Back to Journals » OncoTargets and Therapy » Volume 7
CP-673451, a platelet-derived growth-factor receptor inhibitor, suppresses lung cancer cell proliferation and migration
Authors Xi Y, Chen M, Liu X, Lu Z, Ding Y, Li D, Zhou J
Received 24 February 2014
Accepted for publication 8 April 2014
Published 3 July 2014 Volume 2014:7 Pages 1215—1221
DOI https://doi.org/10.2147/OTT.S62946
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Yuling Xi,1 Ming Chen,1 Xinmin Liu,1 Zhongmin Lu,1 Yi Ding,1 Datong Li2
1First Affiliated Hospital of Xinxiang Medical University, 2Hualan Vaccine, Xinxiang, People's Republic of China
Abstract: Lung cancer is the leading cause of cancer mortality in the world. Although some advances in lung cancer therapy have been made, patient survival is still poor. The platelet-derived growth factor receptors (PDGFRs) and their ligands play critical roles in the regulation of many cancer cell processes, including cell survival and cell motility. Herein, we investigate the anticancer activities of CP-673451, a potent selective inhibitor of PDGFR kinase, in non-small-cell lung cancer (NSCLC) therapy. We found that CP-673451 is effective at suppressing cell viability, inducing cell apoptosis, and inhibiting cell migration and invasion by suppressing the PDGFR downstream signaling pathway in NSCLC cells. Furthermore, CP-673451 is effective at suppressing NSCLC tumor growth in vivo. In summary, our studies suggest that CP-673451 might be a promising therapeutic compound for NSCLC.
Keywords: CP-673451, platelet-derived growth factor receptors (PDGFRs), lung cancer, proliferation, migration
Introduction
Lung cancer is a leading deadly cancer and the most common cause of cancer death worldwide.1–4 Non-small-cell lung cancer (NSCLC) is the most frequent type of lung cancer, accounting for about 85% of total lung cancer.5–8 Although a lot of research has been done on lung cancer, the overall survival rate is still very low.5,6
The platelet-derived growth factor receptors (PDGFRs) and their ligands are involved in proliferation, migration, and invasion signaling in cells.9–13 The ligand binding induces receptor dimerization, enabling autophosphorylation of some tyrosine residues and recruitment of a series of signaling molecules.9,10 It is shown that inhibition of PDGFR signaling disrupts cancer cell survival.13 Moreover, PDGFR overexpression has been observed in metastatic versus nonmetastatic cancer patient samples, and disruption of PDGFR function inhibited the metastatic potential of cancer cells.14 In lung cancer, especially NSCLC, cytoplasmic PDGFR expression by tumors is a negative prognostic indicator.15 Given its critical roles in cell survival and metastatic signaling, PDGFRs have become an attractive therapeutic target in a number of human malignancies, including lung cancer.
CP-673451 is a potent inhibitor of PDGFR kinase, being more than 450-fold more selective for PDGFRβ versus other receptors.16 Although some research has been done to characterize the pharmacology of PDGFRβ inhibition by CP-673451 and its effect on tumor growth, its potential anticancer activity, especially on NSCLC, is far from clear. In this study, we investigated the potential anticancer activities of CP-673451 in NSCLC therapy.
Materials and methods
Materials
CP-673451 (Selleck Chemicals, Houston, TX, USA) was used in all experiments to evaluate its anticancer activities. Rabbit anti-phospho-Akt (Ser473; catalog number 4060), anti-phospho-p70S6K (Thr389; 9205), anti-phospho-S6 (Ser235/236; 2211), anti-phospho-GSK-3β (Ser9; 9336), anti-p70S6K (9202), anti-GSK-3β (9315), anti-PDGFRβ (28E1; 3169), anti-phospho-PDGRα (Tyr849)/PDGFβ (Tyr857 [C43E9]; 3170), anti-phospho-Bad (Ser136 [D25H8]; 4366), anti-β-actin (4967), and secondary horseradish peroxidase-conjugated antibody (7074) (all Cell Signaling Technology, Danvers, MA, USA) were used in Western blot analysis.
