Back to Journals » Neuropsychiatric Disease and Treatment » Volume 12
Cortical spreading depression produces a neuroprotective effect activating mitochondrial uncoupling protein-5
Authors Viggiano E, Monda V, Messina A, Moscatelli F, Valenzano A, Tafuri D, Cibelli G, De Luca B, Messina G, Monda M
Received 24 February 2016
Accepted for publication 29 April 2016
Published 11 July 2016 Volume 2016:12 Pages 1705—1710
DOI https://doi.org/10.2147/NDT.S107074
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Roger Pinder
Emanuela Viggiano,1,2 Vincenzo Monda,1 Antonietta Messina,1 Fiorenzo Moscatelli,3 Anna Valenzano,3 Domenico Tafuri,4 Giuseppe Cibelli,3 Bruno De Luca,1 Giovanni Messina,1,3 Marcellino Monda1
1Department of Experimental Medicine, Section of Human Physiology and Unit of Dietetics and Sports Medicine, Second University of Naples, Naples, 2Department of Medicine, University of Padua, Padua, 3Department of Clinical and Experimental Medicine, University of Foggia, Foggia, 4Department of Motor Sciences and Wellness, University of Naples “Parthenope”, Naples, Italy
Abstract: Depression of electrocorticogram propagating over the cortex surface results in cortical spreading depression (CSD), which is probably related to the pathophysiology of stroke, epilepsy, and migraine. However, preconditioning with CSD produces neuroprotection to subsequent ischemic episodes. Such effects require the expression or activation of several genes, including neuroprotective ones. Recently, it has been demonstrated that the expression of the uncoupling proteins (UCPs) 2 and 5 is amplified during brain ischemia and their expression exerts a long-term effect upon neuron protection. To evaluate the neuroprotective consequence of CSD, the expression of UCP-5 in the brain cortex was measured following CSD induction. CSD was evoked in four samples of rats, which were sacrificed after 2 hours, 4 hours, 6 hours, and 24 hours. Western blot analyses were carried out to measure UCP-5 concentrations in the prefrontal cortices of both hemispheres, and immunohistochemistry was performed to determine the localization of UCP-5 in the brain cortex. The results showed a significant elevation in UCP-5 expression at 24 hours in all cortical strata. Moreover, UCP-5 was triggered by CSD, indicating that UCP-5 production can have a neuroprotective effect.
Keywords: cortical spreading depression, neuroprotective effect, uncoupling protein-5
A Letter to the Editor has been received and published for this article.
Introduction
Cortical spreading depression (CSD) consists of an electrocorticogram (ECoG) depression, induced by several stimuli, propagating over the surface.1 It can be revoked without inducing tissue damage in a normoxic brain.2,3 However, CSD is linked to changes in cerebral blood flow, increased glucose levels, O2 consumption, adenosine triphosphate utilization, and modifications of protein synthesis.4–9 Several studies suggest that CSD is involved in severe neurological diseases such as stroke,10 migraine aura, and epilepsy.11–15
Conversely, a role for CSD in neuroprotection, in particular to subsequent ischemia episodes, was found by Taga et al16 and Matsushima et al.17 However, the neuroprotective action of the CSD is not yet clear. Previous studies have shown that CSD produced epigenetic modifications, causing lysine 4 dimethylation (H3K4) and lysine 9 dimethylation of H3 histone (H3K9).18,19 The epigenetic consequences probably caused alterations in different neuroprotective gene expressions. In fact, CSD increased the expressions of HIF-1, iNOS,20 nNOS,21,22 mitogen-activated protein kinase (MAPK),23 heat shock proteins (hsp27, hsp70),24–28 and heme oxygenase-1 (HO-1).29
These genes are also activated during hypoxia/ischemia and play a pivotal role in neuroprotection during ischemia, suggesting their involvement in CSD-induced neuroprotection.30–34 Recently it was demonstrated that the expression of the UCP-2 and -5 is amplified in human brain ischemic lesions, in vitro, and their expression exerts a long-term effect upon neuron protection35 by reducing the generation of free radicals,36–38 thus reducing their neurotoxic effect.39,40
The aim of this work was to evaluate the expression of UCP-5 in the brain cortex, following CSD induction.
