Back to Journals » Drug Design, Development and Therapy » Volume 20
Copper Homeostasis and Cuproptosis in Neurological Disorders
Authors Liu W ![]() , Xue Y, Cao C, Yang L, Zhang L
, Xue Y, Cao C, Yang L, Zhang L ![]()
Received 8 November 2025
Accepted for publication 15 February 2026
Published 27 February 2026 Volume 2026:20 580005
DOI https://doi.org/10.2147/DDDT.S580005
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Leonidas Panos
Wu Liu,1,* Yan Xue,2,* Chenyin Cao,3,* Liting Yang,2 Lijun Zhang1,*
1School of Basic Medical Sciences, Xianning Medical College, Hubei University of Science and Technology, Xianning, Hubei, 437000, China; 2School of Pharmacy, Hubei Key Laboratory of Diabetes and Angiopathy, Xianning Medical College, Hubei University of Science and Technology, Xianning, Hubei, 437000, China; 3School of Stomatology and Optometry, Hubei University of Science and Technology, Xianning, Hubei, 437000, China
*These authors have contributed equally to this work
Correspondence: Lijun Zhang, School of Basic Medical Sciences, Xianning Medical College, Hubei University of Science and Technology, Xianning, Hubei, 437000, People’s Republic of China, Email [email protected]
Abstract: Neurological disorders such as Alzheimer’s disease (AD) and Parkinson’s disease (PD) pose a serious global public health threat, with complex etiologies involving genetic, environmental, and metabolic factors. Current data indicate that the prevalence of these disorders is rapidly increasing with the aging population, resulting in a growing economic and healthcare burden worldwide. In recent years, the imbalance of copper homeostasis has been increasingly implicated in the pathogenesis of neurological diseases. Copper overload can aggravate neuronal injury by inducing oxidative stress (OS), mitochondrial dysfunction, and protein misfolding, while copper deficiency disrupts the function of copper-dependent enzymes and leads to metabolic abnormalities. The mechanism of cuproptosis, proposed in 2022, describes a novel form of programmed cell death characterized by lipoylated protein aggregation and the loss of Fe-S cluster proteins, offering new insights into copper-related diseases. Multiple studies have demonstrated the crucial role of copper homeostasis and cuproptosis in the onset, progression, and treatment of neurological diseases. This narrative review summarizes the molecular mechanisms involved in copper homeostasis regulation and, on that basis, discusses the role of copper metabolism abnormalities in AD, PD, Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS), multiple sclerosis (MS), Wilson’s disease (WD), Menkes disease (MD), and stroke. Additionally, we highlight the mechanisms of existing copper-regulating drugs and their therapeutic potential in neurological disorders, while pointing out the limitations of current drug development.
Plain Language Summary: Copper homeostasis imbalance plays a critical regulatory role in neurological disorders.Cuproptosis is a unique form of copper-mediated cell death that plays a key role in neuronal injury.Many key questions regarding the differences in copper homeostasis and cuproptosis mechanisms among various neurological disorders remain unresolved.The interplay between copper and other metal ions (such as iron and zinc) in maintaining homeostasis may have important implications in neurological disorders.
Keywords: copper homeostasis, cuproptosis, neurological diseases, copper chelators
Introduction
Neurological disorders, characterized by progressive degeneration of the structure and function of the nervous system, are becoming an increasingly significant global health challenge and burden.1,2 The Global Burden of Disease (GBD) study in 2021 highlighted this phenomenon.3 With the ongoing global aging population, the prevalence of neurological diseases is expected to continue rising, placing an ever-growing strain on medical resources, family care, and public healthcare systems.4,5 The nervous system is a central network that controls sensory, motor, cognitive, and memory functions, and is composed of large numbers of neurons and glial cells.6 Neurofilaments form a cytoskeletal network within neuronal axons, maintaining axonal structural integrity and influencing the velocity of signal conduction.7 Any abnormalities in neuronal structure or signal transmission may lead to impairments in sensory, motor, or cognitive functions.
Therapeutic strategies for neurological diseases face substantial challenges, and a wide range of approaches—including conventional pharmaceuticals, antioxidants, neuroprotective agents, and natural bioactive compounds derived from traditional medicinal plants—are being continuously developed and applied.8 For example, Shrivastava et al designed and synthesized a class of dual-target small-molecule compounds that simultaneously inhibit acetylcholinesterase (AChE) and β‑site amyloid precursor protein cleaving enzyme 1 (BACE1), demonstrating the feasibility of multi-target intervention in Alzheimer’s disease (AD).9
Disruption of copper (Cu) homeostasis has been identified as a key pathogenic mechanism in neurological disorders.10 Numerous studies have shown that dysregulation of copper homeostasis may impact the nervous system through mechanisms such as oxidative stress (OS), mitochondrial dysfunction, neuroinflammation, and protein misfolding.11 The premature aging model, copper sulfate‐induced stress‐induced premature senescence (CuSO4‑SIPS), has also confirmed copper’s critical role in age-related functional decline and the development of age-associated diseases.12 As an essential trace element, copper is indispensable for the normal physiological activities of higher plants and animals.13 Human cells can only maintain optimal bioactivity within a narrow concentration range of copper ions.14 Copper homeostasis is delicately regulated through a complex network of copper-dependent proteins, ensuring precise intracellular distribution. Both copper deficiency and copper overload are detrimental to human health.15
In 2022, Tsvetkov et al first introduced the concept of cuproptosis.16 Unlike apoptosis, pyroptosis, or ferroptosis, cuproptosis is a newly identified form of programmed cell death triggered by copper ion overload.17 Its core mechanism involves excess copper binding to lipoylated proteins in the tricarboxylic acid (TCA) cycle, such as dihydrolipoamide S-acetyltransferase (DLAT) and dihydrolipoamide S-succinyltransferase (DLST), leading to abnormal aggregation of these lipoylated proteins. This disrupts the stability of iron–sulfur (Fe-S) cluster proteins in oxidative phosphorylation (OXPHOS), leading to disulfide bond–dependent aggregation of lipoylated proteins in the TCA cycle, resulting in destabilization or loss of iron-sulfur (Fe-S) cluster proteins and inducing proteotoxic stress.18 Notably, cuproptosis occurs only in cells with active mitochondrial OXPHOS, whereas glycolysis-dependent cells exhibit significant resistance.19
Lutsenko et al summarized and discussed the physiological functions, molecular mechanisms, and associated diseases of copper homeostasis in mammals.20 Meng et al reviewed the mechanisms of copper metabolism and regulation in the AD brain and discussed the involvement of cuproptosis in the pathological processes of AD.21 Peng et al focused on stroke, examining the physiological roles of copper in stroke and its relationships with pathological processes such as cuproptosis and OS.22 Gao et al summarized current knowledge on copper metabolism, mechanisms of cuproptosis, copper-related cell death, and copper-associated neurological diseases.23 Xu et al systematically summarized brain-specific copper homeostasis, detailing the roles of BBB transporters, glia-mediated copper buffering, and neuron-specific copper chaperones in maintaining cerebral copper balance.24 This article provides an in-depth investigation of the mechanisms underlying copper metabolism and cuproptosis, without limitation to a single disease or neurodegenerative disorders, and comprehensively reviews the associations between dysregulation of copper homeostasis and a wide range of neurological diseases, including WD and MD that can manifest with neurological symptoms.
In addition to traditional chemical drugs and certain plant extracts that have shown beneficial effects in interventions for neurological diseases,25,26 novel therapeutics based on the regulation of copper homeostasis have increasingly become a research focus. This article also compiles potential therapeutic agents targeting copper homeostasis mechanisms, with the aim of providing new perspectives for the prevention and treatment of neurological diseases.
Systemic Copper Metabolism
Copper is an essential trace element for nearly all living organisms. According to previous studies, copper is extensively utilized by organisms in various physiological processes mediated by copper-dependent enzymes.27 Due to its potential toxicity, copper requires precise transport and homeostatic regulation28 (Figure 1).
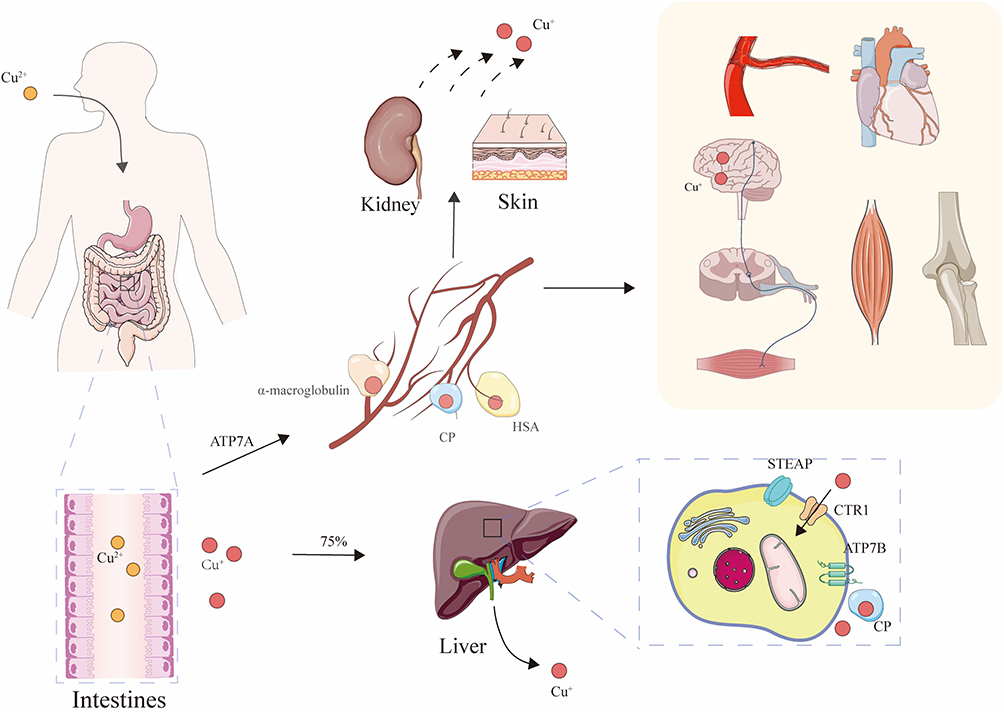 |
Figure 1 Systemic copper metabolism. Dietary Cu(II) is reduced to Cu(I) in the small intestine, where it is absorbed into intestinal epithelial cells. It is then transported into the portal circulation via ATP7A. In the bloodstream, copper binds to soluble carriers such as ceruloplasmin (CP), α2-macroglobulin, and human serum albumin (HSA), which deliver it to specific tissues or organs. Approximately 75% of copper entering the blood is taken up by the liver. In hepatocytes, copper is imported via CTR1. Within the liver, ATP7B has two major functions: it pumps copper into the Golgi apparatus for CP metalation and, under copper overload conditions, relocates to the apical membrane of bile canaliculi to excrete excess copper into bile, thereby maintaining copper homeostasis. Excess copper is mainly excreted via bile, with small amounts eliminated through urine, sweat, and other routes. |
Dietary copper is absorbed in the stomach, duodenum, and small intestine.29 In the intestinal lumen, Cu(II)(Cu2⁺) is reduced to Cu(I)(Cu⁺) by metalloreductases such as six-transmembrane epithelial antigen of the prostate (STEAP)30 and duodenal cytochrome b (DCYTB).31 Cu(I) is then transported into intestinal epithelial cells via the apical high-affinity copper transporter 1 (CTR1/SLC31A1).32 Inside the enterocytes, copper is shuttled by the antioxidant 1 copper chaperone (ATOX1) to the basolateral membrane, where it is exported into the portal circulation by ATPase copper-transporting alpha (ATP7A).33 In the bloodstream, copper binds to soluble carrier proteins such as albumin and α2-macroglobulin for delivery to specific tissues and organs.34
Approximately 75% of the copper entering the bloodstream is taken up by the liver.35 Within hepatocytes, Cu(I) is internalized via CTR1. ATOX1 then delivers copper to ATP7A and ATPase copper-transporting beta (ATP7B) for incorporation into cuproenzymes. The copper chaperone for superoxide dismutase (CCS) transfers copper to superoxide dismutase 1 (SOD1), while the cytochrome c oxidase copper chaperone 17(COX17) delivers copper to cytochrome c oxidase (COX) in the mitochondria. Excess copper is stored by metallothionein (MT) or bound to glutathione (GSH).15,36 ATP7B plays a dual role: it pumps copper into the Golgi apparatus for incorporation into ceruloplasmin (CP), which is then secreted into the bloodstream for systemic distribution, and under conditions of copper excess, ATP7B translocates to the canalicular membrane to excrete surplus copper into the bile, thus regulating systemic copper homeostasis.37
The liver is the primary site for copper storage. Excess copper is mainly excreted via bile, with smaller amounts eliminated through urine, sweat, and desquamated tissues.38 When dietary copper intake is high, copper absorption decreases while excretion increases; the opposite occurs when intake is low.39 When copper levels in peripheral tissues and organs are insufficient to sustain normal physiological function, ATP7B located in the Golgi apparatus mobilizes stored copper from hepatocytes into the bloodstream, where it binds to plasma proteins and enters systemic circulation.40
Cellular Copper Metabolism