Cells and cell culture
Human lung cancer cell lines A549 and H1299 were purchased from the American Type Culture Collection (Manassas, VA, USA), and grown in Roswell Park Memorial Institute medium containing 10% heat-inactivated fetal bovine serum (FBS) with 2 mM L-glutamine and 100 U · mL−1 penicillin–streptomycin in a humidified incubator at 37°C in 5% CO2.
Cell-viability assay
Cells were seeded in triplicate in 96-well plates, treated with CP-673451 for 24,48, or 72 hours, and analyzed using a CellTiter-Glo® assay kit (Promega, Fitchburg, WI, USA) according to the manufacturer’s instructions.
Transwell assay
Briefly, 1 × 105 cells in media supplemented with 1% FBS were plated into the upper chambers of the 24-transwell Boyden chamber wells (Costar®; Sigma-Aldrich, St Louis, MO, USA), and 650 μL media supplemented with 10% FBS was added into lower chambers to use as a chemoattractant. After treatment with CP-673451 for 12 hours, the cells were fixed and stained with 0.1% crystal violet. The nonmigrated cells were wiped off, and the migrated cells were photographed. Then, the cells were lysed with 10% acetic acid, and absorbance was measured at 595 nm.
Wound-healing assay
Cells were seeded in 96-well plates. Once 100% confluence was observed, the cell monolayer was scratched with a pipette tip. After being washed three times, the cells were treated with CP-673451 for 12 hours. Then, the scratches were photographed and the width of scratches measured.
Invasion assay
Briefly, the upper chamber of a 24-transwell Boyden chamber was coated with 1 mg/mL of Matrigel™ (BD Biosciences, San Jose, CA, USA) for 4 hours at 37°C, and then 1 × 105 cells in media supplemented with 1% FBS were plated into the upper chambers of the 24-transwell Boyden chamber wells and 650 μL media supplemented with 10% FBS added into the lower chambers to use as a chemoattractant. After treatment with CP-673451 for 12 hours, the cells were fixed and stained with 0.1% crystal violet. The nonmigrated cells were wiped off, and the migrated cells were photographed. Then, the cells were lysed with 10% acetic acid and absorbance measured at 595 nm.
Fluorescent immunocytochemistry
Cells were seeded on glass coverslips, followed by treatments with compounds. Then, cells were fixed with 4% polyformaldehyde for 30 minutes, permeabilized with 0.1% Triton X-100 for 20 minutes, and blocked with 5% normal serum for 30 minutes. For detection of actin fibers, cells were incubated with tetramethylrhodamine isothiocyanate-labeled phalloidine for 30 minutes. For detection of nuclei, cells were incubated with Hoechst for 10 minutes. Nuclei images were taken with a fluorescent microscope.
Flow-cytometry analysis
Cells treated with CP-673451 were harvested, fixed, and stained with propidium iodide by using a Cycletest™ Plus deoxyribonucleic acid (DNA) reagent kit (BD Biosciences). Then, the cells were analyzed for DNA content with the use of FACSCalibur™ flow cytometry and CellQuest™ Pro software (BD Biosciences).
Western blot analysis
After treatment with CP-673451, the total proteins were extracted with radioimmunoprecipitation assay buffer (0.15 mM NaCl, 0.05 mM Tris-HCl, pH 7.5, 1% Triton, 0.1% sodium dodecyl sulfate, 0.1% sodium deoxycholate, and 1% Nonidet P40). Then, the sample extracts were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis separation, and the proteins were transferred to nitrocellulose membranes. The membranes were blocked for 1 hour with 5% nonfat dry milk in Tris-buffered saline containing 0.05% Tween 20, incubated overnight with primary antibody, washed and incubated with secondary antibody, and visualized by chemiluminescence.