Materials and methods
Animals
Sixteen adult male Sprague Dawley rats (weight 300–350 g) were accommodated in pairs at fixed temperature (22°C±1°C) and moisture (70%), with a 12-hour day/12-hour night cycle. The procedures were in accordance with the Directive of 86/609/EEC. The study was approved by the Human Ethical Review Committee of the Second University of Naples. All protocols respected the Guidelines for Investigation of Experimental CSD in Anesthetized Animals.
Cortical spreading depression
CSD was obtained as described by Viggiano et al.20 All animals were anesthetized using urethane (1 g/kg body weight, intraperitoneally). The body temperature was maintained at 37°C using a thermostated electric plate and was monitored by a rectal probe. Later, the head was positioned on the stereotaxic instrument, and a hole of 3 mm diameter was drilled on the skull, which was made 5 mm lateral to the median line and 4 mm caudal to Bregma. Dura was removed and CSD was led utilizing a chunk of filter paper impregnated with 1 M KCl for 15 minutes; a filter paper impregnated with 0.9% NaCl was used as a control. The sham waves and ECoG were registered using two Ag/AgCl electrodes (1 mm in diameter): the first electrode was placed on the parietal cortex that was medicated with KCl and the second was placed subcutaneously in the neck skin.41 Electrodes were connected to a DC amplifier (Dynograph; Beckman). All animals were sacrificed after 2 hours, 4 hours, 6 hours, or 24 hours of CSD induction. All the brains were swiftly removed. As previously reported,42 to produce the biochemical and morphological data compatible, just the first adjacent sections at brain samples were withdrawn for subsequent Western blot (WB) analyses. In particular, the anterior part of the cortex was dissected by means of a coronal cut anterior to the optical chiasm and frozen in liquid-nitrogen-cooled isopentane for WB analyses. The remainder of the brain was put into 4% formaldehyde for 24 hours, followed by incubation in 30% sucrose for 3 days, and finally frozen at –80°C. The first sections of the exposed part of the brain were cut for WB analyses.
Western blot
The prefrontal cortex from both hemispheres were homogenized using a tight pestle with 7 volumes of lysis buffer (10 mM HEPES pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 12% glycerol, 0.1 mM EGTA, 0.5 mM DTT, and 0.5 mM spermidine) with the addition of a protease inhibitor cocktail (Sigma-Aldrich Co., St Louis, MO, USA). Following centrifugation at 800 g for 15 minutes, the supernatant was recovered and centrifuged at 100,000× g for 15 minutes at 4°C to obtain the cytosolic extracts. The pellet containing mitochondrial proteins was sonicated in a buffer with 1% NP40, 0.1% SDS, 100 μM sodium vanadate, and 0.5% sodium deoxycholate with addition of a protease inhibitor cocktail (Sigma-Aldrich), and centrifuged for 20 minutes at 15,000× g (4°C). The supernatant containing the mitochondrial proteins was recovered and the amount of proteins was determined by the Bio-Rad protein assay (Bio-Rad Laboratories Inc., Hercules, CA, USA). One volume of the loading buffer (β-mercaptoethanol 8.7%, SDS 52.17%, Tris–HCl 0.2 M, bromophenol blue 0.026%, glycerol 17.4%) was added to each sample and denatured at 100°C for 5 minutes. Proteins were divided by 12% SDS-PAGE in Tris buffer. Thereafter, the proteins were shifted to nitrocellulose membranes for electrophoresis in a Tris–glycine buffer for 2 hours at 80 V. Membranes were incubated with an anti-UCP-5 rabbit polyclonal antibody (1:500 dilution; Alpha Diagnostic International, San Antonio, TX, USA) overnight at 4°C, and then incubated with a secondary anti-rabbit-IgG (1:2,500 dilution; Promega Corporation, Fitchburg, WI, USA) for 1 hour at room temperature. Equal loading of samples was checked by Coomassie Blue R-250 (Bio-Rad Laboratories Inc.) staining by cutting the blot into two parts: one was used for the anti-UCP-5 antibody and the other was used for Comassie Blue staining. Signals were revealed by an ECL (enhanced chemiluminescence) system (Pierce, Rockford, IL, USA).
