteoarthritis.57 Compared to adipose tissue that is subcutaneous, the adipose tissue in the infrapatellar fat pad in osteoarthritic patients has higher levels of IL-6 production and also greater tissue fibrosis.55,57 Also, in general, higher levels of IL-6 in the infrapatellar fat pad have been associated with a greater degree of synovial fibrosis.49,58 It has been found that a prostaglandin, PGE2, that is produced by the infrapatellar fat pad is key in inducing the production of not only IL-6 but IL-8 as well.49
IL-17 is another pro-inflammatory cytokine that has been found to be correlated with increased levels of knee osteoarthritis severity.59 Secreted primarily from mast cells and TH17 cells, IL-17 not only results in an increased expression of MMPs but also inhibits chondrocyte proteoglycan formation.2,56 This interleukin also plays a role in the upregulation of various molecules and cytokines such as PGE2, IL-6, and IL-1β.56 IL-17 in the knee has been found to increase both the survival of fibroblasts in the synovium but also other inflammatory cells.60 Synovial fibroblasts are central in the process of synovial fibrosis.4 While it has not been studied yet, IL-17 seems to have a direct impact on fibrotic processes not only in its impact on synovial fibroblast survival but also in its stimulatory effects on molecules like PGE2, IL-6, and IL-1β. IL-17 in other tissues in the body has been found to have a direct impact on fibrosis with some effects including the increase of type I collagen production and also the upregulation of the expression of MMPs.2,61 More research needs to be done to clearly elucidate IL-17ʹs impact on knee fibrosis because it seems to have clear influence on multiple fibrotic processes and factors.
IL-11 is a prominent interleukin that is part of the IL-6 cytokine family. IL-11 is essential in bone formation in healthy joints.62 Decreased amounts of IL-11 are correlated with an earlier onset of osteoarthritis and the large increase in IL-11 in osteoarthritic patients occurs due to the chondrocytes and joint’s effort to attempt to reverse degenerative damage that has taken place.63 While IL-11ʹs role in knee fibrosis has yet to be clearly elucidated, it is well known that IL-11 plays a large role in fibrosis in multiple tissues and organs in the body and is upregulated by TGF-β.2 Further research needs to be done to elucidate the fibrotic pathways of IL-11 to see if there is any overlap with its molecular pathogenesis in osteoarthritis.
When articular cartilage degrades in osteoarthritis, the damaged cartilage secretes a variety of molecules that initiates an inflammatory cascade.4 These molecules cause the release of cytokines that help trigger synovitis, the initiation of the de-differentiation of articular chondrocytes into chondrocytes that have a fibroblastic phenotype, and synovial fibrosis in over half of osteoarthritis patients.4–6 The fact that fibrotic processes such as synovial fibrosis are triggered as a result of osteoarthritic cartilage damage shows that the inflammatory cascades that are involved in osteoarthritis include many molecules that are commonly found in fibrosis pathways as well.2 In osteoarthritis progression, inflammatory cells and the de-differentiated chondrocytes and their secretory products which include fibrocartilage then secrete other cytokines, thereby triggering further synovitis and cartilage damage.4
IL-8 is another interleukin that has potential overlapping roles in both fibrosis and osteoarthritis. In osteoarthritis patients, the infrapatellar fat pad releases several cytokines including IL-8 that are associated with increased synovial fibrosis.49 PGE2 is a key factor in the stimulation and release of not only IL-8 from the infrapatellar fat pad.49 In osteoarthritis, IL-8 is known to cause pathological effects by activating MMP-3 which causes cartilage damage.64 Furthermore, MMP-3 is a marker of synovial inflammation, and high values are associated with progression of cartilage injury in patients with knee OA.65,66 The MMPs play a large role in fibrosis as well. Future studies need to look into IL-8ʹs specific molecular role in synovial fibrosis to see whether there is any similarity with its pathological effect in knee osteoarthritis. For example, studies can look into if there is any involvement of MMPs in IL-8ʹs impact on synovial fibrosis or if molecules like PGE2 are implicated in knee fibrosis.
Anti-Inflammatory Cytokines Involved in Osteoarthritis and Fibrosis
Unlike other interleukins discussed previously, IL-10 is unique because it has been found to have anti-inflammatory effects in joint osteoarthritis.56 IL-10 helps reduce secretion of MMPs, prevents cell death of chondrocytes, and helps promote the formation of both aggrecan type II collagen.56 Lower levels of IL-10 have been correlated with increased patient predisposition to knee osteoarthritis development.67 These protective effects are partly thought to be the result of IL-10ʹs induction of the production of IL-1β antagonist and also MMP inhibitors.56 While IL-10 has been found to have an impact in also reducing fibrosis, more research needs to be done on IL-10ʹs impact on knee fibrosis specifically.2 If IL-10 is indeed involved in knee fibrosis, its antagonistic effects on IL-1β and MMPs might provide a link between the osteoarthritic and fibrotic process of the knee.