Immunohistochemistry analysis
Tumor samples were harvested from mice, trimmed into 5 mm3 pieces, and immediately fixed in 10% neutral buffered formalin, then transferred to 70% ethanol, embedded into paraffin blocks, and sectioned (5 mm thickness). A terminal deoxynucleotidyl transferase deoxyuridine triphosphate nick-end labeling (TUNEL; Promega, Madison, WI, USA) assay was performed according to the manufacturer’s instructions. The nuclei were stained with Hoechst for 20 minutes. The slides were photographed using a fluorescent microscope.
Efficacy studies in mouse xenograft model in vivo
The nude mice were housed under specific pathogen-free conditions. The animal use and experimental protocol were reviewed and approved by the Animal Care and Use Committee of the First Affiliated Hospital of Xinxiang Medical University. A subcutaneous A549 xenograft model in nude mice was used to evaluate the anticancer activity of CP-673451. Briefly, A549 cells were injected into the axillary regions of mice (2 × 106 cells/mouse). When the tumor volumes reached 70 mm3, the mice were randomly assigned to a control group and two CP-673451 groups (n=6 per group): low-dose (20 mg/kg) and high dose (40 mg/kg) groups (vehicle 10% 1-methyl-2-pyrrolidinone and 90% polyethylene glycol 300). These animals were administered intraperitoneally with CP-673451 (20 or 40 mg/kg/day) or with vehicle. During the treatment period, the implanted tumors were measured by caliper once a day in a blind fashion. The animal body weights were also measured at the same time. The tumor volume was calculated as follows: (width × width × length)/2. After treatment, the mice were killed, and the tumors were harvested and analyzed.
Statistics
Student’s t-test and analysis of variance were performed using StatView (SAS Institute, Cary, NC, USA). The data shown are the mean values of triplicate measurements with error bars corresponding to standard deviation.
Results
CP-673451 inhibits PDGFR-mediated signaling pathway in A549 cells
PDGFRs have been suggested as potential targets in cancer therapy. CP-673451 is a known specific inhibitor of PDGFR.16 To explore the roles of CP-673451 in therapeutics for NSCLC, we first examined the effect of CP-673451 on PDGFR-mediated signaling in A549, an NSCLC cell line. As shown in Figure 1, CP-673451 efficiently suppressed the PDGFR downstream signaling pathway. It inhibited phosphorylation of Akt, GSK-3β, p70S6, and S6 in A549 cells in a concentration-dependent manner. In contrast, no significant changes were observed in total protein levels after treatment.
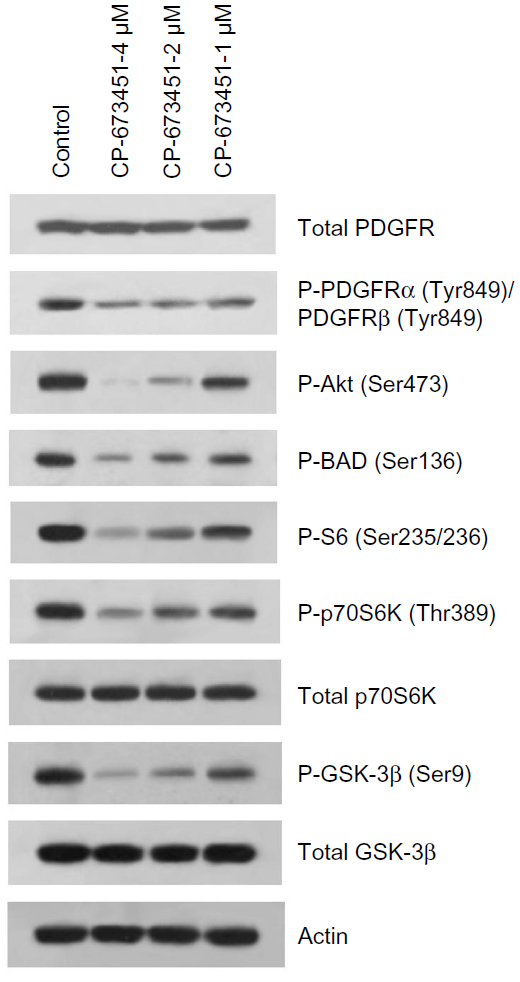 | Figure 1 CP-673451 inhibits the PDGFR-mediated signaling pathway in non-small-cell lung cancer cells. |
CP-673451 is effective at suppressing NSCLC cell viability
PDGFRs and their downstream PI3K/Akt signaling pathways play crucial roles in promoting cancer cell survival.17–23 We next observed the effect of CP-673451 on NSCLC cell viability. The results in Figure 2A show that CP-673451 significantly reduced the viability of NSCLC cell lines A549 and H1299, with a half-maximal inhibitory concentration of 0.49 and 0.61 μM, respectively. Figure 2B shows that CP-673451 reduced the cell viability of these cell lines in a time- and concentration-dependent manner.