Under normal physiological conditions, intracellular copper levels are tightly maintained within a narrow range by a complex network of proteins. This network includes cuproenzymes, copper chaperones, and membrane transporters, which work in coordination to regulate copper uptake, utilization, and efflux at the cellular level (Figure 2). Maintaining optimal copper concentrations is essential for cell survival—copper deficiency disrupts cellular respiration and metabolism, while copper excess impairs cell viability and may lead to cell death.41 Unlike traditional mechanisms of cell death, cuproptosis exhibits a strong dependence on mitochondria, particularly the TCA cycle.42
 |
Figure 2 Intracellular copper metabolism. Cu(II) is reduced to Cu(I) by STEAP on the cell membrane and then enters the cell via CTR1. SLC25A3 transports Cu(I) from the mitochondrial intermembrane space into the mitochondrial matrix and influences superoxide dismutase 1 (SOD1) activity. COX17 transfers Cu(I) to SCO1 and COX11, where COX11 delivers copper to COX1 and SCO1 transfers it to COX2, facilitating the metal center assembly of cytochrome c oxidase (CCO). ATOX1 transports copper and targets it to ATP7A/ATP7B located on the trans-Golgi network (TGN) membrane. ATP7A/ATP7B use ATP hydrolysis to drive transmembrane copper transport, delivering copper into the TGN for the metalation of enzymes such as ceruloplasmin (CP) and extracellular superoxide dismutase 3 (SOD3), which require copper as a cofactor. When cytosolic copper levels rise, ATP7A/ATP7B relocate from the TGN to the plasma membrane or intracellular secretory vesicles to export excess copper. Upon restoration of copper homeostasis, they return to the TGN to resume the transport cycle. ATOX1 can also shuttle copper into the nucleus, where it binds specific DNA promoter sequences to drive gene transcription. In the cytosol, CCS inserts Cu(I) into the copper-binding site of SOD1 and catalyzes the formation of the essential disulfide bond in SOD1. |
Uptake
Cellular copper uptake primarily depends on the CTR1. Under copper-deficient conditions, Specificity Protein 1 (Sp1) is activated and promotes the transcriptional upregulation of the CTR1 gene.43 Conversely, when copper is sufficient, copper ions suppress Sp1 activity and trigger the endocytosis and degradation of CTR1, thereby downregulating its expression and function.44 Mammalian copper homeostasis is regulated through the Cu-Sp1-CTR1 tripartite model.45
However, dietary copper mainly exists in the oxidized form Cu(II), which must first be reduced to Cu(I) by membrane-bound metalloreductases such as STEAP2, STEAP3, and STEAP4,46,47 or by DCYTB,31 before it can be transported into the cell via CTR1. In any tissue expressing STEAP proteins (eg, intestine, liver, heart, prostate), Cu(II) can be reduced to Cu(I) and subsequently absorbed through the CTR1 channel. In intestinal epithelial cells, DCYTB and STEAP enzymes function in concert.48
Divalent metal transporter 1 (DMT1), a member of the proton-coupled metal ion transporter family,49 can mediate Cu(II) uptake when CTR1 is limited or absent, such as in intestinal epithelial cells, though it is less efficient in transporting copper.50 In addition to CTR1 and DMT1, low-affinity Fe(II) transport protein (Fet4p)51,52 and divalent metal ion transporter SMF1 (Smf1p)53 can also transport Cu(II), but due to their low metal specificity, they are collectively referred to as low-affinity transporters.
In summary, Cu(II) is first reduced to Cu(I) by STEAP family proteins or DCYTB at the cell membrane, and then transported into cells via CTR1. In contrast, copper ions entering through DMT1, Fet4p, or Smf1p bypass the redox conversion step and are directly taken up in the Cu(II) form.
Utilization
In the cytoplasm, copper is transported by ATOX1 and targeted to the trans-Golgi network (TGN) membrane-bound transporters ATP7A and ATP7B.54,55 ATP7A and ATP7B are P-type ATPases that contain six metal-binding domains (MBDs) and utilize ATP hydrolysis to drive the transmembrane transport of copper into the TGN.56,57 This process facilitates the metallation of copper-dependent enzymes such as tyrosinase, CP, and superoxide dismutase 3 (SOD3).58 Under normal copper levels, ATP7A/ATP7B are localized to the TGN, where they function in enzyme loading.59 When cytoplasmic copper concentrations rise, ATP7A and ATP7B relocalize to the plasma membrane or intracellular secretory vesicles to export excess copper.58 Once copper levels normalize, they return to the TGN to resume another transport cycle.
ATP7A primarily functions in tissues such as the intestine, placenta, and central nervous system (CNS), where it is responsible for delivering copper into the bloodstream.60 In contrast, ATP7B plays a dominant role in the liver, preventing copper accumulation by excreting it into bile.61 This tissue-specific division of labor ensures proper copper distribution and homeostasis. Mutations in the ATP7A or ATP7B genes result in two distinct genetic disorders: Menkes disease (MD), characterized by copper deficiency,62 and Wilson disease (WD), characterized by copper toxicity.63
Cytochrome c oxidase (COX, also known as complex IV) is a critical enzyme complex in the mitochondrial respiratory chain, essential for electron transfer and oxygen reduction, and plays a vital role in maintaining respiratory function.64 solute carrier family 25 member 3 (SLC25A3) transports copper ions from the mitochondrial intermembrane space (IMS) into the matrix, which is crucial for the metallation of COX and influences the activity of SOD1.65 The copper chaperone COX17 specifically transfers Cu(I) to synthesis of cytochrome c oxidase 1 (SCO1) and cytochrome c oxidase copper chaperone 11 (COX11), which assist in loading the copper centers of COX.66,67
Mitochondrially encoded cytochrome c oxidase subunit I (COX1) and subunit II (COX2) are core components of COX, each containing distinct copper-binding sites—CuB and CuA, respectively.68 COX11 delivers copper to the CuB site on COX1, while SCO1 transfers copper to the CuA site on COX2.69 These two copper sites (CuA and CuB) form the catalytic core of COX,70 playing complementary and essential roles in oxygen reduction and electron transport. Under copper-deficient conditions, cells prioritize mitochondrial copper homeostasis, underscoring its primary importance in overall cellular copper balance.71
In the cytoplasm, CCS receives copper from CTR1 and inserts Cu(I) into the copper-binding site of SOD1, catalyzing the formation of essential disulfide bonds to activate the enzyme.72 SOD1 is an antioxidant enzyme located in both the cytosol and IMS, and CCS also facilitates the maturation of SOD1 within the IMS.73,74
In the nucleus, ATOX1 can also deliver copper ions, where it binds to specific DNA promoter sequences and promotes gene transcription.75
Efflux
When intracellular copper levels become excessive, ATP7A and ATP7B relocate from the TGN to vesicular compartments, which subsequently fuse with the plasma membrane to excrete copper ions via exocytosis into the bile. This represents a major pathway for endogenous copper excretion.37 Once copper concentrations return to physiological levels, these proteins relocalize back to the TGN.76
According to a study by Gioilli et al, small copper carriers (SCCs) also play an important role in copper transport and homeostasis. SCCs can serve as an alternative copper efflux pathway, transferring copper from the liver and intestinal epithelial cells to other cells, and under conditions of copper overload, they may be excreted into the urine.77
Copper Homeostasis Dysregulation
Copper Homeostasis and Oxidative Stress
Reactive oxygen species (ROS) are byproducts of aerobic metabolism. When intracellular ROS levels exceed the cellular antioxidant defense capacity, they induce oxidative stress (OS), leading to oxidative damage of biomolecules and ultimately resulting in cell death.78 Excess copper can increase ROS production, causing OS, while also promoting structural and functional damage to mitochondria. Mitochondrial structural and functional damage, in turn, can feedback to further enhance ROS production, exacerbating oxidative injury.79
Dysregulation of copper homeostasis can exert neurotoxicity by inducing OS. In cell models, part of the mechanism by which copper ion carriers induce astrocyte death involves excessive ROS generation, suggesting a role for OS in copper toxicity.80 The use of the compound copper pyrithione (CPT) can also induce OS-mediated cytotoxicity, inhibiting neurite outgrowth.81 Copper deficiency increases the brain’s susceptibility to OS.82 Additionally, copper may enhance the ability of 6-hydroxydopamine (6-OHDA) to induce OS, further damaging dopaminergic neurons.83
Copper Homeostasis and Inflammatory Response
Nuclear factor kappa‑light‑chain‑enhancer of activated B cells (NF-κB) is a nuclear transcription factor that normally exists in the cytoplasm in an inactive state by binding to inhibitor of NF-κB (IκB) proteins.84 Excess copper can activate the IκB kinase complex, promoting phosphorylation and degradation of IκB proteins and thereby releasing the NF-κB complex. The released p65 subunit is phosphorylated and translocates to the nucleus, where it binds specific DNA sequences to initiate transcription of inflammatory factors such as tumor necrosis factor-alpha (TNF-α), interleukin-1 (IL-1), and interleukin-6 (IL-6).85,86
In BV2 microglial cells, copper can increase ROS levels, activate the NF-κB pathway, induce secretion of inflammatory factors by microglia, and impair mitophagy, ultimately leading to ferroptosis of dopaminergic neurons.85 Copper-binding peptides can mitigate microglial inflammation by inhibiting the NF-κB pathway.87 In addition, ROS-induced OS can activate the NLRP3 inflammasome, promoting caspase-1–dependent secretion of proinflammatory cytokines and inducing pyroptosis.88,89
Copper Homeostasis and Mitochondrial Dysfunction
Mitochondria are double-membrane organelles present in all eukaryotic cells, responsible for aerobic respiration, and cooperate with the cytoskeleton to maintain cell morphology and function.90 They also serve as critical sites for multiple metabolic pathways, including key reactions such as metal ion metabolism.91
In neurons and glial cells, excess copper can accumulate in the mitochondrial matrix, disrupting mitochondrial membrane potential and inhibiting alpha‑ketoglutarate dehydrogenase (KGDH) and pyruvate dehydrogenase (PDH) activities, leading to reduced mitochondrial pyruvate production.92 The use of antioxidants such as dihydrolipoic acid can attenuate this process and the subsequent cell death.92
Additionally, copper can inhibit BNIP3-mediated mitophagy by downregulating the mitochondrial autophagy regulator BCL-2/adenovirus E1B 19kDa interacting protein 3 (BNIP3), which in traumatic brain injury (TBI) models manifests as exacerbated synaptic damage.93 AMP-activated protein kinase (AMPK), a cellular energy sensor, is activated during mitochondrial inhibition to help maintain ATP levels. In the cerebellum of copper-deficient mice, mitochondrial dysfunction leads to energy deficiency, activating AMPK and promoting its phosphorylation, which subsequently phosphorylates acetyl‑CoA carboxylase (ACC) to inhibit fatty acid synthesis and conserve energy.94
Copper Homeostasis and Synaptic Function
When copper is present in the synaptic cleft, it can directly or indirectly modulate the activity of neurotransmitter receptors, thereby affecting excitability.95 In addition, copper may participate in the binding of synaptic vesicles to the cell membrane by regulating the interaction between α-synuclein (α-Syn) and synaptic vesicles.96 Nam et al proposed that the synaptic cleft may contain three elements: Cu(I)/Cu(II), β-amyloid protein (Aβ), and neurotransmitters. Cu(I)/Cu(II) and neurotransmitters released during neuronal excitation are key components for modulating neurotransmitter receptor activation and maintaining signal transduction in the synapse, whereas under pathological conditions, Aβ may interact with these components to contribute to neurodegeneration.97
Mechanism of Cuproptosis
Copper is an essential trace element in organisms, playing irreplaceable roles in maintaining mitochondrial respiration, enzyme activity, and protein function.98 Cells can maintain normal activity only within an extremely narrow range of copper ion concentrations. Experiments using the MC3T3-E1 cell line (derived from the calvaria of a newborn mouse) showed that cells retain high activity only at copper concentrations of approximately 10−7-10−6 M, and even slight increases approach the toxicity threshold.99,100 Dysregulation of copper homeostasis can induce mitochondrial morphological and functional abnormalities, leading to metabolic disorders and irreversible damage to the organism101 (Figure 3).
