Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 21
Critical Biological Functions and Clinical Implications of Epigenetic-Related Candidate Biomarkers in Chronic Obstructive Pulmonary Disease: Integrated Machine Learning Screening and Basic Experimental Validation
Authors Xie J, Huang L, Chen X, Wang X
Received 27 December 2025
Accepted for publication 29 April 2026
Published 7 June 2026 Volume 2026:21 592045
DOI https://doi.org/10.2147/COPD.S592045
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Zijing Zhou
Jianpeng Xie,1,* Linhui Huang,2,* Xin Chen,3 Xilong Wang3
1Huiyu Mingdu Community Health Service Station, Shenzhen Baoan Shiyan People’s Hospital, Shiyan, Baoan District, Shenzhen, 518108, People’s Republic of China; 2Department of Pulmonary and Critical Care Medicine, Hainan General Hospital (Hainan Affiliated Hospital of Hainan Medical University), Haikou, Hainan, People’s Republic of China; 3Department of Pulmonary and Critical Care Medicine, Zhujiang Hospital, Southern Medical University, Guangzhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xilong Wang, Email [email protected] Xin Chen, Email [email protected]
Background: Chronic obstructive pulmonary disease (COPD) is the primary cause of deaths related to respiratory diseases. Epigenetic modifications are crucial in the development of mammals, and any disruption to epigenetic regulation may result in disease.
Methods: We performed differential expression analysis on the GSE19407, GSE11784 and GSE20257 datasets from the Gene Expression Omnibus (GEO) dataset and obtained differentially expressed epigenetic-related genes (DE-ERGs) in COPD. Three machine learning techniques were used to screen the candidate epigenetic-related biomarkers in DE-ERGs, thereby further enhancing the robustness of the analysis framework. Immune infiltration analysis was performed on biomarkers.
Results: A total of 5 biomarkers (HMGN4, CIT, TLE1, TFPT, and UBE2T) were screened utilizing three machine learning algorithms. Immune infiltration analysis showed that the HMGN4 was positively correlated with activated CD4+ T cells and memory B cells and negatively correlated with CD56dim. In quantitative reverse transcription polymerase chain reaction (qRT-PCR) validation, the expression levels of 5 biomarkers were notably higher in COPD than in normal samples.
Conclusion: In summary, we identified 5 epigenetic-related candidate biomarkers that might be involved in COPD progression by bioinformatics techniques, which still require further experimental validation.
Keywords: chronic obstructive pulmonary disease, machine learning, biomarkers, immunoinfiltration
Introduction
Chronic obstructive pulmonary disease (COPD) is a complex condition marked by enduring and advancing restriction of airflow. It has emerged as the third most prevalent cause of mortality globally and is projected to lead to more than 5 million fatalities by 2060.1 With the development of COPD management, the symptoms and further risks of COPD patients have been significantly improved.2 However, few preventative and treatment are available to reduce disease progression and improve survival period and life quality. The existing biomarkers for COPD often lack sufficient sensitivity for early diagnosis and are unable to reflect the complex pathological mechanism of the disease.3 Hence, it is an urgent need for further exploration of novel therapeutic targets in COPD and development of biomarkers with clinical utility.
Epigenetic modifications, such as DNA and RNA methylation, play a critical role in regulating gene transcription programs and ultimately determining cell fate. Recent research on epigenetic modifications has not only shed light on the various regulatory roles of nucleic acids within living organisms but has also showcased their promising uses in clinical practice. Advanced methods, such as loci-specific, genome-wide methylation analysis and single-cell epigenomics, have been employed to detect epigenetic abnormalities in various contexts, including early diagnosis of malignancies, disease prognosis, evaluation of drug responses, and monitoring of complications.4–6 Several epigenome-wide association studies (EWASs) have identified cytosine-phosphate-guanine (CpG) probes related to pulmonary function.7,8 However, most EWASs faced limitations, such as reliance on blood samples and being conducted predominantly in populations of European ancestry. Furthermore, the underlying mechanisms of epigenetic modifications in COPD remain poorly understood due to the absence of a stable short-term model. Exposure to cigarette smoke (CS) in mouse lungs and human lung epithelial cells has been demonstrated to induce a pro-inflammatory gene expression pattern via histone acetylation.9 Emerging evidence suggests that noncoding RNAs, including miRNAs and lncRNAs, may be key regulators of epigenetic and gene transcription. When exposed to cigarette smoke extract, human bronchial epithelial cells exhibited upregulation of miR-101 and miR-144.10 However, CS-induced models may not fully reflect the in vivo functionality of epigenetic modifications in COPD, as COPD is a heterogeneous lung condition arising from gene-environment interactions.
Epigenetic-related genes (ERGs) were defined as a set of genes encoding core epigenetic regulators (DNA/histone mark readers/writers/erasers/remodellers).11 To ensure the reliability and comprehensiveness of the ERG set, we retrieved candidate genes from the EpiFactors database:12 a well-recognized, manually curated database of human epigenetic factors and their complexes, which has been extensively applied to dissect the biological functions of epigenetic modifications in various pathological processes. Various types of machine learning algorithms, such as supervised, unsupervised, semi-supervised and reinforcement learning, in addition to deep learning, which is a member of the machine learning family, can intelligently and comprehensively analyze large amounts of data.13 In this study, we used machine learning algorithms, including the least absolute shrinkage and selection operator (LASSO), Support Vector Machine Recursive Feature Elimination (SVM-RFE), and Boruta analysis to screen the relatively stable and important characteristic genes in COPD. To explore the underlying the regulatory mechanisms, we performed an immune infiltration analysis and constructed potential regulatory network based on the biomarkers. Finally, we investigated the clinical relevance of biomarkers, assessing their utility in disease prediction and as potential drug targets. In general, this study has filled a crucial gap in the field of epigenetics research on COPD. By conducting a comprehensive network-level analysis of ERGs, it addressed the absence of such systematic research and overcame the limitations of traditional statistical methods through machine learning, thereby identifying reliable biomarkers. Moreover, this study bridged the gap between the discovery of epigenetic biomarkers and their functional interpretation, laying a theoretical foundation for translating these insights into clinical diagnosis and treatment of COPD.
Materials and Methods
Data Acquisition
A total of three COPD transcriptome microarray datasets (GSE19407, GSE11784 and GSE20257) were downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/geo/). All three datasets were composed of the transcriptome of small airway epithelium in healthy non-smokers, healthy smokers and smokers with COPD, with the GSE19407 dataset consisting of 105 normal and 22 COPD samples, the GSE11784 dataset composed of 135 normal and 22 COPD samples, and the GSE20257 dataset consisting of 112 normal and 23 COPD samples. The 720 ERGs were downloaded from the EpiFactors database (http://EpiFactors.autosome.ru).
Identification of Differentially Expressed Epigenetic-Related Genes (DE-ERGs) in COPD
Differentially expressed genes (DEGs1, DEGs2, and DEGs3) in the COPD group and normal group were identified in three datasets (GSE19407, GSE11784, and GSE20257) utilizing the “limma” package (version 3.54.0). The statistical significance criterion was |log2FC| > 0.5, and P < 0.05. Volcano maps of all in 3 datasets were generated utilizing the “ggplot2” package (version 3.4.1), and the “pheatmap” package to generate heat maps. In order to obtain the common DEGs, the up-regulated genes and down-regulated genes were intersected, respectively, in the three datasets. Then DEGs and 720 ERGs were further intersected to obtain the DE-ERGs in COPD. The “ggplot2” software package was used to create Venn diagrams.
Enrichment Analysis and Protein-Protein Interaction (PPI) Network Analysis
Gene ontology (GO) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) were utilized to analyze the role of DE-ERGs in biological processes and the biological pathways. GO and KEGG enrichment analyses of DE-ERGs were conducted utilizing the R package “clusterProfiler” (version 4.2.2) and to look for common functions and related pathways between genes, respectively. The main enriched functions and pathways of DE-ERGs were screened at P < 0.05, and the GO-enriched entries and KEGG-enriched entries were visualized using the R language “enrichplot” package (version 1.18.3) and “ggnewscale” package, respectively. The PPI network of DE-ERGs was carried out utilizing the STRING database (http://string.em bl.de/) and visualized utilizing Cytoscape software, which utilized the plug-in cytoHubba to detect node genes in the PPI network (medium confidence = 0.400).
Screening for Biomarkers Using Three Machine Learning Algorithms
Based on the nodal genes screened by the PPI network, we used the “glmnet”, “e1071” and “Boruta” packages for the LASSO, SVM-RFE, and Boruta analysis, respectively. The LASSO model adopted 10-fold cross-validation with α fixed at 1 and finally selected the model corresponding to the maximum λ value (0.0067) within one standard deviation of the minimum cross-validation deviance. SVM-RFE employed the default radial basis kernel function. During the iterative feature elimination process, the importance of features was evaluated through internal cross-validation. After 5-fold external cross-validation and repeating this process 5 times to obtain a stable generalization error estimate, the feature genes corresponding to the minimum average error are selected as the final result. Boruta employed default parameters (ntree = 500, maxRuns = 100), using the expression levels of candidate genes as the feature and the sample grouping as the response variable for feature selection. It screened out the important feature genes by comparing the importance Z scores of the original features with those of the shadow features. Subsequently, the common genes identified by the three machine learning methods were selected as a biomarker. Based on the datasets GSE19407, GSE11784, and GSE20257, ROC curve analyses were carried out on the biomarkers utilizing the “pROC” software package, and the prediction efficiency of biomarkers was verified according to the area under the curve (AUC) values. The R package “ggplot2” was further used to plot the expression box plots of biomarkers in the three datasets.