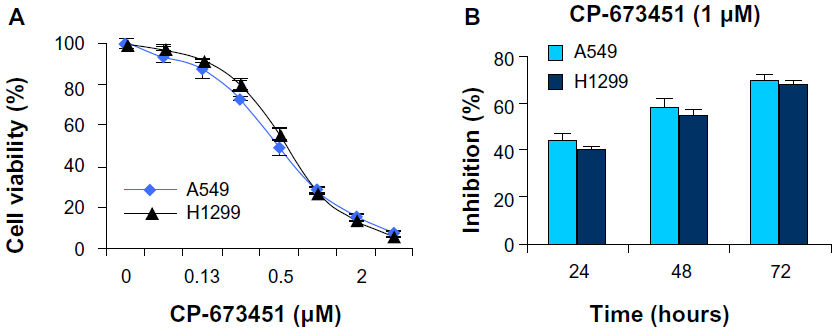 | Figure 2 (A and B) CP-673451 suppresses non-small-cell lung cancer cell viability. |
CP-673451 induces dramatic apoptosis in NSCLC cells
Since the PDGFRs and their downstream signaling pathways play important roles in the suppression of apoptosis,17–23 we next observed CP-673451-induced apoptosis in NSCLC cells. Treatment with CP-673451 caused a dose-dependent increase in sub-G1 DNA content in A549 cells, as measured by flow cytometry (P<0.01). CP-673451 induced 50% apoptotic cell death in A549 and H1299 cell lines at concentrations of 2.4 and 2.1 μM, respectively (Figure 3A). Consistently, CP-673451 treatment dose-dependently induced condensed and fragmented nuclei in A549 cells, suggesting that CP-673451 could significantly induce apoptosis in NSCLC cells (Figure 3B and C).
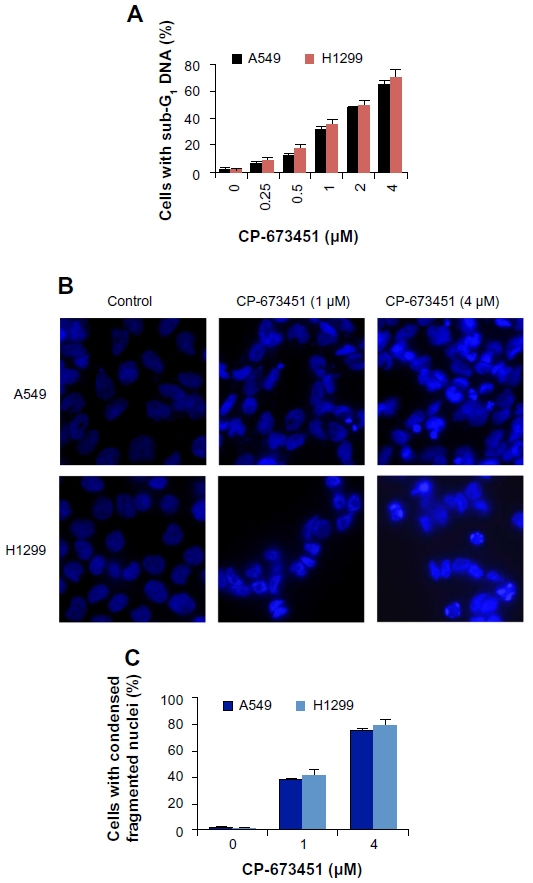 | Figure 3 (A–C) CP-673451 induces apoptosis in non-small-cell lung cancer cells. |
CP-673451 is effective at inhibiting migration and invasion of NSCLC cells by suppression of lamellipodia formation
It is known that PDGFRs and their downstream signaling pathways contribute remarkably to cancer cell migration and invasion by inducing lamellipodia formation at the leading edge of cells.24–26 We next observed the effect of CP-673451 on the migration and invasion of A549 cells. As shown in Figure 4A–D, CP-673451 concentration-dependently inhibited migration of A549 cells in both transwell and wound-healing assays. CP-673451 (25 nM) exerted an inhibition of 56.34%. Furthermore, CP-673451 also efficiently suppressed cancer cell invasion in a dose-dependent manner in a Matrigel-coated transwell model (Figure 4E and F). Due to the significant roles of lamellipodia in cell motility, we next analyzed the influence of CP-673451 on lamellipodia formation, and found CP-673451 dramatically suppressed lamellipodia formation in A549 cells, which at least partially accounted for the motility-inhibitory activity of CP-673451 (Figure 4G and H).