Immunohistochemistry
Brain slices (20 μm) were washed three times for 5 minutes in PBS 0.1 M, pH 7.4, and incubated with 0.6% H2O2 in 50% methanol/PBS for 10 minutes to block endogenous peroxidase. Then, they were washed three times for 5 minutes in PBS, and incubated overnight with an anti-UCP-5 rabbit polyclonal antibody (Upstate Biotechnology Lake Placid, NY, USA) diluted 1:100 in PBS +10% bovine serum (NBS) at 4°C in a humidified box. After second round of rinsing in 0.1 M PBS, slices were incubated with biotinylated anti-rabbit IgG (Vector Laboratories, Burlingame, CA, USA) diluted 1:100 for 1 hour at room temperature, followed by the incubation with the ABC solution (Vector Laboratories) for 45 minutes, as per the manufacturer’s instruction. For signal detection, the slices were rinsed three times for 5 minutes and incubated with a freshly made chromogen/substrate reagent (0.1% DAB/0.1% H2O2) for 10 minutes at room temperature in the dark. Finally, the staining was blocked by the addition of cold PBS, followed by dehydration in alcohol and clearing up in xylene. The slices were then mounted on coverslips with Eukitt mounting medium (Sigma-Aldrich). In control experiments, the slices were incubated in the absence of the primary antibody (mock). Brain slices were analyzed at 10× magnification under a light microscope (Leica CTR500; Leica Microsystems, Wetzlar, Germany), and images were captured by a high-resolution digital camera (Leica 300F).
Statistical analysis
Statistical analyses were done by SPSS 13.0 for Windows (SPSS Inc., Chicago, IL, USA). The results were presented as mean ± SEM. A two-factor analysis of variance was performed to estimate the difference among the ipsilateral and contralateral cortex at different time periods after CSD induction. Multiple comparisons were done with Fisher’s least significant difference (LSD) post hoc test. P<0.05 was deemed significant.
Results
Figure 1 shows the increase of UCP-5 after CSD induction in the ipsilateral cortex at 24 hours, compared with the contralateral cortex (NaCl-treated) considered as mock control. The antibody against UCP-5 presented a major band of 36 kDa. Analysis of variance for UCP-5 protein concentrations (treated vs mock) as a function of time (2 hours, 4 hours, 6 hours, and 24 hours) demonstrated a significant effect of time [F(3, 16) =3.571, P<0.05]. Fisher’s LSD post hoc test showed a significant difference for UCP-5 at 24 hours after CSD in the processed cortex, when compared with mock. No differences in the intensities of the UCP-5 signal were found between the CSD-treated and untreated hemispheres at 2 hours, 4 hours, or 6 hours.
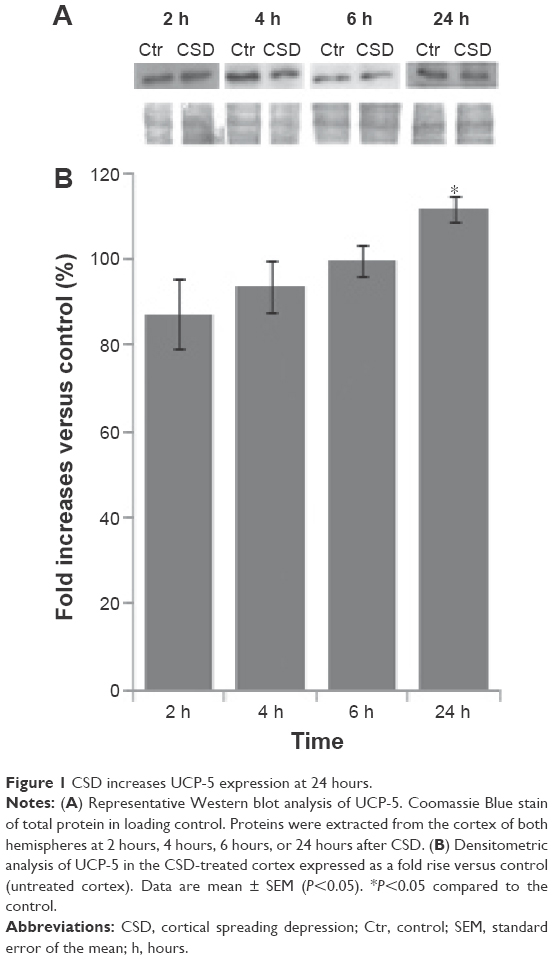 | Figure 1 CSD increases UCP-5 expression at 24 hours. |
Immunohistochemistry was performed to depict the localization of UCP-5 in the cerebral cortex. Morphological data were taken in sections near the marked area for WB sampling, which included analogous cortical regions, especially the motor cortex, prefrontal, and the somatosensory cortex. Densitometric analyses of images at low magnification revealed an increase in UCP-5 signal in the CSD-treated cortex when compared to the contralateral cortex at 24 hours (Figure 2). Moreover, the signal intensity was higher in all cortical strata. No immunostaining was seen in these areas in the mock samples (data not shown).