Transforming Growth Factor-β
The TGF-β superfamily is comprised of more than 40 members and plays a fundamental role in regulating cell proliferation, differentiation, immune responses, and tissue repair in the knee.7 In the primary pathways, TGF-β binds to a type II TGF β transmembrane receptor before recruiting activin receptor-like kinase 5 (ALK5) and/or ALK1 (Figure 2). ALK5 recruitment results in phosphorylation of SMAD2 or SMAD3 while ALK1 prompts phosphorylation of SMAD1, SMAD5, or SMAD8.68 These phosphorylated SMADs bind to SMAD4 and translocate to the nucleus where they control downstream gene transcription with the help of other co-activators and repressors.69 TGF-β generally signals via ALK5 to maintain articular cartilage homeostasis and upregulation through the ALK1 pathway occurs during progression of OA and fibrosis.70 We will focus on these primary pathways; however, there are other SMAD and non-SMAD pathways as well.71
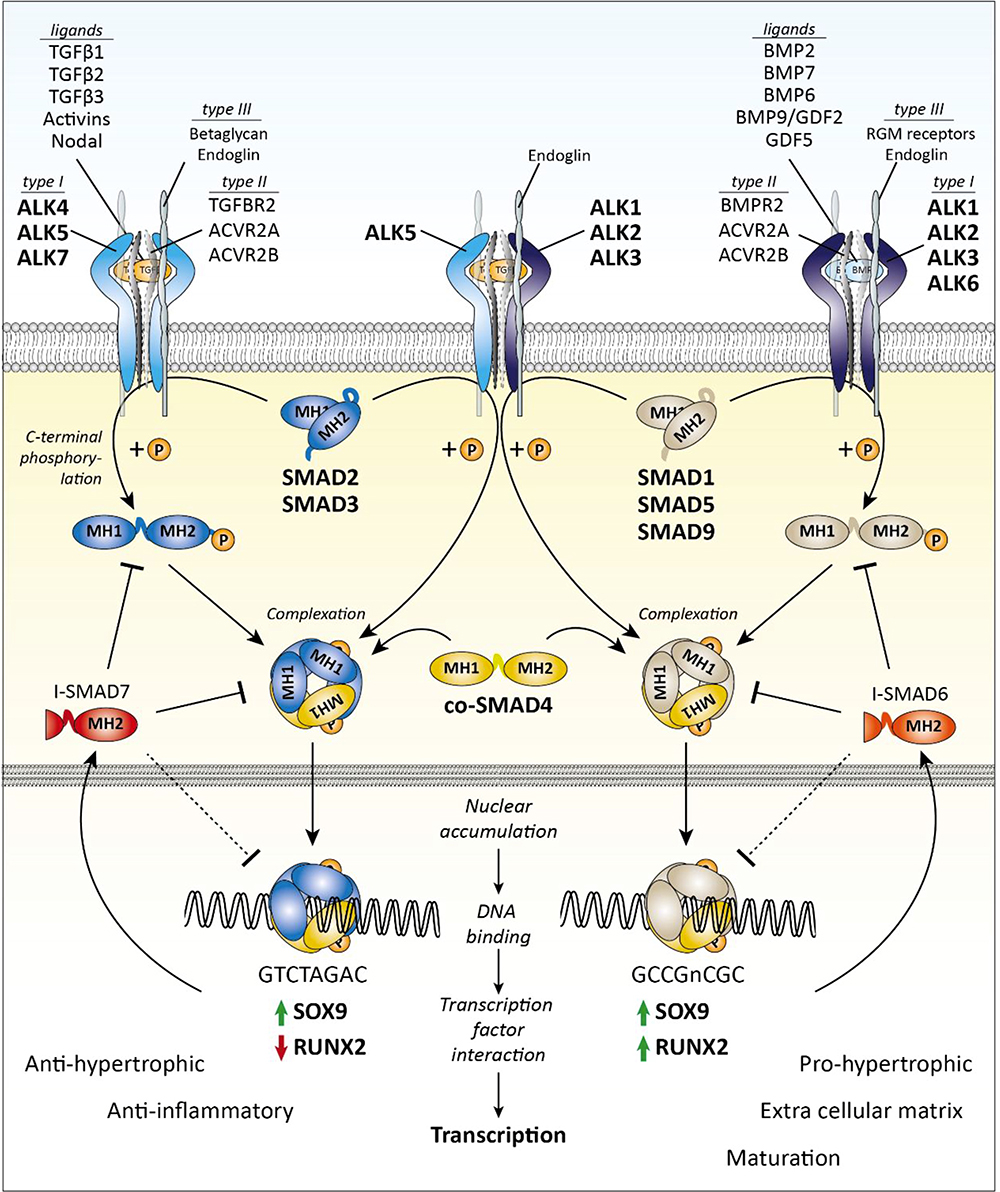 |
Figure 2 Transforming growth factor- β (TGF-β) family SMAD-dependent signaling. Note: Figure reproduced from Thielen NGM, van der Kraan PM, van Caam APM. TGFβ/BMP Signaling Pathway in Cartilage Homeostasis. Cells. 2019; 8(9):969. 10.3390/cells8090969.102 |
Mechanisms Implicated in TGF-β Dysregulation in Osteoarthritis and Fibrosis
Articular cartilage is normally composed of chondrocytes dispersed in a dense extracellular matrix (ECM) and has no intrinsic vasculature or lymphatic supply. These chondrocytes are typically quiescent and produce matrix proteins such as aggrecan as well as primarily type II collagen. The process of chondrocyte maturation and homeostasis is largely controlled by the TGF-β/SMAD pathway which also counteracts catabolic processes.72
Other subgroups of chondrocytes may undergo de-differentiation into a more “fibroblastic” phenotype. This is characterized by increased cell clustering and the production of a lower quality cartilage with decreased type II collagen and increased expression of fibrotic/OA markers such as type I and III collagen as well as α-SMA.72 Articular cartilage is also unique as its maintenance depends on joint loading as this is responsible for increased TGF-β signaling.69 This increased signaling blocks chondrocyte hypertrophy and stimulates the expression of latent TGFβ1 and ALK5 while downregulating the expression of ALK1 to ensure proper cartilage homeostasis. However, studies in bovine cartilage have revealed an age-related, loading-associated loss of protective SMAD2–SMAD3 signaling which makes articular cartilage more vulnerable to OA development in older than in young individuals.69 Overall, we see that in young healthy cartilage, TGF-β exerts a protective affect as it predominantly acts through the ALK5 pathway. With age and other changes to the knee microenvironment, TGF-β can become an inducer of fibrotic/OA symptoms as the catabolic ALK1 pathway begins to take over.69
There are multiple ways that this signaling pathway can be disrupted: OA chondrocytes can become insensitive to TGFβ with age and/or changes in the microenvironment such as injury or inflammation which is accompanied by decreased expression of TGFβ type II receptor on these chondrocytes.69 This compromises chondrocyte function, including the ability to produce aggrecan which is associated with a loss of ALK5 signaling.68 This loss of ALK5 signaling and concomitant increase in ALK1 signaling sets off a chain reaction of events because TGF is more anabolic when signaling through ALK5 and catabolic through ALK1.68 Following subsequent cartilage degradation via the ALK1 pathway, articular chondrocytes undergo changes. This may include the departure from a quiescent state and the acquisition of a hypertrophic phenotype which participates in ECM matrix calcification/degradation signified by the upregulation of type X collagen, MMP-13, and Runx2.4
Another common ground of fibrosis and OA lies in the synovium. The synovium refers to the synovium membrane lining the inside of the joint. The synovium, which is well innervated by a blood and nerve supply, also produces synovial fluid which provides nutrients and oxygen to articular cartilage. Due to the structure of the synovium, it is a common area for not only fibrosis and hyperplasia but also vascularization, inflammation, and osteophyte formation.73 Synovial fibrosis is defined by excess fibroblast proliferation and an imbalance between collagen synthesis and breakdown. Surprisingly, while a lack of TGF-β is responsible for the pathology of articular cartilage, it is the abundance of TGF-β in the synovial tissues that is responsible for the synovial inflammation, fibrosis, and synovial hyperplasia seen in OA.7 TGF-β acts as a chemotactic molecule to recruit fibroblasts and induces production of inflammatory mediators in the synovium.73 Furthermore, as high TGF-β results in flux through the SMAD1/5/8, this also increases fibrogenic differentiation which induces the observed synovial fibrosis and osteophyte formation. Increased TGF-β via the SMAD1/5/8 pathway also increased expression of a CEMIP protein which is considered to play an important role in the pro-fibrotic cascade.74 It does so by regulating de-differentiation of chondrocytes into “chondro-myo-fibroblasts” expressing α-SMA and type III collagen. While TGF-β results in a more persistent version of fibrosis and is needed for its onset, TGF can also potentiate the effects of a more transient fibrosis induced by connective tissue growth factor (CTGF).4 This fibrosis results in greater expression of key fibrotic and OA markers such as PLOD2 and TIMP1. As mentioned, PLOD2 results in pyrinadoline crosslinks that promote collagen stiffness, accumulation, and resistance to breakdown. At the same time, TIMP1 is an inhibitor of MMPs and therefore TIMP upregulation also results in collagen accumulation and together these elements downstream of TGF-β contribute greatly to the synovial fibrosis seen in OA.75 Although less studied, biomolecules such as urotensin II, ADAM12, and PGF2α are also receiving more attention as pro-fibrotic factors that are upregulated in the synovium of OA patients and may be augmented by TGF-β.76