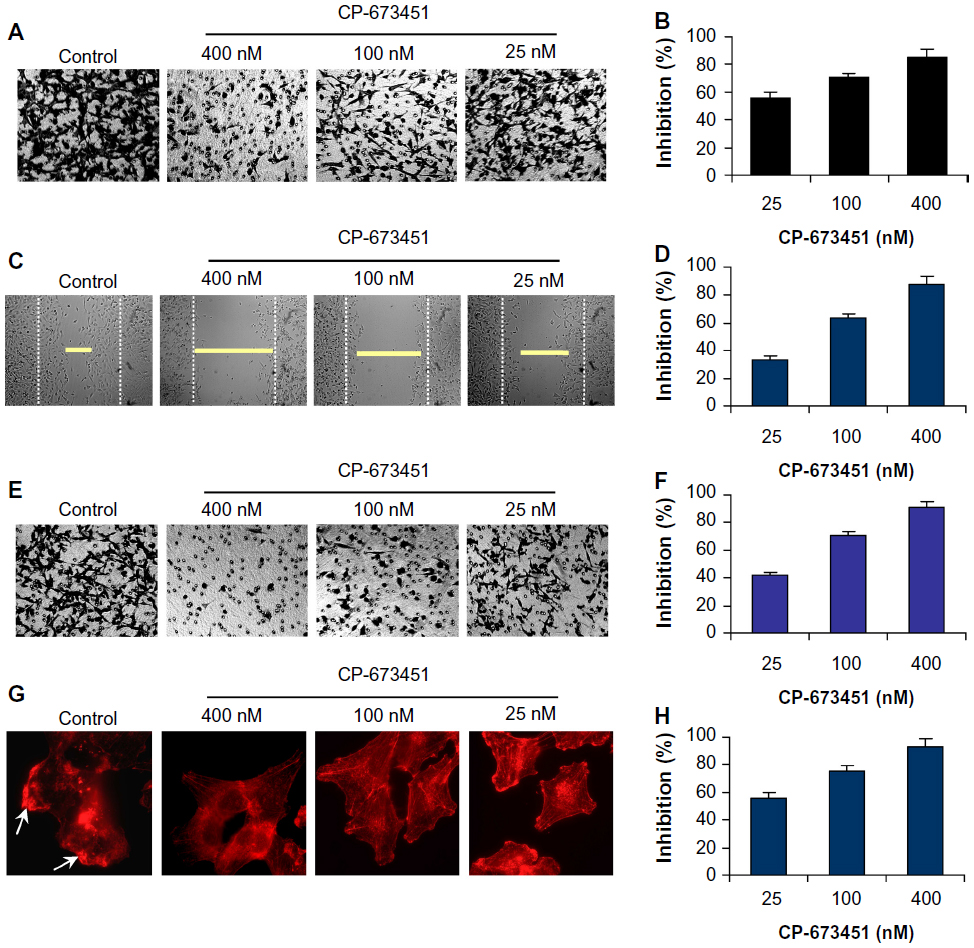 | Figure 4 (A–H) CP-673451 inhibits lamellipodia formation and cell migration and invasion in non-small-cell lung cancer cells. |
CP-673451 suppresses NSCLC tumor growth in vivo
We next examined the effect of CP-673451 on the growth of A549 tumors in nude/nude mice. The results showed that low-dose CP-673451 (20 mg/kg) led to a medium suppression of tumor growth, while high-dose CP-673451 (40 mg/kg) strongly inhibited tumor growth in vivo (Figure 5A). Doses of 20 and 40 mg/kg CP-673451 exerted an inhibition of 42.56% and 78.15% at day 10, respectively. The hematoxylin and eosin staining results showed that CP-673451 induced significant apoptosis in tumors, as shown with a great number of condensed nuclei in the CP-673451-treated group (Figure 5B). This effect was further confirmed by using TUNEL and nuclei-staining assays. In the TUNEL assay, the fluorescent signal in the CP-673451 group was much stronger than in the control group. Nuclei staining showed that CP-673451 induced condensed/fragmented nuclei (Figure 5C). Moreover, CP-673451 showed no significant toxicity in mice, and there was no significant loss of weight (P>0.05) (Figure 5D).