Chromosomal Localization Analysis and GeneMANIA Analysis of Biomarkers
Human chromosome data (UCSC.HG38.Human.CytoBandIdeogram) were imported using the R package “Circos”, and circle maps were drawn to visualize the distribution of biomarkers on chromosomes. Using the online tools on the GeneMANIA website (https://genemania.org/), we predicted the genes associated with biomarkers function, and constructed gene-gene co-expression networks for the biomarkers.
Gene Set Enrichment Analysis (GSEA) Enrichment and Immunoinfiltration Analysis
The genome named “c2.cp.kegg.v7.4.symbols.gmt” was retrieved from the molecular characterization database, and GSEA was performed on the biomarkers to elucidate the significant functional and difference pathway of the biomarkers, using the significance criterion of P < 0.05. The “ssGSEA” package was utilized to evaluate the count of immune cell infiltrations in the GSE19407 dataset, which was then visualized through a stacked plot. Subsequently, an analysis was conducted to compare the differences in each immune cell type between the COPD and control groups (adj.P < 0.05), and expression box plots were drawn using the “ggplot2” package. Finally, Spearman analysis was utilized to observe the association between biomarkers and differentially immune cells.
Construction of Regulatory Network for COPD
We used the staBase (https://rnasysu.com/encori/) database to predict microRNAs (miRNAs) targeting biomarkers, followed by the ChEA3 database (https://maayanlab.cloud/chea3/) to predict transcription factors (TFs) that could regulate biomarkers, and Cytoscape was utilized to create a TF-miRNA-mRNA regulatory network based on the predicted TFs, miRNAs and mRNAs.
Construction of Gene-Disease Network and Gene-Drug Prediction
The DisGeNET database (https://www.disgenet.org/) collected and integrated disease-gene association information from multiple data sources, through which biomarkers-disease association networks were derived. Based on the biomarkers of COPD, the CTD database (https://ctdbase.org/) was utilized to predict potential drug components for the treatment of COPD. The Cytoscape software (version 3.8.2) was utilized for visualizing the gene-drug networks.
Expression of Biomarkers in Clinical Samples
Source of Human Samples
Human induced sputum samples were gathered from a cohort of COPD patients as well as from healthy human volunteers without known cardiopulmonary disease, following the protocol approved by the institutional review board at Zhujiang Hospital (Approval Number: 2023-KY-160-02). Consistent with the global initiative for Chronic Obstructive Lung Disease (GOLD) 2023, the study enrolled participants included individuals aged 40–60 years who had a specialist diagnosis of stable COPD. All participants or their legally authorized representatives provided written informed consent. The control group comprised individuals aged between 40 and 55 years who had no prior history of COPD and had a baseline FEV1/FVC ratio of 0.7 or higher. The characterization details of these participants were shown in Table 1. Induced sputum was conducted with nebulized hypertonic 3% saline for 20 minutes. Albuterol (180 mg via metered dose inhaler) was administered 15 minutes before sputum induction. The sputum samples were dispersed in an equal volume of 0.1% DTT and the supernatant removed after the sample was centrifuged at 1000 × g. The cell pellet was stored at −80°C for subsequent analysis.
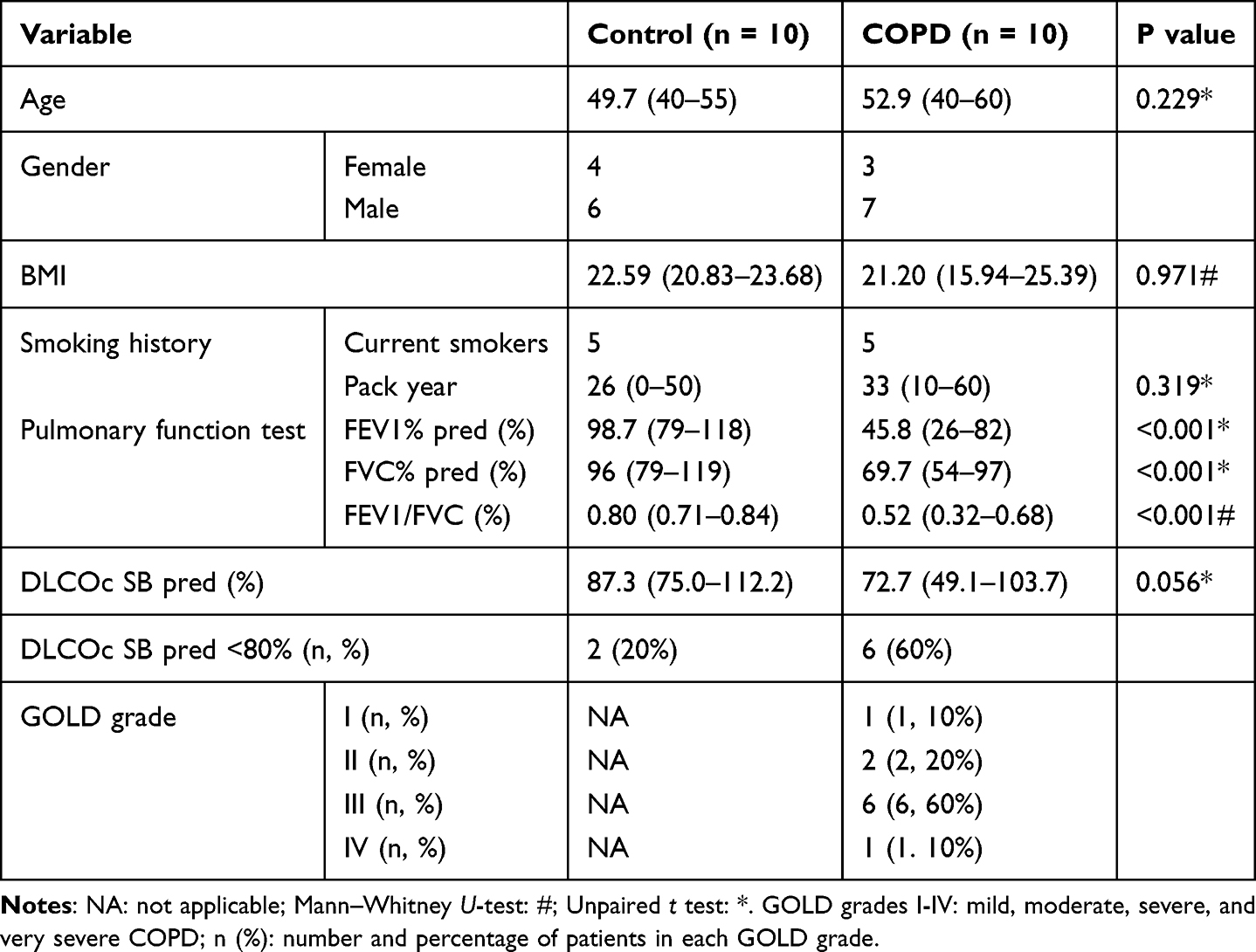 |
Table 1 Characterization Details of Participants |
Quantitative Reverse Transcriptase Polymerase Chain Reaction (qRT-PCR) Analysis
Trizol (Invitrogen, USA) was utilized to extract total RNA, cDNA was synthesized from 1 µg of RNA extracted from sputum samples using the Prime Script RT Kit (Takara, Dalian, China). Real-time PCR (RT-PCR) was carried out utilizing Bio-Rad CFX Connect detector system with SYBR Green universal PCR mix (Takara). Primers were designed and acquired from Sangon Biotech (Shanghai, China). UBE2T, HMGN4, TLE1, CIT and TFPT mRNA expression was normalized to 18S expression.
Fold changes in mRNA expression were calculated utilizing the 2−ΔΔCt method, The following primers were displayed in Table 2.
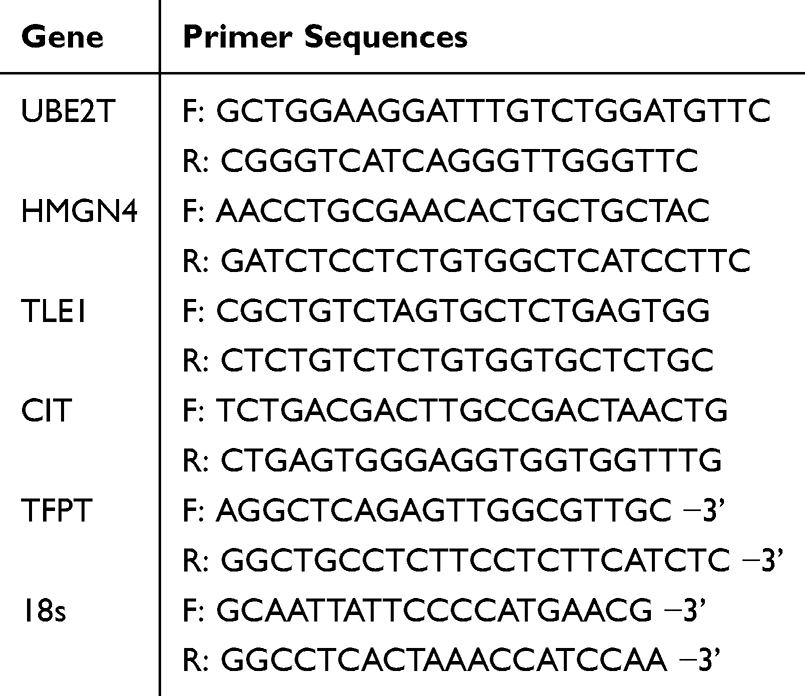 |
Table 2 The Primer Sequences of Biomarkers |
Statistical Analysis
All analyses were performed utilizing R version 4.2.1. The Wilcoxon test was utilized for significance analysis to compare the differences between the two groups of samples. The data were presented as mean ± standard deviation. For comparisons between two independent groups, normally distributed continuous data were analyzed using an unpaired Student’s t-test, whereas non-normally distributed data were assessed with the Mann–Whitney U-test. Normality was verified using the Shapiro–Wilk test (or visual inspection of Q–Q plots, as appropriate). All statistical analyses were performed using GraphPad Prism software. The statistical significance was demonstrated at a level of P < 0.05.