The subchondral bone is the last key region pertaining to OA that we will focus on. Although its role in fibrosis is less pronounced, new research has shown that the articular cartilage and subchondral bone act in concert as one unit controlled by TGF-β.70 Similar to the synovium, an excess of active TGF-β in the osteoblastic cells of subchondral bone is associated with abnormal bone structure and degeneration. This rise of TGF-β in the subchondral bone is, among other factors, a response to abnormal mechanical loading due to aging, injury, or obesity. In response to this abnormal loading, there is an increase in osteoclast activity and bone resorption which leads to release of TGF-β into the bone marrow.70 This occurs at the beginning stage of OA and can be accompanied by the aggregation of mesenchymal stem cells and osteogenic progenitors. This leads to high levels of angiogenesis as well as the formation of osteoid islets and lesions.73 As subchondral bone becomes remodeled and unable to properly distribute mechanical loads, there is reduced support for the overlying articular cartilage leading to its breakdown.
Besides the cartilage, synovium, and subchondral bone, the infrapatellar fat pad (IPFP) has recently gained consideration as an extension of the synovium which also plays a role in fibrosis and OA. The role of the IPFP is to distribute synovial fluid and mechanical force throughout the knee. Although one group has shown that TGF-β is not directly implicated in the IPFP fibrosis that occurs with OA, they did imply an indirect role of TGF-β as other downstream targets of TGF-β are involved. The group found that the IPFP can contribute to synovial fibrosis of OA tissue by increasing collagen synthesis, PLOD2 expression, cell migration, and cell proliferation.6 The extent of collagen and PLOD2 upregulation was correlated with the amount of PGF2 whereas inhibition of TGF-β had no effect. Another group investigated the IPFP 2 weeks following surgery which revealed upregulation of TGF-β, which further implies a role in the high arthrofibrosis rates after surgery.55 Therefore, while the crosstalk between the IPFP and TGF-β requires further exploration, it should not be overlooked in these pathologies.
Vascular Epithelial Growth Factor (VEGF)
Vascular epithelial growth factor (VEGF) family is a group of growth factors that play the primary role in controlling angiogenesis in the human body.77,78 The expression of VEGF is highly connected with tissue hypoxia.79 Expression of VEGF is increased by hypoxia-inducible transcription factors (HIFs) which act as transcription promoters for VEGF genes. Other cytokines, including those previously discussed like TGF-β, increase VEGF expression. Fibroblasts are the primary cells involved in the production and release of VEGF.
The VEGF family contains VEGF-A, VEGF-B, VEGF-C, VEGF-D, VEGF-D, and placental growth factor. The best studied of these growth factors, VEGF-A, is directly implicated in several roles including stimulating endothelial replication and survival, increasing vascular permeability, and increasing the expression of pathways that lead to ECM degradation allowing for endothelial cell migration to the site of angiogenesis. These pathways include activation of urokinase plasminogen activator, tissue plasminogen activator, matrix metalloproteinases and collagenases.80–82 Additionally, VEGF has been shown to increase inflammatory cell migration and ECM deposition.83 The role of VEGF in fibrogenesis has previously been examined hepatic fibrosis, demonstrating increased actions of metalloproteases and resulting matrix stiffness due to VEGF actions.84 Increasing levels of fibrosis conflicts with VEGF’s role in normal wound healing through revascularization.77,83 This may indicate that VEGF’s contribution to the development of fibrosis and osteoarthritis may be due to its dysregulation rather than overexpression.
Mechanisms Implicated in VEGF Dysregulation in Osteoarthritis and Fibrosis
Multiple studies have examined how VEGF dysregulation is related to the development of fibrosis and osteoarthritis of the knee joint. Results from studies examining fibrosis have been mixed. In a study of patient undergoing total knee arthroplasties, Malahias et al examined whether excess scar formation was related to excessive inflammatory responses. When comparing levels of VEGF in synovial fluid and blood plasma between patients with and without knee stiffness, there were significantly higher levels of VEGF in the non-stiff group by post-operative day 2.85 Conversely, Sakamoto et al utilized a mouse-model to study molecular mechanisms of joint stiffness following immobilization and found that VEGF was substantially upregulated after 1 and 2 weeks of immobilization.86 They then examined whether inhibition of VEGF expression through a blockade of HIF signaling, and found that decreased expression of VEGF was correlated with increased range of motion of the knee joint. Emami et al utilized bevacizumab, a monoclonal IgG1 antibody directed at VEGF, to examine the effects of VEGF suppression on range of motion, macroscopic adhesion, and microscopic histopathology following knee surgery in rabbits.87 The low-dose bevacizumab group showed significantly less fibroblasts and ECM deposition while the high dose also demonstrated improved range of motion and decreased macroscopic adhesion.
It has been postulated that the seemingly disparate effects of VEGF on fibrosis are due to differing effects at low and high VEGF concentrations. Yan et al examined the effect of fibroblast suppression on rates of scar adhesion following knee surgery in rabbits. Fibroblast activation was inhibited through the use of the anti-neoplastic hydroxycamptothecin (HCPT).88 They found that VEGF levels were lowest in the high dose HCPT treatment groups and this correlated with decreased scar adhesion; however, they also found that in the high dose HCPT groups, decreased angiogenesis due to low VEGF may have impact the knee’s ability to heal and recover from surgery.