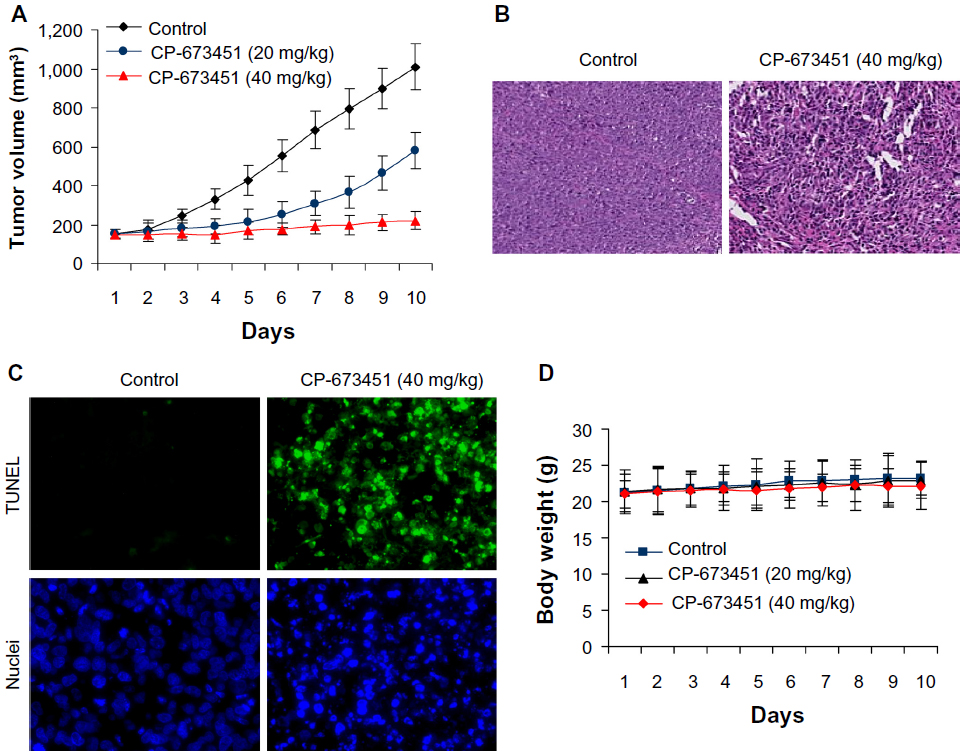 | Figure 5 (A–D) CP-673451 suppresses non-small-cell lung cancer tumor growth in vivo. |
Discussion
Lung cancer is a leading cause of cancer death worldwide, with NSCLC accounting for the majority of all lung cancer cases.1–8 The high mortality rate associated with lung cancer has prompted numerous exhaustive efforts to identify novel therapeutic targets and treatment modalities.5,6 The resistance of many types of cancer to conventional chemotherapies is a major factor undermining successful cancer treatment. It is well known that PDGFRs and their ligands play critical roles in cancer cell migration and proliferation.17–23 One important downstream signaling pathway of PDGFR is the PI3K/Akt pathway, which plays crucial roles in promoting cell survival, inhibiting cell apoptosis, and suppressing cell motility. PDGFR activation and overexpression contributes not only to tumorigenesis and tumor metastasis but also to resistance acquisition to chemotherapy.27,28 Therefore, PDGFR inhibition is a promising therapeutic strategy for cancer, including NSCLC.
