Back to Journals » Clinical Epidemiology » Volume 10
Critical concepts in adaptive clinical trials
Authors Park JJH, Thorlund K ![]() , Mills EJ
, Mills EJ
Received 10 November 2017
Accepted for publication 27 January 2018
Published 23 March 2018 Volume 2018:10 Pages 343—351
DOI https://doi.org/10.2147/CLEP.S156708
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Vera Ehrenstein
Jay JH Park,1 Kristian Thorlund,2,3 Edward J Mills2,3
1Department of Medicine, University of British Columbia, Vancouver, BC, Canada; 2Department of Health Research Methods, Evidence, and Impact (HEI), McMaster University, Hamilton, ON, Canada; 3The Bill and Melinda Gates Foundation, Seattle, WA, USA
Abstract: Adaptive clinical trials are an innovative trial design aimed at reducing resources, decreasing time to completion and number of patients exposed to inferior interventions, and improving the likelihood of detecting treatment effects. The last decade has seen an increasing use of adaptive designs, particularly in drug development. They frequently differ importantly from conventional clinical trials as they allow modifications to key trial design components during the trial, as data is being collected, using preplanned decision rules. Adaptive designs have increased likelihood of complexity and also potential bias, so it is important to understand the common types of adaptive designs. Many clinicians and investigators may be unfamiliar with the design considerations for adaptive designs. Given their complexities, adaptive trials require an understanding of design features and sources of bias. Herein, we introduce some common adaptive design elements and biases and specifically address response adaptive randomization, sample size reassessment, Bayesian methods for adaptive trials, seamless trials, and adaptive enrichment using real examples.
Keywords: adaptive designs, response adaptive randomization, sample size reassessment, Bayesian adaptive trials, seamless trials, adaptive enrichment
Introduction
Clinical trials are needed to determine the effectiveness (and occasionally harms) of therapeutic agents. A new approach to clinical trials, termed adaptive clinical trials, allows for modifying trial design features during the trial. The decision to make a modification is based on the data collected throughout the trial. This is considerably different from traditional trial designs as modifications to the trial design are not generally allowed during the course of enrollment and follow-up of patients, and where the learning about efficacy and safety occurs after the trial is completed. Adaptive designs aim to reduce resources, decrease time to completion, decrease the number of participants required, and/or improve the likelihood of detecting treatment effects in a clinical trial. A recent review of trials registered on ClinicalTrials.gov showed a large increase in the number of adaptive design trials completed since 2006: there were only 10 registered adaptive trials before 2006, but 133 registered from 2006 to 2013 with most trials undertaken in the field of oncology.1 The use of adaptive trials in oncology is primarily for pharmaceutical drug development.2 Reviews of adaptive trials suggest that their use has increased drastically in the past decade, particularly in pharmaceutical research.1,2
The increasing use of adaptive designs in clinical trials should be noted as adaptive designs and traditional trial designs have important differences. Just as there are differences in the planning and conduct of these trials, so too are the reasons and interpretation of bias. Regulatory agencies such as the United States Food and Drug Administration3 and European Medicines Agency4 have recognized the validity of adaptive trial designs and provided guidance on how investigators should consider their regulatory considerations during the presubmission (i.e., planning) and trial stages. However, the understanding and appreciation of adaptive clinical trial features among clinicians and researchers are typically limited.5 To help improve understanding of adaptive designs, herein, we review common adaptive features. We review methods for response adaptive randomization (RAR), sample size reassessment (SSR), Bayesian adaptive methods, seamless design, and adaptive enrichment. We provide real-life examples of clinical trials that have been designed with these adaptive designs.