Studies have also examined the effect of VEGF on the development of osteoarthritis in the knee. A study of the effects of knocking out VEGF expression through microRNA injections in mice found that decreasing VEGF resulted in less excessive synovial angiogenesis and fibrosis.89 They hypothesized that this would delay joint destruction in these mice. An additional study found that in patients with known osteoarthritis, synovial analysis demonstrated that VEGF level was positively correlated with more severely graded osteoarthritis.90 These results were backed by a recent systematic review that found 7 studies examining correlation between VEGF levels in synovial fluid and osteoarthritis severity. In 6 of the 7 studies, there was a significantly positive correlation between osteoarthritis severity and VEGF.91
The relationship between VEGF and OA severity may be due to expression of VEGF in the infrapatellar fat pad (IFP). The IFP has been implicated in the development of OA through other molecular pathways in this paper. When specifically examining VEGF, studies show that patients with OA have larger, more highly vascularized IFPs compared to non-OA patients.58,92 As VEGF is the primary agent for angiogenesis, it is assumed that VEGF is the cause of hypervascularization of the IFP in these patients.
Other studies have examined the effect of blocking VEGF’s actions on the development of knee OA. Agnuside, a small molecule with anti-inflammatory effects, was shown to decrease HIF expression in rat synovial fluid.32 This triggered decreased VEGF expression and was correlated with decreased inflammatory and fibrotic markers.
Other Molecules
Many other molecules and molecular pathways have been studied in the pathophysiology of fibrosis and osteoarthritis of the knee. However, the literature on many of these molecules is limited currently. A selection of these molecules have demonstrated promising early results and will be discussed briefly in this section.
Tumor Necrosis Factors
Tumor necrosis factor alpha (TNF-a) is synthesized by macrophages and T cells and was first noted for its ability to lyse tumor cells.93 More recently, TNF-a has been recognized for the role it plays in the stimulation of inflammatory cytokine cascade and the migration of leukocytes to sites of inflammation.94 TNF-a plays a significant role in the development of rheumatoid arthritis, as high levels lead to the inflammation of the joint and proliferation of fibroblasts and deposition of granulation tissue.95 The role of TNF-a in RA is so significant that TNF-a inhibitors have become a common treatment for persistent severe disease that fails conventional therapy.96 However, little evidence exists that supports its use for OA.
Recent research has examined the potential effects of TNF-a inhibition in knee fibrosis. Salib et al explored the possible role of dysregulation of the inflammatory cascade in the development of fibrosis.97 They found that use of celecoxib, a selective COX-2 inhibitor, decreased expression of inflammatory cytokines including TNF-a and led to increased knee range of motion and decreased collagen expression in rabbits following knee surgery. Furthermore, a case study of a patient with pigmented villonodular synovitis found that use of the TNF-a inhibitor, adalimumab, resulted in decreased knee pain with movement and increased range of flexion.98
Prostaglandins
Prostaglandins, a class of inflammatory cytokines derived from phospholipids and fatty acids, are key products of the cyclooxygenase pathways (COX). Prostaglandins have a variety of physiologic effects, including modulation of vascular constriction and dilation, platelet aggregation, and recruitment of leukocytes to sites of inflammation.99 Many of the current medications used to treat osteoarthritis, including nonsteroidal anti-inflammatory drugs (NSAIDs), inhibit the COX pathways, leading to decreased levels of inflammation through decreased prostaglandins.100 Several studies have examined the role that prostaglandins play in the pathophysiology of fibrosis and osteoarthritis.
In the same study described in the “TNF-a” section, the use of the selective COX-2 inhibitor, celecoxib, resulted in a decrease in prostaglandins in synovial fluid, which was positively correlated with a decrease in collagen expression in the knee joint and increased range of motion.97 This indicates that reduction of prostaglandins may help prevent knee fibrosis; however, there are limited data examining the individual effects of these cytokines.
For knee OA, a study of cultured synoviocytes from the infrapatellar fat pad examined whether inhibitors of the prostaglandin PGF2a had an effect on collagen production and migration and proliferation of synoviocytes, key features of synovial fibrosis.55 They found that use of the PGF2a inhibitor resulted in decreased collagen production and cross-linking, while results for synoviocyte migrations were non-significant.
Conclusion
Review of the molecular mediators involved in OA and fibrosis on the knee highlights the similar pathophysiology of each disease process. Indeed, when reviewing the literature on these subjects, it is difficult to separate the two diseases. Many of the same molecular mediators that increase ECM deposition and scar adhesions in knee fibrosis share pathways with the chronic inflammatory state seen in knee osteoarthritis. By inhibiting and/or modifying the chemical cascade of these physiologic processes, physicians may make breakthroughs in the prevention and treatment of these diseases. Recent studies examining pharmaceuticals that target the molecules in this review have shown promising results, particularly in the in vitro setting.
For collagens and collagen-related molecules, there is clear evidence of their role in the pathology of fibrosis and osteoarthritis. Different pathways, molecules, and receptors have been identified as common targets for modifying collagen remodeling in fibrosis and OA including the lysyl hydroxylase 2 (LH2) enzyme, NOTCH pathway, Wnt/β-catenin pathway, and PI3K/AKT signaling pathway. In in vitro studies, knock-out or alteration of these pathways have been successful in reducing excess or abnormal collagen deposition. However, more research is needed in human trials to identify beneficial therapies.
Research on interleukins in fibrosis and osteoarthritis has provided encouraging data to show that either inhibiting pro-inflammatory or activating anti-inflammatory interleukins may greatly reduce or halt disease develop and progression. IL-1β, IL-6 and IL-8ʹs promotion of knee cartilage degeneration and fibrotic tissue formation make them a potential target for future therapeutics for both OA and fibrosis, while, IL-18, IL-17 and IL-11 appear to better target OA alone. Conversely, given IL-10 anti-inflammatory properties, isolated activation of this interleukin may also have therapeutic benefit.
The cytokine TGF-β, both directly and indirectly, plays a crucial role in the development of OA and fibrosis. Due to the importance of TGF-β in cartilage maintenance, it would be extremely difficult to inhibit it as there would be severe side effects. Therefore, researchers have turned to downstream elements of TGF-β as therapeutic targets. Therapies to alleviate OA and fibrosis at this level may aim to stimulate the ALK5 pathways or inhibit those that act via ALK1. Other inhibitors, such as Asiatic acid, may look to reduce the hypertrophic/fibrotic phenotype switch seen in chondrocytes. Unlike the articular cartilage, the development of OA and fibrosis in the synovium is the result of excessive TGF-β stimulation. Therefore, therapies in this region could focus on increasing TGF-β in the cartilage while inhibiting it in the synovium to prevent its fibrotic side effects. The feasibility of this has been previously shown by administering TGF-β into the cartilage with simultaneous overexpression of the TGF-β inhibitors SMAD6, SMAD7, and LAP in the synovium. Another potential therapy of synovial fibrosis in OA which would also address IPFP pathology would be to block PLOD2. While there are currently no inhibitors for LH2b, the protein product of PLOD2, this is an active area of research.