Results
Determination of DE-ERGs in COPD
The GSE19407, GSE11784, and GSE20257 datasets yielded 2466 DEGs1 (including 2250 up-regulated and 216 down-regulated genes), 2071 DEGs2 (including 1829 up-regulated and 242 down-regulated genes), and 2347 DEGs3 (including 2143 up-regulated and 204 down-regulated genes), respectively. The volcano maps and heat maps showed the differential expression of genes in the three datasets, respectively (Figure 1A and B). A total of 1677 DEGs in COPD were obtained by taking the intersection and then the concatenation of the up-regulated and down-regulated genes of DEGs1, DEGs2, and DEGs3, respectively (Figure 1C). Then, 1677 DEGs and 720 ERGs were taken to intersect to obtain 23 DE-ERGs in COPD (Figure 1D).
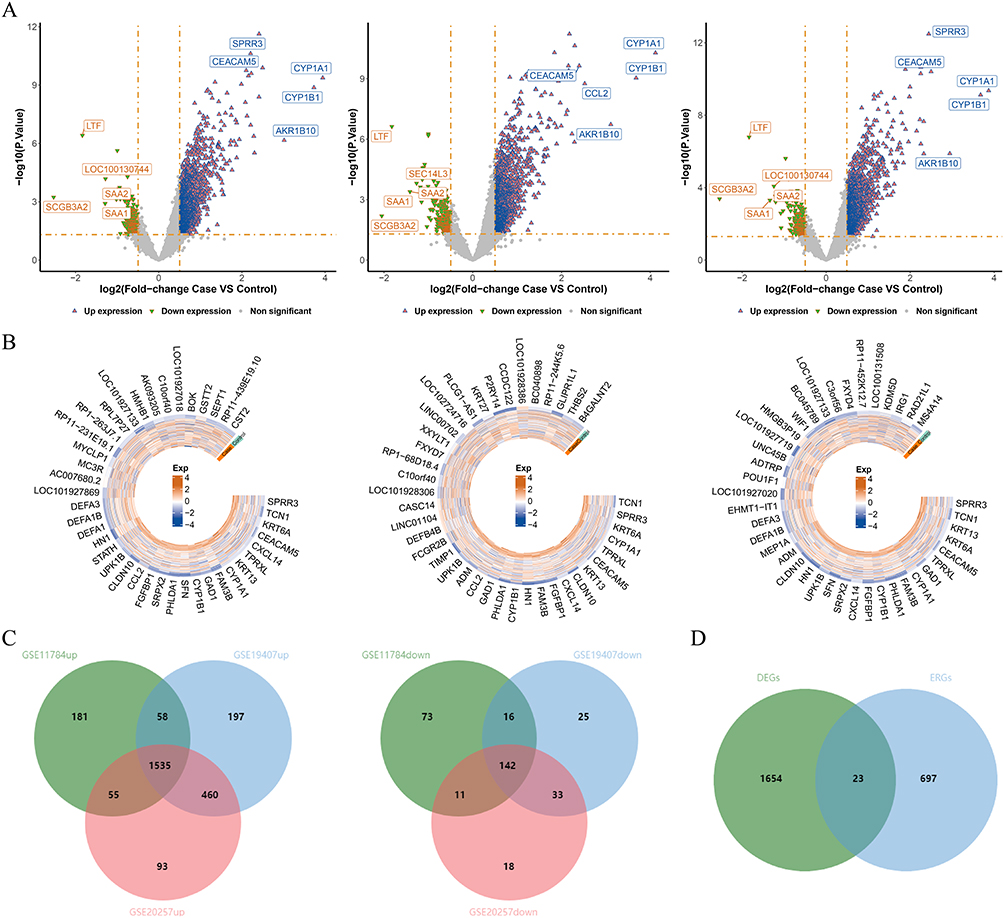 |
Figure 1 Identification of differentially expressed genes (DEGs) in the GSE19407, GSE11784, and GSE20257 datasets. (A) Volcano plot of DEGs in 3 datasets; (B) Heatmap of DEGs in 3 datasets; (C) Venn diagram of up- and down-regulated DEGs in 3 datasets. (D) The overlapping genes between DEGs and ERGs. |
Functional Enrichment and PPI Analysis of DE-ERGs
GO analysis was mainly enriched to the entries of regulation of single stranded viral RNA replication via double stranded DNA intermediate, deoxycytidine deaminase activity and viral RNA genome replication. The top 10 entries were visualized according to the p-value value from smallest to largest, and the results were shown in Figure 2A. KEGG analysis mainly enriched 4 pathways: HIV-1 viral life cycle, FOXO signaling pathway, viral carcinogenesis, and human immunodeficiency virus 1 infection (Figure 2B). The PPI network showed 13 node genes and 10 interacting pairs, with the strongest interactions for APOBEC3A and APOBEC3B (Figure 2C and Table S1).
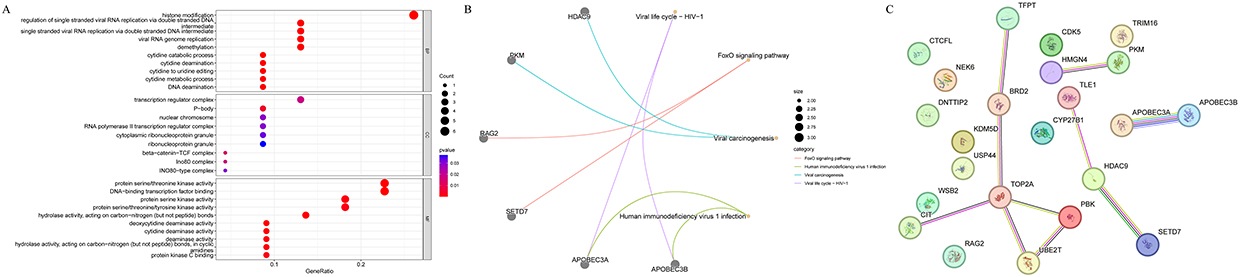 |
Figure 2 Enrichment analysis of differentially expressed epigenetic related genes (DE-ERGs) and construction of protein-protein interaction (PPI) network. (A) Gene Ontology (GO) enrichment terms; (B) Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment pathways; (C) PPI network construction. |
A Total of 5 Biomarkers of COPD Were Screened by Machine Learning Algorithms
Based on the GSE19407 dataset, the 13 node genes were analyzed by machine learning, and 13, 7 and 6 characteristic genes were obtained by SVM-RFE analysis, LASSO analysis, and Boruta analysis, respectively (Figure 3A–D). Subsequently, the genes derived from the three machine learning algorithms were taken as intersections to obtain 5 biomarkers (HMGN4, CIT, TLE1, TFPT, and UBE2T) (Figure 3E). The ROC curves indicated that the AUC values of the biomarkers in three datasets exceeded 0.7 (Figure 4A–C), suggesting that 5 biomarkers had acceptable diagnostic capabilities of COPD. The expression levels of 5 biomarkers were up-regulated in all three datasets (Figure 4D–F).
