Back to Journals » OncoTargets and Therapy » Volume 12
Critical factors in chimeric antigen receptor-modified T-cell (CAR-T) therapy for solid tumors
Received 10 October 2018
Accepted for publication 26 November 2018
Published 24 December 2018 Volume 2019:12 Pages 193—204
DOI https://doi.org/10.2147/OTT.S190336
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjeev K. Srivastava
Lingli Yan, Bainan Liu
Department of Immunology, Zunyi Medical University, Zunyi, Guizhou, China
Abstract: The success of chimeric antigen receptor-modified T-cell (CAR-T) therapy for B-cell lymphocyte malignancies targeting CD19 places it in a rapidly growing field in cancer immunotherapy for both hematological and solid tumors. However, the two types of tumor are quite different in the following respects. Solid tumors are characterized by complex vasculatures and matrix barriers that significantly affect T-cell functions and migration. Moreover, various immunosuppressive molecules expressed in the tumor microenvironment can impede T-cell activation, and the high metabolic rate of tumors competitively suppresses the metabolism of immune cells. All these factors will exert their influences on the development of a cancer, which is a dynamic balance between the host’s immune system and the tumor. At present, solid tumors are treated primarily by surgical resection combined with radiotherapy and chemotherapy, a treatment process that is painful and not always effective. With advantages over traditional treatments, the recently developed CAR-T immunotherapy has been applied and has shown highly promising results. Nevertheless, the complexity of solid tumors presents a great challenge to this technique. This review focuses on elucidating the factors influencing the anti-tumor effects of CAR-T in the specific tumor environment, and hence exploring feasible approaches to overcome them.
Keywords: chimeric antigen receptor-modified T cell, immunotherapy, solid tumor, tumor environment, anti-tumor effects
Introduction
The recognition and killing effect of T cells on tumors plays a central role in anti-tumor immunity. Utilizing the mechanism by which T cells kill tumor cells, scientists have designed protocols that specifically target tumor antigens and simultaneously activate T cells to produce anti-tumor effects. Chimeric antigen receptor (CAR), engineered to be expressed on T cells, is one such approach and has made great progress in cancer therapy, particularly in the treatment of B-cell lymphocyte malignancies.1–5 A typical CAR consists of an ectodomain, a transmembrane domain and an endodomain.6 The ectodomain in this case contains a signal peptide, an antigen recognition region, usually derived from a single-chain variable fragment (scFv) of a monoclonal antibody, and a spacer that connects the antigen recognition region to the transmembrane domain. The transmembrane structure in a CAR is most commonly from CD28, and less commonly from CD3ζ, CD4, CD8 or OX40. The main function of this structure is to provide stability to the CAR, with the transmembrane region from CD28 being more reliable than others in most cases.6–8 The endodomain of a CAR is engineered by a variable number of intracellular signaling molecules. According to the number of signaling molecules in a CAR, CARs have been categorized into four “generations”, which have been reviewed in detail by other groups.9 The evolution of CARs from the first to the fourth generation has encountered many issues in practice, which have been improved gradually. The first generation CAR contained a single signaling structure from CD3ζ or FcεRIγ, accompanied by poor outcomes in most studies because of inadequate proliferation, a short lifespan in vivo and insufficient cytokine products. The second generation CAR added intracellular signaling domains to the first generation CARs from various co-stimulatory molecules, such as CD28, 4-1BB and OX40, which improved the proliferation, cytotoxicity, sustained response and lifespan of CAR-T cells in vivo.6,10 In the third generation CAR, two co-stimulatory molecules were fused to the CD3ζ signaling moiety, with the most common combination of p56 lck+ CD28+ CD3ζ, OX40+ CD28+ CD3ζ or 4-1BB+ CD28+ CD3ζ.8 The third generation CAR can reduce the undesirable anti-inflammatory effects of IL-10,11 but involve the risk of signal leakage and cytokine cascade.12 To optimize the anti-tumor effects of chimeric antigen receptor-modified T cells (CAR-T), the fourth generation CAR has been developed by engineering the second generation CARs with a cytokine expression cassette, which is known as T-cells redirected for universal cytokine-mediated killing (TRUCK). TRUCKs can strengthen T-cell activation and attract innate immune cells to the targeted lesion to eradicate antigen-negative tumor cells by releasing anti-tumor cytokines, thus producing better tumoricidal effects, especially on solid tumors.13 The aforementioned four categories of CARs all have the ability to recognize tumor-associated surface antigens independent of the expression of major histocompatibility complex (MHC) molecules, which results in genetically modified T cells able to recognize tumor cells not being affected by MHC-restricted tumor antigens.
In recent years, early-phase clinical trials of CAR-T for B-cell malignancies have demonstrated promising results, and with Kymriah (Novartis) and Yescarta (Kite Pharma), the first CAR-T therapy products have been approved.14,15 The success has inspired great enthusiasm in the exploration of new innovations in CAR design and manufacture, development and toxicity management. A great deal of attention has also been paid to researching CAR-T therapy and a rapidly growing number of clinical trials on solid tumors is underway.16–18 Nevertheless, it will be more challenging and difficult to translate successful CAR-T therapy to solid tumors than to hematological malignancies because of the differential properties between the two types of tumors. Solid tumors have complicated vasculature matrix barriers and a hostile tumor microenvironment (TME) with many immunosuppressive cells and other inhibitory factors.19,20 Moreover, there is an intricate metabolic competition for nutrients in the TME between tumor-infiltrating T cells and tumor cells that can restrict the anti-tumor effects of T cells.21,22 There is an urgent need to solve these issues in CAR-T therapies for solid tumors to improve the therapeutic effects. The increasing amount of literature in this area enables investigators to keep up with the most recent developments. In this review, we focused on the complicated TME and other factors that significantly hamper the anti-tumor effects of CARs in solid tumors. In addition, constructive suggestions on how to overcome the hurdles are proposed.
Non-parallel clinical evaluation in CAR-T therapy for hematological malignancies and solid tumors
In several clinical evaluations of CAR-T for different solid tumors, the clinical results are inferior to those for hematological malignancies, eg, the disialoganglioside GD2 CAR-T used to evaluate patients with GD2+ neurocytoma, and epidermal growth factor receptor-targeted CAR T-cells used to evaluate patients with non-small-cell lung cancer, and the difference is significant compared with the clinical efficacy of CD19-CAR-T for B-lymphocyte malignancies (Table 1). Therefore, CAR-T therapy targeting solid tumors raises questions that need to be taken into consideration. To improve the therapeutic effect of CAR-T therapy on solid tumors, a series of preclinical trials has explored the structural characteristics of solid tumors and the microenvironment of immunosuppression.
