Back to Journals » Journal of Hepatocellular Carcinoma » Volume 11
CRISPR in Targeted Therapy and Adoptive T Cell Immunotherapy for Hepatocellular Carcinoma
Authors Palaz F ![]() , Ozsoz M
, Ozsoz M ![]() , Zarrinpar A, Sahin I
, Zarrinpar A, Sahin I ![]()
Received 26 December 2023
Accepted for publication 21 May 2024
Published 30 May 2024 Volume 2024:11 Pages 975—995
DOI https://doi.org/10.2147/JHC.S456683
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr David Gerber
Fahreddin Palaz,1,2 Mehmet Ozsoz,3 Ali Zarrinpar,4,5 Ilyas Sahin5,6
1Department of Medicine, Massachusetts General Hospital, Harvard Medical School, Boston, MA, USA; 2Faculty of Medicine, Hacettepe University, Ankara, Turkey; 3Department of Biomedical Engineering, Near East University, Nicosia, Turkey; 4Department of Surgery, College of Medicine, University of Florida, Gainesville, FL, USA; 5University of Florida Health Cancer Center, Gainesville, FL, USA; 6Division of Hematology and Oncology, Department of Medicine, University of Florida, Gainesville, FL, USA
Correspondence: Ilyas Sahin, Email [email protected]
Abstract: Despite recent therapeutic advancements, outcomes for advanced hepatocellular carcinoma (HCC) remain unsatisfactory, highlighting the need for novel treatments. The CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) gene-editing technology offers innovative treatment approaches, involving genetic manipulation of either cancer cells or adoptive T cells to combat HCC. This review comprehensively assesses the applications of CRISPR systems in HCC treatment, focusing on in vivo targeting of cancer cells and the development of chimeric antigen receptor (CAR) T cells and T cell receptor (TCR)-engineered T cells. We explore potential synergies between CRISPR-based cancer therapeutics and existing treatment options, discussing ongoing clinical trials and the role of CRISPR technology in improving HCC treatment outcomes with advanced safety measures. In summary, this review provides insights into the promising prospects and current challenges of using CRISPR technology in HCC treatment, with the ultimate goal of improving patient outcomes and revolutionizing the landscape of HCC therapeutics.
Keywords: CRISPR, hepatocellular carcinoma, HCC, targeted cancer therapy, adoptive T cell immunotherapy, CAR T cell therapy
Introduction
Hepatocellular carcinoma (HCC) constitutes approximately 90% of primary liver cancers, a major global health concern, ranking as the third leading cause of cancer-related mortality worldwide.1 For early-stage HCC, therapeutic options include surgical resection, ablation, and liver transplantation, while transarterial chemoembolization (TACE) or transarterial radioembolization (TARE) is preferred for intermediate-stage cases.2 However, unresectable disease affects a substantial portion of patients, with recurrence impacting up to 70% of those who have undergone tumor resection or ablation within 5 years.3–5 Consequently, over 50% of HCC patients ultimately require systemic therapies, typically during the advanced disease stages.4,6
HCC is resistant to conventional chemotherapy, leading to the use of targeted therapies such as sorafenib or lenvatinib (tyrosine kinase inhibitors, TKIs) as first-line treatments. Second-line therapy options include regorafenib (TKI), cabozantinib (TKI), or ramucirumab (anti-VEGFR2).6,7 Notably, immunotherapy approaches utilizing immune checkpoint inhibitors (ICIs) have shown potent antitumor activity in recent studies.8 Approved immunotherapy regimens now include first-line options like atezolizumab + bevacizumab (anti-PD-L1 + anti-VEGFA) or durvalumab + tremelimumab (anti-PD-L1 + anti-CTLA-4). For advanced HCC in the second-line setting, treatments consist of pembrolizumab (anti-PD-1) monotherapy or the combination of nivolumab + ipilimumab (anti-PD-1 + anti-CTLA-4). Additionally, numerous Phase III clinical trials are currently investigating novel agents and combination therapies.6–9
Despite these notable advancements, outcomes for advanced HCC patients remain suboptimal. For instance, atezolizumab + bevacizumab yields an objective response rate (ORR) of 27% and a median overall survival of 19.2 months, while the novel combination of camrelizumab (anti-PD-1) and rivoceranib (apatinib, a TKI) shows an ORR of 34% and a median overall survival of 22.1 months.10,11 Thus, the exploration of novel targeted therapies and immunotherapeutic approaches remains crucial for further improving the effectiveness of treatment.
CRISPR-Cas (clustered regularly interspaced short palindromic repeats and CRISPR-associated proteins) systems serve as RNA-guided adaptive immune mechanisms utilized by prokaryotes to defend against invading nucleic acids like bacteriophages.12 The effector complex of a CRISPR system comprises one or more Cas proteins and a CRISPR-RNA (crRNA) that guides the effector complex to find and cleave a specific DNA or RNA sequence through base pairing.13 As a bioengineering tool, CRISPR effector complexes can be easily and inexpensively reprogrammed to target different DNA or RNA sequences by simply modifying the crRNA. Therefore, CRISPR-based tools have been widely adopted in biomedical research for various applications, including disease modeling,14 diagnostics,15 drug discovery,16 and gene therapy.17,18 Notably, CRISPR systems have been harnessed in the fields of cancer research, diagnosis, and therapy.19–21 In early clinical trials, CRISPR technology has shown significant promise in the development of chimeric antigen receptor (CAR) T cells and T cell receptor (TCR)-engineered T cells for the treatment of various cancer types.22–25
Despite recent advancements in HCC treatment, current therapies often fail to provide durable remission and are limited by toxicity and the development of resistance.6 The ability of CRISPR to enable precise genetic modifications, particularly through its multiplex targeting capacity, presents an opportunity to reduce the likelihood of resistance, potentially leading to more effective and durable therapeutic strategies either as a potent standalone intervention or in combination with existing treatments. In this review, we focus on the application of CRISPR systems in the management of HCC. We describe the applications of CRISPR systems in targeted cancer therapy and adoptive T cell immunotherapy approaches, including CAR T cells, TCR T cells, tumor-infiltrating lymphocytes (TILs), and cytokine-induced killer (CIK) cells for HCC treatment. We also discuss the potential of combining CRISPR-based therapeutics with current treatment options. Finally, we mention ongoing clinical trials and discuss the promise, potential applications, and challenges of CRISPR systems in HCC treatment, emphasizing their potential for clinical translation and enhanced therapeutic outcomes.
Outline of CRISPR-Based Gene Editing Technologies
Precisely editing human cell DNA is a significant advancement in understanding disease mechanisms and developing new therapies. Early genome editing methods, like meganucleases, zinc finger nucleases (ZFNs), and transcription activator-like effector nucleases (TALENs), relied on protein-DNA interactions. Therefore, adapting these methods for different DNA sequences was labor-intensive and time-consuming.13 CRISPR revolutionized genome editing by using Cas9 nuclease guided by a programmable RNA for precise DNA cleavage, both in vitro and within human cells.26–28 Various CRISPR tools have since emerged and are now widely used in vivo for therapeutic applications.29,30
CRISPR nucleases induce a double-strand break (DSB) at the target DNA site guided by a ~20-nt portion of the crRNA. This target DNA must be adjacent to a protospacer adjacent motif (PAM), typically NGG for commonly used Cas9 from Streptococcus pyogenes (SpCas9). In mammalian cells, DSBs are typically repaired through non-homologous end joining (NHEJ) or microhomology-mediated end joining (MMEJ), resulting in insertion/deletion (indel) mutations at the target DNA site. CRISPR nucleases like Cas9 or Cas12 can disrupt knockout protein-coding genes by introducing frameshift mutations or disrupt non-coding regulatory sequences within the genome (Figure 1A). Homology-directed repair (HDR) is another repair pathway requiring a DNA template, predominantly active in dividing cells and less efficient than error-prone pathways. CRISPR nucleases can use HDR with an external DNA template flanked by homology regions to introduce desired edits in actively dividing cells (Figure 1A).31 To enhance specificity and minimize off-target effects, the double nickase strategy employs two Cas9 nickases (nCas9, an engineered Cas9 that cleaves a single DNA strand that each nick one DNA strand), enabling a controlled double-strand break with higher precision and reduced unintended modifications (Figure 1B). In addition, a catalytically inactive Cas9 (dCas9) fused with transcriptional activator or repressor domains can regulate gene expression transiently, resulting in gene activation (CRISPRa) or inhibition (CRISPRi) (Figure 1C). Combining dCas9 with a transcriptional repressor and DNA methyltransferases enables long-term gene silencing across multiple cell divisions, referred to as the CRISPRoff system (Figure 1D).32 Using dCas9 fused with a DNA demethylase domain, along with gRNA-guided recruitment of transcriptional activators can have the opposite effect, known as CRISPRon.