Common adaptive designs
In this section, we discuss common adaptive designs. To support the comprehension of these common designs, we provide illustrative figures here. Figure 1 demonstrates an RAR design; Figure 2 demonstrates an SSR design and Figure 3 shows a seamless trial design. Figure 4 illustrates an adaptive enrichment trial design with an SSR component. Table 1 summarizes common adaptive designs outlined in the next sections.
 | Figure 1 Response adaptive randomization. Notes: The first interim analysis shows serious toxicity for the high-dose arm and promising results for the medium dose. The RAR design allows the allocation ratio to be changed to zero for the high-dose arm after the first interim analysis, so that patients will no longer be enrolled to this treatment. The allocation ratio for the medium dose, on the other hand, can be increased allowing more patients to be enrolled to this arm. Then, the trial stops after the medium dose demonstrates superiority over the low-dose arm. This example shows how an RAR design can potentially allow for a larger number of patients to be allocated to the superior treatment. Abbreviation: RAR, response adaptive randomization. |
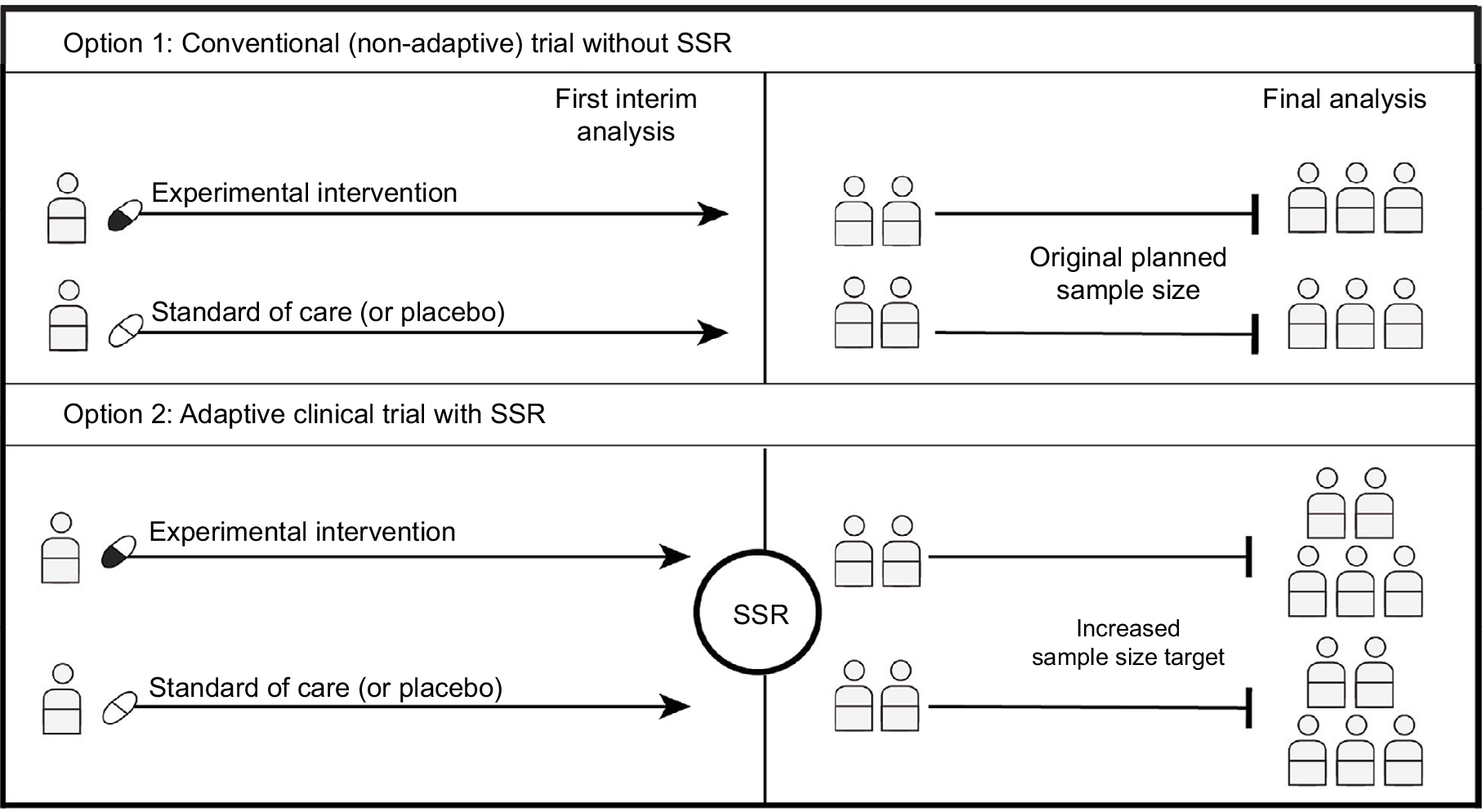 | Figure 2 Sample size reassessment. Notes: If the first interim analysis shows worse results than expected, an SSR can be performed using the interim results. SSR is not permitted in a traditional nonadaptive trial, so even when the original planned sample size is reached, the trial may be underpowered (Option 1). If SSR is permitted, the enrolment target could be increased to ensure that the trial is adequately powered (Option 2). Abbreviation: SSR, sample size reassessment. |
 | Figure 3 Seamless trials. Notes: After first interim analysis, the high-dose arm showing serious toxicity could be discontinued from the trial. Thereafter, the trial transitions seamlessly from the feasibility into the pivotal phase with standard therapy arm being introduced into the trial. |
 | Figure 4 Trial with adaptive enrichment design with SSR. Notes: In this example, the first interim analysis shows the experimental intervention has more promising results on one subgroup of patients (illustrated in gray), but it is not shown to be effective for other patients. The study eligibility criteria could then be modified to investigate the efficacy of the intervention in the gray subgroup (i.e., an adaptive enrichment design) with an SSR ensuring a sufficient sample size for this subgroup. Abbreviation: SSR, sample size reassessment. |
 | Table 1 Summary of common adaptive design features Abbreviation: ASTIN, acute stroke therapy by inhibition of neutrophils |
Response adaptive randomization
Adaptive designs (Figure 1) with RAR allow the treatment allocation ratio to be adapted based on interim analyses over the course of the trial. In RAR, the allocation ratio adapts to favor the treatment arm with more favorable interim results. RAR designs can reduce the overall number of deleterious clinical outcomes observed in a trial and may reduce the overall sample size, without meaningful loss of statistical precision. Probably, the most well-known case of RAR is the “play-the-winner” design.6 In this simple RAR design, one starts with two urns, representing each treatment, containing one ball each to represent an initial 1:1 allocation ratio. Every time a success is observed with one treatment a ball is added to the corresponding urn changing the allocation ratio for the next patient to be enrolled. This RAR design has a high risk of extreme allocation to a treatment with more promising initial results that may occur due to chance. RAR trials generally use more sophisticated rules and apply computational algorithms (e.g., Bayesian