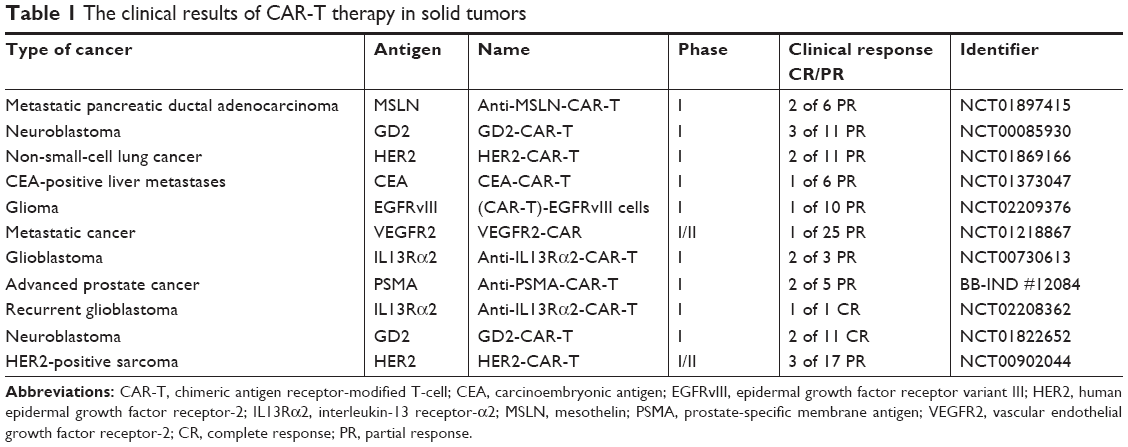 | Table 1 The clinical results of CAR-T therapy in solid tumors |
Solid tumor microenvironment and CAR-T
Unlike the intravenously infused CAR-T co-circulation with cancer cells in hematological cancers, a solid tumor has a hostile TME which presents a major hurdle in the CAR-T exertion effect. The TME is formed with the tumor and progresses spontaneously, which benefits the growth and metastasis of solid tumors. The TME is complex in structure and consists of various elements, wherein the abnormal vasculature and a stromal barrier act as critical physical barriers preventing CAR-T from entering and reacting with the neoplastic tissue. Unmatched chemokines secreted by tumor tissue are not compatible with CAR-T migration into tumor tissue, resulting in CAR-T cells that cannot be effectively migrated. Furthermore, the immunosuppressive effects of the TME also inhibit the effectiveness and persistence of CAR-T.20 The effective destruction of physical barriers and the immunosuppressive elements in the TME enable the CAR-T cell easily to meet the solid tumor (Figure 1).
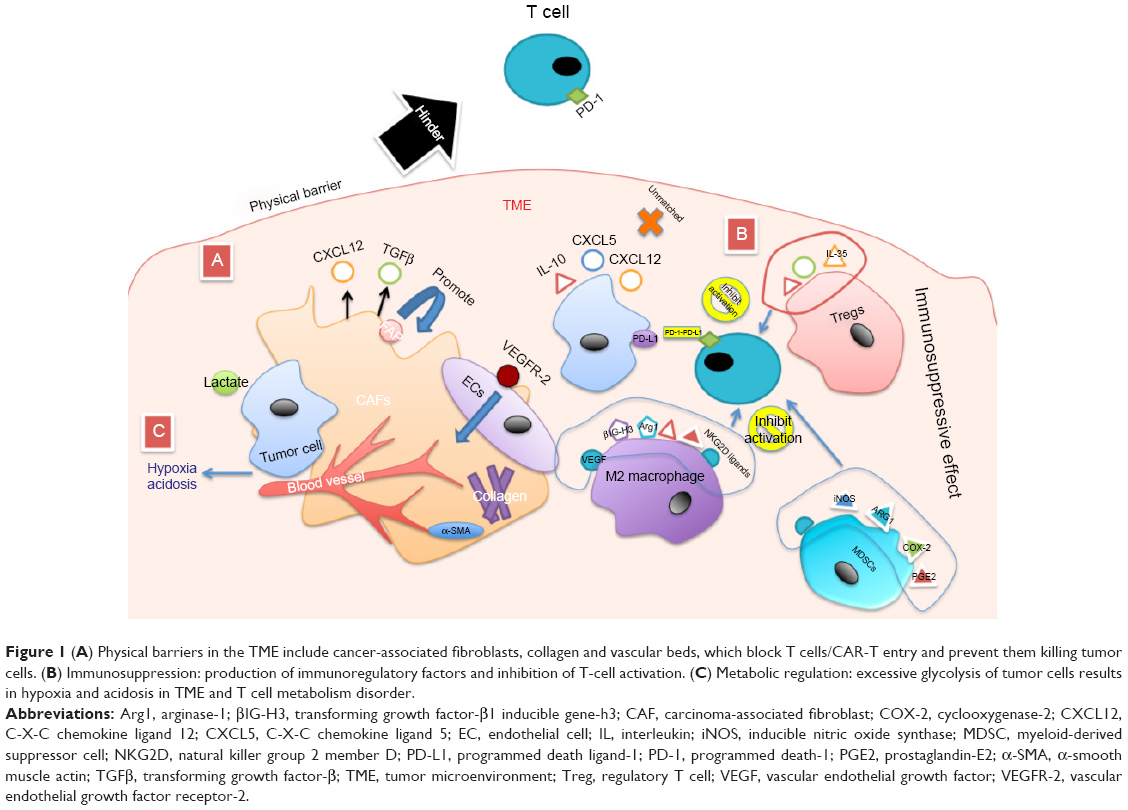 | Figure 1 (A) Physical barriers in the TME include cancer-associated fibroblasts, collagen and vascular beds, which block T cells/CAR-T entry and prevent them killing tumor cells. (B) Immunosuppression: production of immunoregulatory factors and inhibition of T-cell activation. (C) Metabolic regulation: excessive glycolysis of tumor cells results in hypoxia and acidosis in TME and T cell metabolism disorder. |
Vasculature barriers
The formation of a deviant vasculature in neoplastic tissue results in a hypoxic and acidic area supportive of the growth and, especially, metastasis of the tumor,23 and angiogenesis causes the spread of tumor cells to adjacent organs, making it difficult to control the extent of the tumor. Vascular endothelial growth factor A (VEGF-A) binds to vascular endothelial growth factor receptor-2 (VEGFR2) expressed in endothelial cells and makes a contribution to angiogenesis, along with the simultaneously hypoxic and acidic environment.19 These vascular barriers lead to abnormal intratumor vessel beds and stroma, making it difficult for CAR-T to enter the interior of the tumor, which is an enormous obstacle in CAR-T therapy. One appropriate method of therapy is CAR-T combined with anti-angiogenesis monoclonal antibodies. GD2 is a disialoganglioside that is highly expressed on human neuroblastoma (NB) cells, making it an understandable target for GD2+ NB immunotherapy. Bocca et al demonstrated with third generation CAR that GD2-CAR-T, when combined with bevacizumab (BEV; an anti-VEGF-A antibody) in an NB model, exhibited a significant anti-tumor effect, but the clinical effect of GD2-CAR-T cells alone for NB patients had unsatisfactory results. GD2-CAR-T reprogrammed the TME when combined with BEV, reducing the formation of tumor microvessels and relieving hypoxia and acidosis, which improved T-cell migration to the tumor site.24 A clinical trial of VEGF as a target for CAR-T, VEGFR2-CAR, however, was disappointing, with only a partial response in one of the patients who received the treatment and the rest being completely unresponsive (NCT01218867), although the design had promising results in preclinical trials.25 This is the biggest challenge for immunotherapy: the solid tumor in the human body is highly complex in both mechanism and structure, making it difficult to translate successes in animal models to humans.