Base editors comprise a nCas9, a dCas9, or a dCas12 protein combined with either cytidine deaminase and a uracil glycosylase inhibitor (cytosine base editors, CBEs) or adenosine deaminase domains (adenine base editors, ABEs).33 When Cas9 or Cas12 binds to the target DNA site and the crRNA hybridizes with the targeted DNA strand, it forms an R-loop, exposing the complementary DNA strand as single-stranded DNA (ssDNA). Within a small region of this exposed DNA strand, cytidine deaminase or adenosine deaminase catalyzes the deamination of C or A, resulting in the creation of U or I bases, respectively (Figure 1E). Subsequently, the U or I base is repaired to T or G using the cellular DNA repair machinery. Therefore, CBEs and ABEs perform C to T and A to G base editing. Importantly, CBEs can also generate premature stop codons in coding genes. For example, CGA, CAG, and CAA codons can be converted to TGA, TAG, and TAA stop codons.22,34 This capability allows nuclease-free knockout of coding genes, avoiding undesired effects associated with DSB formation using Cas9 nuclease.35 While base editing has facilitated the creation of transition mutations, the scope of the technology has expanded considerably with the advent of the CRISPR prime editing method, which enables the generation of any transition or transversion base conversions, small insertions and deletions, and combinations thereof. This advancement was achieved through the fusion of a Cas9 nickase with a reverse transcriptase (RT) domain, giving rise to prime editors capable of precisely introducing all feasible point mutations and small insertions and deletions (Figure 1F).36
In addition to genomic targeting, CRISPR effectors like Cas13 can efficiently target RNA within human cells. Cas13 enzymes, guided by sequences of around 20–30 nucleotides in length, can locate and cleave various target RNAs, including mRNAs, long non-coding RNAs (lncRNAs), and circular RNAs, enabling effective gene knockdown (Figure 1G). RNA base editors have also been developed by fusing a catalytically inactive Cas13 with adenosine deaminase or cytidine deaminase domains. This enables the conversion of A to I (interpreted as G in translation) and C to U base editing.33
CRISPR-Based Targeted Therapies for HCC
Targeting HCC with CRISPR Demonstrates Antitumor Effects in vitro and in vivo
Numerous preclinical investigations utilizing HCC models have unveiled the potential of CRISPR-based strategies in eliciting various anti-cancer effects (Table 1).37–79 In vitro studies have revealed compelling outcomes, including the inhibition of HCC cell proliferation,73,74 suppression of invasion,70,76 reduced migration,71,72 and the induction of apoptosis.75,78 Further demonstrating the adaptability of these methods, researchers have effectively employed dCas9, targeted at promoter regions to act as a transcriptional repressor by blocking the binding of RNA polymerase. This approach has been utilized to diminish the expression of the long non-coding RNA (lncRNA) SNHG9 that promotes HCC development, thereby achieving significant reductions in cell proliferation, migration, and invasion.72
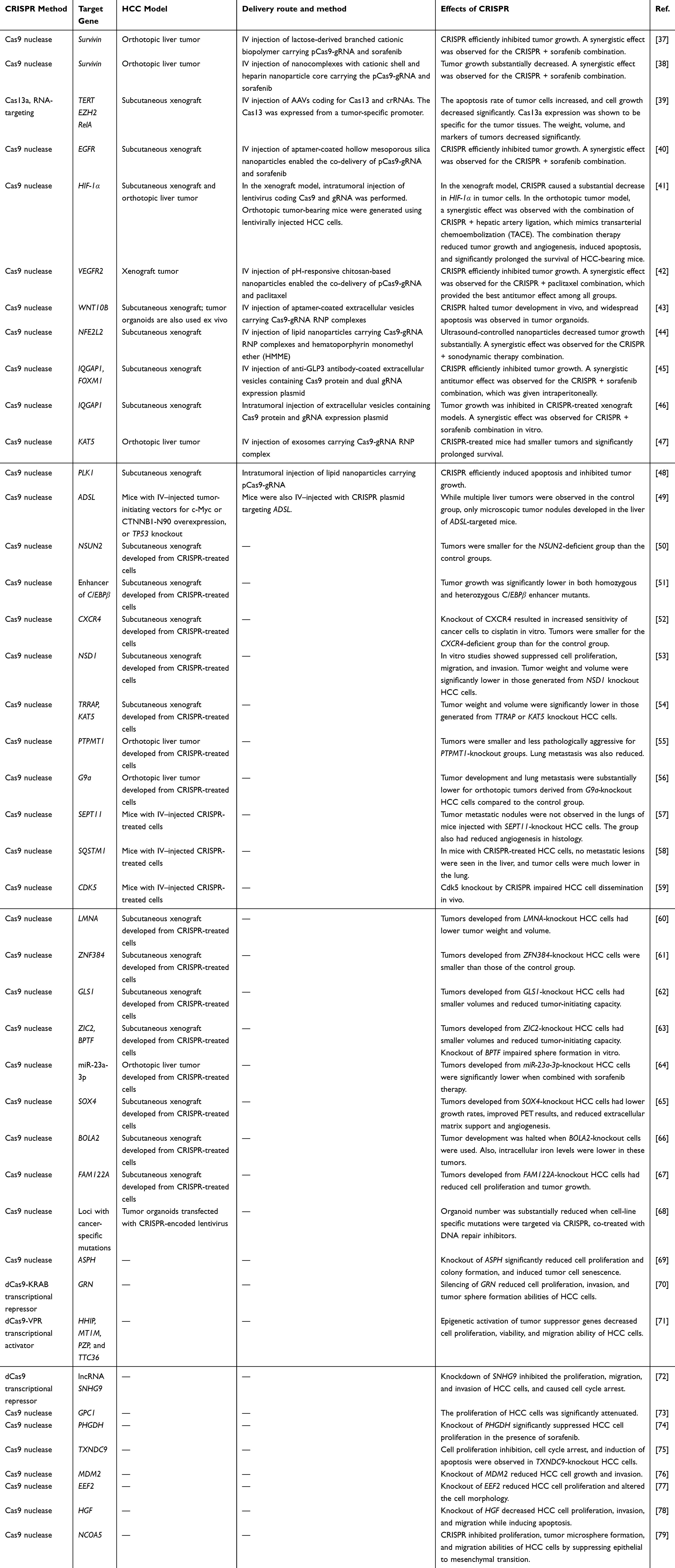 |
Table 1 The Summary of Studies Using CRISPR Tools to Target HCC |
Furthermore, in vivo assessments have provided substantial evidence of CRISPR’s therapeutic efficacy, with studies reporting inhibited tumor growth,37–39 diminished angiogenesis,41,65 reduced tumor-initiating capacity,62,63 and suppressed metastasis.41,56 Moreover, these interventions have been associated with increased survival rates among HCC-bearing mice.41,47 A noteworthy study involved the use of hepatic stellate cell-derived exosomes as carriers to deliver Cas9-gRNA RNP complexes, which are predominantly distributed in the liver, particularly in hepatocytes. In an orthotopic HCC model, this method, utilizing KAT5-targeting gRNA, effectively reduced tumor volumes and significantly improved the survival of tumor-bearing mice.47
Combining CRISPR with Existing Treatment Options Improves Antitumor Effects
The integration of CRISPR-based approaches with conventional treatment modalities has emerged as a promising strategy for enhancing therapeutic outcomes in HCC models. CRISPR-based cancer-targeting methods exhibited synergistic effects when combined with established treatments like sorafenib or TACE, significantly improving treatment efficacy. In this regard, Qi et al devised a delivery system based on a lactose-derived biopolymer with a high affinity for asialoglycoprotein receptors, which are abundant on the surface of HCC cells. Nanoparticles loaded with sorafenib and/or plasmids encoding Cas9 and gRNA targeting Survivin (pCas9-gRNA-Survivin) were intravenously injected. In an orthotopic HCC model, the combination of CRISPR and sorafenib therapy exhibited superior antitumor effects compared to either CRISPR or sorafenib monotherapy.37 Similarly, Nie et al employed nanocomplexes to deliver sorafenib and pCas9-gRNA-Survivin to HCC in an orthotopic tumor model. Upon IV injection of these nanocomplexes, the CRISPR and sorafenib combination therapy yielded the most potent antitumor effect, resulting in a significant reduction in tumor volume.38 He et al utilized extracellular vesicles coated with antibodies specifically targeting glypican-3 (GPC3), a protein abundantly expressed on the surface of HCC cells. These vesicles delivered the Cas9 protein along with a dual gRNA expression plasmid designed to target two genes, IQGAP1 and FOXM1. When combined with intraperitoneal sorafenib treatment, IV injection of extracellular vesicles led to a substantial decrease in the volume of xenograft tumors.45 Another innovative approach involved hollow mesoporous silica nanoparticles coated with tumor-targeting DNA aptamers, specifically binding to epithelial cell adhesion markers (EpCAMs) on HCC cell surfaces.40 These nanoparticles were utilized for the co-delivery of sorafenib along with a CRISPR plasmid targeting EGFR. Systemic administration of aptamer-coated nanoparticles carrying both sorafenib and CRISPR exhibited superior antitumor effects compared to other treatment groups in xenograft tumor-bearing mice.40 Lu et al established orthotopic liver tumors utilizing HCC cells with miR-23a-3p knockout achieved using CRISPR. When combined with sorafenib treatment, their study demonstrated a significant reduction in tumor growth accompanied by an increase in apoptosis.64
While the current treatment strategy for intermediate-stage HCC includes chemoembolization, ongoing clinical trials actively explore avenues to enhance patient survival through the integration of TACE with TKIs or immune checkpoint inhibitors.8 In their study, Liu et al demonstrated the efficacy of combining CRISPR with hepatic artery ligation (HAL), which mimics TACE, in orthotopic tumor-bearing mice. This innovative approach resulted in a substantial reduction in liver tumors developed from HIF-1α knockout cells, leading to improved survival rates in mice receiving the combination therapy compared to those treated with HAL alone. Similar synergistic effects have also been observed in studies combining CRISPR with paclitaxel or cisplatin.42,52 These cumulative findings underscore the potential of combining CRISPR with existing treatment modalities to achieve enhanced antitumor efficacy and ultimately improve patient survival.
Various CRISPR Tools Can Be Used to Target HCC
While the majority of studies employ CRISPR tools for targeted gene knockout, diverse CRISPR approaches have demonstrated effectiveness in targeting HCC. These versatile CRISPR technologies offer precise interventions for tailored HCC treatment. For instance, in one study, the RNA-targeting CRISPR-Cas13 system enabled multiplex knockdown of TERT, EZH2, and RelA mRNAs in HCC cells (Figure 1G), leading to reduced xenograft tumor growth upon IV injection of adeno-associated virus (AAV) carrying Cas13 and crRNAs.39 Furthermore, CRISPR-based transcriptional activators and repressors were applied to target HCC in vitro (Figure 1C and D). Targeting the Granulin (GRN) promoter with dCas9-DNMT3A (for DNA methylation), dCas9-EZH2 (for histone 3 lysine 27 methylation), or dCas9-KRAB (for transcriptional repression) resulted in a substantial reduction in GRN mRNA levels in HCC cells.70 It was observed that These CRISPR-based epigenetic modulators induced de novo CpG DNA methylation in the GRN promoter and histone modifications, leading to gene suppression and subsequently decreased cell proliferation, invasion, and tumor sphere formation ability. Notably, dCas9-KRAB exhibited the most potent antitumor efficiency among the tested epigenetic suppressors.70
The epigenetic silencing of tumor suppressor genes plays a pivotal role in the development of HCC. Sgro et al leveraged the CRISPR activation (CRISPRa) system, which involves dCas9 fused with transcriptional activation domains VP64, p65, and Rta, in combination with MS2 aptamer-containing sgRNA. This combination recruits multiple MS2 coat proteins fused with p65 and HSF1 activator domains to reactivate tumor suppressor genes.71 Targeting HHIP, MT1M, PZP, and TTC36, genes that are significantly downregulated in HCC, the CRISPRa technology enabled highly specific and potent reactivation of these tumor suppressors, surpassing the effectiveness of epigenetic-modifying drugs. As a result, this approach led to a substantial reduction in HCC cell proliferation, viability, and migration.71 These findings underscore the versatility of various CRISPR-based approaches in effectively targeting HCC and hold promise for innovative therapeutic interventions in the field of liver cancer research and treatment.
Reprogrammability and Multiplex Targeting Capacity of CRISPR Enables Personalized and More Potent Cancer Targeting
Utilizing multiple gRNAs, CRISPR systems have the capability to simultaneously target several DNAs or RNAs, potentially leading to more robust antitumor effects and reducing the likelihood of resistance. In HCC cells, the multiplex targeting of TERT, EZH2, and RelA mRNAs using Cas13 resulted in a higher rate of apoptotic cells compared to single mRNA targeting.39 Additionally, the reprogrammability and multiplexing capabilities inherent to CRISPR technology open the door to personalized treatment strategies for HCC. In this context, Jiang et al introduced an innovative CRISPR-based personalized cancer therapy approach. Customized gRNAs were designed to target unique mutations present in cancer cells, identified through DNA sequencing.68 Whole-genome sequencing performed on an HCC cell line defined single nucleotide variations and indels. Multiplex CRISPR targeting, combined with DNA repair inhibitors, effectively suppressed cell proliferation. Notably, the highest rate of apoptosis was observed when CRISPR targeted 8 loci, as opposed to targeting only 4 loci. Another group utilized multiplex CRISPRa to reactivate several epigenetically silenced tumor suppressor genes within HCC cells, achieving potent antitumor effects.71 These findings underscore the potential of CRISPR-based multiplexed and tailored targeting for personalized therapeutic strategies aimed at maximizing antitumor efficacy and minimizing tumor escape.