 |
Figure 3 Mechanism of cuproptosis. Cu(II) is directly imported into the cell and mitochondria by the copper ionophore ELC. In mitochondria, FDX1 catalyzes the reduction of Cu(II) to Cu(I), accompanied by the generation of reactive oxygen species (ROS). FDX1 also interacts with LIAS to promote the lipoylation of DLAT. The lipoylated DLAT proteins interact with Cu(I) and subsequently aggregate. Additionally, Cu(I) disrupts iron–sulfur (Fe–S) cluster proteins, leading to their loss. Together, these effects induce cuproptosis. Cuproptosis causes the accumulation of key tricarboxylic acid (TCA) cycle components such as DLAT, DLST, NADH, and FADH2, reducing their activity and suppressing the electron transport chain (ETC), thereby decreasing ATP production. |
Unlike OS induced by copper overload, cuproptosis occurs primarily in mitochondria and depends on the interaction between copper ions and mitochondrial metabolism.102 Moreover, multiple studies have shown that cuproptosis can proceed even in the presence of antioxidants such as N-acetylcysteine (NAC) or ferrostatin‑1, indicating that ROS are a concomitant rather than an essential driving factor.79,103 ROS accumulation may also activate other programmed cell death pathways, including apoptosis and ferroptosis, but these are only concurrent phenomena and not central mechanisms of cuproptosis.104 Furthermore, classical inhibitors of apoptosis, necrosis, pyroptosis, and ferroptosis (eg, caspase inhibitors, ferrostatin‑1) are ineffective against cuproptosis; only copper chelators or knockout of key genes such as FDX1 and lipoyltransferases can reverse cell fate.105 These findings indicate that cuproptosis is a novel cell death mechanism completely independent of known programmed cell death pathways.
Mitochondrial Lipoylated Protein Aggregation
Under normal physiological conditions, copper uptake is mediated by CTR1, and copper efflux is regulated by ATP7A/ATP7B; however, under high copper load or in the presence of copper ionophores, these regulatory mechanisms fail to maintain copper homeostasis. Excess copper or exogenous copper ion carriers such as Elesclomol (ELC) can induce cuproptosis. ELC forms an Elesclomol-Cu(II) complex that transports Cu(II) from the extracellular space into the cytoplasm and mitochondria. Upon dissociation from the complex, Cu(II) undergoes repeated cycles of binding and release, leading to progressive accumulation of Cu(II) within mitochondria.106,107
In the mitochondrial matrix, Ferredoxin 1 (FDX1), an Fe-S cluster reductase, serves as a critical regulator of cuproptosis and plays a dual role. FDX1 reduces Cu(II) to the more toxic Cu(I), a process that is particularly pronounced in cells highly dependent on OXPHOS.108 FDX1 is an upstream regulator of protein lipoylation. By directly interacting with lipoic acid synthetase (LIAS), FDX1 promotes LIAS-mediated lipoylation of target proteins, increasing the lipoylation levels of TCA cycle-related enzymes such as DLAT, thereby providing a basis for the interaction between copper and these modified proteins and ultimately contributing to the initiation of the cuproptosis signaling pathway.109 Knockout of FDX1 confers resistance to copper ionophore (such as ELC)-induced cell death, indicating that FDX1 is a key upstream regulator of cuproptosis.108 A 2024 study on the drug disulfiram (DSF) showed that inhibition of FDX1 reduces copper ion accumulation and alleviates cuproptosis. Additionally, it mitigates ischemia-reperfusion-induced neuroinflammation through modulation of the heat shock protein 70 (HSP70)/toll-like receptor 4 (TLR4)/NLR family pyrin domain containing 3 (NLRP3) pathway, contributing to brain tissue protection.110
Lipoylation is a highly conserved post-translational modification in which lipoic acid is covalently attached to lysine residues of proteins, primarily occurring on key complexes of the mitochondrial TCA cycle and related metabolic pathways, including DLAT, DLST, dihydrolipoamide branched‑chain transacylase E2 (DBT), and glycine cleavage system protein H (GCSH).111 Excess Cu(I) can bind to certain critical mitochondrial metabolic enzymes, causing their misfolding and aggregation into clumps, thereby obstructing energy metabolism and activating the cellular proteostasis machinery. Ultimately, the inability to clear these damaged proteins leads to mitochondrial instability, collapse of cellular function, and the occurrence of cuproptosis.112,113
In in vitro experiments, FDX1 deletion causes mitochondrial proteins to lose lipoylation modifications and their Cu(II) binding sites, thereby reducing copper toxicity. However, because lipoylation is essential for the function of TCA cycle enzymes, FDX1 deletion impairs TCA metabolism, diminishes mitochondrial respiration and OXPHOS function, and markedly restricts cell growth under low-glucose conditions.109
Loss of Iron-Sulfur Cluster Proteins
In cuproptosis, when a large amount of copper binds to lipoylated proteins, it not only induces aggregation of these proteins but also disrupts the stability of Fe-S cluster proteins, leading to their depletion and inactivation. Since Fe-S cluster proteins are extensively involved in the TCA cycle, electron transport, and nucleic acid repair, their loss exacerbates OXPHOS inefficiency, disrupts electron transport, increases ROS production, and causes widespread metabolic dysfunction.16,114 The loss of Fe-S cluster proteins, together with lipoylated protein aggregation, synergistically triggers mitochondrial proteotoxic stress, further promoting cell death.115
The Relationship Among Copper Homeostasis, Cuproptosis, and Neurological Diseases
Alzheimer’s Disease
Alzheimer’s disease (AD) is one of the most common neurological disorders, clinically characterized by progressive cognitive impairments in visual, language, executive, behavioral, and motor domains.116,117 The formation of extracellular Aβ plaques and neurofibrillary tangles (NFTs) composed of hyperphosphorylated tau protein in the hippocampus and cerebral cortex are central to AD pathogenesis.118 According to the amyloid cascade hypothesis, abnormal processing of amyloid precursor protein (APP) during pathology leads to extracellular accumulation of Aβ, which triggers tau hyperphosphorylation and ultimately NFT formation.119,120 Concurrently, Aβ aggregation induces neuroinflammation, oxidative stress (OS), and synaptic loss, culminating in neuronal death and cognitive decline.121
In AD patients, free copper levels (particularly in serum) are significantly elevated, resulting in diminished antioxidant defenses and mitochondrial dysfunction. Moreover, increased copper levels correlate with the rate of cognitive decline,122,123 and serum copper levels positively associate with AD risk124 (Figure 4).
 |
Figure 4 Copper homeostasis and cuproptosis in Alzheimer’s disease (AD). IFN-γ disrupts copper homeostasis. Excess copper increases free radicals via the Fenton reaction, leading to oxidative stress (OS) and cell death. These radicals interact with Aβ and APP, promoting Aβ aggregation and ROS production. Copper accumulation reduces synaptic proteins (SYP, PSD-95) and neurotransmitters (5-HT, GABA), and inhibits BDNF production through the CREB/BDNF pathway, impairing synaptic plasticity. APP cleavage by α- and γ-secretase produces non-toxic P3, while BACE1 and γ-secretase generate Aβ40 and Aβ42. Copper promotes α-cleavage, whereas copper deficiency favors β-cleavage, worsening AD pathology. Microglia can clear Aβ or Aβ-antibody complexes, but excess copper impairs this function. In the hippocampus, copper elevates NO and LPO levels, damages antioxidant defenses, and disrupts neuronal function. Copper also induces tau hyperphosphorylation and aggregation. |
Copper-induced OS may be one of the primary mechanisms underlying its neurotoxicity. Since copper in the brain can cycle between Cu(II) and Cu(I) oxidation states, it possesses redox activity and can readily catalyze the production of ROS, thereby exacerbating OS in the brain.125 Excess copper generates large amounts of free radicals through the Fenton reaction, inducing oxidative damage and cell death, which leads to neurodegeneration.126 The free radicals produced by the Fenton reaction are not only closely related to OS but can also directly interact with Aβ plaques and APP, promoting Aβ synthesis and aggregation.127 Copper can also bind directly to Aβ plaques, catalyzing ROS generation; these ROS may cause oxidative damage to the Aβ peptides themselves as well as surrounding molecules such as proteins and lipids.128 In the hippocampus, copper treatment significantly elevates levels of nitric oxide (NO) and lipid peroxidation products (LPO), impairs the antioxidant defense system, and consequently damages neural function.129 Copper overload can disrupt critical mitochondrial enzymes such as COX, causing electron transport chain blockage, sharply increasing ROS production, activating cuproptosis, and ultimately inducing neuronal apoptosis or necrosis.130 As the main energy supplier of cells, mitochondrial dysfunction not only increases neurotoxicity but also leads to synaptic loss and memory impairment.131
Hippocampal dysfunction typically occurs early in AD, accompanied by inhibition of long‑term potentiation (LTP) and synaptic dysfunction in patients’ brains. Excessive copper accumulation can downregulate presynaptic protein synaptophysin (SYP), postsynaptic density protein 95 (PSD-95), and neurotransmitters such as 5-hydroxytryptamine (5-HT) and gamma-aminobutyric acid (GABA) in mice.132 SYP expression correlates closely with the number of presynaptic vesicles, while PSD-95 supports and anchors postsynaptic receptors and regulates receptor-associated protein organization.133 5-HT and GABA are key neurotransmitters in the synaptic cleft involved in learning and memory processes.134,135 These four molecules are all involved in higher brain functions within the synaptic system, indicating that copper can simultaneously affect presynaptic regulation, postsynaptic modulation, and neurotransmitter release, leading to synaptic transmission impairments. Moreover, brain-derived neurotrophic factor (BDNF), an important target of cAMP response element-binding protein (CREB),136 regulates synaptic plasticity and participates in neuroprotection and neural regeneration.137 Copper may inhibit the CREB/BDNF signaling pathway, reducing BDNF expression and thereby suppressing synaptic plasticity, which hinders memory formation and consolidation.132
Persistent activation of microglia is closely related to abnormal copper homeostasis and is involved in AD pathology.138 Microglia can phagocytose Aβ or Aβ-antibody complexes,139 but excessive copper ions inhibit this phagocytic function. The inflammatory cytokine interferon-γ (IFN-γ) not only triggers and exacerbates inflammation but also disrupts copper homeostasis by altering the cytoplasmic relocalization of copper transporter ATP7A, increasing copper uptake and upregulating CTR1 expression.140 This process may explain the fluctuating copper homeostasis observed in AD patients.