predictive probability) to ensure that the allocation ratio does not become extreme and converges at a more stable rate.7 In adaptive trials with RAR design, allocation to treatment arm(s) is often reduced based on poor effectiveness or safety at early looks at the data. This affords both ethical benefits as well as additional resources to explore effective intervention arms. Rigorous planning is required to ensure that an arm is not erroneously dropped early. Statistical methods for constraining such error risks to acceptable levels have been developed for adaptive and conventional clinical trials; these methods are discussed in more detail by Bauer et al.8
An example of RAR trial is the ASTIN (Acute Stroke Therapy by Inhibition of Neutrophils) trial.9 In this dose–response placebo-controlled trial, RAR was used to determine the optimal dose of recombinant neutrophil inhibitor factor (rNIF) from 15 doses ranging from 10 to 120 mg for acute ischemic stroke patients.9 The primary outcome was the change in Scandinavian Stroke Scale (ΔSSS), a score representing recovery in acute stroke patients, from baseline to day 90.9 Continual reassessment method was used as the dose–response model and to update the response allocation. Burn-in for response allocation was 200 patients with fixed 15% allocated to placebo and equal allocation to all of rNIF doses. Bayesian posterior estimates of the dose–response curve were used to obtain the effective dose 95% (ED95) and its variance, and after burn-in period, treatment allocation was determined to the dose that minimized the variance of ED95. In order to allow the response allocation to be updated before a patient’s day 90 scores became available, longitudinal modeling was used to estimate the day 90 SSS score based on the patient’s interim scores.9 The protocol prespecified the maximum sample size target of 1,300 patients with minimum 15% of patients being allocated to placebo.9 This trial enrolled a total of 966 patients (26% in the placebo); >40% (the majority) of patients receiving rNIF were allocated to the top three doses (96, 108, and 120 mg) of rNIF.10 Although rNIF was shown to be well tolerated in these patients, this trial was stopped early for futility, as rNIF was shown not to improve recovery from acute ischemic stroke.9
Sample size reassessment
Although the use of adaptive designs has become more popularized recently, the development of confirmatory adaptive design methodology has been popularized over the last three decades.8 SSR (i.e., change in the originally estimated sample size [Figure 2]) has been around for several decades and was a major focus in the early waves of adaptive design methodology development.8 The motivating factor behind SSR development is that a clinical trial may prove to be underpowered, even when the originally planned sample size is reached, due to common incorrect sample size estimation in the planning phase.8 When a trial fails to meet its prespecified endpoints under such a circumstance, simply enrolling more patients is not a valid option since the risk of a false-positive finding will no longer be under control, but SSR can be preplanned to control for the risk of a false positive.11 SSR is not intended to salvage a failed trial, but rather, it minimizes the risk that a trial is under- or overpowered in the first place. SSR may also be used to determine whether to continue or terminate the trial; for instance, if an SSR shows the new sample size required to be too large, the investigators may choose to end the trial early, or switch the objective of the trial from superiority to noninferiority. The required sample size may be adjusted in one or more interim looks at the data depending on the prespecified plan.11 Scenario analyses using analytical approaches or simulations are recommended to inform the preplanning for intended and allowed adjustments.