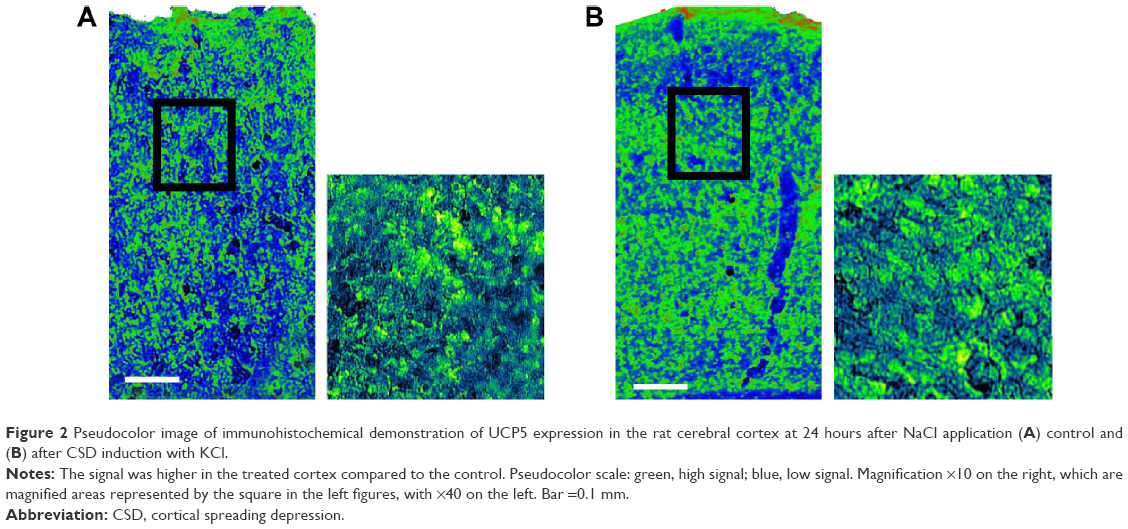 | Figure 2 Pseudocolor image of immunohistochemical demonstration of UCP5 expression in the rat cerebral cortex at 24 hours after NaCl application (A) control and (B) after CSD induction with KCl. |
Discussion
This study shows that CSD increased UCP-5 after 24 hours, suggesting a potential relationship between CSD and neuroprotection at this time. This result is in accordance with previous research showing that CSD increases the ischemic tolerance to temporary focal ischemia long after CSD.43–47
Since CSD has no consequence on cerebral blood flow during the following ischemia, its neuroprotective action might depend on intracellular protective factors against ischemic injuries.43 The mechanisms associated with neuroprotection induced by CSD are not yet clear, though CSD affects the expression of several potentially neuroprotective genes. Moreover, previous studies have reported a dynamic spatiotemporal pattern for gene induction, which may delay the damage to the tissues or, alternatively, mediate neuroprotection, tissue remodeling, and functional compensation.48
Gene expression changed as early as in the first 4 hours after CSD induction.20 In particular, previous studies have demonstrated an increase of iNOS and HIF-1 expression at 1 hour and the modification of AMPK and nNOS expression after 24 hours.42 Therefore, it can be argued that CSD induced neuroprotection through a chain reaction and/or a prolonged effect of early genes.