Furthermore, APP and tau proteins are critical in AD pathogenesis. APP acts as a receptor in the CNS, involved in synaptic growth, neuronal adhesion, axonal transport, and development.141 Under the action of α-secretase and γ-secretase, APP produces a non-toxic P3 fragment. However, cleavage by β-secretase (BACE1) and γ-secretase generates the longer peptides Aβ40 and Aβ42, which form amyloid plaques in the AD brain.142,143 Increased copper levels promote the α-secretase-mediated non-toxic pathway, whereas copper deficiency promotes the BACE1-mediated toxic pathway, accelerating Aβ formation.144 Tau protein, a neuron-specific microtubule-associated protein, normally maintains axonal microtubule stability. In AD, tau becomes hyperphosphorylated and dissociates from microtubules, causing axonal transport deficits and neuronal structural damage, ultimately triggering cell death.145 ROS generation is an important trigger of tau hyperphosphorylation and filament aggregation. Excess copper promotes tau phosphorylation, while treatment with copper chelators can inhibit copper levels and markedly reduce tau phosphorylation.146,147 Studies also suggest that reducing brain copper by limiting dietary copper intake may be a feasible strategy to regulate tau pathology in AD.146
Parkinson’s Disease
Parkinson’s disease (PD) is the second most common neurodegenerative disorder after AD, with rapidly increasing global prevalence and mortality.148 Clinically, it is characterized by bradykinesia, resting tremor, muscle rigidity, postural and gait abnormalities, often accompanied by cognitive impairment.149 Pathologically, PD is marked by progressive loss of dopaminergic neurons in the substantia nigra-striatum region and the presence of α-Syn inclusions in neuronal cytoplasm, forming Lewy bodies and Lewy neurites.150 α-Syn undergoes oligomerization and converts into β-sheet-rich protofibrils, forming cytotoxic amyloid aggregates.151 These aggregates coexist with mitochondrial dysfunction and decreased copper and GSH levels in brain regions, implicating OS and metabolic imbalance in PD pathology progression.152
Studies show that copper levels are decreased in multiple brain regions of PD patients post-mortem, especially in the substantia nigra, as measured by inductively coupled plasma mass spectrometry (ICP-MS).153 Synchrotron X-ray fluorescence microscopy and particle-induced X-ray emission analyses indicate a 55–65% reduction in copper in the substantia nigra and locus coeruleus of PD brains154 (Figure 5). CP, the main copper carrier and ferroxidase in plasma, shows dysregulated function in PD patients. Reduced CP-bound copper may cause iron homeostasis disturbance, worsening neuronal injury.
 |
Figure 5 Copper homeostasis and cuproptosis in Parkinson’s disease (PD). In PD, copper directly interacts with α-synuclein (α-Syn), promoting its aggregation into toxic oligomers. These complexes generate ROS and dopamine oxidation products, impair energy metabolism, and exacerbate neuronal damage. Intracellular GSH chelates copper and reduces its toxicity. When GSH is depleted, free copper increases, leading to elevated DLAT levels and loss of Fe–S cluster proteins, ultimately triggering copper-induced cell death. Copper bound to ceruloplasmin (CP) helps maintain iron homeostasis, which may protect neurons. CTR1 promotes α-Syn phosphorylation, contributing to dopaminergic neuron loss and PD progression. Overexpression of CTR1 may also deplete GSH and induce caspase-3-dependent cell death. Additionally, GSTM1-null and GSTT1-null genotypes may increase PD susceptibility. |
In vitro studies demonstrate that Cu(II) effectively induces spontaneous oligomerization of α-Syn at micromolar concentrations, promoting formation of oligomers and protofibrils.155,156 Binding of α-Syn with Cu(II) produces an N-terminal acetylated α-Syn-Cu(II) complex, exhibiting catechol oxidase and redox activity, catalyzing dopamine oxidation and ROS generation; this activity is further enhanced when membrane-bound.157,158 Additionally, metabolic energy decline is closely linked to disrupted catecholamine metabolism in highly active catecholaminergic neurons (eg, dopaminergic neurons in the substantia nigra pars compacta and noradrenergic neurons in the locus coeruleus), which disrupts redox-active metals such as copper and iron homeostasis.159 These mechanisms suggest α-Syn-Cu(II) exacerbates dopaminergic/noradrenergic neuronal dysfunction via OS and metabolic imbalance, contributing to PD pathology.
CTR1 is the primary high-affinity copper importer. In PD animal models, CTR1 deficiency significantly reduces α-Syn phosphorylation at S129, decreases dopaminergic neuron loss, and improves motor dysfunction.160 Conversely, CTR1 overexpression or excess causes intracellular GSH depletion and caspase-3-dependent cell death, indicating that excessive copper can also induce neuronal injury.161 Decoppering treatment partially restores apoptosis levels and expression of copper death-related proteins,132 supporting a role for copper death mechanisms in PD.
Copper homeostasis is closely linked with GSH metabolism. Zhang et al used the high-efficiency GSH fluorescent probe R13 to find reduced GSH levels in PD mouse brains.162 Clinically, GSH levels are significantly decreased in the substantia nigra of PD patients.163 GSH chelates Cu(I), blocking copper toxicity; however, GSH deficiency increases free Cu(I), disrupting Fe-S cluster proteins and inducing OS and cell death.16 From a genetic perspective, deletion polymorphisms in glutathione S-transferase Mu1 (GSTM1) and Theta1 (GSTT1) are associated with increased PD susceptibility,164 emphasizing the importance of GSH metabolism in defending against copper toxicity. Supplementation with cysteine precursors (eg, 6-OHDA/xanthine induction schemes) enhances cellular GSH levels and restores neuronal vitality.165,166 GSH deficiency may underlie copper-induced neurodegeneration, making modulation of GSH metabolism a potential PD therapeutic strategy.
Recent research links amorphous SOD1 aggregates with PD progression; these new SOD1-containing aggregates are amorphous and spherical, largely devoid of α-Syn but ubiquitin-positive, with copper as a structural and functional cofactor of SOD1, implicating copper deficiency in abnormal SOD1 aggregation.167 Indeed, SOD1 immunoreactivity has been detected in Lewy bodies and Lewy neurites of the substantia nigra and locus coeruleus in PD brains.168
In summary, copper homeostasis imbalance in PD manifests as localized copper deficiency rather than widespread copper toxicity, involving multiple mechanisms including α-Syn aggregation, catecholamine oxidation, GSH deficiency-induced metabolic imbalance, Fe-S protein dysfunction, and SOD1 aggregation. A complex network comprising CTR1, CP, GST, SOD1, and Fe–S cluster proteins underlies these processes. These findings provide a theoretical foundation for PD early diagnosis and therapeutic strategies targeting copper metabolism.