A motivating example of a recently conducted trial with SSR design includes the trial by Casali et al.12 This trial is a two-arm design that assessed the efficacy and safety of imatinib as an adjuvant therapy for localized gastrointestinal stromal tumors and had originally intended to recruit 400 patients over the 5-year period starting December 2004.12 The decision to recruit more patients (908 patients) was made 3 years later after an interim analysis using a significance level of 0.015 showed a survival rate that was higher than expected in the control group.12 With the SSR, this trial showed an improvement of adjuvant imatinib therapy for relapse-free survival that the originally planned sample size was not adequately powered enough to do.12
Bayesian adaptive approaches
Conventional statistics used in clinical trials, such as p-values and confidence intervals, come from the frequentist paradigm of statistics, which is rooted in the philosophy that statistical inferences should be drawn from the probability of observing similar data in a series of subsequent hypothetical examples of the study being conducted. This paradigm is confusing not only to most clinicians, but also to many statisticians. Bayesian statistics is another paradigm of statistics rooted in the philosophy that our future beliefs (e.g., about a treatment effect) are a product of our current belief and newly gathered evidence. In adaptive trials where no clinical trial is ever a replication of itself and adaptations to the design are dependent specifically on our prior beliefs and all recent evidence collected, the Bayesian framework is ideal.
In Bayesian statistics, the prior belief (typically based on limited prior data) is translated into a probability distribution (the “prior distribution”), which is continually updated as data are accumulated (the “posterior distribution”). In the context of adaptive trials, this affords the flexibility to inspect interim results at any time and assess whether adaptations should be made.13 In particular, the updated probability distribution (i.e., the “posterior distribution”) is used to gather probabilities of different outcomes of interest occurring if the trial is continued with the same design. For example, a very low (Bayesian) probability of a treatment being superior can result in a treatment arm being dropped, or at least, inform a change in the allocation ratio to minimize the number of patients exposed to an inferior treatment. Bayesian statistics also have the advantage that the prior information can be varied to reflect anything on the spectrum from optimism to pessimism and from high to low uncertainty. Thus, when clinical trial investigators’ opinions about the likelihood of trial success differ during the trial planning stage, Bayesian statistics provides a way of assessing the degree to which the spectrum of postulated success probabilities impact the actual probability of success upon trial completion. A range of such scenario analyses varying, for example, the target sample size (i.e., Bayesian SSR) can then provide a balanced overview of where the optimal (additional) sample size is likely to be reached. Likewise, scenario analyses’ varying beliefs about the treatment effects can yield a balanced overview of where response adaptive allocation adaptations or treatment terminations for superiority, inferiority, or equivalence should be applied. Such a sensitivity analysis is in accordance with good Bayesian practice, and thus, the quality of any adaptive clinical trials incorporating Bayesian methodology should be appraised by the adequacy of its “prior” sensitivity analyses.