Stromal hindrance
The matrix barrier formed by connective tissue is also an important factor in CAR-T therapy for solid tumors, and is indispensable in the formation of the tumor-suppressive microenvironment. The formation of the fibrous structure induced by cancer-associated stromal cells (CASCs) is the main cause of extracellular matrix (ECM) barrier formation in the tumor environment.26,27 A complex tumor ecosystem is formed from the interaction of heterogeneous tumor cell clones with heterogeneous stromal cells, creating a conducive environment for premalignant cells, thus promoting tumorigenesis and high intratumor heterogeneity.28 These conditions hinder the infiltration of the tumors by CAR-T cells. Carcinoma-associated fibroblasts (CAFs) are the major subset of CASCs and are chosen as a reasonable target antigen for CAR-T therapy. CAFs are identified via the expression of fibroblast-activated protein (FAP), and FAP-secreting transforming growth factor-β (TGFβ) and CXCL12 inhibit the function of effector T cells.29,30 The expression of CAFs in pancreatic ductal adenocarcinoma (PDAC), breast carcinomas and gastric cancer is closely related to progression.31–33 Lo et al proposed that FAP has a crucial role in adaptive immunity by immune-independent but stromal-dependent mechanisms. FAP-CAR-T treatment in the FAP+ tumor model produced an increased intratumoral infiltration of T cells and inhibited the desmoplastic growth and angiogenesis in highly desmoplastic human tumors.27 Moreover, α-smooth muscle actin (α-SMA) positivity is also a critical component of the ECM, and control of high-density expression of α-SMA positivity in pancreatic cancer has shown some positive effects in clinical trials.34 Whether α-SMA positivity can be used as an effective CAR target, or in combination therapy with anti-α-SMA-positive monoclonal antibodies and FAP-CAR-T for tumors with high expression of CFAs, may be a worthwhile research topic. Furthermore, researchers found that T cells in the process of amplification in vivo were unable to break through the matrix barrier into the tumor environment, related to the lack of the expression of the enzyme heparanase (HPSE; degrades heparan sulfate proteoglycans, which are the main components), so they designed an HPSE-modified CAR-T to enhance substrate degradation and increase CAR-T infiltration.35
CAR-T migration
The next step is controlling the accurate orientation of CAR-T into the tumor environment. The effective migration of T cells is called CAR-T homing, during which T cells are accurately transported to the TME. T-cell homing to the TME is affected by the mismatch between chemokine receptor expression on T cells and chemokine secretion by the tumor (eg, CXCL12, CXCL5).36 The migration ability of the CAR-T can be enhanced in two ways: 1) by attacking the barrier effect of the TME: the destruction of the TME structure can not only eliminate the physical barrier but also promote the migration of T cells; for example, studies have shown that anti-VEGFR2 CAR combined with IL-12-cotransduced T-cells can enhance the migration capacity of CAR-T in the mouse model,25 as mentioned already in the section “Vasculature barriers”, and degradation of the ECM can also promote the transendothelial migration of T cells;35 and 2) by designing T cells to express chemokine receptors that match tumor chemokines, so that CAR-T more accurately locates tumor tissue; not only does C-C chemokine ligand 2 (CCL2) drive accumulation of tumor-infiltrating lymphocytes (γδTIL) in tumor tissue, but the expression of CCL2 is also related to the production of endothelial progenitor cells, which cause neovascularization in the TME. CCL is expressed in a variety of tumors,37,38 and this chemotaxis has been applied to CAR-T therapy. Preclinical trials have demonstrated that CAR-T expressed by chemokine receptor CCR2b can cause a rapid chemotaxis response in vivo and in vitro, significantly improving the efficacy of the CAR-T treatment.39 In general, the synergistic effect of CAR-T on the infiltration of tumor tissue was achieved by degrading the ECM and inhibiting the formation of a neovascularization network. However, after CAR-T enters the tumor environment, the abnormal expression of a battery of immunomodulatory molecules leads to immunosuppression, so it is not necessarily effective to infiltrate the tumor tissue alone.
Immunosuppressive effects of T-cell activation inhibitory factor in the TME
The TME mainly consists of numerous suppressive immune cells and molecular factors, which interfere with the CAR-T cells’ fight with the tumor cells. The immunosuppression of tumors by down-regulation of the activation molecules of immune cells and the TME recruiting immunosuppressive cells are crucial factors affecting solid tumor therapy. Even though genetically engineered CAR-T cells with redirected function can recognize tumor antigens without MHC-peptide restriction, divers immunosuppressive factors in the TME present enormous obstacles to CAR-T treatment.40,41 Regulatory T cells (Tregs) inhibit the activation of effector T cells by various pathways in the TME, such as the inhibited effector T cell binding with IL-12 and secreting suppressive cytokines (IL-10, TGFβ, etc).42 M2 macrophages account for the largest proportion of tumor-infiltrating inflammatory cells, mainly participating in tumor growth and metastasis, and innate and adaptive immunity. M2 macrophages produce fibrin-related proteins and matrix connexin TGFβ1-inducible gene-h3 (βIG-H3), and arginase-1 (Arg1) induces the formation of polyamines and proline, thus promoting angiogenesis and ECM precipitation, which means that M2 macrophages can promote the formation of physical barriers in the TME while producing immunosuppression.43 Alternatively, inhibitory receptors of T cells, such as cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4), programmed death ligand-1 (PD-L1) and programmed death-1 (PD-1), function in limiting the potential of CAR-T therapy even though the antigens of CARs are recognized. Overexpression of PD-1/PD-L1 also facilitated Treg suppression of effector T cells,44 except for when checkpoint inhibitor monoclonal antibodies were combined with CAR-T as a solution in some of the trials.45 Suarez et al designed two cassettes of CARs comprising anti-carbonic anhydrase CAR-T (anti-CAXI-CD28z-CAR) with self-inactivating (SIN) bicistronic lentiviral vector and anti-PD-L1 IgG1 or IgG4, which could produce anti-PD-L1 antibodies to decrease both T-cell exhaustion and tumor growth.46 Other immunosuppression molecules of effector T cells, such as T-cell membrane protein-3 (TM-3) or lymphocyte activation gene-3 (LAG-3), can also be used as targets.47 Fourth generation CARs are designed to transfer two transgenes for both CARs and inducible cytokines, which are constructed of functional T cells redirected for universal cytokine-mediated killing (TRUCKs).13,48 TRUCKs are engineered with an additional nuclear factor of the activated T cell (NFAT) for inducible expression of a transgenic cytokine, to reverse the TME and recruit more immune cells to facilitate efforts against cancers of the immune system, mediated by the expression of NFAT/IL-12-CAR-T, and to release IL-12 into target tumor tissue to decrease side effects and improve the local function of CAR-T.13 With IL-18 TRUCKs, IL-18 polarizes CAR-T cells toward T-bet high FoxO1low, which decreases the percentage of Tregs and M2 macrophages in the TME, providing a promising strategy in CAR-T therapy for large solid tumors.49 Target immunosuppression molecular designed CAR-T include TGFβ-receptor-CAR-T50 and natural killer group 2 member D (NKG2D-CAR-T),51 to recognize NKG2D ligands expressed on Tregs and myeloid-derived suppressor cells (MDSCs).52,53 Other immunosuppression molecules, such as MDSCs, tumor-associated macrophages, immature dendritic cells and tumor-associated neutrophils, also affect the normal activation of T cells even if they can successfully enter tumor tissue, and hence, reversing the immunosuppressive effect is the key to successful CAR-T therapy.