Here, we investigated the potential anticancer activities of CP-673451, a PDGF-specific inhibitor, in NSCLC therapy. Our studies show that CP-673451 could efficiently suppress NSCLC cell viability, induce cell apoptosis, and inhibit lamellipodia formation, cell migration, and invasion. All these effects are at least partially attributed to the inhibition of PDGFR and subsequently its downstream signaling pathway in NSCLC cells by CP-673451 treatment. Importantly, CP-673451 is effective at suppressing NSCLC tumor growth in vivo.
The anticancer activity of CP-673451 has been poorly reported, and the detailed underlying anticancer mechanisms are far from clear. The antiangiogenic activity of CP-673451 was previously reported.1 However, there have been no reports about its antimigration and anti-invasion activities so far. Here, for the first time, we report that CP-673451 exerted strong inhibition on actin reorganization and lamellipodia formation, a driving resource for cell motility. Subsequently, we found CP-673451 has strong antimigration and anti-invasion activities in NSCLC cells. We found CP-673451, as a specific PDGF specific inhibitor, could strongly inhibit PDGFR-mediated signaling pathways, which can be used to explain, at least partially, its activities of cell-viability suppression, cell-apoptosis induction, lamellipodia inhibition, and cell migration and invasion suppression. Our study is also a systemic study of anticancer activities of CP-673451, including its antiproliferation, antimotility, and apoptosis-induction activities in NSCLC cells, a high-mortality cancer type worldwide. In summary, our studies suggest that CP-673451 might be a potential therapeutic agent in NSCLC, and might shed new light on NSCLC therapy.
Disclosure
The authors report no conflicts of interest in this work.
References
Couraud S, Zalcman G, Milleron B, Morin F, Souquet PJ. Lung cancer in never smokers – a review. Eur J Cancer. 2012;48(9):1299–1311. | |
Gibb H, Haver C, Gaylor D, et al. Utility of recent studies to assess the National Research Council 2001 estimates of cancer risk from ingested arsenic. Environ Health Perspect. 2011;119(3):284–290. | |
Ginsberg MS, Grewal RK, Heelan RT. Lung cancer. Radiol Clin North Am. 2007;45(1):21–43. | |
Webb JD, Simon MC. Novel insights into the molecular origins and treatment of lung cancer. Cell Cycle. 2010;9(20):4098–4105. | |
Novaes FT, Cataneo DC, Ruiz Junior RL, Defaveri J, Michelin OC, Cataneo AJ. Lung cancer: histology, staging, treatment and survival. J Bras Pneumol. 2008;34(8):595–600. | |
Vansteenkiste J, Dooms C, De Leyn P. Early stage non-small-cell lung cancer: challenges in staging and adjuvant treatment: evidence-based staging. Ann Oncol. 2010;21 Suppl 7:vii189–vii195. | |
Langer CJ, Besse B, Gualberto A, Brambilla E, Soria JC. The evolving role of histology in the management of advanced non-small-cell lung cancer. J Clin Oncol. 2010;28(36):5311–5320. | |
Youlden DR, Cramb SM, Baade PD. The International Epidemiology of Lung Cancer: geographical distribution and secular trends. J Thorac Oncol. 2008;3(8):819–831. | |
Hermanson M, Funa K, Hartman M, et al. Platelet-derived growth factor and its receptors in human glioma tissue: expression of messenger RNA and protein suggests the presence of autocrine and paracrine loops. Cancer Res. 1992;52(11):3213–3219. | |