Huntington’s Disease
Huntington’s disease (HD) is a progressive neurodegenerative disorder with a genetic origin. The most common symptoms include loss of energy and initiative, poor perseverance and work quality, impaired judgment, decreased self-care ability, and emotional blunting; about half of studied patients exhibit emotional symptoms such as depression, anxiety, and irritability.169 HD is an autosomal dominant hereditary disease caused by CAG repeat expansion mutations in the gene encoding huntingtin (HTT) protein located on the short arm of chromosome 4.170 The N-terminus of the HTT protein contains a polyglutamine (polyQ) tract, and when the CAG repeats expand, a structurally defective mutated huntingtin (mHTT) protein is produced.171 This mHTT protein is prone to misfolding and forms β-sheet-rich aggregates and inclusions in the neuronal cytoplasm and nucleus.172 These abnormal aggregates disrupt cellular protein homeostasis, axonal transport, transcription, translation, mitochondrial, and synaptic functions.173,174 Diagnosis is typically confirmed by detecting the number of CAG repeats (≥36 repeats, with ≥40 repeats being fully pathogenic) in the HTT gene of patients exhibiting clinical features.175
Researchers have found abnormally high concentrations of copper in the striatum of both HD patients and HD mouse models, suggesting that copper may be involved in HD pathogenesis.176 In Drosophila models, copper ions increase the formation of mHTT aggregates and enhance their neurotoxicity,177 while copper chelators inhibit mHTT aggregate formation, further supporting the link between copper and HD pathogenesis.178 Copper ions promote mHTT aggregate formation by binding to mHTT monomers and oligomers, lowering the nucleation energy barrier for aggregation.176 In vitro studies show that copper ions selectively bind to the N-terminal fragment of HTT protein containing 17–68 glutamine residues, while other metal ions such as Fe3⁺ and Zn2⁺ show no significant binding affinity.178 Excess copper in the brain inhibits key mitochondrial and energy metabolism dehydrogenases, including lactate dehydrogenase (LDH)179 and succinate dehydrogenase (SDH),92 leading to energy metabolism imbalance. Neurons highly depend on lactate released by astrocytes as their primary energy substrate;180 thus, copper inhibition of LDH directly disrupts lactate metabolism, further weakening neuronal energy supply and exacerbating HD neurodegeneration. Additionally, in Drosophila HD models, copper ions increase mHTT aggregate formation and enhance accumulation of sulfur-positive Aβ structures within mHTT aggregates, representing the toxic aggregation of mHTT and altering autophagy in the brain.177 Studies also show that copper ions directly bind mHTT, increasing its aggregation propensity and β-sheet structure, thereby enhancing its cytotoxicity in neurons.176
Mutant HTT disrupts nuclear pore proteins in the nuclear pore complex (NPC), impairing nucleocytoplasmic transport and causing cellular toxicity.181 Meanwhile, mHTT interacts with the mitochondrial protein dynamin-related protein 1 (DRP1), promoting mitochondrial fission, resulting in mitochondrial dysfunction and synaptic protein sequestration.182 Moreover, compared to healthy controls, levels of several ubiquitin-proteasome system (UPS) enzymes, including ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzyme (E2), and ubiquitin ligase (E3), are altered in HD patients. Increased expression of these enzymes may affect proteasome-dependent degradation of mHTT or influence its solubility, aggregation, or assembly, thereby directly or indirectly impacting cellular pathways and stress responses involved in HD pathogenesis.183 The UPS plays a crucial role in clearing aggregated mHTT protein.184 Additionally, UPS participates in the translation and regulation of copper transport proteins and copper chaperones; however, since UPS contains copper-dependent enzyme components, it is itself influenced by copper levels.184,185
Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS) is a common neurological disorder characterized by the simultaneous degeneration of upper motor neurons in the motor cortex and lower motor neurons in the spinal cord and brainstem.186 Most ALS cases (~90%) are sporadic without a family history, while a minority (~10%) represent rare autosomal dominant familial amyotrophic lateral sclerosis (FALS).187 The exact etiology of sporadic amyotrophic lateral sclerosis (SALS) remains unclear; however, current consensus suggests it is a complex multistep disease triggered by a combination of genetic susceptibility, environmental exposures, and aging factors.188 The primary cause of FALS is mutations in the SOD1 gene, which encodes a metalloprotein that forms a highly stable homodimer by binding copper and zinc ions. SOD1 mutations cause OS, mitochondrial dysfunction, electron transport disruption, and protein aggregation.189,190
It is widely accepted that accumulation of misfolded conformers, oligomers, and aggregates of SOD1 in motor neurons is a core pathological mechanism of ALS. Due to disulfide bond disruption, SOD1 oligomers exhibit enhanced pro-oxidant activity and toxicity.191 Mutant SOD1 reduces protein folding stability by impairing metal binding and disulfide bond formation, leading to misfolding, aggregation, and eventual cytotoxicity.192 Copper is not only central to SOD1 enzymatic activity but is also crucial for its structural integrity and stability; abnormal copper metabolism may contribute to ALS pathogenesis by affecting SOD1 function or aggregation.193,194 One of the earliest pathological features in mutant SOD1 is loss of metal at copper-binding sites.195 Copper deficiency is common in mutant SOD1 cells, and aggregates are generally copper-deficient.196 Moreover, copper levels in cerebrospinal fluid (CSF) from ALS patients are lower than controls, especially in spinal-onset ALS, where copper concentration significantly decreases.197
Overexpression of COX in adult mice carrying mutant SOD1 results in impaired copper transport in the late stages of life, suggesting that age-related copper deficiency may be an important factor in the onset of SOD1-induced ALS.198 Deficiency of metals such as copper and zinc promotes the exposure of hydrophobic residues in SOD1, which facilitates the formation and stabilization of toxic SOD1 oligomers. These oligomers exhibit high toxicity and pro-oxidant activity, representing a key pathological process leading to neuronal damage.199 In addition, the disruption of copper binding and disulfide bond formation in SOD1 may contribute to the onset and progression of ALS.200 When SOD1 forms disulfide bonds before binding metal ions, it loses the ability to be recognized and activated by CCS, thereby blocking the copper supply from CCS and hindering the maturation of the disulfide bond in SOD1.201
Conversely, abnormally elevated free copper levels may act as immune stimuli. Misallocated copper ions generate large amounts of free radicals via Fenton reactions, directly causing oxidative damage and inducing microglial release of pro-inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α) and interleukin-1 beta (IL-1β).194 Excess copper also activates NF-κB in astrocytes, amplifying inflammatory responses and further damaging motor neurons.194 Indeed, in SOD1-ALS models, both copper deficiency (leading to protein dysfunction) and copper overload (triggering toxic inflammation) coexist and synergistically drive disease progression.194
Mitochondrial dysfunction has been observed in ALS mouse models and patients,202 representing a key hallmark of ALS neuropathology. Aberrant interaction between CCS and mutant SOD1 results in increased copper binding to SOD1, reducing copper transfer to mitochondria and causing accumulation of unstable SOD1, limiting ROS clearance, and ultimately leading to motor neuron toxicity.203 Overexpression of CCS accelerates disease progression in SOD1(G93A) transgenic mice (Gly93Ala mutation in the copper/zinc superoxide dismutase 1 gene), significantly reducing COX activity in late-stage disease.204 Furthermore, soluble misfolded mutant SOD1 deposits on mitochondrial intermembrane and outer membranes in ALS animals, which is a key mechanism underlying mitochondrial dysfunction.205
Multiple Sclerosis
Multiple sclerosis (MS) is an autoimmune inflammatory disease of the CNS and a leading cause of neurological disability in young adults.206 MS is characterized by neuronal lesions formed in the brain and spinal cord, resulting in a range of clinical symptoms including visual impairment, limb weakness, sensory abnormalities, and ataxia.207 Key factors triggering MS include neuroinflammation, oligodendrocyte death, subsequent OS, and axonal demyelination.208 Cuprizone, a copper chelator, is used to induce an MS mouse model in which microglial activation, demyelination, and mitochondrial damage have been observed.209
Significantly elevated copper concentrations have been detected in the serum and CSF of MS patients. Upregulation of copper transport proteins in the CNS may contribute to astrocyte-mediated demyelination. Moreover, under neuroinflammatory conditions, copper homeostasis alterations mediated by tropomyosin receptor kinase B (TrkB) and upregulation of copper transporter CTR1 play critical roles in MS. These processes affect astrocyte copper uptake and release, ultimately inducing demyelination.210 Additionally, increased expression of transient receptor potential melastatin 2 (TRPM2) has been observed in the cuprizone model. TRPM2 gene promotes cuprizone-induced demyelination, synaptic loss, microglial NLRP3 inflammasome activation, and production of pro-inflammatory cytokines, indicating a key role for TRPM2 in MS-associated neuroinflammation.211 However, excessively low divalent copper ion concentrations inhibit TRPM2 channel activity irreversibly,212 thereby contributing to MS pathological features.
SOD1 is an antioxidant and anti-inflammatory enzyme. Studies by Mezzaroba et al, Arakawa et al, and Rasoul et al have shown that OS induced by SOD1 deficiency plays an important role in tissue damage during MS progression.213–215 However, excessive copper accumulation in vivo can cause copper toxicity and generate ROS under Fenton reaction conditions, leading to OS.216 Therefore, copper is currently believed to contribute to MS by inducing oxidative damage, which plays a key role in the pathogenesis of demyelination and neurodegeneration.215
Wilson Disease
Wilson disease (WD) is a non-traditional neurological disorder caused by various mutations in the ATP7B gene. It is characterized by impaired copper excretion into bile, leading to copper accumulation in the liver, while copper buildup in the brain may cause neuropsychiatric symptoms.217 The most common neurological manifestations of WD are movement disorders, including tremor, ataxia, dystonia, parkinsonism, and chorea.218,219 To date, over 600 pathogenic variants in ATP7B have been identified, with the most common being single nucleotide missense and nonsense mutations, followed by insertions/deletions, and more rarely, splice site mutations.220
ATP7B plays a dual critical role in the liver: on one hand, it transports copper to the TGN, where copper binds to CP and is excreted via bile; on the other hand, its dysfunction directly causes impaired copper excretion, leading to abnormal copper deposition in target organs.221 When ATP7B is inactivated in WD patients, excess copper cannot enter bile and instead overflows into the bloodstream, depositing in various tissues including the brain. The accumulation of free copper triggers neurological lesions, manifesting as neurological symptoms and psychiatric disorders.222,223 Simultaneously, ATP7B dysfunction causes loosely bound non-CP copper in the blood to bind to albumin and attack erythrocyte membranes, leading to coombs-negative hemolysis and potential hemolytic crises, ultimately resulting in anemia.224
Copper toxicity is considered the main cause of organ damage in WD patients. When cellular copper levels reach a critical threshold and exceed the binding capacity of MT and GSH, OS is induced, resulting in free radical damage to proteins, lipids, and nucleic acid structures. This ultimately causes cellular damage and membrane rupture.126 Additionally, mitochondrial dysfunction has been observed in the livers of WD patients.225 Protein thiols are key targets of mitochondrial copper toxicity; copper accumulation in the liver attacks protein thiols, leading to mitochondrial dysfunction and subsequent hepatocellular injury in WD patients.226 GSH is a critical intracellular molecule protecting protein thiols from oxidation. Compared with liver mitochondria, brain mitochondria have lower total GSH levels, rendering them more sensitive to copper.226 Moreover, significantly elevated copper levels can be detected in nearly all brain regions of WD patients.227 Therefore, excessive copper accumulation may be a central pathological mechanism underlying the neurological damage and psychiatric abnormalities observed in WD.
Structural brain magnetic resonance imaging (MRI) scans of WD patients reveal marked changes in normal brain structures, including widespread lesions in the midbrain, globus pallidus, putamen, pons, cerebellum, and thalamus, as well as cortical atrophy.228 In the early disease stages, Alzheimer-type I and II astrocytes, along with morphologically abnormal astrocytes, can be observed in the basal ganglia of WD patients, representing typical neuropathological features of WD.229 Studies in WD mouse models have shown activated microglia and astrocytes in the striatum and corpus callosum regions, closely associated with demyelination and neuronal loss.230 Furthermore, astrocytes protect neurons from copper toxicity by sequestering excess copper ions.229 However, as the disease progresses, brain parenchymal copper levels increase significantly, astrocyte damage worsens, weakening their protective function, which further impairs neuronal physiology and ultimately leads to neuronal death.231
Menkes Disease
Menkes disease (MD) is an X-linked recessive genetic disorder of copper metabolism caused by mutations in the copper transporter gene ATP7A. It primarily manifests as growth retardation and severe neurological impairments during infancy.232 ATP7A plays a critical role in the TGN, facilitating intracellular copper transport.233 In MD, mutations in ATP7A impair copper export function on the basolateral membrane of intestinal epithelial cells, leading to defective copper transport in the intestine, resulting in abnormally low copper levels in blood, kidney, liver, and brain, while copper accumulates abnormally within intestinal epithelial cells.126 Furthermore, COX deficiency has been observed in the brains of MD patients, which may be related to mutations in the SCO1 and SCO2 genes, essential for copper metallation of the CuA site.234 Therefore, mitochondrial dysfunction caused by reduced COX activity may contribute to neuronal degeneration in MD patients.