The growth factor VEGF is well known for its angiogenic properties, though its role in development of OA and fibrosis appears to be more closely due to its effects on metalloproteinases and fibroblasts. The data on VEGFs effect on knee fibrosis are mixed, with many studies showing that increased VEGF is correlated with increased fibrosis, while others show the opposite. This contrast seems to be a product of varying VEGF concentrations and should be a point of future study. For knee OA however, the relationship between elevated VEGF and worsening OA is clear and may be a reasonable therapeutic target.
The results of studies on other molecules, such as TNF-a and prostaglandins are limited, but the existing data are promising that these molecules and their pathways may have value in the treatment of OA and fibrosis. Future research should continue to assess the impact these molecules have on the pathogenesis of OA and fibrosis.
Disclosure
Dr Frank A Petrigliano reports IP royalties, Paid presenter or speaker from Exactech, Inc, Paid presenter or speaker from Stryker, outside the submitted work.
References
1. Ouyang X, Ghani A, Mehal WZ. Inflammasome biology in fibrogenesis. Biochim Biophys Acta. 2013;1832(7):979–988. doi:10.1016/j.bbadis.2013.03.020
2. Usher KM, Zhu S, Mavropalias G, Carrino JA, Zhao J, Xu J. Pathological mechanisms and therapeutic outlooks for arthrofibrosis. Bone Res. 2019;7:9. doi:10.1038/s41413-019-0047-x
3. Liu-Bryan R, Terkeltaub R. Emerging regulators of the inflammatory process in osteoarthritis. Nat Rev Rheumatol. 2015;11(1):35–44. doi:10.1038/nrrheum.2014.162
4. Rim YA, Ju JH. The role of fibrosis in osteoarthritis progression. Life. 2020;11(1). doi:10.3390/life11010003
5. Mathiessen A, Conaghan PG. Synovitis in osteoarthritis: current understanding with therapeutic implications. Arthritis Res Ther. 2017;19(1):18. doi:10.1186/s13075-017-1229-9
6. Remst DFG, Blaney Davidson EN, van der Kraan PM. Unravelling osteoarthritis-related synovial fibrosis: a step closer to solving joint stiffness. Rheumatology. 2015;54(11):1954–1963. doi:10.1093/rheumatology/kev228
7. Charlier E, Deroyer C, Ciregia F, et al. Chondrocyte dedifferentiation and osteoarthritis (OA). Biochem Pharmacol. 2019;165:49–65. doi:10.1016/j.bcp.2019.02.036
8. Von Der Mark K, Gauss V, Von Der Mark H, MÜLler P. Relationship between cell shape and type of collagen synthesised as chondrocytes lose their cartilage phenotype in culture. Nature. 1977;267(5611):531–532. doi:10.1038/267531a0
9. Stoop R, Kraan PMVD, Buma P, et al. Type II collagen degradation in spontaneous osteoarthritis in C57BL/6 and BALB/c mice. Arthritis Rheum. 1999;42(11):2381–2389. doi:10.1002/1529-0131(199911)42:11<2381::AID-ANR17>3.0.CO;2-E
10. Li C, Luo J, Xu X, et al. Single cell sequencing revealed the underlying pathogenesis of the development of osteoarthritis. Gene. 2020;757:144939. doi:10.1016/j.gene.2020.144939
11. Gay S, Müller PK, Lemmen C, Remberger K, Matzen K, Kühn K. Immunohistological study on collagen in cartilage-bone metamorphosis and degenerative osteoarthrosis. Klin Wochenschr. 1976;54(20):969–976. doi:10.1007/bf01468947
12. Hosseininia S, Weis MA, Rai J, et al. Evidence for enhanced collagen type III deposition focally in the territorial matrix of osteoarthritic hip articular cartilage. Osteoarthr Cartil. 2016;24(6):1029–1035. doi:10.1016/j.joca.2016.01.001
13. Velasco J, Li J, DiPietro L, Stepp MA, Sandy JD, Plaas A. Adamts5 deletion blocks murine dermal repair through CD44-mediated aggrecan accumulation and modulation of Transforming Growth Factor β1 (TGFβ1) signaling. J Biol Chem. 2011;286(29):26016–26027. doi:10.1074/jbc.M110.208694
14. Karsdal MA, Nielsen SH, Leeming DJ, et al. The good and the bad collagens of fibrosis - their role in signaling and organ function. Adv Drug Deliv Rev. 2017;121:43–56. doi:10.1016/j.addr.2017.07.014
15. Bayram B, Limberg AK, Salib CG, et al. Molecular pathology of human knee arthrofibrosis defined by RNA sequencing. Genomics. 2020;112(4):2703–2712. doi:10.1016/j.ygeno.2020.03.004
16. Georgiev T, Ivanova M, Kopchev A, et al. Cartilage oligomeric protein, matrix metalloproteinase-3, and Coll2-1 as serum biomarkers in knee osteoarthritis: a cross-sectional study. Rheumatol Int. 2018;38(5):821–830. doi:10.1007/s00296-017-3887-y
17. Posey KL, Coustry F, Hecht JT. Cartilage oligomeric matrix protein: cOMPopathies and beyond. Matrix Biol. 2018;71–72:161–173. doi:10.1016/j.matbio.2018.02.023
18. Zelenski NA, Leddy HA, Sanchez-Adams J, et al. Collagen VI regulates pericellular matrix properties, chondrocyte swelling, and mechanotransduction in articular cartilage. Arthritis Rheumatol. 2015;67(5):1286–1294. doi:10.1002/art.39034
19. Christensen SE, Coles JM, Zelenski NA, et al. Altered trabecular bone structure and delayed cartilage degeneration in the knees of collagen VI null mice. PLoS One. 2012;7(3):e33397. doi:10.1371/journal.pone.0033397
20. Remst DFG, Blaney Davidson EN, Vitters EL, et al. Osteoarthritis-related fibrosis is associated with both elevated pyridinoline cross-link formation and lysyl hydroxylase 2b expression. Osteoarthr Cartil. 2013;21(1):157–164. doi:10.1016/j.joca.2012.10.002
21. Piersma B, Bank RA, Adams J. Collagen cross-linking mediated by lysyl hydroxylase 2: an enzymatic battlefield to combat fibrosis. Essays Biochem. 2019;63(3):377–387. doi:10.1042/EBC20180051
22. Bank RA, Verzijl N, Lafeber FP, Tekoppele JM. Putative role of lysyl hydroxylation and pyridinoline cross-linking during adolescence in the occurrence of osteoarthritis at old age. Osteoarthritis Cartilage. 2002;10(2):127–134. doi:10.1053/joca.2001.0487
23. Kohn A, Dong Y, Mirando AJ, et al. Cartilage-specific RBPjκ-dependent and -independent notch signals regulate cartilage and bone development. Development. 2012;139(6):1198–1212. doi:10.1242/dev.070649
24. Liu Z, Chen J, Mirando AJ, et al. A dual role for NOTCH signaling in joint cartilage maintenance and osteoarthritis. Sci Signal. 2015;8(386):ra71. doi:10.1126/scisignal.aaa3792
25. Takamatsu A, Ohkawara B, Ito M, et al. Verapamil protects against cartilage degradation in osteoarthritis by inhibiting Wnt/β-catenin signaling. PLoS One. 2014;9(3):e92699. doi:10.1371/journal.pone.0092699
26. Lietman C, Wu B, Lechner S, et al. Inhibition of Wnt/β-catenin signaling ameliorates osteoarthritis in a murine model of experimental osteoarthritis. JCI Insight. 2018;3(3). doi:10.1172/jci.insight.96308
27. Wang M, Lessard SG, Singh P, et al. Knee fibrosis is associated with the development of osteoarthritis in a murine model of tibial compression. J Orthop Res. 2020;39(5):1030–1040.