The survival of the tumor depends on a suitable environment, and when the environment is in a steady state it is beneficial for tumor progression. Once this balance is broken, the tumor cells are more easily exposed, making them easier to recognize and kill. Correcting the body’s internal environment to a normal state is exactly what we need to achieve and verify. When we explore effective treatments, in addition to trying to destroy the TME, accurately identifying and killing tumor cells are also crucial elements in CAR-T therapy.
Target selection
The ectodomain of CAR-T cells consists of a tumor-associated antigen (TAA) recognition region derived from a monoclonal antibody (usually an scFv of an antibody), which provides a signal peptide to transfer the recognized protein signal to the cellular endoplasmic reticulum. This design can eliminate the requirements of both the MHC restriction and the antigen presentation of the antigen-presenting cell (APC) when the T-cell receptor (TCR) recognizes the target antigen; therefore, choosing an optimal target is a critical factor in the implementation of CAR strategies. A number of targets have been selected in the solid tumor CAR-T strategy (Figure 2). However, owing to the large variety of solid tumors, TAAs expressed on solid tumors are mostly heterogeneous and change frequency with the tumor development stage, which means that the choice of an optimal target antigen has many restrictions that should be considered. In contrast, some TAAs are expressed in normal cells and tissues. The affinity of the tumor-targeting antigen TCR should also be considered to affect normal tissue that expresses the same antigen, which is called on-target/off-tumor toxicity.
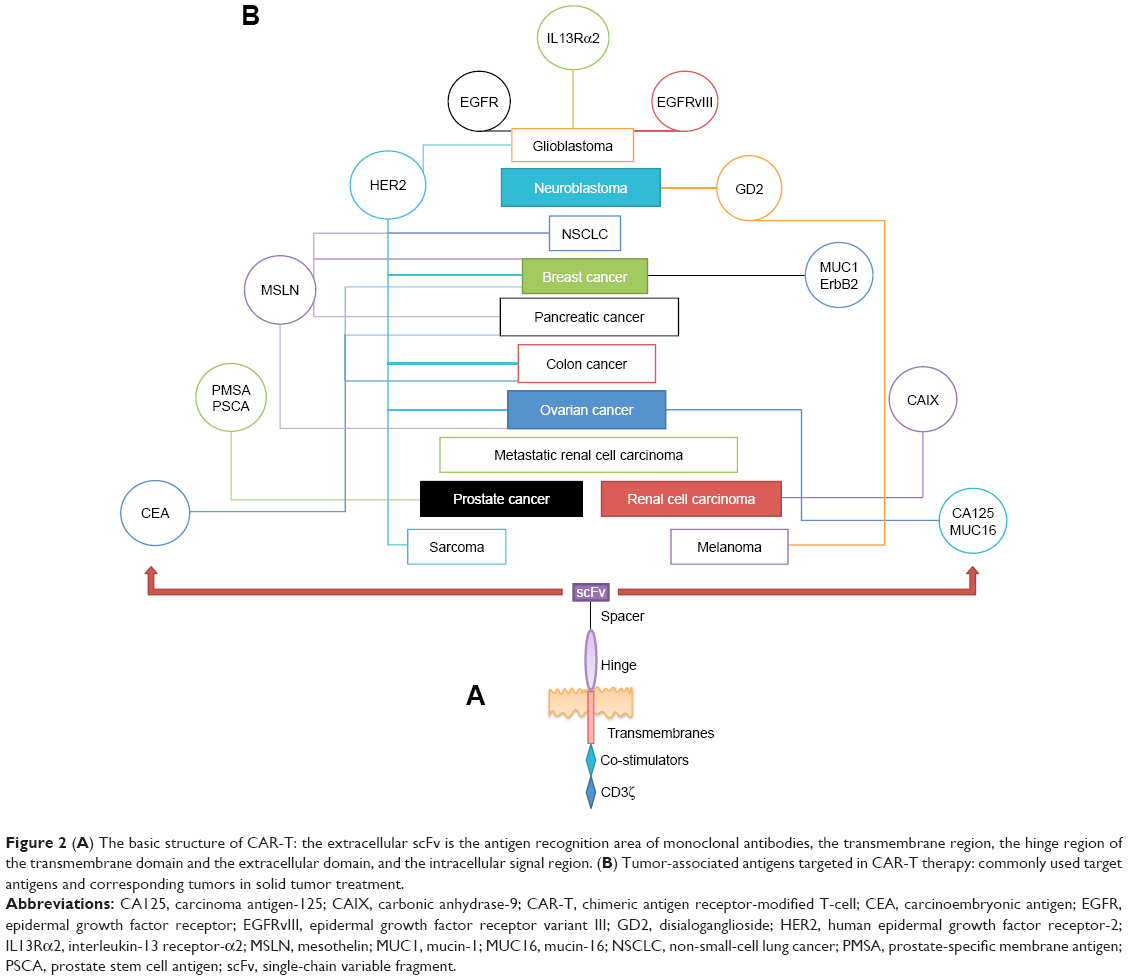 | Figure 2 (A) The basic structure of CAR-T: the extracellular scFv is the antigen recognition area of monoclonal antibodies, the transmembrane region, the hinge region of the transmembrane domain and the extracellular domain, and the intracellular signal region. (B) Tumor-associated antigens targeted in CAR-T therapy: commonly used target antigens and corresponding tumors in solid tumor treatment. |
Design of the optimal target
Tumor antigens are divided into tumor-specific antigens (TSAs), which are characterized by expression on the tumor cells only, and TAAs, which are expressed on tumor tissue and normal tissue in differing quantities. TSAs are an ideal target antigen choice; however, the number of options is limited. In addition to the down-regulation of tumor antigens leading to immune escape, the intratumor heterogeneous subpopulation of tumor cells54 is composed of barriers to the functioning of single-target-CAR-T. To address this issue, in a promising protocol called targeting multiple antigens of CAR-T therapy, Anurathapan et al simultaneously targeted the CARs mucin-1 and prostate stem cell antigen, which produced a superior anti-tumor effect and increased the intensity of targeting expression to improve the efficacy of T-cell killing.55 Other protocols such as dual-specific CAR-T targeted CD20 and human epidermal growth factor receptor-2 (HER2), which are tandem antigens where the conventional scFv is replaced by two variable heavy chains of different scFvs contained in the nanobody. This scheme is for antigen loss or negative expression of an antigenic marker, and the tandem CAR prepared with the nanobody not only solves the problem of capacity limitation of the scFv region but also forms a preset affinity.56 Combination antigen CARs are used in dual-antigen expression tumors to avoid the on-target/off-tumor effect, in which the targeting of multiple antigens could avoid an attack on normal tissues that also express tumor antigens.57 Further research has investigated how to select the best matching antigen as a dual target. Orentas et al found that defined pairwise related antigens can be overexpressed on the tumor surface, and when chosen as the target and compared by bioinformatics using statistical tools, the chosen optimal antigen pair acts as a stringency control on the expression level of normal tissues, thus avoiding damage to normal tissues and vital organs.58 Moreover, for the trans-signaling CAR-T strategy, dissociation activates the signal of CD3ζ and co-stimulates the signal of CD28 in two kinds of CARs assembled from two different scFvs. The therapeutic effect of enhanced CAR-T as a dual-specificity treatment against on-target toxicity and the immune escape of the tumor,59 therefore, is dependent on selection of the appropriate target, which plays a critical role in therapy efficacy.