Some genes regulated by CSD are also involved in oxidative stress. In particular, CSD decreases superoxide ion (O2−) expression and increases hydrogen peroxide (H2O2) and superoxide dismutatase gene expression after 1 hour, thus suggesting an increase in O2− to H2O2 dismutation.49 Expression of UCP-5 is augmented in human brain ischemic lesions in vitro, and its expression has a long-term effect on neuroprotection,35,38 because it decreases the mitochondrial membrane potential and in turn reduces reactive oxygen species production.35 The decrease in O2− can be determined by the increased activity of both superoxide dismutatase and UCP-5.
There have been several studies on the cortical electroencephalogram in stroked animals subjected to neuroprotective hypothermia. Hypothermia has pleiotropic effects on brain physiology, which may be necessary for the effective protection of the brain after a stroke. The beneficial effects of hypothermia have been attributed to diminished excitotoxicty, neuroinflammation, apoptosis, free-radical production, seizure activity, blood–brain barrier disruption, blood vessel leakage, and/or cerebral thermopooling.50
These represent potential mechanisms by which CSD may induce neuroprotection, because they lead to a decrease in radical oxygen production that is usually involved in tissue damage.39,40
Conclusion
We suggest a role of UCP5 in the neuroprotection induced by CSD. However, other studies a long time after CSD induction are necessary to confirm this data and to study the potential role of the other UCPs, such as UCP-2 and UCP-4.
Disclosure
The authors report no conflicts of interest in this work.
References
Leao AA. The slow voltage variation of cortical spreading depression of activity. Electroencephalogr Clin Neurophysiol. 1951;3(3):315–321. | ||
Nedergaard M, Hansen AJ. Spreading depression is not associated with neuronal injury in the normal brain. Brain Res. 1988;449(1–2):395–398. | ||
Monda M, Pittman QJ. Cortical spreading depression blocks prostaglandin E1 and endotoxin fever in rats. Am J Physiol. 1993;264(2 pt 2):R456–R459. | ||
Bures J, Buresova O. Spreading depression as a research instrument in neurophysiology. Actual Neurophysiol (Paris). 1962;4:107–124. | ||
Messina G, Palmieri F, Monda V, et al. Exercise causes muscle GLUT4 translocation in an insulin-independent manner. Biol Med (Aligarh). 2015;1:2. | ||
Di Bernardo G, Messina G, Capasso S, et al. Sera of overweight people promote in vitro adipocyte differentiation of bone marrow stromal cells. Stem Cell Res Ther. 2014;5(1):4. | ||
Viggiano A, Vicidomini C, Monda M, et al. Fast and low-cost analysis of heart rate variability reveals vegetative alterations in non-complicated diabetic patients. J Diabetes Complications. 2009;23(2):119–123. | ||
Esposito M, Serpe FP, Diletti G, et al. Serum levels of polychlorinated dibenzo-p-dioxins, polychlorinated dibenzofurans and polychlorinated biphenyls in a population living in the Naples area, southern Italy. Chemosphere. 2014;94:62–69. | ||
Mies G, Paschen W. Regional changes of blood flow, glucose, and ATP content determined on brain sections during a single passage of spreading depression in rat brain cortex. Exp Neurol. 1984;84(2):249–258. | ||
Fabricius M, Fuhr S, Bhatia R, Boutelle M, Hashemi P, Strong AJ. Cortical spreading depression and peri-infarct depolarization in acutely injured human cerebral cortex. Brain. 2006;129(pt 3):778–790. | ||
Lauritzen M, Dreier JP, Fabricius M, Hartings JA, Graf R, Strong AJ. Clinical relevance of cortical spreading depression in neurological disorders: migraine, malignant stroke, subarachnoid and intracranial hemorrhage, and traumatic brain injury. J Cereb Blood Flow Metab. 2011;31(1):17–35. | ||