Seamless trials
Traditionally, an intervention tested in humans is evaluated in three conceptual phases: Phase I tests tolerability of the intervention in healthy individuals, Phase II primarily tests tolerable dose ranges in individuals with the health condition of interest, and Phase III primarily tests the efficacy and safety of the intervention in individuals with the health condition of interest. In addition, some investigations may continue on to Phase IV trials where patients are tested in a setting corresponding more to a real-world clinical setting compared with the often much controlled setting of the Phase III trial. With conventional randomized clinical trials (RCTs), the knowledge from each phase can be used only once the phase is finished, and the evaluation of an intervention pauses between the phases of the clinical trials. Seamless studies are a type of design that allows for immediate continuation from one phase to the subsequent phase. Because Phase II and III are similar in their clinical settings, seamless designs are most commonly used to seamlessly combine Phase II and III trials.14 A seamless Phase II/III RCT can be feasible even with significant changes to the design across phases, but does require overlap in some essential components. For example, if a Phase II trial examines the tolerability of five doses and three of these are terminated at the end of the trial, a Phase III trial can seamlessly continue with enrollment of the two remaining doses (treatment arms) and likely also add additional arms such as a placebo group. Seamless trials require that early data can be rapidly analyzed and interpreted to inform decisions about seamless transition into the subsequent phase.14 Seamless designs, therefore, have the capacity to significantly reduce time from first Phase II randomized patients to reaching a definitive conclusion about efficacy and safety by the completion of the Phase III trial.14
For a motivating example of an adaptive seamless design trial, we refer to the seamless II/III trial on 9-valent human papillomavirus (HPV) vaccine by Joura et al.15 The overall goal of this trial was to compare the efficacy and safety of 9-valent HPV vaccine against quadrivalent vaccine, the gold standard for HPV vaccine.15 This seamless design started as a Phase II trial with the goal of identifying an optimal formulation of 9-valent HPV vaccine between three different formulations with varying doses (low, medium, and high) based on their immunogenicity (the ability of the vaccine to provoke an immune response and produce HPV-neutralizing antibiotics).15 The interim analysis of 1,240 patients based on immunological endpoints resulted in selecting the medium dose vaccine, which was thereafter seamlessly tested in Phase III in this adaptive trial. Data from 310 patients who received the medium dose in the first phase were concatenated with the data from the subsequent 13,578 patients enrolled in the next phase (1:1 ratio 9-valent HPV vaccine vs quadrivalent vaccine).15 Collectively, these were used to test the noninferiority of 9-valent HPV vaccine vs quadrivalent vaccine for both efficacy (e.g., prevention of infections and disease) and safety (e.g., adverse events to vaccine).15
Continuing seamlessly from the feasibility to the confirmatory phase saved time and resources to achieve the overall goal of testing for efficacy and safety of 9-valent HPV vaccine to the quadrivalent vaccine. In this study, patients who received the selected vaccine dose formulation of 9-valent HPV vaccine (i.e., medium dose) in the early phase could continue into the subsequent phase and followed up for efficacy and safety, reducing the overall sample size required to conduct these trials separately.16 Time and resources were saved, as the trial infrastructure for patient recruitment and follow-up could be tested and established in the feasibility phase.