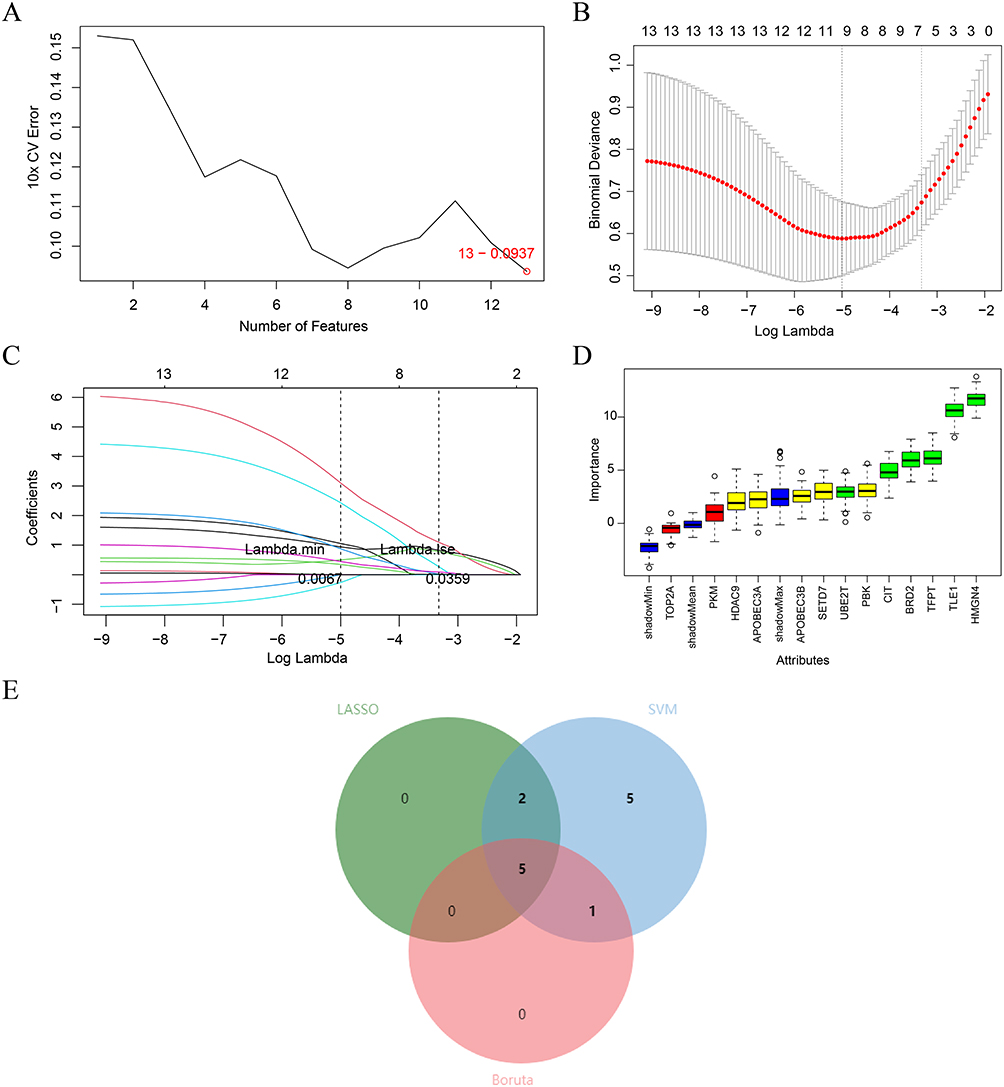 |
Figure 3 Construction of Machine Learning Diagnostic Models. (A-D) Feature genes screening in the (A) support vector machine recursive feature elimination (SVM-RFE); (B-C) least absolute shrinkage and selection operator (LASSO); and (D) Boruta algorithms. (E) Three machine learning algorithms selected feature genes and the Venn diagram of their intersection. |
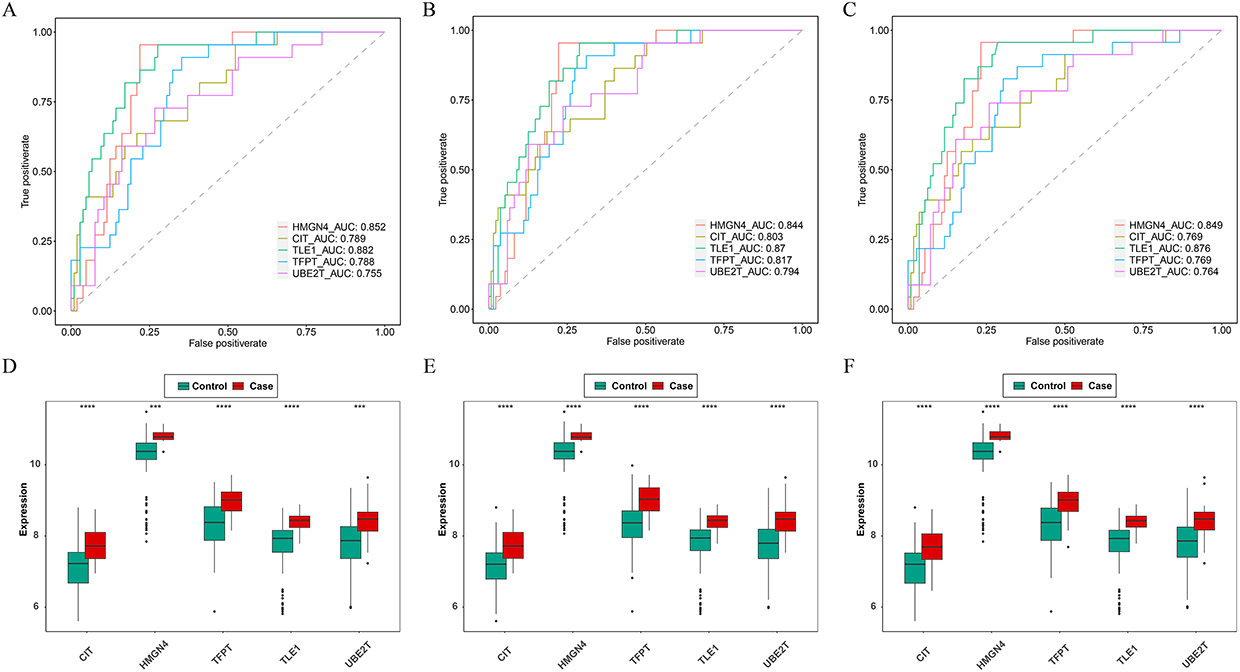 |
Figure 4 Receiver operating characteristic (ROC) analysis and expression validation of five key genes in the GSE19407, GSE11784, and GSE20257 datasets. (A-C) ROC curve of key genes in three datasets; (D-F) Validation of expression of key genes in three datasets. *** P < 0.001, **** P < 0.0001. |
Chromosomal Localization and GeneMANIA Network Analysis of Biomarkers
The circle diagram showed the localization of the 5 biomarkers in the chromosomes, UBE2T, HMGN4, TLE1, CIT, and TFPT were localized on 1, 6, 9, 12, and 19 chromosomes, respectively (Figure 5A). The GeneMANIA network map showed a total of 20 genes associated with biomarker function, with HES1, FANCL and SRAI had the stronger interactions with these biomarkers (Figure 5B).
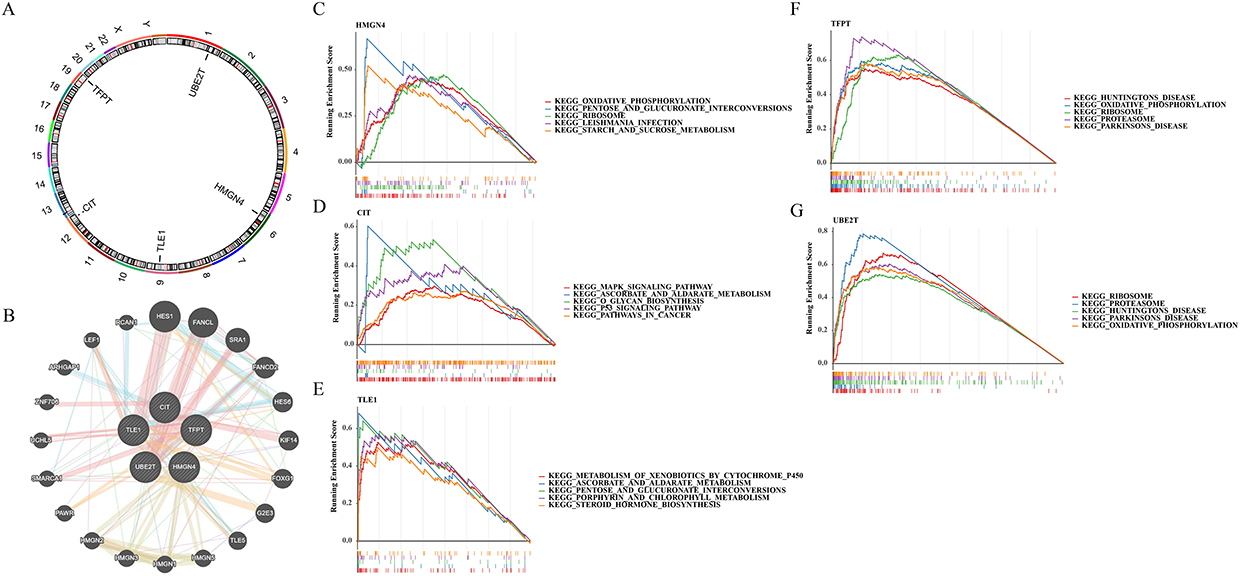 |
Figure 5 Chromosomal localization, GeneMANIA network, and Gene set enrichment analysis (GSEA) enrichment analysis of key genes. (A) Chromosomal distribution of key genes; (B) Construction of GeneMANIA Network for key genes; (C-G) GSEA analysis enrichment of key genes. |
GSEA Analysis of 5 Biomarkers
After GSEA analysis of the 5 biomarkers, we found that the HMGN4 was enriched in total 84 pathways, mainly enriched in oxidative_phosphorylation, pentose and glucuronate interconversions, etc. CIT was enriched 58 pathways, mainly enriched in MAPK signaling pathway, ascorbate and aldarate_metabolism. TLE1 was enriched in 72 pathways, mainly in MAPK signaling pathway, ascorbate and aldarate metabolism, etc. TLE1 was enriched in 72 pathways, mainly in metabolism of xenobiotics by cytochrome p450, ascorbate and aldarate metabolism, etc. TFPT was enriched in 91 pathways, mainly in Huntingtons disease, oxidative_phosphorylation. TFPT was enriched in 91 pathways, mainly in Huntingtons disease, oxidative phosphorylation and other pathways. UBE2T was enriched in 81 pathways, mainly in ribosome, proteasome and other pathways. We selected the top 5 pathways for display (Figure 5C–G).