Monda M, Messina G, Mangoni C, De Luca B. Resting energy expenditure and fat-free mass do not decline during aging in severely obese woman. Clin Nutr. 2008;27(4):657–659. | ||
Messina G, Vicidomini C, Viggiano A, et al. Enhanced parasympathetic activity of sportive women is paradoxically associated to enhanced resting energy expenditure. Auton Neurosci. 2012;169(2):102–106. | ||
Messina G, Dalia C, Tafuri D, et al. Orexin-A controls sympathetic activity and eating behavior. Front Psychol. 2014;5:997. | ||
De Luca V, Viggiano E, Messina G, et al. Peripheral amino acid levels in schizophrenia and antipsychotic treatment. Psychiatry Investig. 2008;5(4):203–208. | ||
Taga K, Patel PM, Drummond JC, Cole DJ, Kelly PJ. Transient neuronal depolarization induces tolerance to subsequent forebrain ischemia in rats. Anesthesiology. 1997;87(4):918–925. | ||
Matsushima K, Schmidt-Kastner R, Hogan MJ, Hakim AM. Cortical spreading depression activates trophic factor expression in neurons and astrocytes and protects against subsequent focal brain ischemia. Brain Res. 1998;807(1–2):47–60. | ||
Passaro D, Rana G, Piscopo M, Viggiano E, De Luca B, Fucci L. Epigenetic chromatin modifications in the cortical spreading depression. Brain Res. 2010;1329:1–9. | ||
Rana G, Donizetti A, Virelli G, et al. Cortical spreading depression differentially affects lysine methylation of H3 histone at neuroprotective genes and retrotransposon sequences. Brain Res. 2012;1467:113–119. | ||
Viggiano E, Ferrara D, Izzo G, et al. Cortical spreading depression induces the expression of iNOS, HIF-1alpha, and LDH-A. Neuroscience. 2008;153(1):182–188. | ||
Caggiano AO, Kraig RP. Neuronal nitric oxide synthase expression is induced in neocortical astrocytes after spreading depression. J Cereb Blood Flow Metab. 1998;18(1):75–87. | ||
Shen PJ, Gundlach AL. Prolonged induction of neuronal NOS expression and activity following cortical spreading depression (SD): implications for SD- and NO-mediated neuroprotection. Exp Neurol. 1999;160(2):317–332. | ||
Chow AK, Thompson CS, Hogan MJ, Banner D, Sabourin LA, Hakim AM. Cortical spreading depression transiently activates MAP kinases. Brain Res Mol Brain Res. 2002;99(1):75–81. | ||
Kobayashi S, Harris VA, Welsh FA. Spreading depression induces tolerance of cortical neurons to ischemia in rat brain. J Cereb Blood Flow Metab. 1995;15(5):721–727. | ||
Monda M, Messina G, Scognamiglio I, et al. Short-term diet and moderate exercise in young overweight men modulate cardiocyte and hepatocarcinoma survival by oxidative stress. Oxid Med Cell Longev. 2014;2014:131024. | ||
Rinaldi B, Guida F, Furiano A, et al. Effect of prolonged moderate exercise on the changes of nonneuronal cells in early myocardial infarction. Neural Plast. 2015;2015:265967. | ||
Plumier JC, David JC, Robertson HA, Currie RW. Cortical application of potassium chloride induces the low-molecular weight heat shock protein (Hsp27) in astrocytes. J Cereb Blood Flow Metab. 1997;17(7):781–790. | ||
Urbach A, Bruehl C, Witte OW. Microarray-based long-term detection of genes differentially expressed after cortical spreading depression. Eur J Neurosci. 2006;24(3):841–856. | ||
Koistinaho J, Pasonen S, Yrjänheikki J, Chan PH. Spreading depression-induced gene expression is regulated by plasma glucose. Stroke. 1999;30(1):114–119. | ||
Lu A, Tang Y, Ran R, Clark JF, Aronow BJ, Sharp FR. Genomics of the periinfarction cortex after focal cerebral ischemia. J Cereb Blood Flow Metab. 2003;23(7):786–810. | ||