Adaptive enrichment
Adaptive enrichment refers to a modification of the trial eligibility criteria.17 If an interim analysis shows that prespecified patient subgroups have different responses, the eligibility criteria can be modified to include only the favorable group moving forward.17 Here, an SSR could also be performed separately to modify the sample size requirement for each subgroup. Vice versa, if new external evidence surfaces during the course of the trial that a broader population than that defined by the trial’s eligibility criteria might benefit, the trial may also be enriched by broadening the eligibility criteria.
There have been different proposals of adaptive enrichment design trials.18 With the emergence of the precision medicine era and its emphasis on biomarkers, there have been increasing calls for examination of biomarker subgroup effects. The I-SPY2 and BATTLE adaptive trials are two examples where enrichment was an integral part of the design. I-SPY 2 is a recently completed (large) Phase II trial that evaluated 12 neoadjuvant therapies’ efficacies against 10 biomarker signatures.19,20 The treatment(s) with a high predictive probability of being effective in patients with their corresponding biomarker signature continued onto the subsequent Phase III trial. The BATTLE trial is another biomarker-driven enrichment adaptive trial.21 At the first stage, BATTLE had a goal of identifying predictive biomarkers that could be used to randomize advanced non-small cell lung cancer (NSCLC) patients into one of four possible treatment arms. On the basis of the first stage, BATTLE-2 then ensued with an emphasis of evaluating targeting therapies for NSCLC with KRAS mutations.
A review by Rosenblum and Hanley22 provides a good motivating example where an adaptive enrichment design can be an important consideration for design of Phase III trial using results from a completed Phase II clinical trial for stroke interventions (i.e., MISTIE trial).23,24 In the MISTIE trial, there were two subgroups of interest: small and large intraventricular hemorrhage (IVH) patients (small IVH patients were defined with IVH level ≤10 mL, and large IVH patients were defined with IVH level >10 mL at baseline).24 The results of the MISTIE trial showed stronger evidence that small IVH patients benefit from treatment, whereas the treatment effect was not clear for large IVH patients. As one of the motivating factors for an adaptive enrichment design is to enroll subgroups of patients with greater potential of benefits, Rosenblum and Hanley22 performed clinical trial simulations using the results from the MISTIE trial and compared the sample size required to achieve the desired power and type I error for a Phase III trial that is designed with a standard design versus with an adaptive enrichment design. In the standard design, interim analyses were allowed where the entire trial (i.e., for both subgroups) could be stopped early, and in the investigated adaptive enrichment design, interim analyses could be used only to enroll subgroup of patients with promising results (e.g., small IVH patients).22 The estimated number of patients for the subgroup with favorable Phase II results was larger in the adaptive enrichment design when compared with the standard design (598 versus 491), but the overall population required was shown to be less for the adaptive enrichment design (1,148 versus 1,473) with fewerpatients being recruited for the subgroup with less promising treatment effects (550 versus 982).22
Advantages and limitations of adaptive designs
Adaptive designs can be more cost and time efficient and can enhance patient protection. Multiple interim analyses are allowed in adaptive designs, so if a trial demonstrates unanticipated results, it can be stopped early. Interim analyses can also be used to adjust the allocation probability such that fewer patients are exposed to an experiment treatment(s) with inferior efficacy or safety. If an inferior efficacy or smaller treatment effect is observed, terminating the entire trial will save resources, or dropping the inferior treatment arm(s) will act to enhance patient protection as well. Adaptive designs can improve the chance of conducting a successful trial because SSRs, as they are preplanned, can be performed without a penalty. If an SSR shows a smaller treatment effect than the magnitude that was anticipated, without a modification, the trial can be underpowered under the originally planned sample size. In such a case, if the new required sample size is too large, the trial can be terminated. If the new required sample size is reasonable, the trial can be continued with the larger recruitment target.