Tumor-Specific Delivery, Expression, and Activation Methods Enable More Effective and Specific Targeting of HCC
Ensuring the precision and safety of CRISPR applications in vivo is crucial. Tumor-specific delivery methods play a pivotal role in achieving efficient targeting of CRISPR tools to the tumor microenvironment, thereby enhancing antitumor activity, elevating local concentrations of CRISPR components, and ensuring the safety of genome editing. In this regard, Zhang et al harnessed EpCAM-targeting aptamer-coated silica nanoparticles loaded with sorafenib and an EGFR-targeting CRISPR plasmid to specifically target HCC cells in vivo. It was shown that the nanoparticles predominantly accumulated in tumor tissue with significantly higher concentrations compared to control groups, contributing to the safety and efficacy of the treatment.40 Similarly, Zhuang et al employed extracellular vesicles coated with the TLS11a DNA aptamer, which has a specific affinity for HCC cells,80 to deliver Cas9-gRNA ribonucleoprotein (RNP) complexes to tumor tissue. This approach facilitated highly efficient and specific delivery of CRISPR cargo to HCC cells within tumor organoids and xenograft tumor-bearing mice in vivo. It induced widespread apoptosis in tumor organoids and resulted in a significant reduction in tumor size in mice.43 Another strategy involved the use of extracellular vesicles containing anti-GPC3 antibodies, enabling the specific delivery of Cas9 protein and dual sgRNA-encoding plasmids to HCC cells in xenograft tumor-bearing mice. Systemic administration of these extracellular vesicles successfully demonstrated their accumulation within xenograft tumors.45
The overexpression of the asialoglycoprotein receptor (ASGPR) on the surface of HCC cells provides an opportunity for selective binding to galactose residues, facilitating endocytosis. Researchers have leveraged this receptor to improve the delivery of therapeutic agents, including CRISPR plasmids. To achieve efficient and specific delivery, researchers conjugated β-galactose-carrying lactobionic acid to chitosan nanoparticles. These nanoparticles served as carriers for paclitaxel and CRISPR plasmids targeting VEGFR.42 Moreover, the water solubility of chitosan is increased at acidic pH, promoting the controlled release of nanoparticle content within the tumor microenvironment. This two-stage control system facilitated the accumulation of nanoparticles in xenograft tumor tissue, leading to a significant reduction in tumor volume upon systemic administration. This approach highlights the effectiveness of tumor-specific delivery methods.42 Another ASGPR-specific targeting approach involved the use of lactose-derived branched cationic biopolymer. This enabled the efficient delivery of CRISPR plasmids to HCC within an orthotopic liver tumor model upon systemic administration.37 Nie et al utilized nanocomplexes consisting of a cationic shell with high transfection efficiency and a negatively charged heparin core to deliver CRISPR plasmid to orthotopic liver tumors. After systemic administration, the nanocomplexes exhibited prolonged enrichment within the liver of mice, demonstrating the safe and efficient targeting of the liver in vivo.38
Cancer-specific promoters represent a valuable strategy for ensuring tumor-specific CRISPR targeting. In this regard, Jiang et al employed an AAV vector for in vivo Cas13 and crRNA delivery in xenograft-bearing mice. A cancer-specific promoter known as the decoy minimal protomer was used, which enables Cas13 expression only in the presence of high levels of NF-κB, a cancer-associated transcription factor overexpressed in many cancer types.39 After systemic administration, while AAVs were detected in several organs, Cas13 expression was exclusively observed in xenograft tumor tissue derived from HCC cells. This approach exemplifies the capability of tumor-specific expression methods to enhance safety and precision in CRISPR-based therapies.39
Sonodynamic therapy (SDT) relies on the generation of reactive oxygen species (ROS) to induce cancer cell death and represents a promising approach for HCC treatment due to its low cost, non-invasiveness, and high tissue-penetrating depth. In a study by Yin et al, it was observed that NFE2L2 expression is activated immediately after SDT, leading to tumor growth promotion and reduced treatment efficiency. To address this issue, the researchers utilized an FDA-approved lipid nanoparticle system for the delivery of a sonosensitizer called hematoporphyrin monomethyl ether (HMME) and a Cas9-gRNA RNP complex targeting the NFE2L2 gene. Tumor-localized ultrasound application facilitated the production of abundant ROS by HMME, which also induced endosomal rupture and the release of Cas9-gRNA complexes into the cytoplasm. These complexes subsequently translocated into the nucleus to knock out NFE2L2. Systemic administration of the lipid nanoparticle system resulted in a high concentration of ROS combined with CRISPR-mediated NFE2L2 knockout within xenograft tumors. This dual approach led to a significant reduction in tumor volume. Importantly, spatial control of ultrasound stimulation ensured the specific action of CRISPR within tumor tissue, minimizing off-target effects in unrelated tissues.44 These studies underscore the importance of tumor-specific delivery, activation, and expression methods in enhancing the efficiency and safety of CRISPR-based targeting for HCC treatment.
CRISPR-Based Adoptive T Cell Therapies in HCC and Ongoing Clinical Trials
The complex immune landscape of the liver plays a dual role in both immune surveillance and immune tolerance. It contains various immune cell types, such as Kupffer cells, natural killer (NK) cells, dendritic cells, CD4+ T cells, and CD8+ T cells, which collectively serve to detect and combat pathogens circulating in the bloodstream. However, the liver also maintains an immunosuppressive environment to tolerate harmless substances like food antigens and baseline microbial products, which is crucial for normal liver function and immune homeostasis. This immunosuppressive state also plays a role in facilitating organ transplantation.81 In cases of chronic inflammatory conditions like hepatitis B (HBV) or hepatitis C (HCV) infections, excessive alcohol consumption, and Metabolic dysfunction-Associated Fatty Liver Disease (MAFLD), continuous recruitment of immune cells and chronic inflammation can lead to the initiation of tumorigenesis in the liver. During this process, immunosuppressive mechanisms may come into play, leading to the inhibition and exhaustion of T cells, resulting in the failure of halting de novo tumorigenesis events or metastases from different organs.82 As cancer cells proliferate, a process known as epithelial-to-mesenchymal transition occurs, contributing to the creation of a tumor microenvironment that further suppresses the immune response. This suppression occurs through various mechanisms, including the production of anti-inflammatory cytokines and the overexpression of immune checkpoint molecules like PD-L1. Given these challenges, the use of immunotherapy approaches for enhancing the function of existing host immune cells and targeting HCC by utilizing ex vivo activated and expanded T cells hold great promise to halt cancer progression.7
Adoptive cancer immunotherapy is a potent approach based on enhancing the ability of immune cells to target cancer, with significant improvements have been achieved over the last decade. This method involves the ex vivo sensitization and expansion of autologous or allogeneic lymphocytes, which are then infused into patients. Key strategies within adoptive immunotherapy include the use of chimeric antigen receptor (CAR)-expressing T cells, tumor antigen-specific T cell receptor (TCR)-engineered T cells, cytokine-induced killer (CIK) cells, tumor-infiltrating lymphocytes (TILs), lymphokine-activated killer (LAK) cells, and NK cells. While adoptive T cell therapies have primarily been employed for hematologic malignancies, researchers have increasingly focused on their application in solid tumors, with liver cancer, including hepatocellular carcinoma (HCC), being a prominent target.83 Recent clinical trials in this context have shown promising results.9,84,85
While adoptive T cell therapies have shown promise in treating solid tumors like HCC, there are significant challenges that need to be addressed to enhance their safety and efficacy. One major challenge is T cell exhaustion, which can occur due to the immunosuppressive nature of the tumor microenvironment. This exhaustion is often characterized by the overexpression of immune checkpoint proteins like PD-1 on T cells, which can render them ineffective in targeting cancer cells.86 Another concern in adoptive T cell therapies, particularly those involving T cell receptor (TCR)-engineered T cells, is the potential for unintended dimeric complex formation between endogenous and recombinant TCRs. This can result in unpredictable antigen-binding specificities and potential safety issues.87 Additionally, the delivery of chimeric antigen receptors (CARs) and TCRs to T cells using lentiviruses can lead to random integration into the genome. This randomness can result in variable CAR and TCR expression levels and may raise safety concerns.88 CRISPR-based tools offer promising solutions to address these challenges. For instance, CRISPR can be used to knock out the PD-1 gene in T cells, preventing T cell exhaustion and enhancing their effectiveness in targeting cancer cells. It can also be employed to knock out endogenous TCR-coding genes, eliminating the risk of unintended dimeric complex formation (Figure 2). Furthermore, CRISPR can enable the precise insertion of TCR or CAR expression cassettes into defined loci in the T cell genome. This approach ensures safer and more stable expression of recombinant receptors, potentially improving the overall safety and efficacy of adoptive T cell therapies.19
Knockout of Immune Checkpoint Proteins Alleviates T Cell Exhaustion and Enhances the Efficacy of CAR T Cell Therapy
To improve the effectiveness of CAR T cell therapy for HCC, researchers have explored CRISPR-based strategies to disrupt the PDCD1 gene, which codes for PD-1, in CAR T cells, thereby mitigating T cell exhaustion and enhancing antitumor activity. In a study, researchers observed a significant upregulation of PD-L1 in HCC cells when cocultured with glypican-3 (GPC3)-targeted CAR T cells. To counteract T cell exhaustion, PD-1 was disrupted in CAR T cells using Cas9 with two gRNAs targeting PDCD1.89 This intervention resulted in the potent killing activity of GPC3-targeted CAR T cells against PD-L1-expressing HCC cell lines in vitro. Additionally, PD-1 disruption augmented the antitumor efficacy, persistence, infiltration, and pro-inflammatory cytokine production of GPC3-CAR T cells in a subcutaneous HCC xenograft model, leading to reduced tumor volume, prolonged presence of peripheral blood T cells post-infusion, increased CAR T cell infiltration in tumor tissue, and elevated serum levels of IFN-γ and IL-2. However, a more comprehensive approach involved the disruption of both PD-1 and endogenous TCR to ensure the safe and efficient use of GPC3-CAR T cells, as PD-1 knockout CAR T cells could potentially express autoreactive TCRs, posing a risk of autoimmune adverse effects (Figure 2A).90 In another study, researchers harvested CIK cells from the peripheral blood of HCC patients, and electroporated Cas9-gRNA RNPs into CIK cells to knockout PD-1 followed by lentiviral delivery of the human telomerase reverse transcriptase (hTERT) gene.91 In vitro investigations demonstrated that hTERT expression in CIK cells led to an increase in telomere length and significantly improved the persistence of these engineered CIK cells. Furthermore, the knockout of PD-1 enhanced the IFN-γ secretion capacity and antitumor efficacy of CIK cells (Table 2). Adjuvant immunotherapy using autologous CIK cells has demonstrated a significant increase in recurrence-free and overall survival rates among patients with HCC.92 Leveraging CRISPR-based methods presents an exciting opportunity to enhance CIK cell therapies further, potentially contributing to improved survival outcomes in HCC patients.