Immune Infiltration Analysis of Biomarkers
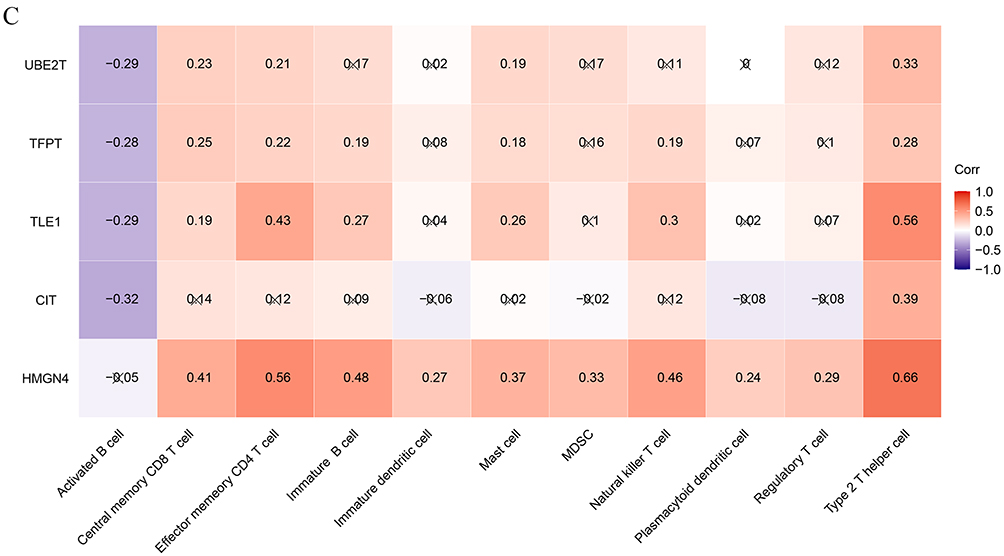
The stacked plot indicated that the count of 28 immune cells between the COPD and control groups in the GSE19407 dataset (Figure 6A). The box plot showed that the 11 immune infiltrating cells such as activated B cell, central memory CD8 T cell, effector memory CD4 T cell, etc. were significantly different between the groups (Figure 6B). Overall, the 5 candidate biomarkers (UBE2T, TFPT, TLE1, CIT, and HMGN4) exhibited predominantly positive correlations with most immune cells. Notably, HMGN4 showed no significant correlation with activated B cells, but had significant positive correlations with the other 10 types of immune cells. Among them, the correlations with type 2 T helper cells and effector memory CD4 T cells were the strongest (cor > 0.5, P < 0.05). Additionally, activated B cell had significant negative correlations with the other 4 candidate markers except for HMGN4 (Figure 6C). These results suggest that these 5 candidate biomarkers may participate in the immune disorder process of COPD by regulating the infiltration and activation status of immune cells and have potential immunoregulatory significance. Figure 6 Immunoinfiltration analysis. (A) Percentage of immune cell infiltration in normal and COPD samples; (B) Differences in immune cell infiltration between normal and COPD groups; ns, not significant; * P < 0.05. (C) Correlation analysis of key genes and immune cells. Red indicates positive correlation, blue indicates negative correlation; results marked with “×” are not statistically significant. Figure 6 continued.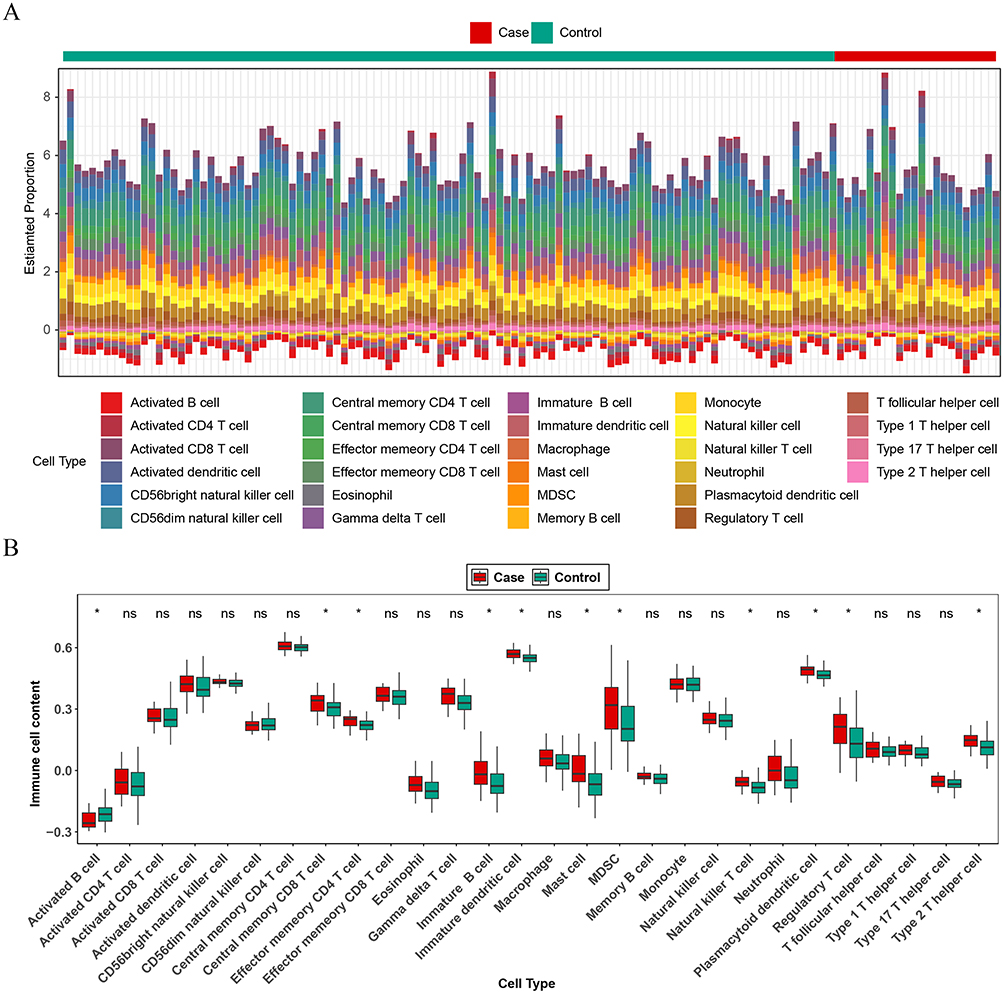
TF-miRNA-mRNA Regulatory Network of Biomarkers
To further explore the regulatory mechanisms of biomarkers, miRNAs and TFs targeting biomarkers were predicted. We predicted 20, 9, 2, and 3 miRNA for CIT, HMGN4, TLE1, and UBE2T, respectively, but no miRNAs were predicted for the TFPT. The TF-miRNA-mRNA regulatory network showed 127 nodes with 167 action pairs, and CTCF could simultaneously regulate the expression of UBE2T and TLE1 (Figure 7A and Table S2). Figure 7 Construction of Key Gene Related Networks. (A) Transcription factor (TF)-microRNA (miRNA)-mRNA regulatory network; (B) Gene disease association network; (C) Drug-gene interaction. Figure 7 continued.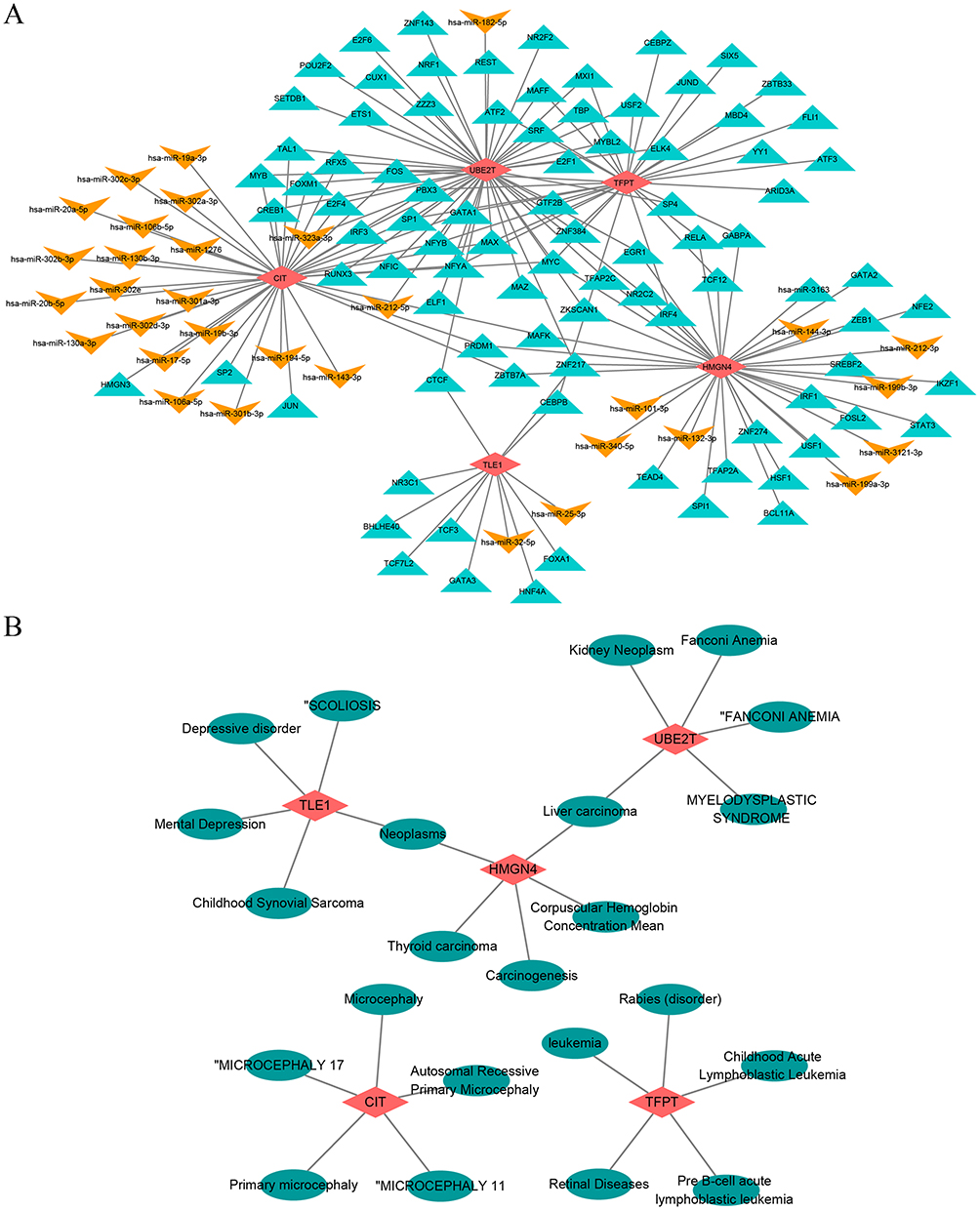
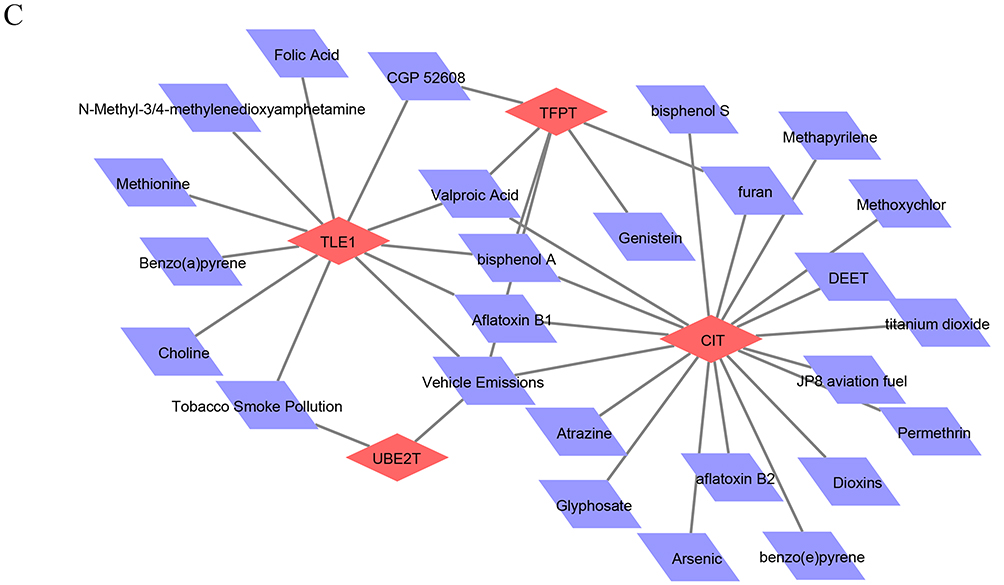
Gene-Disease Association Networks and Gene-Drug Prediction Networks for Biomarkers
The gene-disease network had 28 nodes and 25 edges, and we selected the TOP5 to construct the network and display it, and showed that both the TLE1 and the HMGN4 predicted neoplasms, and both HMGN4 and UBE2T predicted liver carcinoma (Figure 7B). The gene-drug network demonstrated 30 nodes and 37 action pairs, with 18, 6, 13, and 2 potential drug components predicted for the CIT, TFPT, TLE1, and UBE2T, respectively. Among the drugs associated with the development of COPD, bisphenol a and valproic acid were predicted by CIT, TFPT and TLE1, tobacco smoke pollution by both TLE1 and UBE2T, and furan by CIT and TFPT (Figure 7C).
Validation of the Expression for Biomarkers in Clinical Samples
To further verify the difference in the expression of biomarkers between normal and COPD samples, we collected clinical samples for qRT-PCR experiments. The results showed that the expression of 5 biomarkers (HMGN4, CIT, TLE1, TFPT, and UBE2T) were significantly higher in COPD samples than in normal samples, which were consistent with the analysis results in public databases, thus further improving the stability and reliability of our findings (Figure 8).
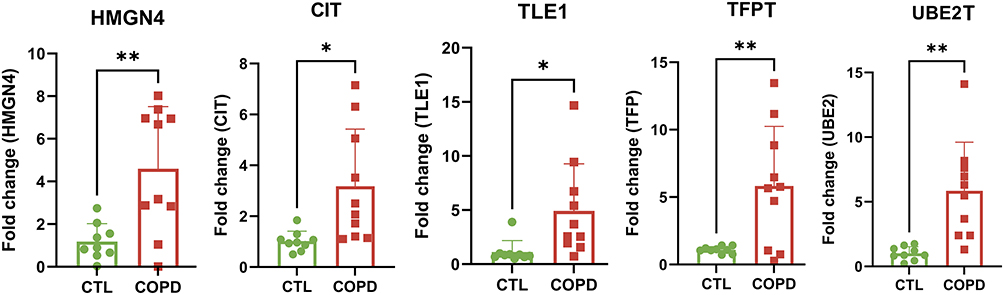 |
Figure 8 Validation of the expression for biomarkers in clinical samples. * P < 0.05; ** P < 0.01. |
Discussion
With a global prevalence exceeding 10%, COPD ranks as the third most significant cause of mortality worldwide.14 Due to the lack of typical early symptoms and reliable biomarkers, once COPD is diagnosed, most patients are at intermediate or advanced stages, often missing the optimal treatment window. Recent study suggested that epigenetic modifications were associated with reduced lung function and inflammatory response.15,16 Therefore, seeking for effective and novel biomarkers linked to epigenetic modification has considerable promise, potentially providing targets of diagnosis and therapy in COPD, thereby enhancing clinical outcomes.