Tseveleki V, Rubio R, Vamvakas SS, et al. Comparative gene expression analysis in mouse models for multiple sclerosis, Alzheimer’s disease and stroke for identifying commonly regulated and disease-specific gene changes. Genomics. 2010;96(2):82–91. | ||
Buga AM, Margaritescu C, Scholz CJ, Radu E, Zelenak C, Popa-Wagner A. Transcriptomics of post-stroke angiogenesis in the aged brain. Front Aging Neurosci. 2014;6:44. | ||
Buga AM, Scholz CJ, Kumar S, et al. Identification of new therapeutic targets by genome-wide analysis of gene expression in the ipsilateral cortex of aged rats after stroke. PLoS One. 2012;7(12):e50985. | ||
Buga AM, Sascau M, Pisoschi C, Herndon JG, Kessler C, Popa-Wagner A. The genomic response of the ipsilateral and contralateral cortex to stroke in aged rats. J Cell Mol Med. 2008;12(6B):2731–2753. | ||
Nakase T, Yoshida Y, Nagata K. Amplified expression of uncoupling proteins in human brain ischemic lesions. Neuropathology. 2007;27(5):442–447. | ||
Arsenijevic D, Onuma H, Pecqueur C, et al. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat Genet. 2000;26(4):435–439. | ||
Viggiano A, Nicodemo U, Viggiano E, et al. Mastication overload causes an increase in O2-production into the subnucleus oralis of the spinal trigeminal nucleus. Neuroscience. 2010;166(2):416–421. | ||
Horvath TL, Diano S, Barnstable C. Mitochondrial uncoupling protein 2 in the central nervous system: neuromodulator and neuroprotector. Biochem Pharmacol. 2003;65(12):1917–1921. | ||
Breimer LH. Repair of DNA damage induced by reactive oxygen species. Free Radic Res Commun. 1991;14(3):159–171. | ||
Dean RT, Gieseg S, Davies MJ. Reactive species and their accumulation on radical-damaged proteins. Trends Biochem Sci. 1993;18(11):437–441. | ||
Gold L, Back T, Arnold G, et al. Cortical spreading depression-associated hyperemia in rats: involvement of serotonin. Brain Res. 1998;783(2):188–193. | ||
Viggiano E, Viggiano D, Viggiano A, De Luca B, Monda M. Cortical spreading depression increases the phosphorylation of AMP-activated protein kinase in the cerebral cortex. Neurochem Res. 2014;39(12):2431–2439. | ||
Kawahara N, Ruetzler CA, Klatzo I. Protective effect of spreading depression against neuronal damage following cardiac arrest cerebral ischaemia. Neurol Res. 1995;17(1):9–16. | ||
Matsushima K, Hakim AM. Transient forebrain ischemia protects against subsequent focal cerebral ischemia without changing cerebral perfusion. Stroke. 1995;26(6):1047–1052. | ||
Yanamoto H, Mizuta I, Nagata I, Xue J, Zhang Z, Kikuchi H. Infarct tolerance accompanied enhanced BDNF-like immunoreactivity in neuronal nuclei. Brain Res. 2000;877(2):331–344. | ||
Yanamoto H, Xue JH, Miyamoto S, et al. Spreading depression induces long-lasting brain protection against infarcted lesion development via BDNF gene-dependent mechanism. Brain Res. 2004;1019(1–2):178–188. | ||
Otori T, Greenberg JH, Welsh FA. Cortical spreading depression causes a long-lasting decrease in cerebral blood flow and induces tolerance to permanent focal ischemia in rat brain. J Cereb Blood Flow Metab. 2003;23(1):43–50. | ||
Küry P, Schroeter M, Jander S. Transcriptional response to circumscribed cortical brain ischemia: spatiotemporal patterns in ischemic vs. remote non-ischemic cortex. Eur J Neurosci. 2004;19(7):1708–1720. | ||
Viggiano A, Viggiano E, Valentino I, Monda M, Viggiano A, De Luca B. Cortical spreading depression affects reactive oxygen species production. Brain Res. 2011;1368:11–18. | ||
Joseph C, Buga AM, Vintilescu R, et al. Prolonged gaseous hypothermia prevents the upregulation of phagocytosis-specific protein annexin 1 and causes low-amplitude EEG activity in the aged rat brain after cerebral ischemia. J Cereb Blood Flow Metab. 2012;32(8):1632–1642. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.