 |
Table 2 CRISPR-Mediated Adoptive T Cell Immunotherapy Studies in HCC |
In an ongoing Phase I clinical trial (NCT04417764) initiated in 2019, researchers are investigating a combination therapy approach for advanced HCC. This trial combines transarterial chemoembolization (TACE) with infusions of autologous T cells that have undergone PD-1 knockout using CRISPR-Cas9 technology. The treatment regimen involves one TACE procedure followed by three or more cycles of CRISPR-Cas9-mediated PD-1 knockout T cell infusions at 4-week intervals. During each cycle, a total of 1 to 3×109 engineered T cells are administered via percutaneous infusion into the peripheral tumor.95 Another phase I clinical trial (NCT04842812) focuses on patients with advanced solid tumors, including liver cancer. In this trial, tumor-infiltrating lymphocytes (TILs) are harvested from patients and subjected to CRISPR-Cas9-mediated knockout of the PD-1 gene. Additionally, the TILs are engineered to express single-chain fragment variables (scFvs) targeting immune checkpoint proteins PD-1 and CTLA-4, as well as chimeric antigen receptors (CARs) against various antigens, including GPC3. The resulting PD-1 knockout CAR/TILs, which secrete anti-PD-1/CTLA-4 scFvs and carry CARs against multiple antigens, are administered systemically or locally at a dose of 1 to 10×108 cells per kilogram for each treatment, with at least three cycles of treatment planned.96 Furthermore, researchers have identified an intracellular immune checkpoint protein Cytokine-induced SH2 (CISH) protein, which negatively regulates T cell function. Knockout of the CISH gene has been shown to enhance the efficacy of adoptive TIL therapy for gastrointestinal cancers.97 In an associated phase I/II clinical trial (NCT04426669), investigators aim to utilize CISH-knockout (CISH-KO) TILs as a therapeutic approach for patients with metastatic gastrointestinal epithelial cancers that have not responded to any first-line therapy (Table 3).98 These studies and ongoing clinical trials represent promising developments in the field of adoptive T cell therapy for HCC and underscore the potential of CRISPR-based strategies to enhance the safety and efficacy of these treatments.
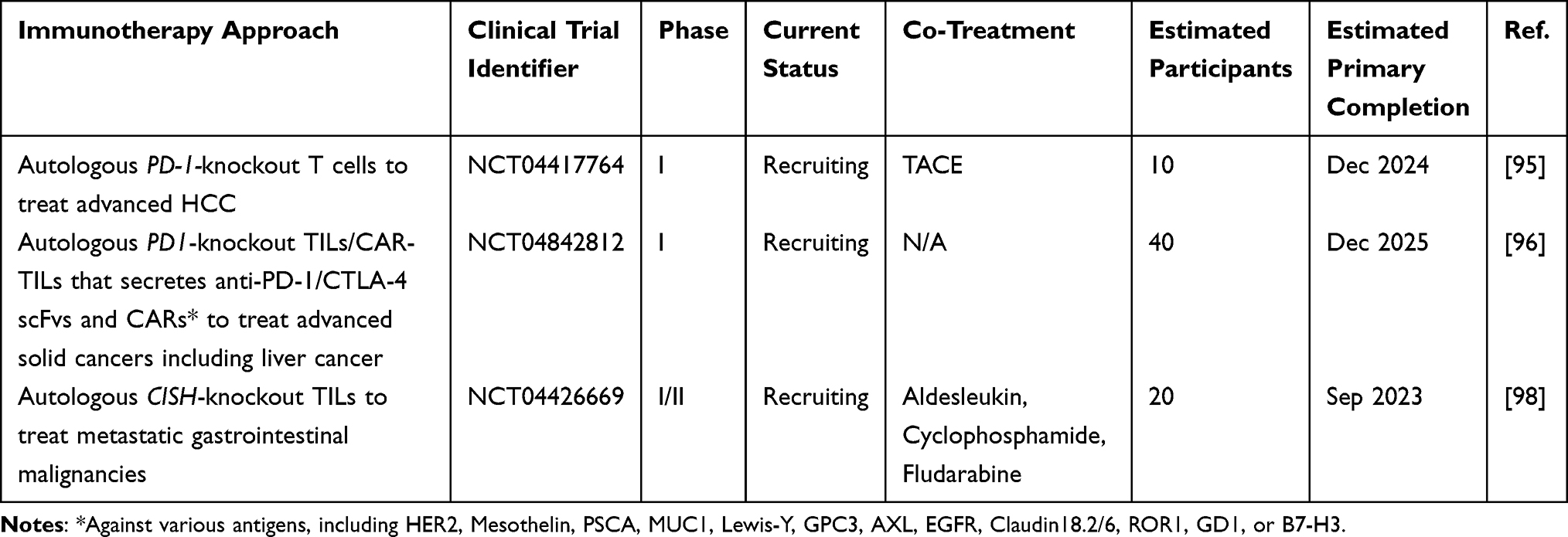 |
Table 3 Clinical Trials Using CRISPR in Adoptive Cell Therapy for HCC |
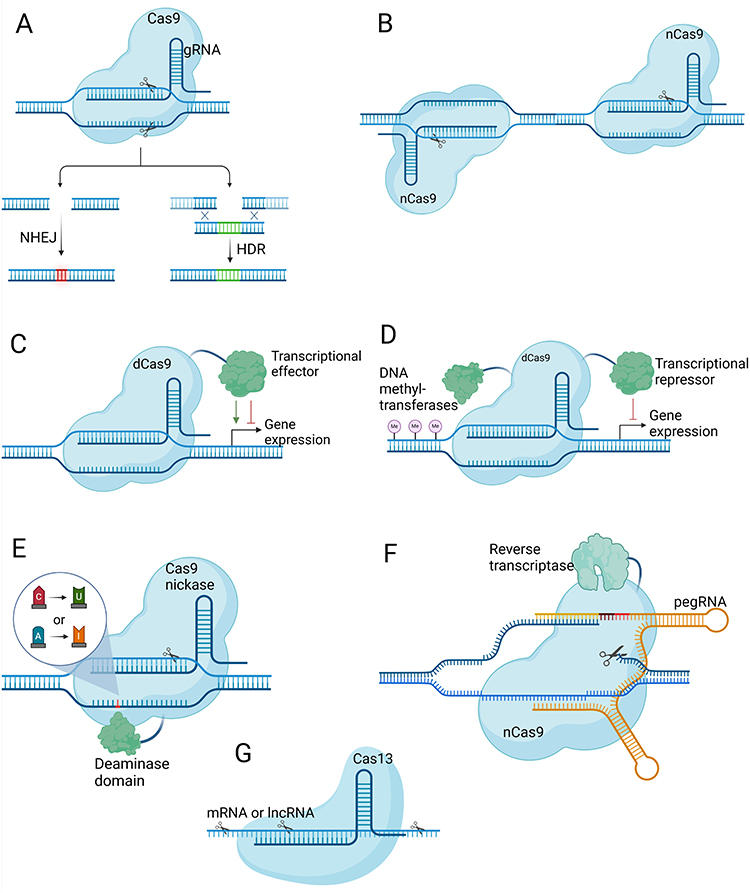 |
Figure 1 Overview of CRISPR-based gene editing technologies. Created with BioRender.com. (A) CRISPR-mediated approaches for gene disruption (NHEJ) and precise editing (HDR). (B) Double nickase strategy employing two Cas9 nickases for controlled and precise DSBs, minimizing off-target effects. (C) CRISPRa and CRISPRi systems for transient regulation of gene expression using deactivated Cas9 and transcription activator/inhibitor domains. (D) CRISPRoff system for durable gene repression by combining deactivated Cas9 with DNA methyltransferase domains and a transcriptional repressor domain. (E) Cytosine and adenine base editing systems that alter DNA bases without inducing double-strand breaks. (F) Prime editing method enabling precise installation of single base mutations and small indels using a Cas9 nickase and reverse transcriptase. (G) RNA-targeting Cas13 system for the manipulation of RNA molecules in cells. |
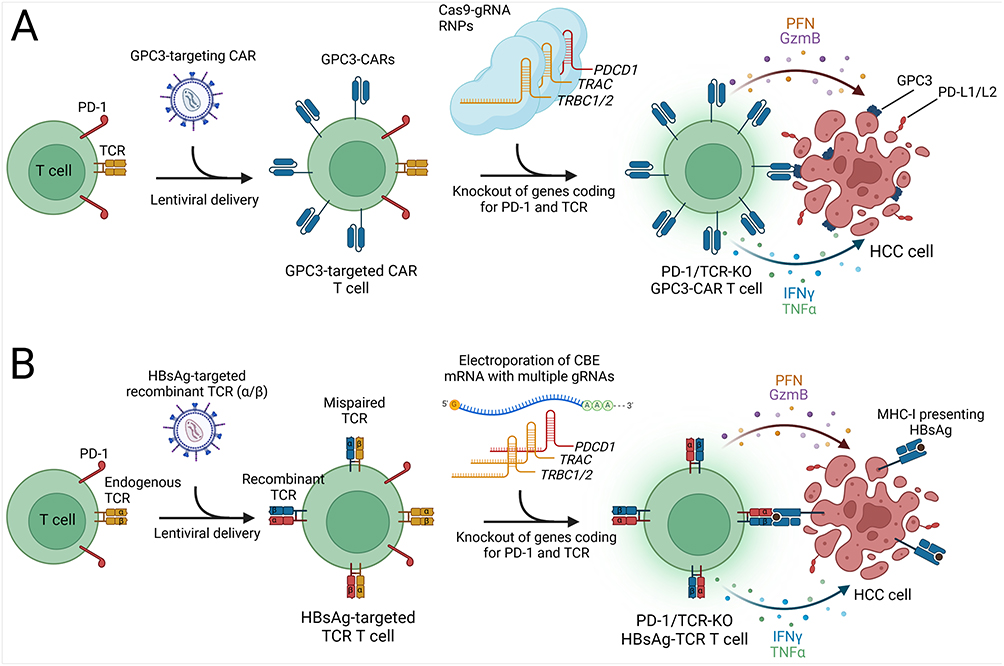 |
Figure 2 CRISPR-based enhancements for CAR T and TCR T cell therapies in HCC. Created with Biorender.com. (A) Autologous T cells are transduced with a lentivirus carrying a GPC3-targeting CAR expression cassette, followed by electroporation of Cas9-gRNA RNPs to knockout genes encoding the PD-1 and TCR α and β chains. These modifications prevent T cell exhaustion and enhance specificity. The engineered CAR T cells effectively bind GPC3 on HCC cells via GPC3-targeted CARs, triggering a release of cytotoxic proteins (perforin, granzyme B) and pro-inflammatory cytokines (IFN-γ, TNF-α), which collectively contribute to the targeted destruction of tumor cells. (B) Development of Hepatitis B surface antigen (HBsAg)-targeted TCR T cells involves lentiviral delivery of recombinant TCR α and β chains to T cells. The presence of endogenous TCR chains can lead to the formation of mispaired TCRs with unpredictable antigen specificity, posing a potential safety risk. For enhancing safety and efficacy, the cytosine base editor (CBE) is introduced as mRNA to perform knockout of endogenous TCR genes and PD-1. The specific interaction between the HBsAg-targeted TCR and HBsAg presented by MHC-I on HCC cells activates the TCR T cells, promoting robust antitumor activity. |
Targeting Endogenous TCRs for Recombinant TCR T Cell Therapy
Cytosine base editors offer a valuable tool for generating premature stop codons via single-base editing (Figure 1E).22,99 This approach offers several advantages, including the ability to achieve gene knockout without inducing DSBs, thereby preventing the formation of large insertions or deletions and complex genomic rearrangements that can occur with traditional Cas9-based gene disruption methods.35 In the context of adoptive T cell therapy, preventing the expression of endogenous T cell receptor (TCR) chains is crucial to avoid unpredictable pairings with exogenously delivered recombinant TCR α and β chains, which can lead to mispairing of TCRs, which reduces the cell surface expression of recombinant TCR and potentially generates self-targeting TCRs. To address this challenge, Preece et al utilized cytosine base editing to disrupt the endogenous TCR β chain, encoded by the TRBC1/2 genes (Figure 2B). Recombinant TCR and gRNA expression cassettes targeting hepatitis B surface antigen (HBsAg) were introduced via lentivirus delivery, followed by the electroporation of Base Editor 3 (BE3) mRNA into T cells. This approach resulted in a significant reduction in the number of T cells expressing the endogenous TCR, with the majority of cells carrying the recombinant TCR. To further purify the cell population, a magnetic bead-mediated depletion process was conducted to remove any remaining endogenous TCR-expressing cells, resulting in a homogeneous population in which approximately 95% of the T cells expressed the recombinant TCR while lacking the endogenous TCR. In vitro analyses using a 3D microfluidic device, which included HBsAg-expressing HCC cells, demonstrated that T cells equipped with the recombinant TCR effectively killed cancer cells and exhibited elevated levels of inflammatory cytokines, including IFN-γ, TNF-α, and IL-2, compared to control groups. Although the incorporation of a disulfide bond between recombinant TCR chains reduced the likelihood of mispairing with endogenous TCR chains in this study, targeting multiple genes, including TRAC (TCR α chain) and PDCD1 (PD-1), in addition to TRBC1/2 (TCR β chain), represents a promising and comprehensive strategy to further improve the safety and efficacy of TCR T cell therapy for HCC (Figure 2B).23
CRISPR Enables Site-Specific Genome Integration of CAR in T Cells
CD105, also known as endoglin, is highly expressed on the surface of cancer cells and endothelial cells within the tumor microenvironment, playing a key role in angiogenesis. Mo et al utilized CRISPR-Cas9-mediated HDR to precisely insert the anti-CD105 nanobody expression cassette into the AAVS1 locus of T cells (Figure 1A). This approach allowed for the stable and controlled expression of the anti-CD105 CAR on the surface of T cells. The engineered CD105-CAR T cells, administered twice to mice with xenograft HCC tumors, significantly reduced tumor size and improved the survival of the mice. Furthermore, CD105-CAR T cells effectively inhibited the growth of HCC patient-derived xenografts in vivo. Importantly, these engineered T cells were well-tolerated, with no fever, bleeding, or increased IL-6 levels observed in the mice. The CAR cassette can also be inserted into the TRAC locus using CRISPR-Cas9, enabling stable expression of CAR with the simultaneous knockout of TRAC.100
Discussion and Future Perspectives
The emergence of CRISPR-based therapies in the context of hepatocellular carcinoma represents a significant advancement in the field of cancer treatment. Promising results have emerged from preclinical studies, and early-phase clinical trials are currently in progress. However, as we move toward practical implementation, it is essential to acknowledge and address important considerations and challenges.
In translating CRISPR-based cancer therapies from bench to bedside, several critical challenges must be addressed to realize their potential in clinical settings. Regulatory hurdles are particularly significant, as current frameworks are not fully adapted to the novel complexities introduced by gene editing technologies.101 Additionally, the development of efficient and safe delivery methods remains a pivotal concern. These delivery systems must ensure targeted delivery to cancer cells while minimizing systemic exposure to reduce adverse effects.102 More research is also needed to develop more precise CRISPR systems that can enhance targeting accuracy, thereby reducing the likelihood of off-target effects and improving therapeutic outcomes.103 Furthermore, the long-term safety and efficacy of these therapies need to be rigorously evaluated through extended follow-up in clinical trials to monitor for potential delayed adverse effects and ensure sustained therapeutic benefits.25 Addressing these challenges is essential for the successful integration of CRISPR therapies into routine clinical practice for treating cancers, including HCC.