In this study, common DEGs were found in three datasets of COPD patients and normal samples, with 1535 up-regulated genes and 142 down-regulated genes, respectively. Among these DEGs, 23 genes were associated with epigenetic modifications. To understand the biological function of 23 DE-ERGs, GO and KEGG analyses were performed. Using GO enrichment analysis, this study identified that DE-ERGs were primarily associated with “regulation of single stranded viral RNA replication via double stranded DNA intermediate”. This may suggest that COPD could increase the risk and worsen clinical outcomes of RNA virus infection. Cigarette smoke (CS) exposure, the leading cause of COPD, impairs antiviral responses and increased viral replication in bronchial epithelial cells.17 Exacerbations of the disease, marked by sudden decline in lung function, are related to RNA virus infections, such as rhinovirus and respiratory syncytial virus (RSV),18 contributing to COPD progression.19 The GO analysis also predicted the highest scoring molecular function as “deoxycytidine deaminase activity”.20 Deoxycytidine kinase was found upregulated in lungs of COPD patients, possibly due to hypoxia, and may contribute to increased apoptosis.21 Detecting genes modifying deoxycytidine in sputum might serve as predictive and pharmacodynamic biomarkers for COPD patients.22 The present study further performed KEGG pathway enrichment analysis, which identified that the DE-ERGs were associated with “HIV-1 viral life cycle” and “FOXO signaling pathway”. HIV-1 infection serves as a significant risk factor of COPD. HIV-1 virus infection enhances the retention of CD8+ T cells within the airway mucosa and induces the expression of transforming growth factor-β1 (TGFβ1) in airway epithelial cells,23,24 which might be implicated as a mechanism of COPD development. Alterations in the microbiota of various body sites in HIV patients with COPD may indirectly promote the progression of chronic bronchitis and emphysema.25,26 Additionally, the FOXO signaling plays a critical role in extensive airway remodeling, a hallmark of COPD.27 Activation of the FOXO signaling pathway of the proteasome system is involved in the atrophy of COPD peripheral muscle cells, primarily through enhanced autophagy and oxidative stress.28,29 Inhibition of the FOXO signaling pathway has been shown to protect lung function decline and exert anti-pulmonary fibrosis effect.30 Collectively, these enrichments suggest that epigenetic-related biomarkers may influence COPD progression through these pathways, though direct causal links remain to be validated.
Biomarkers associated with epigenetic modification in COPD were screened through bioinformatics analyses, including HMGN4, CIT, TLE1, TFPT and UBE2T. HMGN4 performs its biological functions by facilitating gene transcription through its binding with nucleosomes. Despite being discovered as a novel epigenetic regulator in 2001, its biological roles have been scarcely investigated.31 A recent study has revealed a positive association between HMGN4 and STAT3, which promoted cell proliferation and tumor progression.32 However, there are few studies of HMGN4 in COPD. In our research, we observed elevated expression of HMGN4 in COPD samples compared to normal controls, which suggests a potential role for HMGN4 in COPD progression. CIT, a crucial component of the midbody, is indispensable for cytokinesis.33 As a Rho effector, CIT was shown to directly phosphorylate eNOS-Thr497, associated with vascular dysfunction.34 Overexpression of CIT may potentially contribute to the increased risk of cardiovascular diseases, which are prevalent comorbidities in COPD patients. TLE1 acted as a corepressor by interacting with various DNA-binding transcription factors, inhibiting the nuclear factor-κB pathway linking with chronic inflammation,35,36 and regulating the Wnt pathway inducing lung regeneration in COPD.37,38 TFPT had been recognized as a molecular fusion partner of TCF3 and is implicated in processes like cell proliferation and the induction of programmed cell death.39 Moreover, studies had shown that TCF3 was highly expressed in lung cancer, suggesting that TFPT played a role in lung tissue diseases.40 UBE2T facilitated the repair of DNA interstrand crosslinks and stalled replication forks.41 Studies have identified UBE2T as a major oncogene in lung cancer,42 and its high expression in COPD samples might promote disease progression. In summary, the upregulation of these biomarkers in COPD likely reflects their potential involvement in driving disease processes, albeit further mechanistic studies are required.