Affinity of CARs for antigens
While selecting the optimal target, the affinity of the scFv for the target antigen should also be considered. With the on-target/off-tumor toxic effect, that is, when CAR-T recognizes and attacks normal tissue expressing the tumor antigen,60 the effects on these organs and tissues are predictable. As previously reported, ERBB2 (HER2)-CAR-T treatment in an HER2+ breast cancer patient resulted in rapid death, where ERBB2-CAR-T binding with the low expression of HER2+ on lung epithelial cells resulted in cross-reaction toxicity.61 Therefore, aside from the selection of dual-specific CARs mentioned in the previous section (which affected only tumor tissues that expressed both dual-specific antigens but not normal tissues that expressed a single antigen), we can also control the degree of affinity of the scFv of CARs for the targeted antigen.62 In light of this, we constructed a CAR-T that can accurately distinguish between the antigen density in normal tissues and that in tumor tissues, selecting a suitable “killing range” that does not attack normal tissues but can maximize the ability to target tumor tissues.63 One method is to design CARs with different magnitudes of affinity based on the affinity range of TCRs recognizing the peptide–major histocompatibility complex (pMHC), and then determining the correct antigen densities of CAR-T as expressed on the tumor cells compared to the same antigen on normal tissues when the antigens are recognized, to find an optimal critical value for CAR affinity.64 Other researchers have introduced safety switches, by inserting an inducible caspase-9 suicide gene into CAR-T to control the safe activation of CAR-T and induce apoptosis at high levels of the expressed transgene.65 The suicide switches may not act fast enough to prevent cross-reaction toxicity at an early stage.66 “On-switch CAR” is a complementary protocol in which CARs are gradually activated to an optimal therapeutic level. The protocol is designed with a dual-input signal (small molecule and antigen dimerization) and a split signal pattern that is small molecule dependent for CAR-T activation.66 Richman et al tried to change the affinity of the GD-2 CAR-targeted antigen via single amino acid substitution in the scFv and searched for the optimal degree of target antigen affinity to avoid fatal encephalitis caused by the high affinity of the CARs, an appropriate treatment plan.67 The purpose of these protocols is to find the most suitable affinity index for a CAR-T therapy, adjusting the optimal range to kill as many tumor cells as possible while minimizing the attack on normal tissues. These strategies cannot completely eliminate the tumor cells, but they are aiming for a minimum threshold for tumor cell survival, gradually decreasing the tumor cells to a non-lethal range, and inhibiting tumor expansion and spread to achieve a state in which tumor cells coexist with the body’s healthy cells.
When the growth environment of the tumor has been destroyed, the best target selected and the optimal degree of target affinity adjusted, we also need to consider how CAR-T can increase in stability and continue to produce the desired effect after entering the internal environment. Adoptive T cells are different from endogenous T cells, and the tumor’s high-depletion metabolism competitively inhibits the metabolism of T cells. These are problems that we need to solve.
CAR-T kinetics
It is well known that structure affects function, and so it is with the design of CARs. T-cell activation kinetics are driven by the formation of TCR-pMHC immune synapses.59 However, the formation of CAR-T immunological synapses has not been precisely defined; this concept in CAR-T design is optimized based on the mechanism of activation and exertion of T cells in vivo and the dynamic metabolic mechanism of T cells.
CAR-T kinetics and immune synapses
The in vivo activation kinetics of T cells in recognizing pMHCs presented by APCs are divided into two modes: immunokinase (immediately) and immune synapse (stabilized).68 In addition, there are three phases: the first phase is the rapid transit of T cells to the pMHC, usually lasting from 30 minutes to 8 hours; in the second stage, T cells maintain stable binding to APCs and produce cytokines, and this phase is the stage through the immune synapse, which lasts for 12 hours; in the third stage, T cells recover their motility and start to divide.69 The concept of immunological synapses was first proposed by Allen et al as the area of contact between T cells and APCs, including the interaction of T cells with APCs and the role of related adhesion molecules, leading to T-cell activation and proliferation.70 The stage of the immune synapse is usually considered to be the stage of T-cell activation.71 When T cells receive stimulation signals that reach a precise threshold, they switch to the second stage, causing T-cell activation.72 The kinetics of CAR-T activation related to the immune synapse formation that bonds CAR to the target and the crucial anti-tumor effect of CAR-T are dependent on their ability to form effective immune synapses with tumor targets. Comparing the synapse formation of CAR with the endogenous immune synapse formed by the TCR, the CAR can have a faster recognition rate than the TCR; in the long term, despite their different structures, CAR expression can be induced by further activation of the TCR signal, which may be associated with a long-lasting effect.73 There are also researchers using TCR-specific identification capabilities (to identify peptides that degrade a cell protein and the entire protein group, to accurately distinguish and identify the mutated proteins in tumor tissues) to construct a TCR–CAR frame that maintains the original TCR characteristics.74 When combined with the TCR and CAR effects on T-cell engineering, this can be used to adjust the CAR-T cytotoxicity response to the desired level, depending on the disease state.75
Subsets of T cells for CAR-T
The selection of an optimal T-cell subset as the material for preparing CAR-T can enhance the effect of the therapy. With regard to endogenous T cells, after receiving the pMHC signal, naive T cells differentiate into T memory cell subsets, of which T stem cell memory (TSCM) and T central memory (TCM) cells account for only 2%–4%. TSCMs exhibit stem cell-like properties and have a greater capacity for self-renewal and long-term survival compared to other activated subpopulations. TCMs lose CCR7 expression after receiving twice the level of antigenic stimuli and eventually differentiate into terminally differentiated effector T cells; they have superior engraftment and persistence to other memory subsets. TSCMs and TCMs exhibit better expansion, persistence and anti-tumor effects than other T-cell subsets in treating HIV infection,76–78 and T-cell subsets have shown some efficacy in hematological tumors when used in CAR-T therapy.79 Solid tumors have increased the requirements for the expansion and maintenance of the T-cell effect in killing tumors. In a test comparing the activation and effect of different T-cell subsets, such as GD2-CAR-T and CD19-CAR-T, it was found that TSCM was the best for expansion and maintainability.80 This demonstrated that the correct choice of T-cell subsets enhances the therapeutic effects of CAR-T, but regulating the activation of T cells and the balance of T cells in the tumor environment according to the mechanism of activation of endogenous T cells are points of concern.