Addressing the immunogenicity of CRISPR-based therapeutics is imperative for their successful translation into clinical applications. The development of adaptive immune responses against CRISPR-associated proteins, such as Cas9, which often originate from common bacterial pathogens, represents a significant hurdle.104 This immunogenicity can compromise the safety and efficacy of CRISPR-based interventions, potentially leading to adverse outcomes similar to those observed in past gene therapy trials. Strategies to circumvent these immune responses include engineering CRISPR components to reduce immunogenicity and utilizing delivery tools with limited immune visibility.105 Furthermore, leveraging in silico methods combined with empirical testing can help predict and mitigate potential immunogenic hotspots within the CRISPR proteins.106 Continuous monitoring and evaluation of immune responses in preclinical and clinical settings are crucial. This approach includes assessing both the cell-mediated and humoral immune responses to CRISPR components and their delivery vectors.107 Regulatory frameworks should also adapt to these evaluations, ensuring that immunogenicity assessments are an integral part of the development pipeline for CRISPR-based therapies.
CRISPR-based therapies hold a pivotal promise in the context of HCC, primarily due to their precision in targeting genes directly involved in cancer progression and in modulating immune checkpoints in adoptive T cells. This allows for highly tailored therapeutic strategies. However, the use of CRISPR technology also rises some ethical and safety concerns, such as the risk of inadvertently editing germline cells, which could lead to heritable genetic changes.108 To mitigate this risk, it is essential to implement rigorous control measures and utilize specific delivery methods that exclusively target cancer cells while avoiding germline cells during the development and execution of in vivo CRISPR treatments.109 Additionally, nuclease-mediated genome editing, a key component of CRISPR, may unintentionally create large indels at targeted DNA sites, edit off-target DNA regions, and cause chromosomal translocations, inversions, and truncations, presenting significant safety concerns.35,110–112 However, the field has made notable progress in addressing these issues. One approach to enhance precision involves the utilization of engineered high-fidelity Cas9 variants and improved gRNAs that exhibit reduced off-target editing, thereby mitigating the risk of unintended genetic alterations.113,114 Alternatively, optimized cytosine base editors (CBEs) with reduced off-target and genotoxic effects can be employed to introduce premature stop codons, effectively knocking out coding genes (Figure 1E).115–117 This method is regarded as a safer alternative to Cas9-mediated gene disruption, as CBEs introduce a significantly lower level of DNA double-strand breaks (DSBs). This approach has been recently applied in a preclinical study for TCR T cell development for HCC and in a clinical study for CAR T cell therapy in T-cell acute lymphoblastic leukemia.22,93 Additionally, the CRISPRoff system, capable of establishing a robust and enduring epigenetic memory spanning hundreds of cell divisions, can be harnessed to suppress the expression of specific genes in T cells, including immune checkpoint regulators (Figure 1D).32 Moreover, the rigorous implementation of unbiased, genome-wide methods for assessing off-target effects is imperative to ensure the safe application of CRISPR technologies before their translation into clinical practice.118,119 These advancements underscore the ongoing efforts to mitigate the ethical and safety concerns associated with CRISPR-based interventions in the HCC treatment landscape.
The combination of CRISPR-based therapies with established treatments like sorafenib and TACE in both preclinical investigations and clinical trials for HCC offers exciting prospects.37,38,41,95 Encouragingly, these studies have demonstrated synergistic effects, suggesting the potential for improved therapeutic outcomes in HCC patients. However, the intricacies of integrating diverse modalities, including considerations related to timing, dosage, and patient selection, necessitate rigorous exploration through clinical trials to ensure both safety and efficacy. Future research endeavors may emphasize the integration of CRISPR-based targeted cancer therapy approaches with immune checkpoint inhibitors, such as atezolizumab (anti-PD-L1) or pembrolizumab (anti-PD-1). Furthermore, researchers might prioritize the development of CRISPR-engineered CAR T and TCR T cell immunotherapies for HCC, given the highly promising preclinical results (Table 2). These therapies are more likely to transition into clinical practice compared to CRISPR-based strategies that directly target tumor cells (Table 3).
To optimize the accessibility and effectiveness of CAR T cell immunotherapy, advancing CRISPR technology for the development of universal CAR T cells is essential. Through multiplex gene editing, targeting specific genes such as TRAC and beta-2 microglobulin (B2M), T cells can be engineered for allogeneic use, substantially reducing the risk of graft-versus-host disease (GvHD).99,120,121 Such advancements could not only decrease production costs and enhance the availability of CAR T cell therapies but also expedite their integration into clinical practice, thus extending this advanced treatment to a wider range of patients.120–122 Successfully implementing these strategies for HCC treatment has the potential to dramatically transform the therapeutic outlook for this challenging cancer, markedly improving patient outcomes.
One of the most important challenges in HCC treatment lies in the immunosuppressive microenvironment, which hampers the infiltration and activation of T cells within the tumor.123 CRISPR-based immunotherapies aim to overcome this obstacle by targeting immune checkpoints like PD-1 and CISH. Additionally, an innovative approach involves engineering CAR T cells to secrete single-chain variable fragments (scFvs) that bind and inhibit immune checkpoint proteins, such as PD-1 and CTLA-4.124 This strategy prevents the suppression of both CAR T cells and endogenous T cells in the tumor microenvironment and is currently undergoing clinical evaluation for advanced solid tumors, including HCC.96 While these approaches have shown promise, the complex interplay of immune cells, cytokines, and tumor cells within the microenvironment necessitates further investigation.125 In addition, it is imperative to conduct thorough investigations into resistance mechanisms against adoptive T cell therapies and potential undesired outcomes, including antigen modulation in cancer cells and CAR-related toxicities.126
Delivery tools for CRISPR components to tumor tissues in vivo and T cells ex vivo are another critical area of development.109,127 In the context of targeted cancer therapy, the critical objective is to ensure that these components effectively reach their designated targets within the liver, particularly at the tumor site, as this is fundamental for both safety and therapeutic efficacy. Innovative delivery systems designed specifically for tumor targeting, such as nanoparticles and extracellular vesicles, demonstrate promise in this regard (Table 1). However, it is imperative to subject these technologies to comprehensive assessments of their clinical feasibility and safety profiles.
CRISPR-based functional genomic screens conducted both in vitro and in vivo have paved the way for the discovery of novel genes and gene regulatory elements that hold essential roles in cancer development, tumor immunology, and immune cell function.128–130 CRISPR screens are useful for investigating tumor-specific processes, including hypoxia, immune evasion, effects of cytokines, and DNA damage. Notably, several studies performing CRISPR screens in HCC cells have identified potential therapeutic targets to impede tumor progression.49,54,55,74 Similarly, CRISPR screens in T cells have unveiled novel genes associated with T cell proliferation, activation, and antitumor activity.131–133 These discoveries can be leveraged through CRISPR-based interventions to enhance adoptive T cell therapies. Therefore, future research focusing on CRISPR screening in HCC cells or organoids to identify novel therapeutic targets, alongside investigations in T cells to unveil factors that can enhance adoptive T cell immunotherapy, represent areas of significant interest and potential advancement.