To further explore the biological processes involved in biomarkers, GSEA analysis was performed, we observed increased enrichment of oxidative phosphorylation for HMGN4, TFPT and UBE2T in COPD patients, which was supported by previous studies demonstrating upregulated expression of the mitochondrial electron transport chain and oxidative phosphorylation in CS-induced mice and COPD patients.43,44 Enhanced oxidative phosphorylation leads to increased ROS, important to aged-related comorbidities, such as lung cancer, bronchiectasis, periodontitis, diabetes, and so on.45–47 These findings aligned with our results, suggesting that our discoveries were accurate as well as providing further evidence and a theoretical basis for COPD pathogenesis.
Studies have shown that COPD patients exhibited abnormal immune responses, which could lead to airway inflammation and lung damage in COPD.48 So, immune infiltration analysis was performed, we identified 11 types immune infiltration cells in COPD samples and normal samples, including activated B cell, central memory CD8 T cell, effector memory CD4 T cell, and so on. Further analysis revealed that TLE1 and HMGN4 exhibited stronger positive correlation with effector memory CD4 T cell and immature B cell, and numerous immunohistochemical studies have shown increased volume fractions of CD4+ T cells, B cells in the small airways and parenchyma of COPD patients.49,50 Furthermore, type 2 T helper cells showed a significant and strong positive correlation with all 5 candidate biomarkers, which was highly consistent with the conclusion from previous studies that the imbalance of T helper cell cytokines (including Th1/Th2 and Th17/T regulatory cells) is involved in the pathophysiological process of COPD.51 Additionally, HMGN4 showed a significant positive correlation with 10 immune cell subtypes other than activated B cells, including effector memory CD4 T cells and natural killer T cells. Yang et al’s research in hepatocellular carcinoma also reported a similar positive correlation between HMGN4 and T cells, further supporting the results of this study. These observational correlations suggest that the identified epigenetic-related candidate genes may be indirectly associated with the dysregulation of immune cell subpopulations in COPD, which warrants further mechanistic validation.
However, most current studies on epigenetics in COPD remain descriptive or retrospective, lacking systematic screening of epigenetic-related biomarkers using machine learning-based bioinformatic strategies with large-scale datasets. To address this gap, this study leveraged the GEO public database to identify candidate epigenetic biomarkers associated with lung function decline and inflammatory response in COPD through an integrated machine learning framework. These findings provide a foundation for future experimental validation and may inform the development of diagnostic or prognostic tools for COPD. However, several limitations should be acknowledged in the present study. However, this study is an exploratory research primarily based on bioinformatics analysis and preliminary experimental validation. The conclusions drawn only reflect correlations rather than definite causal relationships, and the underlying regulatory mechanisms as well as clinical translational value still require further clarification through more in-depth mechanistic investigations and large-sample validation in subsequent studies. Although the qRT-PCR verification in this study was based on a limited sample size, the results were statistically significant and consistent with those from public databases. Future studies will need to conduct further verification of gene expression levels and clinical applicability through larger-scale multi-center cohorts. In addition, the dataset from GEO database used in this study lacks information on pulmonary function, laboratory findings, and radiologic data, limiting the scope of our conclusions.
Conclusion
To sum up, this study screened five epigenetic-related candidate genes (HMGN4, CIT, TLE1, TFPT and UBE2T) for COPD using bioinformatics analysis, which were subsequently validated in a small cohort of COPD patients. Based on GEO database expression profiles and functional annotation, these genes were found to be associated with virus infections, DNA modifications, FOXO signaling, and oxidative phosphorylation. These candidate biomarkers might contribute to the alterations of immune cell subpopulations, including increased type 2 T helper cell and effector memory CD4 T cell, and decreased activated B cell, etc. Given the exploratory nature of this study and the limited validation sample size, these genes should be regarded as candidate biomarkers rather than validated diagnostic or therapeutic targets. Further investigations with larger cohorts and mechanistic studies are warranted to validate their clinical value and biological functions in COPD.
Abbreviations
COPD, Chronic obstructive pulmonary disease; ERGs, Epigenetic-related genes; DE-ERGs, Differentially expressed epigenetic-related genes; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; qRT-PCR, quantitative reverse transcriptase polymerase chain reaction; DEGs, Differentially expressed genes; EWASs, Epigenome-wide association studies; CpG, Cytosine-phosphate-guanine; SVM-RFE, Support vector machine recursive feature elimination; LASSO, Least absolute shrinkage and selection operator; AUC, Area under the curve; GOLD, Chronic Obstructive Lung Disease; TGFβ1, Transforming growth factor-β1.
Data Sharing Statement
The datasets supporting the conclusions of this article are available in the open-access GEO database (https://www.ncbi.nlm.nih.gov/geo/), [GSE19407, GSE11784, and GSE20257].
Ethics Approval and Consent to Participate
This study was performed in line with the principles of the Declaration of Helsinki. Approval was granted by the institutional review board at Zhujiang Hospital (Approval Number: 2023-KY-160-02).
Acknowledgments
We would like to sincerely thank the authors for their scientific contribution.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation of China [grant number: 82470030] and the National Natural Science Foundation of China [grant number: 82070038].
Disclosure
The authors report no conflicts of interest in this work.
References
1. Christenson SA, Smith BM, Bafadhel M, Putcha N. Chronic obstructive pulmonary disease. Lancet. 2022;399(10342):2227–19. doi:10.1016/S0140-6736(22)00470-6
2. Labaki WW, Rosenberg SR. Chronic obstructive pulmonary disease. Ann Intern Med. 2020;173(3):ITC17–ITC32. doi:10.7326/AITC202008040
3. Zhu Z, Zeng Z, Song B, Chen H, Zeng H. Identification of diagnostic biomarkers and immune cell profiles associated with COPD integrated bioinformatics and machine learning. J Cell & Mol Med. 2024;28(18):e70107. doi:10.1111/jcmm.70107
4. Xi Y, Lin Y, Guo W, et al. Multi-omic characterization of genome-wide abnormal DNA methylation reveals diagnostic and prognostic markers for esophageal squamous-cell carcinoma. Signal Transduct Target Ther. 2022;7(1):53. doi:10.1038/s41392-022-00873-8
5. Oliver J, Garcia-Aranda M, Chaves P, et al. Emerging noninvasive methylation biomarkers of cancer prognosis and drug response prediction. Semin Cancer Biol. 2022;83:584–595. doi:10.1016/j.semcancer.2021.03.012
6. Christiansen C, Potier L, Martin TC, et al. Enhanced resolution profiling in twins reveals differential methylation signatures of type 2 diabetes with links to its complications. EBioMedicine. 2024;103:105096. doi:10.1016/j.ebiom.2024.105096
7. Qiu W, Baccarelli A, Carey VJ, et al. Variable DNA methylation is associated with chronic obstructive pulmonary disease and lung function. Am J Respir Crit Care Med. 2012;185(4):373–381. doi:10.1164/rccm.201108-1382OC
8. Imboden M, Wielscher M, Rezwan FI, et al. Epigenome-wide association study of lung function level and its change. Eur Respir J. 2019;54(1):1900457. doi:10.1183/13993003.00457-2019
9. Chung S, Sundar IK, Hwang JW, et al. NF-kappaB inducing kinase, NIK mediates cigarette smoke/TNFalpha-induced histone acetylation and inflammation through differential activation of IKKs. PLoS One. 2011;6(8):e23488. doi:10.1371/journal.pone.0023488
10. Hassan F, Nuovo GJ, Crawford M, et al. MiR-101 and miR-144 regulate the expression of the CFTR chloride channel in the lung. PLoS One. 2012;7(11):e50837. doi:10.1371/journal.pone.0050837
11. Wang J, Shi A, Lyu J. A comprehensive atlas of epigenetic regulators reveals tissue-specific epigenetic regulation patterns. Epigenetics. 2023;18(1):2139067. doi:10.1080/15592294.2022.2139067
12. Medvedeva YA, Lennartsson A, Ehsani R, et al. EpiFactors: a comprehensive database of human epigenetic factors and complexes. Database. 2015;2015:bav067. doi:10.1093/database/bav067