T-cell metabolism
T cells exist in three phases in the body, namely the initial resting, activation and memory stages,81 with each state having its own metabolic characteristics. Metabolism in the activation phase is significantly enhanced, and T cells are activated via TCR binding with pMHC, ie, CD4+/CD8+ naive T cells transform to effector T cells (T-helper cells or CD8+ cytotoxic T lymphocytes) when they recognize an antigen and receive co-stimulatory signals. These processes rely mainly on glycolysis to meet their metabolic needs.22,82 In the case of tumors with sufficient oxygen, even if oxidative phosphorylation is possible, the tumors still prefer to use glycolysis to generate energy, the so-called Warburg effect.83 Tumor expansion requires even more energy and exerts competitive inhibition on the effective activation of T cells.21 Moreover, up-regulation of PD-L1 in tumors and the TME causes PD-1 to inhibit Akt phosphorylation by inhibiting PI3K activation mediated by CD28 molecules.84 The design of CAR-T related to the metabolic processes of T cells, with the second generation of CAR-T with CD28/4-BB1 co-stimulatory signals, added two kinds of co-stimulators to CARs, increasing the functional effects while stabilizing proliferation and accelerating the accumulation of T cells.85 Alternatively, following adoptive T-cell (ATC) therapy in melanoma patients, the expression of GPI and PGAM4 in the tumor tissues of non-responsive patients was elevated, and IRF1 (a key transcription factor in the interferon-γ signaling pathway) was down-regulated in high-glycolytic melanoma. These phenomena are significantly associated with the development of immunosuppression and high glycolysis. Competitive glycolytic metabolism is the core role of immunosuppression, and it is also an important factor affecting the efficacy of ATC therapy.86 It is possible that using glycolysis inhibitors combined with 4-1BB/CD28-CAR could significantly improve the efficacy of CAR-T therapy.
CAR-T and traditional therapy
Traditional therapy such as surgery, radiotherapy and chemotherapy is still the main treatment for tumors, but chemotherapy tolerance and recurrence and metastasis after surgery remain the major obstacles to traditional therapies. CAR-T therapy first showed great success in the treatment of hematological malignancies. A study confirmed that the use of CAR-T as an intravenous injection can help to inhibit the proliferation and metastasis of tumor cells through the circulatory system.87 In addition, combining chemotherapy with CAR-T therapy has been investigated, since chemotherapeutic drugs can eliminate or inhibit immunosuppressive factors and promote an anti-tumor immune response.88,89
Conclusion
CAR-T therapy has undergone long-term research and exploration and is an active field of discovery for the treatment of solid tumors. Solid tumors are complex in structure, which is the primary obstacle to the effectiveness of CAR-T therapy. The complex matrix barriers, vasculature and tumors in the TME cause abnormal expression of immunomodulatory factors, thereby inhibiting the activation and effects of T cells. To mitigate these issues, we must first determine which molecules play a key role in the TME and then use them as targets to destroy the tumor environment, relieving the immunosuppression of T-cell activation induced via abnormal expression of immunomodulatory factors as a complementary strategy. Destroying the TME, increasing the infiltration capacity of CAR-T and optimizing the chemokine receptor that matches the CAR expression with the tumor has a synergistic effect. The second factor is that the expression of tumor antigens is not constant. In the process of tumor growth and metastasis, the expression of tumor antigens will vary or be lost altogether. With complex and unpredictable regulatory mechanisms, this has become the most difficult barrier for CAR-specific recognition. Since CARs have selectivity in target recognition, a multiple-target protocol is used to increase the targeting accuracy while adjusting the affinity of CARs to identified targets, to prevent CARs from producing the on-target/off-tumor effect, yet still allowing for recognition of tumor antigens. After meeting these conditions, we need to focus on whether CAR-T can successfully activate, proliferate and kill tumor cells after entering the body. To address this problem, one must select the appropriate subset as the basis for CAR, first according to the potential effects of the various subsets of T cells and then according to the stimulation signals required for the activation and the recognition of pMHC by endogenous T cells. The mechanism of the formation of immune synapses to optimize the structure of the CAR-T is equivalent to the CAR-T simulating the process of normal T-cell activation in the body, then interfering with the glycolytic pathway of highly metabolic tumors and removing the competitive inhibitory effect of the tumor on immune cell metabolism. Considering these factors, we have been exploring the optimal solution for CAR-T tumor therapy.
Future research directions
The study of immunotherapy will eventually result in clinical application, and CAR-T therapy has shown a satisfactory clinical effect in hematological cancers. Although research on CAR-T therapy on solid tumors shows positive therapeutic effects in animal models, in clinical treatment settings it has always been ranked as a failure (unresponsive), and with the occurrence of cytokine release syndrome (CRS), tumor lysis syndrome, and on-target/off-tumor and other toxic effects, it has not become a conventional clinical medication. Improving the therapeutic effect of CAR-T and minimizing side effects are problems worthy of further study. In addition to the solutions described in this review, we need to pay attention to the problems that the carrier design of CAR-T will also affect its safety, and CAR-T’s sorting and training mode also affects its performance. The CAR design can also be used for cells such as natural killer cells that have a direct killing effect in anti-tumor immunity, that is, the so-called CAR-NK cells. The effectiveness of CAR technology lies in the expression of specific molecules on the surface of the cell to activate intracellular signals simultaneously, and whether this method can be used in cells other than immune cells, for example, in the design of a tumor cell vaccine, is also a promising research direction. CAR-T therapy alone does not have satisfactory effects on complex solid tumors, but it may be more effective in combination with checkpoint inhibitors, monoclonal antibodies, vaccines, etc. In addition, because CAR-T therapy is expensive, not all patients can afford treatment, so we need to try to find a way to reduce the cost of production so that T-cell gene engineering therapy can reach more patients.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (grant number 81560276) and the Foundation for Returnees from the Department of Human Resources and Social Security of Guizhou Province of China (grant number [2014]08).
Disclosure
The authors report no conflicts of interest in this work.