The normal physiological functions of copper-dependent enzymes are impaired in MD patients. Dopamine-β-hydroxylase (DBH), a key enzyme in noradrenergic neurons responsible for converting dopamine to norepinephrine, shows decreased activity.235 This reduction leads to a characteristic neurochemical pattern in patients’ plasma and CSF, namely elevated dopamine and its metabolites and decreased norepinephrine and its metabolites.235–237 Clinically, this pattern can be used for newborn screening and early diagnosis of MD.235 In infants with MD, significantly increased levels of L-DOPA (L-3,4-dihydroxyphenylalanine, a precursor of catecholamines) have been detected in blood and CSF, indicating reduced DBH activity.238 Since DBH catalyzes the copper-dependent conversion of dopamine to norepinephrine, its dysfunction further reflects the copper deficiency-associated neurochemical disturbances in MD patients.239
Lysyl oxidase (LOX) is another copper-dependent enzyme catalyzing the initial oxidative deamination step in the crosslinking of collagen and elastin.240 In MD patients, LOX enzymatic activity is significantly decreased due to defective copper transport, leading to connective tissue abnormalities.241 Related studies show that reduced plasma copper levels and copper transport defects in MD patients not only decrease LOX catalytic activity but also downregulate the mRNA expression of LOX and its substrate pro-elastin.242,243
Occipital horn syndrome (OHS) is a mild variant of WD caused by ATP7A gene defects, characterized mainly by connective tissue abnormalities, including characteristic bony projections (occipital horns) at the attachment of the occipital muscles.244 This phenotype is closely associated with decreased copper-dependent LOX activity and can result in typical kinky hair, vascular tortuosity, and peripheral arterial aneurysms.245 Due to residual ATP7A function (~30%) that retains partial copper transport, neurological symptoms in OHS patients are relatively mild.246 Their serum copper and CP levels are lower than normal but higher than in classic WD, while elevated catecholamine levels in CSF indicate insufficient DBH activity consistent with the neurochemical abnormalities of classic MD.236
Stroke
Stroke is a common neurological disorder caused by acute focal injury to the CNS. It ranks as the second leading cause of death worldwide among adults, second only to ischemic heart disease, and is the third leading cause of disability.247 Stroke is typically classified as ischemic or hemorrhagic, with ischemic stroke being more prevalent.248 Elevated plasma copper levels increase susceptibility to stroke.249 Plasma copper is positively associated with a higher risk of ischemic stroke but inversely associated with hemorrhagic stroke risk.250 Another meta-analysis showed that serum copper levels were significantly higher in ischemic stroke patients compared to controls.251 These studies collectively indicate that copper homeostasis imbalance is a potential risk factor for stroke.
Copper imbalance influences stroke through pathways including vascular regeneration, hyperlipidemia, OS, maturation of neuroprotective peptides, and inflammatory responses.
Endothelial progenitor cells (EPCs) are bone marrow–derived cells circulating in the blood that can differentiate into endothelial cells and participate in angiogenesis and tissue repair. Studies have shown that EPCs play a crucial role in the recovery process after ischemic brain injury by promoting vascular reperfusion, inhibiting apoptosis and inflammation in the ischemic penumbra, thereby improving neurological function.252 However, copper overload significantly impairs EPC migration, adhesion, and tube formation by upregulating thrombospondin-1 (TSP-1) expression, while also weakening their antioxidant defenses. This leads to suppressed cerebral microvascular angiogenesis post-ischemia and exacerbates brain injury after stroke.253,254
Additionally, a retrospective study based on the 2011–2016 NHANES database found that in women, serum copper levels positively correlated with total cholesterol and low-density lipoprotein low‑density lipoprotein (LDL). Each 1 µg/dL increase in copper was associated with approximately 0.11 mg/dL and 0.09 mg/dL increases in total cholesterol and LDL, respectively, suggesting copper may indirectly elevate stroke risk by increasing blood lipid levels. However, as this was a cross-sectional study, prospective research is needed to validate this mechanism.255
Copper also plays a critical role as an essential cofactor of SOD1, maintaining its antioxidant function. Dysregulated copper homeostasis may lead to reduced SOD1 activity or abnormal aggregation, enhancing OS and exacerbating neural damage.193,194 Conversely, SOD1 overexpression in ischemia-reperfusion animal models significantly reduces superoxide anion production and overall ROS levels, promoting neural stem cell survival and partially mitigating ischemic stroke damage.256
Furthermore, copper is vital for the maturation of multiple neuropeptides, especially through the peptidylglycine α-amidating monooxygenase (PAM)-catalyzed C-terminal amidation pathway. Copper deficiency reduces PAM enzyme activity, resulting in neuropeptides such as neuropeptide Y (NPY) and corticotropin-releasing hormone (CRH) failing to form structurally mature C-terminal amidated forms.257,258 These peptides are crucial in neuroprotection and inflammation regulation following stroke. NPY has been shown to exert anti-inflammatory effects, reduce infarct volume, and promote neural regeneration,259,260 whereas CRH mediates stress-related inflammatory responses, modulating neuroinflammation during post-stroke pathology.261
Drugs Related to Neurological Diseases
For human copper homeostasis and cuproptosis, therapeutic drugs for neurological diseases are continuously being developed, and several agents with potential efficacy require further investigation. These drugs mainly include copper chelators, copper ionophores, copper nanoparticles, and natural compounds (Table 1).
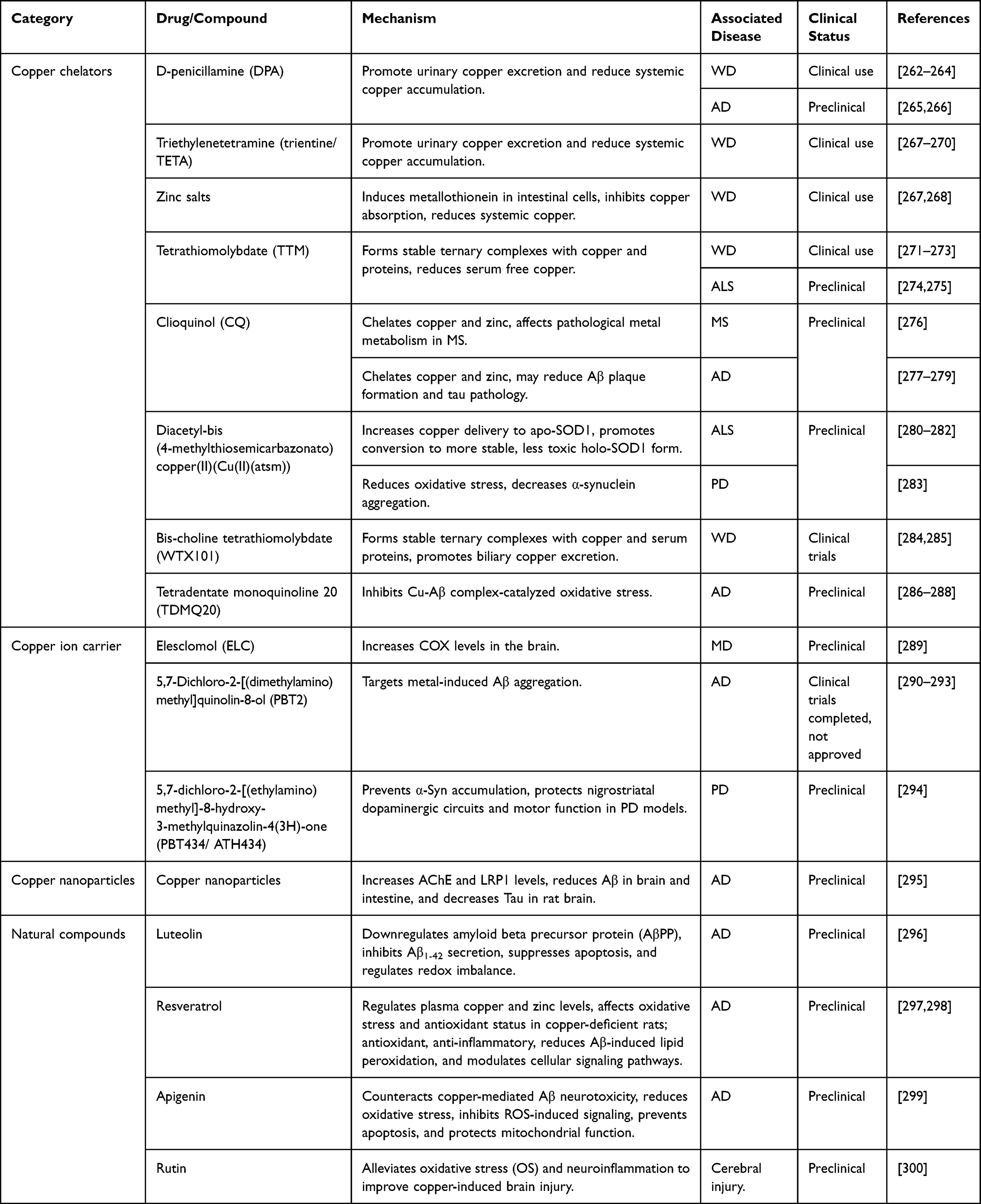 |
Table 1 Drugs Related to Nervous System Diseases |
Copper Chelators
Copper chelators can bind excess copper ions, blocking their participation in redox reactions and thereby reducing ROS production and protein aggregation.301 Determining the optimal timing for intervention with copper chelators is a major challenge, as the development and permeability of the blood-brain barrier (BBB) directly determine whether copper ions are sequestered in the bloodstream or delivered into neurons.302 Most classical chelators have limited BBB penetration, preventing them from reaching brain tissue to exert their effects.303 For example, D-penicillamine (DPA) poorly crosses the BBB, cannot directly modulate intracellular copper homeostasis, and its therapeutic efficacy is therefore limited.304 Efforts to enhance BBB penetration by increasing compound hydrophobicity may lead to increased toxicity or altered clearance, creating a trade-off between hydrophilicity and hydrophobicity.
In addition, clioquinol (CQ) and its derivative 5,7-Dichloro-2-[(dimethylamino)methyl]quinolin-8-ol (PBT2), although able to penetrate the BBB due to their chemical properties, lack selectivity for copper ions. These chelators cannot distinguish between copper bound to essential metalloproteins and toxic copper associated with Aβ, resulting in uneven copper distribution in the nervous system and potentially exacerbating neural damage. Consequently, CQ and PBT2 have not achieved the expected efficacy in clinical trials.305 Therefore, developing copper chelators with high selectivity and good BBB permeability remains a significant challenge in the treatment of neurological diseases. Future research should focus on optimizing the molecular structures of chelators to enhance copper ion selectivity and BBB penetration, thereby achieving more effective therapeutic outcomes.