13. Sarker IH. Machine learning: algorithms, real-world applications and research directions. SN Computer Sci. 2021;2(3):160. doi:10.1007/s42979-021-00592-x
14. GBDCRD C, Kendrick PJ, Paulson KR. Prevalence and attributable health burden of chronic respiratory diseases, 1990-2017: a systematic analysis for the global burden of disease study 2017. Lancet Respir Med. 2020;8(6):585–596. doi:10.1016/S2213-2600(20)30105-3
15. Ito K, Ito M, Elliott WM, et al. Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N Engl J Med. 2005;352(19):1967–1976. doi:10.1056/NEJMoa041892
16. Gunes Gunsel G, Conlon TM, Jeridi A, et al. The arginine methyltransferase PRMT7 promotes extravasation of monocytes resulting in tissue injury in COPD. Nat Commun. 2022;13(1):1303. doi:10.1038/s41467-022-28809-4
17. Wang Y, Ninaber DK, Faiz A, et al. Acute cigarette smoke exposure leads to higher viral infection in human bronchial epithelial cultures by altering interferon, glycolysis and GDF15-related pathways. Respir Res. 2023;24(1):207. doi:10.1186/s12931-023-02511-5
18. Seemungal T, Harper-Owen R, Bhowmik A, et al. Respiratory viruses, symptoms, and inflammatory markers in acute exacerbations and stable chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2001;164(9):1618–1623. doi:10.1164/ajrccm.164.9.2105011
19. Ritchie AI, Definition WJA. Causes, pathogenesis, and consequences of chronic obstructive pulmonary disease exacerbations. Clin Chest Med. 2020;41(3):421–438. doi:10.1016/j.ccm.2020.06.007
20. Pham P, Bransteitter R, Petruska J, Goodman MF. Processive AID-catalysed cytosine deamination on single-stranded DNA simulates somatic hypermutation. Nature. 2003;424(6944):103–107. doi:10.1038/nature01760
21. Weng T, Karmouty-Quintana H, Garcia-Morales LJ, et al. Hypoxia-induced deoxycytidine kinase expression contributes to apoptosis in chronic lung disease. FASEB J. 2013;27(5):2013–2026. doi:10.1096/fj.12-222067
22. Tessema M, Tassew DD, Yingling CM, et al. Identification of novel epigenetic abnormalities as sputum biomarkers for lung cancer risk among smokers and COPD patients. Lung Cancer. 2020;146:189–196. doi:10.1016/j.lungcan.2020.05.017
23. Crothers K, Huang L, Goulet JL, et al. HIV infection and risk for incident pulmonary diseases in the combination antiretroviral therapy era. Am J Respir Crit Care Med. 2011;183(3):388–395. doi:10.1164/rccm.201006-0836OC
24. Corleis B, Cho JL, Gates SJ, et al. Smoking and human immunodeficiency virus 1 infection promote retention of cd8(+) t cells in the airway mucosa. Am J Respir Cell Mol Biol. 2021;65(5):513–520. doi:10.1165/rcmb.2021-0168OC
25. Yang L, Dunlap DG, Qin S, et al. Alterations in oral microbiota in hiv are related to decreased pulmonary function. Am J Respir Crit Care Med. 2020;201(4):445–457. doi:10.1164/rccm.201905-1016OC
26. Lozupone C, Cota-Gomez A, Palmer BE, et al. Widespread colonization of the lung by Tropheryma whipplei in HIV infection. Am J Respir Crit Care Med. 2013;187(10):1110–1117. doi:10.1164/rccm.201211-2145OC
27. Wagner C, Uliczka K, Bossen J, et al. Constitutive immune activity promotes JNK- and FoxO-dependent remodeling of Drosophila airways. Cell Rep. 2021;35(1):108956. doi:10.1016/j.celrep.2021.108956
28. Guo Y, Gosker HR, Schols AM, et al. Autophagy in locomotor muscles of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;188(11):1313–1320. doi:10.1164/rccm.201304-0732OC
29. Pomies P, Blaquiere M, Maury J, Mercier J, Gouzi F, Hayot M. Involvement of the foxo1/murf1/atrogin-1 signaling pathway in the oxidative stress-induced atrophy of cultured chronic obstructive pulmonary disease myotubes. PLoS One. 2016;11(8):e0160092. doi:10.1371/journal.pone.0160092
30. Zhang M, Wang W, Liu K, Jia C, Hou Y, Bai G. Astragaloside IV protects against lung injury and pulmonary fibrosis in COPD by targeting GTP-GDP domain of RAS and downregulating the RAS/RAF/FoxO signaling pathway. Phytomedicine. 2023;120:155066. doi:10.1016/j.phymed.2023.155066
31. Birger Y, Ito Y, West KL, Landsman D, Bustin M. HMGN4, a newly discovered nucleosome-binding protein encoded by an intronless gene. DNA Cell Biol. 2001;20(5):257–264. doi:10.1089/104454901750232454
32. Mou J, Xu X, Wang F, Kong W, Chen J, Ren J. HMGN4 plays a key role in STAT3-mediated oncogenesis of triple-negative breast cancer. Carcinogenesis. 2022;43(9):874–884. doi:10.1093/carcin/bgac056
33. Hanicinec V, Brynychova V, Rosendorf J, et al. Gene expression of cytokinesis regulators PRC1, KIF14 and CIT has no prognostic role in colorectal and pancreatic cancer. Oncol Lett. 2021;22(2):598. doi:10.3892/ol.2021.12859
34. Seo J, Cho DH, Lee HJ, et al. Citron Rho-interacting kinase mediates arsenite-induced decrease in endothelial nitric oxide synthase activity by increasing phosphorylation at threonine 497: mechanism underlying arsenite-induced vascular dysfunction. Free Radic Biol Med. 2016;90:133–144. doi:10.1016/j.freeradbiomed.2015.11.020
35. Ramasamy S, Saez B, Mukhopadhyay S, et al. Tle1 tumor suppressor negatively regulates inflammation in vivo and modulates NF-kappaB inflammatory pathway. Proc Natl Acad Sci U S A. 2016;113(7):1871–1876. doi:10.1073/pnas.1511380113
36. Lee KY, Ho SC, Chan YF, et al. Reduced nuclear factor-kappaB repressing factor: a link toward systemic inflammation in COPD. Eur Respir J. 2012;40(4):863–873. doi:10.1183/09031936.00146811
37. Chodaparambil JV, Pate KT, Hepler MR, et al. Molecular functions of the TLE tetramerization domain in Wnt target gene repression. EMBO J. 2014;33(7):719–731. doi:10.1002/embj.201387188
38. Conlon TM, John-Schuster G, Heide D, et al. Publisher Correction: inhibition of LTbetaR signalling activates WNT-induced regeneration in lung. Nature. 2021;589(7842):E6. doi:10.1038/s41586-020-03087-6
39. Franchini C, Fontana F, Minuzzo M, Babbio F, Privitera E. Apoptosis promoted by up-regulation of TFPT (TCF3 fusion partner) appears p53 independent, cell type restricted and cell density influenced. Apoptosis. 2006;11(12):2217–2224. doi:10.1007/s10495-006-0195-5
40. Sun Y, Sun J, Ying K, et al. EP300 regulates the SLC16A1-AS1-AS1/TCF3 axis to promote lung cancer malignancies through the Wnt signaling pathway. Heliyon. 2024;10(6):e27727. doi:10.1016/j.heliyon.2024.e27727
41. Machida YJ, Machida Y, Chen Y, et al. UBE2T is theE2in the Fanconi anemia pathway and undergoes negative autoregulation. Mol Cell. 2006;23(4):589–596. doi:10.1016/j.molcel.2006.06.024
42. Yu H, Xiang P, Pan Q, Huang Y, Xie N, Zhu W. Ubiquitin-conjugating enzyme e2t is an independent prognostic factor and promotes gastric cancer progression. Tumour Biol. 2016;37(9):11723–11732. doi:10.1007/s13277-016-5020-3
43. Agarwal AR, Zhao L, Sancheti H, Sundar IK, Rahman I, Cadenas E. Short-term cigarette smoke exposure induces reversible changes in energy metabolism and cellular redox status independent of inflammatory responses in mouse lungs. Am J Physiol Lung Cell Mol Physiol. 2012;303(10):L889–898. doi:10.1152/ajplung.00219.2012
44. Yang M, Kohler M, Heyder T, et al. Long-term smoking alters abundance of over half of the proteome in bronchoalveolar lavage cell in smokers with normal spirometry, with effects on molecular pathways associated with COPD. Respir Res. 2018;19(1):40. doi:10.1186/s12931-017-0695-6
45. Lonni S, Chalmers JD, Goeminne PC, et al. Etiology of non-cystic fibrosis bronchiectasis in adults and its correlation to disease severity. Ann Am Thorac Soc. 2015;12(12):1764–1770. doi:10.1513/AnnalsATS.201507-472OC
46. Eke PI, Dye BA, Wei L, Thornton-Evans GO, Genco RJ. Cdc Periodontal disease surveillance workgroup: James Beck GDRP. prevalence of periodontitis in adults in the united states: 2009 and 2010. J Dent Res. 2012;91(10):914–920. doi:10.1177/0022034512457373
47. Ahlqvist E, Storm P, Karajamaki A, et al. Novel subgroups of adult-onset diabetes and their association with outcomes: a data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol. 2018;6(5):361–369. doi:10.1016/S2213-8587(18)30051-2
48. Qi Y, Yan Y, Tang D, et al. Inflammatory and Immune Mechanisms in COPD: current status and therapeutic prospects. J Inflamm Res. 2024;17:6603–6618. doi:10.2147/JIR.S478568
49. Booth S, Hsieh A, Mostaco-Guidolin L, et al. A single-cell atlas of small airway disease in chronic obstructive pulmonary disease: a cross-sectional study. Am J Respir Crit Care Med. 2023;208(4):472–486. doi:10.1164/rccm.202303-0534OC
50. Meng ZJ, Wu JH, Zhou M, et al. Peripheral blood CD4+ T cell populations by CD25 and Foxp3 expression as a potential biomarker: reflecting inflammatory activity in chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2019;14:1669–1680. doi:10.2147/COPD.S208977
51. Yu Y, Zhao L, Xie Y, et al. Th1/Th17 cytokine profiles are associated with disease severity and exacerbation frequency in COPD patients. Int J Chronic Obstr. 2020;15:1287–1299. doi:10.2147/COPD.S252097
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.