References
Levine BL, Miskin J, Wonnacott K, Keir C. Global manufacturing of CAR T cell therapy. Mol Ther Methods Clin Dev. 2017;4:92–101. | ||
Maude SL, Teachey DT, Porter DL, Grupp SA. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood. 2015;125(26):4017–4023. | ||
Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385(9967):517–528. | ||
Maher J. Clinical immunotherapy of B-cell malignancy using CD19-targeted CAR T-cells. Curr Gene Ther. 2014;14(1):35–43. | ||
Makita S, Yoshimura K, Tobinai K. Clinical development of anti-CD19 chimeric antigen receptor T-cell therapy for B-cell non-Hodgkin lymphoma. Cancer Sci. 2017;108(6):1109–1118. | ||
Zhang C, Liu J, Zhong JF, Zhang X. Engineering CAR-T cells. Biomark Res. 2017;5:22. | ||
Dotti G, Gottschalk S, Savoldo B, Brenner MK. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol Rev. 2014;257(1):107–126. | ||
Muhammad N, Mao Q, Xia H. CAR T-cells for cancer therapy. Biotechnol Genet Eng Rev. 2017;33(2):190–226. | ||
Zhao Y, Wang QJ, Yang S, et al. A herceptin-based chimeric antigen receptor with modified signaling domains leads to enhanced survival of transduced T lymphocytes and antitumor activity. J Immunol. 2009;183(9):5563–5574. | ||
Oldham RAA, Medin JA. Practical considerations for chimeric antigen receptor design and delivery. Expert Opin Biol Ther. 2017;17(8):961–978. | ||
Hombach AA, Heiders J, Foppe M, Chmielewski M, Abken H. OX40 costimulation by a chimeric antigen receptor abrogates CD28 and IL-2 induced IL-10 secretion by redirected CD4(+) T cells. Oncoimmunology. 2012;1(4):458–466. | ||
Campana D, Schwarz H, Imai C. 4-1BB chimeric antigen receptors. Cancer J. 2014;20(2):134–140. | ||
Chmielewski M, Abken H. TRUCKs: the fourth generation of CARs. Expert Opin Biol Ther. 2015;15(8):1145–1154. | ||
Yip A, Webster RM. The market for chimeric antigen receptor T cell therapies. Nat Rev Drug Discov. 2018;17(3):161–162. | ||
Hartmann J, Schüßler-Lenz M, Bondanza A, Buchholz CJ. Clinical development of CAR T cells-challenges and opportunities in translating innovative treatment concepts. EMBO Mol Med. 2017;9(9):1183–1197. | ||
Sun M, Shi H, Liu C, et al. Construction and evaluation of a novel humanized HER2-specific chimeric receptor. Breast Cancer Res. 2014;16(3):R61. | ||
Thomas S, Straathof K, Himoudi N, Anderson J, Pule M. An optimized GD2-targeting retroviral cassette for more potent and safer cellular therapy of neuroblastoma and other cancers. PLoS One. 2016;11(3):e0152196. | ||
Liu X, Zhang N, Shi H. Driving better and safer HER2-specific CARs for cancer therapy. Oncotarget. 2017;8(37):62730–62741. | ||
de Palma M, Biziato D, Petrova TV. Microenvironmental regulation of tumour angiogenesis. Nat Rev Cancer. 2017;17(8):457–474. | ||
Becker JC, Andersen MH, Schrama D, Thor Straten P. Immune-suppressive properties of the tumor microenvironment. Cancer Immunol Immunother. 2013;62(7):1137–1148. | ||
Chang CH, Qiu J, O’Sullivan D, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. 2015;162(6):1229–1241. | ||
Siska PJ, Rathmell JC. T cell metabolic fitness in antitumor immunity. Trends Immunol. 2015;36(4):257–264. | ||
Böhme I, Bosserhoff AK. Acidic tumor microenvironment in human melanoma. Pigment Cell Melanoma Res. 2016;29(5):508–523. | ||
Bocca P, di Carlo E, Caruana I, et al. Bevacizumab-mediated tumor vasculature remodelling improves tumor infiltration and antitumor efficacy of GD2-CAR T cells in a human neuroblastoma preclinical model. Oncoimmunology. 2017;7(1):e1378843. | ||
Chinnasamy D, Yu Z, Theoret MR, et al. Gene therapy using genetically modified lymphocytes targeting VEGFR-2 inhibits the growth of vascularized syngenic tumors in mice. J Clin Invest. 2010;120(11):3953–3968. | ||
Costa A, Kieffer Y, Scholer-Dahirel A, et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell. 2018;33(3):463–479. | ||
Lo A, Wang LS, Scholler J, et al. Tumor-promoting desmoplasia is disrupted by depleting FAP-expressing stromal cells. Cancer Res. 2015;75(14):2800–2810. | ||
Ye B, Stary CM, Li X, et al. Engineering chimeric antigen receptor-T cells for cancer treatment. Mol Cancer. 2018;17(1):32. | ||
Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480(7378):480–489. | ||
Paulsson J, Micke P. Prognostic relevance of cancer-associated fibroblasts in human cancer. Semin Cancer Biol. 2014;25:61–68. | ||
Yan Y, Wang LF, Wang RF. Role of cancer-associated fibroblasts in invasion and metastasis of gastric cancer. World J Gastroenterol. 2015;21(33):9717–9726. | ||
Pan B, Liao Q, Niu Z, Zhou L, Zhao Y. Cancer-associated fibroblasts in pancreatic adenocarcinoma. Future Oncol. 2015;11(18):2603–2610. | ||
Fu Z, Song P, Li D, et al. Cancer-associated fibroblasts from invasive breast cancer have an attenuated capacity to secrete collagens. Int J Oncol. 2014;45(4):1479–1488. | ||
Sinn M, Denkert C, Striefler JK, et al. α-Smooth muscle actin expression and desmoplastic stromal reaction in pancreatic cancer: results from the CONKO-001 study. Br J Cancer. 2014;111(10):1917–1923. | ||
Caruana I, Savoldo B, Hoyos V, et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med. 2015;21(5):524–529. | ||
Feig C, Jones JO, Kraman M, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A. 2013;110(50):20212–20217. | ||
Lança T, Costa MF, Gonçalves-Sousa N, et al. Protective role of the inflammatory CCR2/CCL2 chemokine pathway through recruitment of type 1 cytotoxic γδ T lymphocytes to tumor beds. J Immunol. 2013;190(12):6673–6680. | ||
Chen X, Wang Y, Nelson D, et al. CCL2/CCR2 regulates the tumor microenvironment in HER-2/neu-driven mammary carcinomas in mice. PLoS One. 2016;11(11):e0165595. | ||
Craddock JA, Lu A, Bear A, et al. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother. 2010;33(8):780–788. | ||
Beatty GL, Moon EK. Chimeric antigen receptor T cells are vulnerable to immunosuppressive mechanisms present within the tumor microenvironment. Oncoimmunology. 2014;3(11):e970027. | ||
Wang D, Dubois RN. Immunosuppression associated with chronic inflammation in the tumor microenvironment. Carcinogenesis. 2015;36(10):1085–1093. | ||
Ward-Hartstonge KA, Kemp RA. Regulatory T-cell heterogeneity and the cancer immune response. Clin Transl Immunology. 2017;6(9):e154. | ||
Murray PJ, Wynn TA. Obstacles and opportunities for understanding macrophage polarization. J Leukoc Biol. 2011;89(4):557–563. | ||
Zhou Q, Munger ME, Highfill SL, et al. Program death-1 signaling and regulatory T cells collaborate to resist the function of adoptively transferred cytotoxic T lymphocytes in advanced acute myeloid leukemia. Blood. 2010;116(14):2484–2493. | ||
Blake SJ, Ching AL, Kenna TJ, et al. Blockade of PD-1/PD-L1 promotes adoptive T-cell immunotherapy in a tolerogenic environment. PLoS One. 2015;10(3):e0119483. | ||
Suarez ER, Chang Dek, Sun J, et al. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget. 2016;7(23):34341–34355. | ||