In addressing the transition of CRISPR therapies from bench to bedside, it is imperative to consider the regulatory challenges inherent in such innovative treatments. The evolving regulatory landscape requires developers to navigate complex approval processes that ensure safety and efficacy while adapting to the rapid advancements in gene editing technologies.
The field of CRISPR-based therapies for HCC holds tremendous potential. As ongoing clinical trials generate more data and regulatory approvals are pursued, these therapies may emerge as valuable additions to the repertoire of liver cancer treatments. However, it is essential to approach these advancements with caution, prioritizing patient safety and ethical considerations. In conclusion, CRISPR-based therapies for hepatocellular carcinoma are poised to bring significant improvements to cancer treatment. While challenges do exist, the application of scientific rigor, collaborative efforts, and continuous research endeavors will undoubtedly shape the promising future of HCC therapy. The incorporation of CRISPR technology into clinical practice has the potential to enhance patient outcomes and represents an exciting frontier in the fight against HCC.
Funding
The authors received no specific funding for this work.
Disclosure
The authors declare no competing interests in this work.
References
1. Sung H, Ferlay J, Siegel RL, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. Ca a Cancer J Clinicians. 2021;71(3):209–249. doi:10.3322/caac.21660
2. Llovet JM, Kelley RK, Villanueva A, et al. Hepatocellular carcinoma. Nature Reviews Disease Primers. 2021;7(1):6. doi:10.1038/s41572-020-00240-3
3. Yang JD, Hainaut P, Gores GJ, Amadou A, Plymoth A, Roberts LR. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol. 2019;16(10):589–604. doi:10.1038/s41575-019-0186-y
4. Llovet JM, Montal R, Sia D, Finn RS. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat Rev Clin Oncol. 2018;15(10):599–616. doi:10.1038/s41571-018-0073-4
5. Bruix J, Sherman M. Management of hepatocellular carcinoma: an update. Hepatology. 2011;53(3):1020–1022. doi:10.1002/hep.24199
6. Yang C, Zhang H, Zhang L, et al. Evolving therapeutic landscape of advanced hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2023;20(4):203–222. doi:10.1038/s41575-022-00704-9
7. Sangro B, Sarobe P, Hervás-Stubbs S, Melero I. Advances in immunotherapy for hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2021;18(8):525–543. doi:10.1038/s41575-021-00438-0
8. Llovet JM, Castet F, Heikenwalder M, et al. Immunotherapies for hepatocellular carcinoma. Nat Rev Clin Oncol. 2022;19(3):151–172. doi:10.1038/s41571-021-00573-2
9. Ozer M, Goksu SY, Akagunduz B, George A, Sahin I. Adoptive Cell Therapy in Hepatocellular Carcinoma: a Review of Clinical Trials. Cancers. 2023;15(6):1808.
10. Cheng AL, Qin S, Ikeda M, et al. Updated efficacy and safety data from IMbrave150: atezolizumab plus bevacizumab vs. sorafenib for unresectable hepatocellular carcinoma. J Hepatol. 2022;76(4):862–873. doi:10.1016/j.jhep.2021.11.030
11. Qin S, Chan SL, Gu S, et al. Camrelizumab plus rivoceranib versus sorafenib as first-line therapy for unresectable hepatocellular carcinoma (CARES-310): a randomised, open-label, international Phase 3 study. Lancet. 2023. doi:10.1016/S0140-6736(23)00961-3
12. Barrangou R, Fremaux C, Deveau H, et al. CRISPR Provides Acquired Resistance Against Viruses in Prokaryotes. Science. 2007;315(5819):1709–1712. doi:10.1126/science.1138140
13. Liu G, Lin Q, Jin S, Gao C. The CRISPR-Cas toolbox and gene editing technologies. Molecular Cell. 2022;82(2):333–347. doi:10.1016/j.molcel.2021.12.002
14. Geurts MH, Clevers H. CRISPR engineering in organoids for gene repair and disease modelling. Nature Reviews Bioengineering. 2023;1(1):32–45. doi:10.1038/s44222-022-00013-5
15. Kaminski MM, Abudayyeh OO, Gootenberg JS, Zhang F, Collins JJ. CRISPR-based diagnostics. Nat Biomed Eng. 2021;5(7):643–656. doi:10.1038/s41551-021-00760-7
16. Fellmann C, Gowen BG, Lin P-C, Doudna JA, Corn JE. Cornerstones of CRISPR–Cas in drug discovery and therapy. Nat Rev Drug Discov. 2017;16(2):89–100. doi:10.1038/nrd.2016.238
17. Kan MJ, Doudna JA. Treatment of Genetic Diseases With CRISPR Genome Editing. JAMA. 2022;328(10):980–981. doi:10.1001/jama.2022.13468
18. Chavez M, Chen X, Finn PB, Qi LS. Advances in CRISPR therapeutics. Nat Rev Nephrol. 2023;19(1):9–22. doi:10.1038/s41581-022-00636-2
19. Katti A, Diaz BJ, Caragine CM, Sanjana NE, Dow LE. CRISPR in cancer biology and therapy. Nat Rev Cancer. 2022;22(5):259–279. doi:10.1038/s41568-022-00441-w
20. Awwad SW, Serrano-Benitez A, Thomas JC, Gupta V, Jackson SP. Revolutionizing DNA repair research and cancer therapy with CRISPR–Cas screens. Nat Rev Mol Cell Biol. 2023;24(7):477–494. doi:10.1038/s41580-022-00571-x
21. Palaz F, Kalkan AK, Can Ö, et al. CRISPR-Cas13 System as a Promising and Versatile Tool for Cancer Diagnosis, Therapy, and Research. ACS Synth. Biol. 2021;10(6):1245–1267. doi:10.1021/acssynbio.1c00107
22. Chiesa R, Georgiadis C, Syed F, et al. Base-Edited CAR7 T Cells for Relapsed T-Cell Acute Lymphoblastic Leukemia. N Engl J Med. 2023;389(10):899–910. doi:10.1056/NEJMoa2300709
23. Stadtmauer EA, Fraietta JA, Davis MM, et al. CRISPR-engineered T cells in patients with refractory cancer. Science. 2020;367(6481):eaba7365. doi:10.1126/science.aba7365
24. Ottaviano G, Georgiadis C, Gkazi SA, et al. Phase 1 clinical trial of CRISPR-engineered CAR19 universal T cells for treatment of children with refractory B cell leukemia. Sci, trans med. 2022;14(668):eabq3010. doi:10.1126/scitranslmed.abq3010
25. Lu Y, Xue J, Deng T, et al. Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer. Nature Med. 2020;26(5):732–740. doi:10.1038/s41591-020-0840-5
26. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A Programmable Dual-RNA–Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science. 2012;337(6096):816–821. doi:10.1126/science.1225829
27. Gasiunas G, Barrangou R, Horvath P, Siksnys V. Cas9–crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci. 2012;109(39):E2579–E2586. doi:10.1073/pnas.1208507109
28. Cong L, Ran FA, Cox D, et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science. 2013;339(6121):819–823. doi:10.1126/science.1231143
29. Anzalone AV, Koblan LW, Liu DR. Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nature Biotechnol. 2020;38(7):824–844. doi:10.1038/s41587-020-0561-9
30. Raguram A, Banskota S, Liu DR. Therapeutic in vivo delivery of gene editing agents. Cell. 2022;185(15):2806–2827. doi:10.1016/j.cell.2022.03.045
31. Pickar-Oliver A, Gersbach CA. The next generation of CRISPR–Cas technologies and applications. Nat Rev Mol Cell Biol. 2019;20(8):490–507. doi:10.1038/s41580-019-0131-5
32. Nuñez JK, Chen J, Pommier GC, et al. Genome-wide programmable transcriptional memory by CRISPR-based epigenome editing. Cell. 2021;184(9):2503–2519.e17. doi:10.1016/j.cell.2021.03.025
33. Porto EM, Komor AC, Slaymaker IM, Yeo GW. Base editing: advances and therapeutic opportunities. Nat Rev Drug Discov. 2020;19(12):839–859. doi:10.1038/s41573-020-0084-6
34. Kuscu C, Parlak M, Tufan T, et al. CRISPR-STOP: gene silencing through base-editing-induced nonsense mutations. Nature Methods. 2017;14(7):710–712. doi:10.1038/nmeth.4327
35. Kosicki M, Tomberg K, Bradley A. Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nature Biotechnol. 2018;36(8):765–771. doi:10.1038/nbt.4192
36. Chen PJ, Liu DR. Prime editing for precise and highly versatile genome manipulation. Nat Rev Genet. 2023;24(3):161–177. doi:10.1038/s41576-022-00541-1
37. Qi Y, Liu Y, Yu B, et al. A Lactose-Derived CRISPR/Cas9 Delivery System for Efficient Genome Editing In Vivo to Treat Orthotopic Hepatocellular Carcinoma. Adv. Sci. 2020;7(17):2001424. doi:10.1002/advs.202001424
38. Nie -J-J, Liu Y, Qi Y, et al. Charge-reversal nanocomolexes-based CRISPR/Cas9 delivery system for loss-of-function oncogene editing in hepatocellular carcinoma. J Control Release. 2021;333:362–373. doi:10.1016/j.jconrel.2021.03.030
39. Gao J, Luo T, Lin N, Zhang S, Wang J. A New Tool for CRISPR-Cas13a-Based Cancer Gene Therapy. Molecular Therapy - Oncolytics. 2020;19:79–92. doi:10.1016/j.omto.2020.09.004
40. Zhang B-C, Luo B-Y, Zou -J-J, et al. Co-delivery of Sorafenib and CRISPR/Cas9 Based on Targeted Core–Shell Hollow Mesoporous Organosilica Nanoparticles for Synergistic HCC Therapy. ACS Appl. Mater. Interfaces. 2020;12(51):57362–57372. doi:10.1021/acsami.0c17660
41. Liu Q, Fan D, Adah D, et al. CRISPR/Cas9‑mediated hypoxia inducible factor‑1α knockout enhances the antitumor effect of transarterial embolization in hepatocellular carcinoma. Oncol Rep. 2018;40(5):2547–2557. doi:10.3892/or.2018.6667
42. Zhang B-C, P-Y W, Zou -J-J, et al. Efficient CRISPR/Cas9 gene-chemo synergistic cancer therapy via a stimuli-responsive chitosan-based nanocomplex elicits anti-tumorigenic pathway effect. Chem Eng J. 2020;393:124688. doi:10.1016/j.cej.2020.124688
43. Zhuang J, Tan J, Wu C, et al. Extracellular vesicles engineered with valency-controlled DNA nanostructures deliver CRISPR/Cas9 system for gene therapy. Nucleic Acids Res. 2020;48(16):8870–8882. doi:10.1093/nar/gkaa683
44. Yin H, Sun L, Pu Y, et al. Ultrasound-Controlled CRISPR/Cas9 System Augments Sonodynamic Therapy of Hepatocellular Carcinoma. ACS Cent. Sci. 2021;7(12):2049–2062. doi:10.1021/acscentsci.1c01143
45. He C, Jaffar Ali D, Qi Y, et al. Engineered extracellular vesicles mediated CRISPR-induced deficiency of IQGAP1/FOXM1 reverses sorafenib resistance in HCC by suppressing cancer stem cells. J Nanobiotechnol. 2023;21(1):154. doi:10.1186/s12951-023-01902-6
46. He C, Jaffar Ali D, Xu H, et al. Epithelial cell -derived microvesicles: a safe delivery platform of CRISPR/Cas9 conferring synergistic anti-tumor effect with sorafenib. Exp. Cell. Res. 2020;392(2):112040. doi:10.1016/j.yexcr.2020.112040
47. Wan T, Zhong J, Pan Q, Zhou T, Ping Y, Liu X. Exosome-mediated delivery of Cas9 ribonucleoprotein complexes for tissue-specific gene therapy of liver diseases. Sci Adv. 2022;8(37):eabp9435. doi:10.1126/sciadv.abp9435
48. Li C, Yang T, Weng Y, et al. Ionizable lipid-assisted efficient hepatic delivery of gene editing elements for oncotherapy. Bioact. Mater. 2022;9:590–601. doi:10.1016/j.bioactmat.2021.05.051
49. Jiang T, Sánchez‐Rivera FJ, Soto‐Feliciano YM, et al. Targeting the De Novo Purine Synthesis Pathway Through Adenylosuccinate Lyase Depletion Impairs Liver Cancer Growth by Perturbing Mitochondrial Function. Hepatology. 2021;74(1):458.