Adaptive designs are not without limitations and biases. An adaptive trial requires thorough planning and transparent reporting. Preplanning of modifications and statistical analyses requires extensive efforts that may be complicated and difficult to plan or execute. The overall results can be more difficult to interpret. Incorrectly planned and/or executed adaptive designs can introduce operational bias and statistical bias that may be difficult for a reader to determine. A trial modification can be confounding and make it more difficult to interpret the trial results. Particularly, in trials with SSR and seamless designs, the sample size and the trial duration could also be greater due to the modification(s). For adaptive enrichment trials, generalizability is a potential issue; in trials that end up modifying the eligibility criteria to a particular subgroup, there is generalizability only to the subgroup population, not to the larger disease population. As well, it can be difficult to recruit the subgroup with promising results, so these adaptive designs may be more logistically complicated to conduct. If there is a long delay in time before outcomes of interest can be measured, it is important to consider that there could be insufficient information available at an interim analysis to make an appropriate modification to the eligibility criteria, patient recruitment, and allocation ratio.
Adaptive designs can also introduce investigator-driven bias that is difficult to detect as changes may be post-hoc decided based on results, rather than preplanned based on arising circumstances. The systematic review by Mistry et al2 on adaptive design methods in oncology trials found that 91% (49/54 papers) stated their adaptive methods to be predetermined; however, it is probable that this review missed a large proportion of existing adaptive designs in their trials, as only 4% (2/54 papers) explicitly stated their trials as being “adaptive”. It is possible that there may be a higher number of existing adaptive trials that may have made post-hoc adaptations to their study design than what was reported in the paper by Mistry et al.2 Therefore, for an adaptive trial, it is critical to have an independent data monitoring committee that can adhere to the protocol, make per-protocol adaptations during the trial, and avoid post-hoc adaptations that may compromise the scientific validity of the trial. Moreover, while there are potential ethical advantages of adaptive trial designs (e.g., RAR that may expose fewer participants to ineffective treatments), it is important to acknowledge the potential ethical disadvantages such as potential for less transparency and potential concerns about unbinding if without appropriate firewalls.25
Conclusion
Adaptive designs are an increasingly common approach to evaluate interventions and offer many advantages over better-known traditional designs. Their utility is limited, however, if clinicians or other readers are unfamiliar with underlying concepts. Herein, we have reviewed common critical concepts at an introductory level to assist clinicians and readers.
Acknowledgment
This work was supported by the Bill and Melinda Gates Foundation. The article contents are the sole responsibility of the authors and may not necessarily represent the official views of the Bill and Melinda Gates Foundation or other agencies that may have supported the primary data studies used in the present work.
Author contributions
All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Hatfield I, Allison A, Flight L, Julious SA, Dimairo M. Adaptive designs undertaken in clinical research: a review of registered clinical trials. Trials. 2016;17(1):150. | ||
Mistry P, Dunn JA, Marshall A. A literature review of applied adaptive design methodology within the field of oncology in randomised controlled trials and a proposed extension to the CONSORT guidelines. BMC Med Res Methodol. 2017;17(1):108. | ||
United States Food and Drug Administration. Adaptive Designs for Medical Device Clinical Studies Guidance for Industry and Food and Drug Administration Staff. United States Department of Health and Human Services; 2016. | ||