D-Penicillamine
D-penicillamine (DPA) is a thiol-containing α‑amino thiol and a degradation product of penicillin with high affinity for copper ions. In Drosophila melanogaster experiments, DPA significantly extended the survival of flies exposed to Cu(II), reversed Cu(II)-induced changes in oxidative stress (OS) markers—such as restoring total thiol (T-SH) and GSH levels—and reduced copper-induced alterations in AChE activity, demonstrating certain neuroprotective effects.306
DPA is an effective clinical treatment for WD. Both DPA and triethylenetetramine (trientine/TETA) mobilize copper in the liver and other tissues and promote its urinary excretion, reversing hepatic, neurological, and psychiatric symptoms in most WD patients.263,267 However, a systematic meta-analysis found that although DPA is effective, it is associated with a relatively higher incidence of adverse reactions compared with zinc therapy. In clinical practice, trientine or zinc salts may be considered as alternative or adjunct therapies.268
DPA may also have potential effects in AD. A small-scale clinical trial showed that DPA could reduce OS markers in the serum of AD patients but did not significantly improve cognitive function or slow clinical progression.265 In animal models, intranasally administered DPA carriers demonstrated possible effects on Aβ deposition and improved cognitive performance, although extensive further validation is still required.266
Triethylenetetramine
Triethylenetetramine (trientine/TETA) is a polyamine-structured copper chelator that forms stable complexes with copper ions through its four nitrogen atoms, effectively reducing systemic copper levels. It is an oral drug used for the treatment of WD.269 Trientine acts similarly to DPA but has fewer side effects and a lower risk of neurological deterioration. It promotes urinary copper excretion and inhibits intestinal copper absorption.270,307 Compared with DPA for restoring copper balance, trientine is particularly suitable for patients who are intolerant to penicillamine.308 Additionally, trientine can suppress in vivo inflammatory responses induced by hepatic radiofrequency ablation (RFA).309
Tetrathiomolybdate
Tetrathiomolybdate (TTM) has potent anti-copper properties. TTM forms stable ternary complexes with copper and proteins, reducing serum free copper levels.271 By rapidly lowering serum copper, TTM can restore normal copper homeostasis within weeks without causing abnormal elevations in serum copper levels.271 TTM is an effective clinical treatment for WD, significantly improving neurological symptoms, and the risk of neurological deterioration in WD patients treated with TTM is markedly lower than with TETA.272,273 TTM may cause adverse effects such as anemia, leukopenia, and elevated transaminase levels, but these are generally reversible with dose adjustment or temporary discontinuation.10
There is experimental evidence that TTM has therapeutic effects in ALS animal models. TTM can remove copper ions from copper-thiolate clusters such as SOD1, reducing the amount of SOD1 aggregates. It prolongs survival in familial ALS mouse models (SOD1(G93A)), mitigates loss of motor neurons and axons, and prevents skeletal muscle atrophy.274,275 In addition, TTM effectively lowers spinal cord copper levels and inhibits lipid peroxidation (LPO), thereby suppressing SOD1 enzymatic activity in SOD1(G93A) mice.275 However, there is currently insufficient clinical data to demonstrate clear efficacy in ALS patients.
Clioquinol
Clioquinol (CQ) is a hydrophobic small molecule capable of crossing the BBB and exhibiting moderate affinity for copper and zinc ions, showing therapeutic potential in several neurological disease models.
CQ’s chelation of copper and zinc effectively reduces spinal white matter damage and behavioral deficits in MS mouse models, markedly decreases microglial activation in experimental autoimmune encephalomyelitis mice, improves clinical symptoms, and lowers the incidence of spinal demyelination.276 In AD studies, CQ reduces ROS generation and LPO in yeast cells, decreases the toxicity of Aβ42 (an isoform of Aβ), and restores GSH homeostasis disrupted by Aβ42 through regulation of Yes-associated protein 1 (YAP1), thereby protecting cells from OS.277 In AD mouse models, CQ treatment increases soluble Aβ levels and reduces Aβ deposition in the brain.278 Intracellular Cu(I) accumulation is significantly reduced, and CQ also lowers endogenous phosphorylated tau in primary cortical neurons of mice.279
Diacetyl-Bis(4-Methylthiosemicarbazonato)copper(II)
Diacetyl-bis(4-methylthiosemicarbazonato)copper(II)(Cu(II)(atsm)) is an inflammation-modulating compound with potential therapeutic effects for ALS and PD.282,310
In ALS, treatment with Cu(II)(atsm) after symptom onset is less effective than pre-symptomatic administration, but it can still improve motor function and survival.281 In SOD1(G37R) mice (where glycine at residue 37 in SOD1 is replaced with arginine), Cu(II)(atsm) enhances copper delivery to metal-free (apo) SOD1, promoting its conversion to a more stable and less toxic holo-SOD1 form,280 inhibiting SOD1 activity and reducing aggregation of mutant SOD1 protein. Improvements in motor function and survival in SOD1(G37R) mice support Cu(II)(atsm) as a potential ALS therapeutic.281
In PD models, Cu(II)(atsm) exhibits neuroprotective effects associated with reduced OS and decreased α-Syn aggregation.283 Cu(II)(atsm) also reduces Aβ levels in APP-CHO cells (Chinese hamster ovary cells overexpressing APP).311 Additionally, Cu(II)(atsm) serves as a tracer for positron emission tomography (PET) imaging. In animal models, Cu(II)(atsm) demonstrates neuroprotective potential, possibly by improving mitochondrial function and reducing OS to protect dopaminergic neurons.312 However, its clinical efficacy requires further validation.
Bis-Choline Tetrathiomolybdate
Bis-choline tetrathiomolybdate (WTX101) is a first-in-class oral copper-protein chelator for the treatment of WD that is currently undergoing clinical trials. WTX101 can directly remove excess copper from hepatic copper stores and form stable ternary complexes with copper and proteins such as serum albumin in the circulation, promoting biliary copper excretion.284
In a Phase II open-label study, WTX101 demonstrated the potential to rapidly reduce serum non‑ceruloplasmin‑bound copper (NCC) and improve certain pathological markers.285
Tetradentate Monoquinolines
Tetradentate monoquinoline 20 (TDMQ20) acts on the cholinergic system to regulate copper homeostasis in the brains of AD patients. TDMQ20 improves memory and cognitive function in AD rat models by inhibiting OS catalyzed by Cu‑Aβ complexes.286,287 TDMQ20 efficiently crosses the BBB and is a potential therapeutic candidate for AD.288
Copper Ion Carriers
Elesclomol
Elesclomol (ELC) is a mitochondria-targeting copper ionophore and an inducer of OS. ELC targets mitochondria by forming a complex with copper and transporting it into the organelle, where ELC-Cu(II) is reduced to Cu(I), triggering ROS production.313 Studies have shown that ELC has potential to improve MD, as it delivers copper to mitochondria and increases COX levels in the brain, mitigating detrimental neurological lesions and improving survival in mottled brindled mice, a severe MD mouse model.289
5,7-Dichloro-2-[(Dimethylamino)methyl]-8-Hydroxyquinoline
The CQ derivative 5,7-Dichloro-2-[(dimethylamino)methyl]quinolin-8-ol (PBT2) is similar to CQ, targeting metal-induced Aβ aggregation, but is more effective as a zinc/copper ionophore and has improved BBB permeability and solubility,290 showing potential benefits in AD treatment. In AD mouse models, PBT2 inhibits copper-induced Aβ accumulation, reduces interstitial Aβ levels and tau phosphorylation, and restores cognitive function.291,292 PBT2 has undergone Phase II clinical trials in both AD and HD, demonstrating good safety but without clear clinical efficacy, indicating the need for further investigation.293,305
Additionally, in PD animal models, 5,7-dichloro-2-[(ethylamino)methyl]-8-hydroxy-3-methylquinazolin-4(3H)-one (PBT434/ATH434) prevents α-Syn accumulation and protects the nigrostriatal dopaminergic circuitry and motor function.294,314
Copper Nanoparticles
Nanoparticle delivery systems can cross the BBB and have the potential to transport DPA into the brain, preventing Aβ1-42 accumulation and reducing metal ion buildup in CNS diseases.315 Copper nanoparticles increase AChE and low-density lipoprotein (LDL) receptor-related protein 1 (LRP1) levels, reduce Aβ in the brain and gut, and decrease tau protein in the rat brain.295
However, in animal studies, high intake of copper nanoparticles severely affects hepatic drug metabolism in rats by inhibiting the expression of multiple cytochrome P450 (CYP450) enzymes, increasing the risk of drug-drug interactions.316 The combination of copper oxide nanoparticles (CuONPs) and furan synergistically enhances cardiovascular toxicity in zebrafish embryos.317
For neurological diseases, the optimal type of copper nanoparticles and their mechanisms of action remain unclear. Copper nanoparticle-based drugs for clinical use in neurological disorders are yet to be developed.
Natural Compounds
Natural compounds are a treasure trove for humans, with diverse and often mysterious biological effects. Many medicinal plants and their natural constituents possess antioxidant, free radical–scavenging, and neuroprotective properties, demonstrating significant efficacy in preventing copper-induced neurotoxicity.
Luteolin, a plant flavonoid extracted from Artemisia species, exhibits neuroprotective effects in neurological diseases and traumatic brain injury (TBI).296 Luteolin exerts its effects by downregulating amyloid beta precursor protein (AβPP) expression, inhibiting Aβ1-42 secretion, suppressing apoptosis, and modulating redox imbalance.318 Another study showed that coordination and transfer of divalent copper ions significantly enhance luteolin’s free radical scavenging efficiency and antioxidant activity.319
Apigenin, a low-toxicity flavonoid, antagonizes copper-mediated Aβ neurotoxicity and provides neuroprotection by alleviating OS, inhibiting ROS-induced signaling pathways, preventing apoptosis, and preserving mitochondrial function.299
Rutin, a glycoside and polyphenol, can cross the BBB and exhibits antioxidant properties. Rutin mitigates copper-induced brain injury—including cortical perforation layers and neuronal degeneration—by reducing OS and neuroinflammation.300
Resveratrol, a natural polyphenol found in various plants, modulates plasma copper and zinc levels and influences OS and antioxidant status in copper-deficient rats.320 Resveratrol improves memory and neuroinflammation through antioxidant, anti-inflammatory effects, reduction of Aβ-induced LPO, and regulation of cellular signaling pathways.297 Supplementation with resveratrol has shown some effects on cognitive and functional decline in AD patients.298 Additionally, resveratrol regulates cellular proteostasis by upregulating autophagy, thereby attenuating CuSO4-induced senescence.12 Another study reported that resveratrol effectively alleviates OS and hepatic-renal injury induced by CuONPs.321
Conclusion and Future Perspectives
Copper is an essential element involved in oxidative metabolism and is indispensable for brain cell function. The storage and utilization of copper affect tissues and organs throughout the body. Copper-dependent protein networks—including copper reductases, copper chaperones, and membrane transporters—play critical regulatory roles in maintaining intracellular copper homeostasis. Disruption of copper homeostasis contributes significantly to the pathogenesis of various neurological disorders. Both copper overload and copper deficiency can affect the nervous system through multiple mechanisms, including the induction of oxidative stress (OS), mitochondrial dysfunction, interference with protein folding, and disturbance of metabolic balance, ultimately leading to neuronal dysfunction and death.
In recent years, the discovery of cuproptosis has expanded our understanding of copper-related pathological mechanisms. Unlike classical forms of cell death such as apoptosis, necrosis, or ferroptosis, cuproptosis is characterized by the aberrant binding of copper ions to lipoylated proteins, resulting in protein aggregation and loss of iron–sulfur cluster proteins, which trigger mitochondrial destabilization and metabolic collapse.
Copper dyshomeostasis is closely associated with numerous neurological diseases, including Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS), multiple sclerosis (MS), Wilson disease (WD), Menkes disease (MD), and stroke. The pathogenic mechanisms of copper metabolism abnormalities vary considerably among different diseases, encompassing both the toxic effects of copper overload and enzyme dysfunction or metabolic imbalance caused by copper deficiency. These two opposing states often coexist in different stages or regions of the same disease.