28. Liu N, Fu D, Yang J, et al. Asiatic acid attenuates hypertrophic and fibrotic differentiation of articular chondrocytes via AMPK/PI3K/AKT signaling pathway. Arthritis Res Ther. 2020;22(1). doi:10.1186/s13075-020-02193-0
29. Fujita T, Azuma Y, Fukuyama R, et al. Runx2 induces osteoblast and chondrocyte differentiation and enhances their migration by coupling with PI3K-Akt signaling. J Cell Biol. 2004;166(1):85–95. doi:10.1083/jcb.200401138
30. Kita K, Kimura T, Nakamura N, Yoshikawa H, Nakano T. PI3K/Akt signaling as a key regulatory pathway for chondrocyte terminal differentiation. Genes Cells. 2008;13(8):839–850. doi:10.1111/j.1365-2443.2008.01209.x
31. Zhang L, Huang Z, Xing R, et al. Increased HIF-1α in knee osteoarthritis aggravate synovial fibrosis via fibroblast-like synoviocyte pyroptosis. Oxid Med Cell Longev. 2019;2019. doi:10.1155/2019/6326517
32. Zhang L, Li X, Zhang H, et al. Agnuside alleviates synovitis and fibrosis in knee osteoarthritis through the inhibition of HIF-1 α and NLRP3 inflammasome. Mediators Inflamm. 2021;2021:1–11. doi:10.1155/2021/5534614
33. Schminke B, Frese J, Bode C, Goldring MB, Miosge N. Laminins and nidogens in the pericellular matrix of chondrocytes: their role in osteoarthritis and chondrogenic differentiation. Am J Pathol. 2016;186(2):410–418. doi:10.1016/j.ajpath.2015.10.014
34. Zhang X, Wu S, Zhu Y, Chu C-Q. Exploiting joint-resident stem cells by exogenous SOX9 for cartilage regeneration for therapy of osteoarthritis. Front Med. 2021;8(100). doi:10.3389/fmed.2021.622609
35. Cucchiarini M, Thurn T, Weimer A, Kohn D, Terwilliger EF, Madry H. Restoration of the extracellular matrix in human osteoarthritic articular cartilage by overexpression of the transcription factorSOX9. Arthritis Rheum. 2007;56(1):158–167. doi:10.1002/art.22299
36. Hattori T, Müller C, Gebhard S, et al. SOX9 is a major negative regulator of cartilage vascularization, bone marrow formation and endochondral ossification. Development. 2010;137(6):901–911. doi:10.1242/dev.045203
37. Vasheghani F, Monemdjou R, Fahmi H, et al. Adult cartilage-specific peroxisome proliferator-activated receptor gamma knockout mice exhibit the spontaneous osteoarthritis phenotype. Am J Pathol. 2013;182(4):1099–1106. doi:10.1016/j.ajpath.2012.12.012
38. Vaamonde-Garcia C, Malaise O, Charlier E, et al. 15-deoxy-Δ-12, 14-prostaglandin J2 acts cooperatively with prednisolone to reduce TGF-β-induced pro-fibrotic pathways in human osteoarthritis fibroblasts. Biochem Pharmacol. 2019;165:66–78. doi:10.1016/j.bcp.2019.03.039
39. Sin A, Tang W, Wen CY, Chung SK, Chiu KY. The emerging role of endothelin-1 in the pathogenesis of subchondral bone disturbance and osteoarthritis. Osteoarthr Cartil. 2015;23(4):516–524. doi:10.1016/j.joca.2014.11.002
40. Dar Q-A, Schott EM, Catheline SE, et al. Daily oral consumption of hydrolyzed type 1 collagen is chondroprotective and anti-inflammatory in murine posttraumatic osteoarthritis. PLoS One. 2017;12(4):e0174705. doi:10.1371/journal.pone.0174705
41. Crowley DC, Lau FC, Sharma P, et al. Safety and efficacy of undenatured type II collagen in the treatment of osteoarthritis of the knee: a clinical trial. Int J Med Sci. 2009;6(6):312–321. doi:10.7150/ijms.6.312
42. Zhang L, Xing R, Huang Z, Zhang N, Li X, Wang P. Inhibition of synovial macrophage pyroptosis alleviates synovitis and fibrosis in knee osteoarthritis. Mediators Inflamm. 2019;2019:1–11. doi:10.1155/2019/2165918
43. Zhang H, Cai D, Bai X. Macrophages regulate the progression of osteoarthritis. Osteoarthritis Cartilage. 2020;28(5):555–561. doi:10.1016/j.joca.2020.01.007
44. An S, Hu H, Li Y, Hu Y. Pyroptosis plays a role in osteoarthritis. Aging Dis. 2020;11(5):1146–1157. doi:10.14336/ad.2019.1127
45. Zhao LR, Xing RL, Wang PM, et al. NLRP1 and NLRP3 inflammasomes mediate LPS/ATP‑induced pyroptosis in knee osteoarthritis. Mol Med Rep. 2018;17(4):5463–5469. doi:10.3892/mmr.2018.8520
46. Ma Z, Huang Z, Zhang L, et al. Vanillic acid reduces pain-related behavior in knee osteoarthritis rats through the inhibition of NLRP3 inflammasome-related synovitis. Front Pharmacol. 2020;11:599022. doi:10.3389/fphar.2020.599022
47. Li Y, Jiang JM, Yang DH, Wang FL, Mao ZX. [Determination of the concentrations of interleukin-18 and other cytokines in the synovial fluid in patients with osteoarthritis]. Nan Fang Yi Ke Da Xue Xue Bao. 2009;29(4):729–731. Chinese.