European Medicines Agency Committee for Medicinal Products for Human Use (CHMP). Reflection paper on methodological issues in confirmatory clinical trials with flexible design and analysis plan. European Medicines Agency; 2006. | ||
Meurer WJ, Legocki L, Mawocha S, et al. Attitudes and opinions regarding confirmatory adaptive clinical trials: a mixed methods analysis from the Adaptive Designs Accelerating Promising Trials into Treatments (ADAPT-IT) project. Trials. 2016;17:373. | ||
Bartlett RH, Roloff DW, Cornell RG, Andrews AF, Dillon PW, Zwischenberger JB. Extracorporeal circulation in neonatal respiratory failure: a prospective randomized study. Pediatrics. 1985;76(4):479–487. | ||
Saville BR, Connor JT, Ayers GD, Alvarez J. The utility of Bayesian predictive probabilities for interim monitoring of clinical trials. Clin Trials. 2014;11(4):485–493. | ||
Bauer P, Bretz F, Dragalin V, Konig F, Wassmer G. Twenty-five years of confirmatory adaptive designs: opportunities and pitfalls. Stat Med. 2016;35(3):325–347. | ||
Krams M, Lees KR, Hacke W, Grieve AP, Orgogozo JM, Ford GA; ASTIN Study Investigators. Acute Stroke Therapy by Inhibition of Neutrophils (ASTIN): an adaptive dose-response study of UK-279,276 in acute ischemic stroke. Stroke. 2003;34(11):2543–2548. | ||
Grieve AP, Krams M. ASTIN: a Bayesian adaptive dose-response trial in acute stroke. Clin Trials. 2005;2(4):340–351. | ||
Bauer P, Koenig F. The reassessment of trial perspectives from interim data-a critical view. Stat Med. 2006;25(1):23–36. | ||
Casali PG, Le Cesne A, Poveda Velasco A, et al. Time to definitive failure to the first tyrosine kinase inhibitor in localized GI stromal tumors treated with imatinib as an adjuvant: A European Organisation for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group intergroup randomized trial in collaboration with the Australasian Gastro-Intestinal Trials Group, UNICANCER, French Sarcoma Group, Italian Sarcoma Group, and Spanish Group for Research on Sarcomas. J Clin Oncol. 2015;33(36):4276–4283. | ||
Bolstad WM, Curran JM. Introduction to Bayesian Statistics. John Wiley & Sons, Inc., Hoboken, New Jersey; 2016. | ||
Cuffe RL, Lawrence D, Stone A, Vandemeulebroecke M. When is a seamless study desirable? Case studies from different pharmaceutical sponsors. Pharm Stat. 2014;13(4):229–237. | ||
Joura EA, Giuliano AR, Iversen OE, et al; Broad Spectrum HPV Vaccine Study. A 9-valent HPV vaccine against infection and intraepithelial neoplasia in women. N Engl J Med. 2015;372(8):711–723. | ||
Chen YH, Gesser R, Luxembourg A. A seamless phase IIB/III adaptive outcome trial: design rationale and implementation challenges. Clin Trials. 2015;12(1):84–90. | ||
Simon N, Simon R. Adaptive enrichment designs for clinical trials. Biostatistics. 2013;14(4):613–625. | ||
Antoniou M, Jorgensen AL, Kolamunnage-Dona R. Biomarker-guided adaptive trial designs in Phase II and Phase III: a methodological review. PLoS One. 2016;11(2):e0149803. | ||
Park JW, Liu MC, Yee D, et al. Adaptive randomization of neratinib in early breast cancer. N Engl J Med. 2016;375(1):11–22. | ||
Rugo HS, Olopade OI, DeMichele A, et al. Adaptive randomization of veliparib-carboplatin treatment in breast cancer. N Engl J Med. 2016;375(1):23–34. | ||
Kim ES, Herbst RS, Wistuba II, et al. The BATTLE trial: personalizing therapy for lung cancer. Cancer Discov. 2011;1(1):44–53. | ||
Rosenblum M, Hanley DF. Adaptive enrichment designs for stroke clinical trials. Stroke. 2017;48(7):2021–2025. | ||
Morgan T, Zuccarello M, Narayan R, Keyl P, Lane K, Hanley D. Preliminary findings of the minimally-invasive surgery plus rtPA for intracerebral hemorrhage evacuation (MISTIE) clinical trial. Acta Neurochir Suppl. 2008;105:147–151. | ||
Hanley DF, Thompson RE, Muschelli J, et al. Safety and efficacy of minimally invasive surgery plus alteplase in intracerebral haemorrhage evacuation (MISTIE): a randomised, controlled, open-label, phase 2 trial. Lancet Neurol. 2016;15(12):1228–1237. | ||
Legocki LJ, Meurer WJ, Frederiksen S, et al. Clinical trialist perspectives on the ethics of adaptive clinical trials: a mixed-methods analysis. BMC Med Ethics. 2015;16:27. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.