50. Sun Z, Xue S, Zhang M, et al. Aberrant NSUN2-mediated m5C modification of H19 lncRNA is associated with poor differentiation of hepatocellular carcinoma. Oncogene. 2020;39(45):6906–6919. doi:10.1038/s41388-020-01475-w
51. Xiong L, Wu F, Wu Q, et al. Aberrant enhancer hypomethylation contributes to hepatic carcinogenesis through global transcriptional reprogramming. Nat Commun. 2019;10(1):335. doi:10.1038/s41467-018-08245-z
52. Wang X, Zhang W, Ding Y, Guo X, Yuan Y, Li D. CRISPR/Cas9-mediated genome engineering of CXCR4 decreases the malignancy of hepatocellular carcinoma cells in vitro and in vivo. Oncol Rep. 2017;37(6):3565–3571. doi:10.3892/or.2017.5601
53. Zhang S, Zhang F, Chen Q, Wan C, Xiong J, Xu J. CRISPR/Cas9-mediated knockout of NSD1 suppresses the hepatocellular carcinoma development via the NSD1/H3/Wnt10b signaling pathway. J Exp Clin Cancer Res. 2019;38(1):467. doi:10.1186/s13046-019-1462-y
54. Kwan S-Y, Sheel A, Song C-Q, et al. Depletion of TRRAP Induces p53-Independent Senescence in Liver Cancer by Down-Regulating Mitotic Genes. Hepatology. 2020;71(1):275–290. doi:10.1002/hep.30807
55. Bao MH-R, Yang C, Tse AP-W, et al. Genome-wide CRISPR-Cas9 knockout library screening identified PTPMT1 in cardiolipin synthesis is crucial to survival in hypoxia in liver cancer. Cell Rep. 2021;34(4):108676. doi:10.1016/j.celrep.2020.108676
56. Wei L, Chiu DK-C, Tsang FH-C, et al. Histone methyltransferase G9a promotes liver cancer development by epigenetic silencing of tumor suppressor gene RARRES3. J Hepatol. 2017;67(4):758–769. doi:10.1016/j.jhep.2017.05.015
57. Fu L, Wang X, Yang Y, et al. Septin11 promotes hepatocellular carcinoma cell motility by activating RhoA to regulate cytoskeleton and cell adhesion. Cell Death Dis. 2023;14(4):280. doi:10.1038/s41419-023-05726-y
58. Lu J, Ding Y, Zhang W, et al. SQSTM1/p62 Knockout by Using the CRISPR/Cas9 System Inhibits Migration and Invasion of Hepatocellular Carcinoma. Cells. 2023;12(9):1238.
59. Ardelt MA, Fröhlich T, Martini E, et al. Inhibition of Cyclin‐Dependent Kinase 5: a Strategy to Improve Sorafenib Response in Hepatocellular Carcinoma Therapy. Hepatology. 2019;69(1):634.
60. Liu H, Li D, Zhou L, et al. LMNA functions as an oncogene in hepatocellular carcinoma by regulating the proliferation and migration ability. J Cell & Mol Med. 2020;24(20):12008–12019. doi:10.1111/jcmm.15829
61. He L, Fan X, Li Y, et al. Overexpression of zinc finger protein 384 (ZNF 384), a poor prognostic predictor, promotes cell growth by upregulating the expression of Cyclin D1 in Hepatocellular carcinoma. Cell Death Dis. 2019;10(6):444. doi:10.1038/s41419-019-1681-3
62. Li B, Cao Y, Meng G, et al. Targeting glutaminase 1 attenuates stemness properties in hepatocellular carcinoma by increasing reactive oxygen species and suppressing Wnt/beta-catenin pathway. EBioMedicine. 2019;39:239–254. doi:10.1016/j.ebiom.2018.11.063
63. Zhu P, Wang Y, He L, et al. ZIC2-dependent OCT4 activation drives self-renewal of human liver cancer stem cells. J Clin Invest. 2015;125(10):3795–3808. doi:10.1172/JCI81979
64. Lu Y, Chan Y-T, Tan H-Y, et al. Epigenetic regulation of ferroptosis via ETS1/miR-23a-3p/ACSL4 axis mediates sorafenib resistance in human hepatocellular carcinoma. J Exp Clin Cancer Res. 2022;41(1):3. doi:10.1186/s13046-021-02208-x
65. Tsai C-N, S-C Y, Lee C-W, et al. SOX4 activates CXCL12 in hepatocellular carcinoma cells to modulate endothelial cell migration and angiogenesis in vivo. Oncogene. 2020;39(24):4695–4710. doi:10.1038/s41388-020-1319-z
66. Luo J, Wang D, Zhang S, et al. BolA family member 2 enhances cell proliferation and predicts a poor prognosis in hepatocellular carcinoma with tumor hemorrhage. J Cancer. 2019;10(18):4293–4304. doi:10.7150/jca.31829
67. Zhou Y, Shi W-Y, He W, et al. FAM122A supports the growth of hepatocellular carcinoma cells and its deletion enhances Doxorubicin-induced cytotoxicity. Exp. Cell. Res. 2020;387(1):111714. doi:10.1016/j.yexcr.2019.111714
68. Jiang J, Chen Y, Zhang L, et al. i-CRISPR: a personalized cancer therapy strategy through cutting cancer-specific mutations. Mol Cancer. 2022;21(1):164. doi:10.1186/s12943-022-01612-x
69. Iwagami Y, Huang CK, Olsen MJ, et al. Aspartate β‐hydroxylase modulates cellular senescence through glycogen synthase kinase 3β in hepatocellular carcinoma. Hepatology. 2016;63(4).
70. Wang H, Guo R, Du Z, et al. Epigenetic Targeting of Granulin in Hepatoma Cells by Synthetic CRISPR dCas9 Epi-suppressors. Mol Ther Nucleic Acids. 2018;11:23–33. doi:10.1016/j.omtn.2018.01.002
71. Sgro A, Cursons J, Waryah C, et al. Epigenetic reactivation of tumor suppressor genes with CRISPRa technologies as precision therapy for hepatocellular carcinoma. Clin epigenetics. 2023;15(1):73. doi:10.1186/s13148-023-01482-0
72. Ye S, Ni Y. lncRNA SNHG9 Promotes Cell Proliferation, Migration, and Invasion in Human Hepatocellular Carcinoma Cells by Increasing GSTP1 Methylation, as Revealed by CRISPR-dCas9. Original Research. Front Mol Biosci. 2021;8. doi:10.3389/fmolb.2021.649976
73. Cheng F, Hansson VC, Georgolopoulos G, Mani K. Attenuation of cancer proliferation by suppression of glypican-1 and its pleiotropic effects in neoplastic behavior. Glypican-1; TCGA; bladder carcinoma; hepatocellular carcinoma; glioma. Oncotarget. 2023;14(1).
74. Wei L, Lee D, Law C-T, et al. Genome-wide CRISPR/Cas9 library screening identified PHGDH as a critical driver for Sorafenib resistance in HCC. Nat Commun. 2019;10(1):4681. doi:10.1038/s41467-019-12606-7
75. Chen D, Zou J, Zhao Z, et al. TXNDC9 promotes hepatocellular carcinoma progression by positive regulation of MYC-mediated transcriptional network. Cell Death Dis. 2018;9(11):1110. doi:10.1038/s41419-018-1150-4
76. Wang W, Hu B, Qin -J-J, et al. A novel inhibitor of MDM2 oncogene blocks metastasis of hepatocellular carcinoma and overcomes chemoresistance. Genes Dis. 2019;6(4):419–430. doi:10.1016/j.gendis.2019.06.001
77. Pott LL, Hagemann S, Reis H, et al. Eukaryotic elongation factor 2 is a prognostic marker and its kinase a potential therapeutic target in HCC. Oncotarget. 2017;8(7).
78. Lee HK, Lim HM, Park S-H, Nam MJ. Knockout of Hepatocyte Growth Factor by CRISPR/Cas9 System Induces Apoptosis in Hepatocellular Carcinoma Cells. Journal of Personalized Medicine. 2021;11(10):983.
79. He J, Zhang W, Li A, Chen F, Luo R. Knockout of NCOA5 impairs proliferation and migration of hepatocellular carcinoma cells by suppressing epithelial-to-mesenchymal transition. Biochem. Biophys. Res. Commun. 2018;500(2):177–183. doi:10.1016/j.bbrc.2018.04.017
80. Shangguan D, Meng L, Cao ZC, et al. Identification of Liver Cancer-Specific Aptamers Using Whole Live Cells. Anal. Chem. 2008;80(3):721–728. doi:10.1021/ac701962v
81. Kubes P, Jenne C. Immune Responses in the Liver. Ann Rev Immunol. 2018;36(1):247–277. doi:10.1146/annurev-immunol-051116-052415
82. Zheng M, Tian Z. Liver-Mediated Adaptive Immune Tolerance. Front Immunol. 2019;10:2525. doi:10.3389/fimmu.2019.02525
83. Wang V, Gauthier M, Decot V, Reppel L, Bensoussan D. Systematic Review on CAR-T Cell Clinical Trials Up to 2022: academic Center Input. Cancers. 2023;15(4):1003.