Heldin CH, Westermark B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev. 1999;79(4):1283–1316. | |
Betsholtz C, Karlsson L, Lindahl P. Developmental roles of platelet-derived growth factors. Bioessays. 2001;23(6):494–507. | |
Yarden Y, Escobedo JA, Kuang WJ, et al. Structure of the receptor for platelet-derived growth factor helps define a family of closely related growth factor receptors. Nature. 1986;323(6085):226–232. | |
Corless CL, Schroeder A, Griffith D, et al. PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensitivity to imatinib. J Clin Oncol. 2005;23(23):5357–5364. | |
MacDonald TJ, Brown KM, LaFleur B, et al. Expression profiling of medulloblastoma: PDGFRA and the RAS/MAPK pathway as therapeutic targets for metastatic disease. Nat Genet. 2001;29(2):143–152. | |
Donnem T, Al-Saad S, Al-Shibli K, Andersen S, Busund LT, Bremnes RM. Prognostic impact of platelet-derived growth factors in non-small cell lung cancer tumor and stromal cells. J Thorac Oncol. 2008;3(9):963–970. | |
Roberts WG, Whalen PM, Soderstrom E, et al. Antiangiogenic and antitumor activity of a selective PDGFR tyrosine kinase inhibitor, CP-673,451. Cancer Res. 2005;65(3):957–966. | |
Dahia PL, Marsh DJ, Zheng Z, et al. Somatic deletions and mutations in the Cowden disease gene, PTEN, in sporadic thyroid tumors. Cancer Res. 1997;57(21):4710–4713. | |
Gimm O, Perren A, Weng LP, et al. Differential nuclear and cytoplasmic expression of PTEN in normal thyroid tissue and benign and malignant epithelial thyroid tumors. Am J Pathol. 2000;156(5):1693–1700. | |
Ringel MD, Hayre N, Saito J, et al. Overexpression and overactivation of Akt in thyroid carcinoma. Cancer Res. 2001;61(16):6105–6111. | |
García-Rostán G, Costa AM, Pereira-Castro I, et al. Mutation of the PIK3CA gene in anaplastic thyroid cancer. Cancer Res. 2005;65(22):10199–10207. | |
Ha HT, Lee JS, Urba S, et al. A phase II study of imatinib in patients with advanced anaplastic thyroid cancer. Thyroid. 2010;20(9):975–980. | |
Zhang J, Wang P, Dykstra M, et al. Platelet-derived growth factor receptor-α promotes lymphatic metastases in papillary thyroid cancer. J Pathol. 2012;228(2):241–250. | |
Kim MJ, Kim SK, Park HJ, et al. PDGFRA promoter polymorphisms are associated with the risk of papillary thyroid cancer. Mol Med Rep. 2012;5(5):1267–1270. | |
Oka N, Nakahara S, Takenaka Y, et al. Galectin-3 inhibits tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by activating Akt in human bladder carcinoma cells. Cancer Res. 2005;65(17):7546–7553. | |
Moses SA, Ali MA, Zuohe S, et al. In vitro and in vivo activity of novel small-molecule inhibitors targeting the pleckstrin homology domain of protein kinase B/AKT. Cancer Res. 2009;69(12):5073–5081. | |
Kundra V, Escobedo JA, Kazlauskas A, et al. Regulation of chemotaxis by the platelet-derived growth factor receptor-β. Nature. 1994;367(6462):474–476. | |
Garofalo M, Di Leva G, Romano G, et al. miR-221&222 regulate TRAIL resistance and enhance tumorigenicity through PTEN and TIMP3 downregulation. Cancer Cell. 2009;16(6):498–509. | |
Garofalo M, Romano G, Di Leva G, et al. EGFR and MET receptor tyrosine kinase-altered microRNA expression induces tumorigenesis and gefitinib resistance in lung cancers. Nat Med. 2011;18(1):74–82. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.