Turnis ME, Andrews LP, Vignali DA. Inhibitory receptors as targets for cancer immunotherapy. Eur J Immunol. 2015;45(7):1892–1905. | ||
Li J, Li W, Huang K, et al. Chimeric antigen receptor T cell (CAR-T) immunotherapy for solid tumors: lessons learned and strategies for moving forward. J Hematol Oncol. 2018;11(1):22. | ||
Chmielewski M, Abken H. CAR T cells releasing IL-18 convert to T-Bethigh FoxO1low effectors that exhibit augmented activity against advanced solid tumors. Cell Rep. 2017;21(11):3205–3219. | ||
Chang ZL, Lorenzini MH, Chen X, et al. Rewiring T-cell responses to soluble factors with chimeric antigen receptors. Nat Chem Biol. 2018;14(3):317–324. | ||
Weiss T, Weller M, Guckenberger M, Sentman CL, Roth P. NKG2D-based CAR T cells and radiotherapy exert synergistic efficacy in glioblastoma. Cancer Res. 2018;78(4):1031–1043. | ||
Nausch N, Galani IE, Schlecker E, Cerwenka A. Mononuclear myeloid-derived “suppressor” cells express RAE-1 and activate natural killer cells. Blood. 2008;112(10):4080–4089. | ||
Zhang T, Sentman CL. Cancer immunotherapy using a bispecific NK receptor fusion protein that engages both T cells and tumor cells. Cancer Res. 2011;71(6):2066–2076. | ||
Fisher R, Pusztai L, Swanton C. Cancer heterogeneity: implications for targeted therapeutics. Br J Cancer. 2013;108(3):479–485. | ||
Anurathapan U, Chan RC, Hindi HF, et al. Kinetics of tumor destruction by chimeric antigen receptor-modified T cells. Mol Ther. 2014;22(3):623–633. | ||
de Munter S, Ingels J, Goetgeluk G, et al. Nanobody based dual specific CARs. Int J Mol Sci. 2018;19(2):E403:403. | ||
Roybal KT, Rupp LJ, Morsut L, et al. Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell. 2016;164(4):770–779. | ||
Orentas RJ, Sindiri S, Duris C, et al. Paired expression analysis of tumor cell surface antigens. Front Oncol. 2017;7:173. | ||
Lanitis E, Poussin M, Klattenhoff AW, et al. Chimeric antigen receptor T Cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol Res. 2013;1(1):43–53. | ||
Curran KJ, Pegram HJ, Brentjens RJ. Chimeric antigen receptors for T cell immunotherapy: current understanding and future directions. J Gene Med. 2012;14(6):405–415. | ||
Morgan RA, Yang JC, Kitano M, et al. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18(4):843–851. | ||
Gross G, Eshhar Z. Therapeutic potential of T cell chimeric antigen receptors (CARs) in cancer treatment: counteracting off-tumor toxicities for safe CAR T cell therapy. Annu Rev Pharmacol Toxicol. 2016;56:59–83. | ||
Caruso HG, Hurton LV, Najjar A, et al. Tuning sensitivity of CAR to EGFR density limits recognition of normal tissue while maintaining potent antitumor activity. Cancer Res. 2015;75(17):3505–3518. | ||
Park S, Shevlin E, Vedvyas Y, et al. Micromolar affinity CAR T cells to ICAM-1 achieves rapid tumor elimination while avoiding systemic toxicity. Sci Rep. 2017;7(1):14366. | ||
Gargett T, Brown MP. The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front Pharmacol. 2014;5:235. | ||
Wu CY, Roybal KT, Puchner EM, Onuffer J, Lim WA. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science. 2015;350(6258):aab4077. | ||
Richman SA, Nunez-Cruz S, Moghimi B, et al. High-affinity GD2-specific CAR T cells induce fatal encephalitis in a preclinical neuroblastoma model. Cancer Immunol Res. 2018;6(1):36–46. | ||
Dustin ML. Modular design of immunological synapses and kinapses. Cold Spring Harb Perspect Biol. 2009;1(1):a002873. | ||
Scholer A, Hugues S, Boissonnas A, Fetler L, Amigorena S. Intercellular adhesion molecule-1-dependent stable interactions between T cells and dendritic cells determine CD8+ T cell memory. Immunity. 2008;28(2):258–270. | ||
Allen PM, Matsueda GR, Evans RJ, et al. Identification of the T-cell and Ia contact residues of a T-cell antigenic epitope. Nature. 1987;327(6124):713–715. | ||
Finetti F, Onnis A, Baldari CT. Regulation of vesicular traffic at the T cell immune synapse: lessons from the primary cilium. Traffic. 2015;16(3):241–249. | ||
Henrickson SE, Mempel TR, Mazo IB, et al. T cell sensing of antigen dose governs interactive behavior with dendritic cells and sets a threshold for T cell activation. Nat Immunol. 2008;9(3):282–291. | ||
Davenport AJ, Jenkins MR, Cross RS, et al. CAR-T cells inflict sequential killing of multiple tumor target cells. Cancer Immunol Res. 2015;3(5):483–494. | ||
Walseng E, Köksal H, Sektioglu IM, et al. A TCR-based chimeric antigen receptor. Sci Rep. 2017;7(1):10713. | ||
Davenport AJ, Cross RS, Watson KA, et al. Chimeric antigen receptor T cells form nonclassical and potent immune synapses driving rapid cytotoxicity. Proc Natl Acad Sci U S A. 2018;115(9):E2068–E2076. | ||
Flynn JK, Gorry PR. Stem memory T cells (TSCM)-their role in cancer and HIV immunotherapies. Clin Transl Immunology. 2014;3(7):e20. | ||
Sommermeyer D, Hudecek M, Kosasih PL, et al. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia. 2016;30(2):492–500. | ||
Klebanoff CA, Scott CD, Leonardi AJ, et al. Memory T cell-driven differentiation of naive cells impairs adoptive immunotherapy. J Clin Invest. 2016;126(1):318–334. | ||
Turtle CJ, Hanafi LA, Berger C, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. 2016;126(6):2123–2138. | ||
Schmueck-Henneresse M, Omer B, Shum T, et al. Comprehensive approach for identifying the T cell subset origin of CD3 and CD28 antibody-activated chimeric antigen receptor-modified T cells. J Immunol. 2017;199(1):348–362. | ||
Kaech SM, Wherry EJ, Ahmed R. Effector and memory T-cell differentiation: implications for vaccine development. Nat Rev Immunol. 2002;2(4):251–262. | ||
Almeida L, Lochner M, Berod L, Sparwasser T. Metabolic pathways in T cell activation and lineage differentiation. Semin Immunol. 2016;28(5):514–524. | ||
Warburg O. On respiratory impairment in cancer cells. Science. 1956;124(3215):269–270. | ||
Parry RV, Chemnitz JM, Frauwirth KA, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. 2005;25(21):9543–9553. | ||
van der Stegen SJ, Hamieh M, Sadelain M. The pharmacology of second-generation chimeric antigen receptors. Nat Rev Drug Discov. 2015;14(7):499–509. | ||
Cascone T, Mckenzie JA, Mbofung RM, et al. Increased tumor glycolysis characterizes immune resistance to adoptive T cell therapy. Cell Metab. 2018;27(5):977–987. | ||
Zhang Q, Wang H, Li H, et al. Chimeric antigen receptor-modified T Cells inhibit the growth and metastases of established tissue factor-positive tumors in NOG mice. Oncotarget. 2017;8(6):9488–9499. | ||
Alizadeh D, Trad M, Hanke NT, et al. Doxorubicin eliminates myeloid-derived suppressor cells and enhances the efficacy of adoptive T-cell transfer in breast cancer. Cancer Res. 2014;74(1):104–118. | ||
Siegler EL, Kim YJ, Chen X, et al. Combination cancer therapy using chimeric antigen receptor-engineered natural killer cells as drug carriers. Mol Ther. 2017;25(12):2607–2619. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.