84. Shi D, Shi Y, Kaseb AO, et al. Chimeric Antigen Receptor-Glypican-3 T-Cell Therapy for Advanced Hepatocellular Carcinoma: results of Phase I Trials. Clin Cancer Res. 2020;26(15):3979–3989. doi:10.1158/1078-0432.Ccr-19-3259
85. Sangro B, Borad MJ, Hausner PF, et al. LBO12 - Data from the third dose cohort of an ongoing study with ADP-A2AFP SPEAR T cells. J Hepatol. 2020;73:S122. doi:10.1016/S0168-8278(20)30761-3
86. Chow A, Perica K, Klebanoff CA, Wolchok JD. Clinical implications of T cell exhaustion for cancer immunotherapy. Nat Rev Clin Oncol. 2022;19(12):775–790. doi:10.1038/s41571-022-00689-z
87. van Loenen MM, de Boer R, Amir AL, et al. Mixed T cell receptor dimers harbor potentially harmful neoreactivity. Proc Natl Acad Sci. 2010;107(24):10972–10977. doi:10.1073/pnas.1005802107
88. Chan JD, Lai J, Slaney CY, Kallies A, Beavis PA, Darcy PK. Cellular networks controlling T cell persistence in adoptive cell therapy. Nat Rev Immunol. 2021;21(12):769–784. doi:10.1038/s41577-021-00539-6
89. Guo X, Jiang H, Shi B, et al. Disruption of PD-1 Enhanced the Anti-tumor Activity of Chimeric Antigen Receptor T Cells Against Hepatocellular Carcinoma. Front Pharmacol. 2018;9. doi:10.3389/fphar.2018.01118
90. Rupp LJ, Schumann K, Roybal KT, et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep. 2017;7(1):737. doi:10.1038/s41598-017-00462-8
91. Huang K, Sun B, Luo N, Guo H, Hu J, Peng J. Programmed Death Receptor 1 (PD1) Knockout and Human Telomerase Reverse Transcriptase (hTERT) Transduction Can Enhance Persistence and Antitumor Efficacy of Cytokine-Induced Killer Cells Against Hepatocellular Carcinoma. Med Sci Monit. 2018;24:4573–4582. doi:10.12659/msm.910903
92. Lee JH, Lee J-H, Lim Y-S, et al. Adjuvant Immunotherapy With Autologous Cytokine-Induced Killer Cells for Hepatocellular Carcinoma. Gastroenterology. 2015;148(7):1383–1391.e6. doi:10.1053/j.gastro.2015.02.055
93. Preece R, Pavesi A, Gkazi SA, et al. CRISPR-Mediated Base Conversion Allows Discriminatory Depletion of Endogenous T Cell Receptors for Enhanced Synthetic Immunity. Mol Ther Methods Clin Dev. 2020;19:149–161. doi:10.1016/j.omtm.2020.09.002
94. Mo F, Duan S, Jiang X, et al. Nanobody-based chimeric antigen receptor T cells designed by CRISPR/Cas9 technology for solid tumor immunotherapy. Signal Transduction and Targeted Therapy. 2021;6(1):80. doi:10.1038/s41392-021-00462-1
95. TACE Combined With PD-1 Knockout Engineered T Cell in Advanced Hepatocellular Carcinoma. Available from: https://clinicaltrials.gov/study/NCT04417764.
96. Engineered TILs/CAR-TILs to Treat Advanced Solid Tumors. Available from: https://clinicaltrials.gov/study/NCT04842812.
97. Palmer DC, Webber BR, Patel Y, et al. Internal checkpoint regulates T cell neoantigen reactivity and susceptibility to PD1 blockade. Med. 2022;3(10):682–704.e8. doi:10.1016/j.medj.2022.07.008
98. A Study of Metastatic Gastrointestinal Cancers Treated With Tumor Infiltrating Lymphocytes in Which the Gene Encoding the Intracellular Immune Checkpoint CISH Is Inhibited Using CRISPR Genetic Engineering. Available from: https://clinicaltrials.gov/study/NCT04426669.
99. Webber BR, Lonetree C-L, Kluesner MG, et al. Highly efficient multiplex human T cell engineering without double-strand breaks using Cas9 base editors. Nat Commun. 2019;10(1):5222. doi:10.1038/s41467-019-13007-6
100. Eyquem J, Mansilla-Soto J, Giavridis T, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543(7643):113–117. doi:10.1038/nature21405
101. Anliker B. Regulatory Considerations for Clinical Trial Applications with CRISPR-Based Medicinal Products. CRISPR J. 2022;5(3):364–376. doi:10.1089/crispr.2021.0148
102. Xu X, Liu C, Wang Y, et al. Nanotechnology-based delivery of CRISPR/Cas9 for cancer treatment. Adv. Drug Delivery Rev. 2021;176:113891. doi:10.1016/j.addr.2021.113891
103. Guo C, Ma X, Gao F, Guo Y. Off-target effects in CRISPR/Cas9 gene editing. Front Bioeng Biotechnol. 2023;11:43157. doi:10.3389/fbioe.2023.1143157
104. Crudele JM, Chamberlain JS. Cas9 immunity creates challenges for CRISPR gene editing therapies. Nat Commun. 2018;9(1):3497. doi:10.1038/s41467-018-05843-9
105. Kenjo E, Hozumi H, Makita Y, et al. Low immunogenicity of LNP allows repeated administrations of CRISPR-Cas9 mRNA into skeletal muscle in mice. Nat Commun. 2021;12(1):7101. doi:10.1038/s41467-021-26714-w
106. Ewaisha R, Anderson KS. Immunogenicity of CRISPR therapeutics—Critical considerations for clinical translation. Front Bioeng Biotechnol. 2023;11:38596. doi:10.3389/fbioe.2023.1138596
107. Pierce Eric A, Aleman Tomas S, Jayasundera Kanishka T, et al. Gene Editing for CEP290-Associated Retinal Degeneration. N Engl J Med. 2024. doi:10.1056/NEJMoa2309915
108. Brokowski C, Adli M. CRISPR Ethics: moral Considerations for Applications of a Powerful Tool. J Mol Biol. 2019;431(1):88–101. doi:10.1016/j.jmb.2018.05.044
109. Madigan V, Zhang F, Dahlman JE. Drug delivery systems for CRISPR-based genome editors. Nat Rev Drug Discov. 2023;22(11):875.
110. Leibowitz ML, Papathanasiou S, Doerfler PA, et al. Chromothripsis as an on-target consequence of CRISPR–Cas9 genome editing. Nature Genet. 2021;53(6):895–905. doi:10.1038/s41588-021-00838-7
111. Cullot G, Boutin J, Toutain J, et al. CRISPR-Cas9 genome editing induces megabase-scale chromosomal truncations. Nat Commun. 2019;10(1):1136. doi:10.1038/s41467-019-09006-2
112. Turchiano G, Andrieux G, Klermund J, et al. Quantitative evaluation of chromosomal rearrangements in gene-edited human stem cells by CAST-Seq. Cell Stem Cell. 2021;28(6):1136–1147.e5. doi:10.1016/j.stem.2021.02.002
113. Kleinstiver BP, Pattanayak V, Prew MS, et al. High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529(7587):490–495. doi:10.1038/nature16526
114. Akcakaya P, Bobbin ML, Guo JA, et al. In vivo CRISPR editing with no detectable genome-wide off-target mutations. Nature. 2018;561(7723):416–419. doi:10.1038/s41586-018-0500-9
115. Fiumara M, Ferrari S, Omer-Javed A, et al. Genotoxic effects of base and prime editing in human hematopoietic stem cells. Nature Biotechnol. 2023:7–15.
116. Neugebauer ME, Hsu A, Arbab M, et al. Evolution of an adenine base editor into a small, efficient cytosine base editor with low off-target activity. Nature Biotechnol. 2023;41(5):673–685. doi:10.1038/s41587-022-01533-6
117. Lam DK, Feliciano PR, Arif A, et al. Improved cytosine base editors generated from TadA variants. Nature Biotechnol. 2023;41(5):686–697. doi:10.1038/s41587-022-01611-9
118. Kim D, Kang B-C, Kim J-S. Identifying genome-wide off-target sites of CRISPR RNA–guided nucleases and deaminases with Digenome-seq. Nature Protocols. 2021;16(2):1170–1192. doi:10.1038/s41596-020-00453-6
119. Zou RS, Liu Y, Gaido OER, et al. Improving the sensitivity of in vivo CRISPR off-target detection with DISCOVER-Seq+. Nature Methods. 2023;20(5):706–713. doi:10.1038/s41592-023-01840-z
120. Liu X, Zhang Y, Cheng C, et al. CRISPR-Cas9-mediated multiplex gene editing in CAR-T cells. Cell Res. 2017;27(1):154–157. doi:10.1038/cr.2016.142
121. Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin Cancer Res. 2017;23(9):2255–2266. doi:10.1158/1078-0432.Ccr-16-1300
122. Dimitri A, Herbst F, Fraietta JA. Engineering the next-generation of CAR T-cells with CRISPR-Cas9 gene editing. Mol Cancer. 2022;21(1):78. doi:10.1186/s12943-022-01559-z
123. Nishida N. Role of Oncogenic Pathways on the Cancer Immunosuppressive Microenvironment and Its Clinical Implications in Hepatocellular Carcinoma. Cancers. 2021;13(15):3666.
124. Rafiq S, Yeku OO, Jackson HJ, et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nature Biotechnol. 2018;36(9):847–856. doi:10.1038/nbt.4195
125. Guizhen Z, Guanchang J, Liwen L, et al. The tumor microenvironment of hepatocellular carcinoma and its targeting strategy by CAR-T cell immunotherapy. Front Endocrinol. 2022;13:869. doi:10.3389/fendo.2022.918869
126. Shah NN, Fry TJ. Mechanisms of resistance to CAR T cell therapy. Nat Rev Clin Oncol. 2019;16(6):372–385. doi:10.1038/s41571-019-0184-6
127. Song X, Liu C, Wang N, et al. Delivery of CRISPR/Cas systems for cancer gene therapy and immunotherapy. Adv. Drug Delivery Rev. 2021;168:158–180. doi:10.1016/j.addr.2020.04.010
128. Buquicchio FA, Satpathy AT. Interrogating immune cells and cancer with CRISPR-Cas9. Trends in Immunology. 2021;42(5):432–446. doi:10.1016/j.it.2021.03.003
129. Bock C, Datlinger P, Chardon F, et al. High-content CRISPR screening. Nat Rev Method Primers. 2022;2(1):8. doi:10.1038/s43586-021-00093-4
130. Shi H, Doench JG, Chi H. CRISPR screens for functional interrogation of immunity. Nat Rev Immunol. 2023;23(6):363–380. doi:10.1038/s41577-022-00802-4
131. Dong MB, Wang G, Chow RD, et al. Systematic Immunotherapy Target Discovery Using Genome-Scale In Vivo CRISPR Screens in CD8 T Cells. Cell. 2019;178(5):1189–1204.e23. doi:10.1016/j.cell.2019.07.044
132. Gurusamy D, Henning AN, Yamamoto TN, et al. Multi-phenotype CRISPR-Cas9 Screen Identifies p38 Kinase as a Target for Adoptive Immunotherapies. Cancer Cell. 2020;37(6):818–833.e9. doi:10.1016/j.ccell.2020.05.004
133. Mamedov MR, Vedova S, Freimer JW, et al. CRISPR screens decode cancer cell pathways that trigger γδ T cell detection. Nature. 2023;621(7977):188–195.
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.