48. Wang Y, Xu D, Long L, Deng X, Tao R, Huang G. Correlation between plasma, synovial fluid and articular cartilage Interleukin-18 with radiographic severity in 33 patients with osteoarthritis of the knee. Clin Exp Med. 2014;14(3):297–304. doi:10.1007/s10238-013-0251-8
49. Eymard F, Pigenet A, Citadelle D, et al. Induction of an inflammatory and prodegradative phenotype in autologous fibroblast-like synoviocytes by the infrapatellar fat pad from patients with knee osteoarthritis. Arthritis Rheumatol. 2014;66(8):2165–2174. doi:10.1002/art.38657
50. Futani H, Okayama A, Matsui K, et al. Relation between interleukin-18 and PGE2 in synovial fluid of osteoarthritis: a potential therapeutic target of cartilage degradation. J Immunother. 2002;25 Suppl 1:S61–S64. doi:10.1097/00002371-200203001-00009
51. Solbak N, Achari Y, Chung M, Shrive NG, Hart DA, Frank CB. Normal sheep synovium has similar appearances and constitutive expression of inflammatory cytokines within and between knee joints: a baseline histological and molecular analysis. Connect Tissue Res. 2014;55(2):156–163. doi:10.3109/03008207.2014.880427
52. Jenei-Lanzl Z, Meurer A, Zaucke F. Interleukin-1β signaling in osteoarthritis - chondrocytes in focus. Cell Signal. 2019;53:212–223. doi:10.1016/j.cellsig.2018.10.005
53. Kapoor M, Martel-Pelletier J, Lajeunesse D, Pelletier JP, Fahmi H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat Rev Rheumatol. 2011;7(1):33–42. doi:10.1038/nrrheum.2010.196
54. Yuan GH, Tanaka M, Masuko-Hongo K, et al. Characterization of cells from pannus-like tissue over articular cartilage of advanced osteoarthritis. Osteoarthritis Cartilage. 2004;12(1):38–45. doi:10.1016/j.joca.2003.08.004
55. Bastiaansen-Jenniskens YM, Wei W, Feijt C, et al. Stimulation of fibrotic processes by the infrapatellar fat pad in cultured synoviocytes from patients with osteoarthritis: a possible role for prostaglandin f2α. Arthritis Rheum. 2013;65(8):2070–2080. doi:10.1002/art.37996
56. Wojdasiewicz P, Poniatowski ŁA, Szukiewicz D. The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of osteoarthritis. Mediators Inflamm. 2014;2014:561459. doi:10.1155/2014/561459
57. Eymard F, Pigenet A, Citadelle D, et al. Knee and hip intra-articular adipose tissues (IAATs) compared with autologous subcutaneous adipose tissue: a specific phenotype for a central player in osteoarthritis. Ann Rheum Dis. 2017;76(6):1142–1148. doi:10.1136/annrheumdis-2016-210478
58. Favero M, El-Hadi H, Belluzzi E, et al. Infrapatellar fat pad features in osteoarthritis: a histopathological and molecular study. Rheumatology. 2017;56(10):1784–1793. doi:10.1093/rheumatology/kex287
59. Liu Y, Peng H, Meng Z, Wei M. Correlation of IL-17 level in synovia and severity of knee osteoarthritis. Med Sci Monit. 2015;21:1732–1736. doi:10.12659/msm.893771
60. Snelling SJ, Bas S, Puskas GJ, et al. Presence of IL-17 in synovial fluid identifies a potential inflammatory osteoarthritic phenotype. PLoS One. 2017;12(4):e0175109. doi:10.1371/journal.pone.0175109
61. Chan DD, Xiao W, Li J, de la Motte CA, Sandy JD, Plaas A. Deficiency of hyaluronan synthase 1 (Has1) results in chronic joint inflammation and widespread intra-articular fibrosis in a murine model of knee joint cartilage damage. Osteoarthr Cartil. 2015;23(11):1879–1889. doi:10.1016/j.joca.2015.06.021
62. Kuo CL, Liu ST, Chang YL, Wu CC, Huang SM. Zac1 regulates IL-11 expression in osteoarthritis. Oncotarget. 2018;9(65):32478–32495. doi:10.18632/oncotarget.25980
63. Tuerlings M, van Hoolwerff M, Houtman E, et al. RNA sequencing reveals interacting key determinants of osteoarthritis acting in subchondral bone and articular cartilage: identification of IL11 and CHADL as attractive treatment targets. Arthritis Rheumatol. 2021;73(5):789–799. doi:10.1002/art.41600
64. Koh SM, Chan CK, Teo SH, et al. Elevated plasma and synovial fluid interleukin-8 and interleukin-18 may be associated with the pathogenesis of knee osteoarthritis. Knee. 2020;27(1):26–35. doi:10.1016/j.knee.2019.10.028
65. Sun S, Bay-Jensen AC, Karsdal MA, et al. The active form of MMP-3 is a marker of synovial inflammation and cartilage turnover in inflammatory joint diseases. BMC Musculoskelet Disord. 2014;15(1):93. doi:10.1186/1471-2474-15-93
66. Georgiev T, Ivanova M, Velikova T, Stoilov R. Serum levels of matrix metalloproteinase-3 as a prognostic marker for progression of cartilage injury in patients with knee osteoarthritis. Acta Reumatol Port. 2020;45(3):207–213.
67. Barker T, Rogers VE, Henriksen VT, Trawick RH, Momberger NG, Lynn Rasmussen G. Circulating IL-10 is compromised in patients predisposed to developing and in patients with severe knee osteoarthritis. Sci Rep. 2021;11(1):1812. doi:10.1038/s41598-021-81382-6
68. Blaney Davidson EN, Van der kraan PM, van den Berg WB. TGF-beta and osteoarthritis. Osteoarthritis Cartilage. 2007;15(6):597–604. doi:10.1016/j.joca.2007.02.005
69. van der Kraan PM, Blaney Davidson EN, van den Berg WB. A role for age-related changes in TGFbeta signaling in aberrant chondrocyte differentiation and osteoarthritis. Arthritis Res Ther. 2010;12(1):201. doi:10.1186/ar2896
70. van der Kraan PM. The changing role of TGFβ in healthy, ageing and osteoarthritic joints. Nat Rev Rheumatol. 2017;13(3):155–163. doi:10.1038/nrrheum.2016.219
71. Zhen G, Cao X. Targeting TGFβ signaling in subchondral bone and articular cartilage homeostasis. Trends Pharmacol Sci. 2014;35(5):227–236. doi:10.1016/j.tips.2014.03.005
72. Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425(6958):577–584. doi:10.1038/nature02006
73. Shen J, Li S, Chen D. TGF-β signaling and the development of osteoarthritis. Bone Res. 2014;2(1):14002. doi:10.1038/boneres.2014.2
74. Blom AB, van Lent PL, Holthuysen AE, et al. Synovial lining macrophages mediate osteophyte formation during experimental osteoarthritis. Osteoarthritis Cartilage. 2004;12(8):627–635. doi:10.1016/j.joca.2004.03.003
75. Deroyer C, Charlier E, Neuville S, et al. CEMIP (KIAA1199) induces a fibrosis-like process in osteoarthritic chondrocytes. Cell Death Dis. 2019;10(2):103. doi:10.1038/s41419-019-1377-8
76. Remst DFG. Lysyl hydroxylase 2b the X factor in OA-related synovial fibrosis. The cause of joint stiffness in OA?; 2015.
77. Kajdaniuk D, Marek B, Borgiel-Marek H, Kos-Kudła B. Vascular endothelial growth factor (VEGF) - part 1: in physiology and pathophysiology. Endokrynol Pol. 2011;62(5):444–455.
78. Ferrara N. Vascular endothelial growth factor. Eur J Cancer. 1996;32a(14):2413–2422. doi:10.1016/s0959-8049(96)00387-5
79. Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9(6):669–676. doi:10.1038/nm0603-669
80. Rifkin DB, Moscatelli D, Bizik J, et al. Growth factor control of extracellular proteolysis. Cell Differ Dev. 1990;32(3):313–318. doi:10.1016/0922-3371(90)90045-x
81. Pepper MS, Montesano R. Proteolytic balance and capillary morphogenesis. Cell Differ Dev. 1990;32(3):319–327. doi:10.1016/0922-3371(90)90046-y
82. Ferrara N. VEGF: an update on biological and therapeutic aspects. Curr Opin Biotechnol. 2000;11(6):617–624. doi:10.1016/s0958-1669(00)00153-1
83. Bosch U, Zeichen J, Skutek M, Haeder L, van Griensven M. Arthrofibrosis is the result of a T cell mediated immune response. Knee Surg Sports Traumatol Arthrosc. 2001;9(5):282–289. doi:10.1007/s001670100218
84. Schuppan D, Ruehl M, Somasundaram R, Hahn EG. Matrix as a modulator of hepatic fibrogenesis. Semin Liver Dis. 2001;21(3):351–372. doi:10.1055/s-2001-17556
85. Malahias MA, Birch GA, Zhong H, et al. Postoperative serum cytokine levels are associated with early stiffness after total knee arthroplasty: a prospective cohort study. J Arthroplasty. 2020;35(6s):S336–s347. doi:10.1016/j.arth.2020.02.046
86. Sakamoto J, Origuchi T, Okita M, et al. Immobilization-induced cartilage degeneration mediated through expression of hypoxia-inducible factor-1alpha, vascular endothelial growth factor, and chondromodulin-I. Connect Tissue Res. 2009;50(1):37–45. doi:10.1080/03008200802412454
87. Emami MJ, Jaberi FM, Azarpira N, Vosoughi AR, Tanideh N. Prevention of arthrofibrosis by monoclonal antibody against vascular endothelial growth factor: a novel use of bevacizumab in rabbits. Orthop Traumatol Surg Res. 2012;98(7):759–764. doi:10.1016/j.otsr.2012.05.020
88. Yan L, Sun Y, Li X, et al. The effect of hydroxycamptothecin on wound healing following reduction of the knee intra-articular adhesion in rabbits. Cell Biochem Biophys. 2015;73(1):221–227. doi:10.1007/s12013-015-0593-9
89. Ko JY, Lee MS, Lian WS, et al. MicroRNA-29a counteracts synovitis in knee osteoarthritis pathogenesis by targeting VEGF. Sci Rep. 2017;7(1):3584. doi:10.1038/s41598-017-03616-w
90. Kim HR, Lee JH, Kim KW, Kim BM, Lee SH. The relationship between synovial fluid VEGF and serum leptin with ultrasonographic findings in knee osteoarthritis. Int J Rheum Dis. 2016;19(3):233–240. doi:10.1111/1756-185x.12486
91. Boffa A, Merli G, Andriolo L, Lattermann C, Salzmann GM, Filardo G. Synovial fluid biomarkers in knee osteoarthritis: a systematic review and quantitative evaluation using BIPEDs criteria. Cartilage. 2020;13:82S–103S. doi:10.1177/1947603520942941
92. Belluzzi E, Macchi V, Fontanella CG, et al. Infrapatellar fat pad gene expression and protein production in patients with and without osteoarthritis. Int J Mol Sci. 2020;21(17):6016. doi:10.3390/ijms21176016
93. Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci U S A. 1975;72(9):3666–3670. doi:10.1073/pnas.72.9.3666
94. Roach DR, Bean AG, Demangel C, France MP, Briscoe H, Britton WJ. TNF regulates chemokine induction essential for cell recruitment, granuloma formation, and clearance of mycobacterial infection. J Immunol. 2002;168(9):4620–4627. doi:10.4049/jimmunol.168.9.4620
95. Scott DL, Wolfe F, Huizinga TW. Rheumatoid arthritis. Lancet. 2010;376(9746):1094–1108. doi:10.1016/s0140-6736(10)60826-4
96. Aletaha D, Smolen JS. Diagnosis and management of rheumatoid arthritis: a review. JAMA. 2018;320(13):1360–1372. doi:10.1001/jama.2018.13103
97. Salib CG, Reina N, Trousdale WH, et al. Inhibition of COX-2 pathway as a potential prophylaxis against arthrofibrogenesis in a rabbit model of joint contracture. J Orthop Res. 2019;37(12):2609–2620. doi:10.1002/jor.24441
98. Kobak S. Intraarticular Adalimumab in a patient with pigmented villonodular synovitis. Rheumatol Int. 2011;31(2):251–254. doi:10.1007/s00296-009-1185-z
99. Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31(5):986–1000. doi:10.1161/atvbaha.110.207449
100. Berenbaum F. Osteoarthritis as an inflammatory disease (osteoarthritis is not osteoarthrosis!). Osteoarthr Cartil. 2013;21(1):16–21. doi:10.1016/j.joca.2012.11.012
101. Shang X, Böker KO, Taheri S, Hawellek T, Lehmann W, Schilling AF. The Interaction between microRNAs and the Wnt/β-Catenin Signaling Pathway in Osteoarthritis. International Journal of Molecular Sciences. 2021; 22(18):9887. doi:10.3390/ijms22189887
102. Thielen NGM, van der Kraan PM, van Caam APM. TGFβ/BMP Signaling Pathway in Cartilage Homeostasis. Cells. 2019; 8(9):969. doi:10.3390/cells8090969
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.