Copper death (cuproptosis) modulators hold potential for alleviating neurotoxicity and protecting neurons. Developing more precise molecular-targeted modulators—such as drugs affecting copper transporters, copper chaperones, or mitochondrial copper utilization—could provide neuroprotection by intervening in the core regulatory mechanisms of cuproptosis. This approach may form part of future therapeutic strategies but faces challenges, including poor BBB penetration, insufficient targeting, and the risk of inhibiting essential metalloprotein enzymes. Combining modulators with targeted delivery systems, such as brain-targeted nanoparticles, may enhance their efficacy and safety in the central nervous system (CNS), enabling more precise treatment. In the context of neurological drug development, all preclinical experiments on cuproptosis modulators should be conducted according to GLP standards to ensure scientific rigor and reliability of experimental design and data analysis. The research workflow should encompass key steps, including cell and animal models, drug administration, assessment of copper homeostasis, and measurement of cuproptosis activity, thereby providing a high-quality evidence base for the subsequent development of cuproptosis-targeted therapeutics.
Abbreviations
Cu, copper; CuSO4‑SIPS, copper sulfate‐induced stress‐induced premature senescence; GBD, Global Burden of Disease; AchE, acetylcholinesterase; BACE1, β‑site amyloid precursor protein cleaving enzyme 1; AD, Alzheimer’s disease; TCA, tricarboxylic acid; DLAT, dihydrolipoamide S-acetyltransferase; DLST, dihydrolipoamide S-succinyltransferase; OXPHOS, oxidative phosphorylation; Fe-S, iron-sulfur; STEAP, six-transmembrane epithelial antigen of the prostate; CTR1/SLC31A1, high-affinity copper transporter 1; DCYTB, duodenal cytochrome b; ATOX1, antioxidant 1 copper chaperone; ATP7A, ATPase copper-transporting alpha; ATP7B, ATPase copper-transporting beta; CCS, copper chaperone for superoxide dismutase; SOD1, superoxide dismutase 1; COX17, cytochrome c oxidase copper chaperone 17; MT, metallothionein; GSH, glutathione; CP, ceruloplasmin; Sp1, specificity protein 1; DMT1, divalent metal transporter 1; Smf1p, divalent metal ion transporter SMF1; Fet4p, low-affinity Fe(II) transport protein; TGN, trans-Golgi network; MBDs, metal-binding domains; SOD3, superoxide dismutase 3; MD, Menkes disease; WD, Wilson disease; CNS, central nervous system; COX, cytochrome c oxidase; SLC25A3, solute carrier family 25 member 3; IMS, intermembrane space; SCO1, synthesis of cytochrome c oxidase 1; COX11, cytochrome c oxidase copper chaperone 11; COX1, mitochondrially encoded cytochrome c oxidase subunit I; COX2, mitochondrially encoded cytochrome c oxidase subunit II; SCC, small copper carrier; NAC, N-acetylcysteine; ELC, Elesclomol; FDX1, Ferredoxin 1; LIAS, lipoic acid synthetase; DSF, disulfiram; HSP70, heat shock protein 70; TLR4, toll-like receptor 4; NLRP3, NLR family pyrin domain containing 3; DBT, dihydrolipoamide branched‑chain transacylase E2; GCSH, glycine cleavage system protein H; UPS, ubiquitin-proteasome system; OS, oxidative stress; ROS, reactive oxygen species; CPT, copper pyrithione; 6-OHDA, 6-hydroxydopamine; NF-κB, nuclear factor kappa‑light‑chain‑enhancer of activated B cells; IκB, inhibitor of NF-κB; TNF-α, tumor necrosis factor-alpha; IL-1, interleukin-1; IL-6, interleukin-6; KGDH, alpha‑ketoglutarate dehydrogenase; PDH, pyruvate dehydrogenase; BNIP3, BCL-2/adenovirus E1B 19kDa interacting protein 3; TBI, traumatic brain injury; AMPK, AMP-activated protein kinase; ACC, acetyl‑CoA carboxylase; α-Syn, α-synuclein; Aβ, β-amyloid protein; NO, nitric oxide; LPO, lipid peroxidation; APP, amyloid precursor protein; LTP, long‑term potentiation; SYP, synaptophysin; PSD-95, postsynaptic density protein 95; 5-HT, 5-hydroxytryptamine; GABA, gamma-aminobutyric acid; BDNF, brain-derived neurotrophic factor; CREB, cAMP response element-binding protein; IFN-γ, interferon-γ; PD, Parkinson’s disease; ICP-MS, inductively coupled plasma mass spectrometry; GSTM1, glutathione S-transferase Mu1; GSTT1, glutathione S-transferase Theta1; HD, Huntington’s disease; HTT, huntingtin; polyQ, polyglutamine; mHTT, mutated huntingtin; LDH, lactate dehydrogenase; SDH, succinate dehydrogenase; NPC, nuclear pore complex; DRP1, dynamin-related protein 1; E1, ubiquitin-activating enzyme; E2, ubiquitin-conjugating enzyme; E3, ubiquitin ligase; ALS, amyotrophic lateral sclerosis; FALS, familial amyotrophic lateral sclerosis; SALS, sporadic amyotrophic lateral sclerosis; CSF, cerebrospinal fluid; TNF‑α, tumor necrosis factor‑alpha; IL‑1β, interleukin‑1 beta; MS, multiple sclerosis; TrkB, tropomyosin receptor kinase B; TRPM2, transient receptor potential melastatin 2; MRI, magnetic resonance imaging; DBH, dopamine-β-hydroxylase; L-DOPA, L-3,4-dihydroxyphenylalanine; LOX, lysyl oxidase; OHS, occipital horn syndrome; EPCs, endothelial progenitor cells; TSP‑1, thrombospondin-1; LDL, low-density lipoprotein; PAM, peptidylglycine α-amidating monooxygenase; NYP, neuropeptide Y; CRH, corticotropin-releasing hormone; BBB, blood-brain barrier; DPA, D-penicillamine; trientine/TETA, triethylenetetramine; CQ, clioquinol; YAP1, Yes-associated protein 1; TTM, tetrathiomolybdate; PBT2, 5,7-Dichloro-2-[(dimethylamino)methyl]quinolin-8-ol; Cu(II)(atsm), diacetyl-bis(4-methylthiosemicarbazonato)copper(II); PET, positron emission tomography; RFA, radiofrequency ablation; WTX101, bis-choline tetrathiomolybdate; TDMQs, tetradentate monoquinolines; TDMQ20, tetradentate monoquinoline20; PBT434, 5,7-dichloro-2-[(ethylamino)methyl]-8-hydroxy-3-methylquinazolin-4(3H)-one; CYP450, cytochrome P450; CuONPs, copper oxide nanoparticles; AβPP, amyloid beta precursor protein.
Acknowledgment
This work was strongly supported by the Natural Science Foundation of Hubei Province of China (2023AFB455).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Natural Science Foundation of Hubei Province of China (2023AFB455), the Foundation of Hubei Educational Committee (D20232803), the Hubei Provincial Natural Science Foundation and Xianning Innovation and development project (Grant Number: 2025AFD403), Hubei University of Science and Technology Horizontal Research Project (Grant Number: 2023HX188) and the Scientific Innovation Team of Hubei University of Science and Technology (Grant Number: 2023T11).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Scheltens P, De Strooper B, Kivipelto M, et al. Alzheimer’s disease. Lancet. 2021;397(10284):1577–32. doi:10.1016/s0140-6736(20)32205-4
2. Ben-Shlomo Y, Darweesh S, Llibre-Guerra J, Marras C, San Luciano M, Tanner C. The epidemiology of Parkinson’s disease. Lancet. 2024;403(10423):283–292. doi:10.1016/s0140-6736(23)01419-8
3. GBD 2021 Nervous System Disorders Collaborators. Global, regional, and national burden of disorders affecting the nervous system, 1990-2021: a systematic analysis for the global burden of disease study 2021. Lancet Neurol. 2024;23(4):344–381. doi:10.1016/s1474-4422(24)00038-3
4. Su D, Cui Y, He C, et al. Projections for prevalence of Parkinson’s disease and its driving factors in 195 countries and territories to 2050: modelling study of global burden of disease Study 2021. BMJ. 2025;388:e080952. doi:10.1136/bmj-2024-080952
5. Zhu J, Cui Y, Zhang J, et al. Temporal trends in the prevalence of Parkinson’s disease from 1980 to 2023: a systematic review and meta-analysis. Lancet Healthy Longev. 2024;5(7):e464–e479. doi:10.1016/s2666-7568(24)00094-1
6. Rasband MN. Glial contributions to neural function and disease. Mol Cell Proteomics. 2016;15(2):355–361. doi:10.1074/mcp.R115.053744
7. Sharma P, Giri A, Tripathi PN. Emerging trends: neurofilament biomarkers in precision neurology. Neurochem Res. 2024;49(12):3208–3225. doi:10.1007/s11064-024-04244-3
8. Tripathi PN, Lodhi A, Rai SN, et al. Review of pharmacotherapeutic targets in Alzheimer’s disease and its management using traditional medicinal plants. Degener Neurol Neuromuscul Dis. 2024;14:47–74. doi:10.2147/dnnd.S452009
9. Shrivastava SK, Nivrutti AA, Bhardwaj B, et al. Drug reposition-based design, synthesis, and biological evaluation of dual inhibitors of acetylcholinesterase and β-Secretase for treatment of Alzheimer’s disease. J Mol Struct. 2022;1262:132979. doi:10.1016/j.molstruc.2022.132979
10. Zhong G, Wang X, Li J, et al. Insights into the role of copper in neurodegenerative diseases and the therapeutic potential of natural compounds. Curr Neuropharmacol. 2024;22(10):1650–1671. doi:10.2174/1570159x22666231103085859
11. Wang Y, Li D, Xu K, Wang G, Zhang F. Copper homeostasis and neurodegenerative diseases. Neural Regen Res. 2025;20(11):3124–3143. doi:10.4103/nrr.Nrr-d-24-00642
12. Matos L, Gouveia AM, Almeida H. Resveratrol attenuates copper-induced senescence by improving cellular proteostasis. Oxid Med Cell Longev. 2017;2017:3793817. doi:10.1155/2017/3793817
13. Festa RA, Thiele DJ. Copper: an essential metal in biology. Curr Biol. 2011;21(21):R877–883. doi:10.1016/j.cub.2011.09.040
14. Yang Y, Wu J, Wang L, Ji G, Dang Y. Copper homeostasis and cuproptosis in health and disease. MedComm. 2024;5(10):e724. doi:10.1002/mco2.724
15. Chen L, Min J, Wang F. Copper homeostasis and cuproptosis in health and disease. Signal Transduct Target Ther. 2022;7(1):378. doi:10.1038/s41392-022-01229-y
16. Tsvetkov P, Coy S, Petrova B, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. 2022;375(6586):1254–1261. doi:10.1126/science.abf0529
17. Zhao R, Sukocheva O, Tse E, et al. Cuproptosis, the novel type of oxidation-induced cell death in thoracic cancers: can it enhance the success of immunotherapy? Cell Commun Signal. 2024;22(1):379. doi:10.1186/s12964-024-01743-2
18. Cobine PA, Brady DC. Cuproptosis: cellular and molecular mechanisms underlying copper-induced cell death. Mol Cell. 2022;82(10):1786–1787. doi:10.1016/j.molcel.2022.05.001
19. Xiong C, Ling H, Hao Q, Zhou X. Cuproptosis: p53-regulated metabolic cell death? Cell Death Differ. 2023;30(4):876–884. doi:10.1038/s41418-023-01125-0
20. Lutsenko S, Roy S, Tsvetkov P. Mammalian copper homeostasis: physiological roles and molecular mechanisms. Physiol Rev. 2025;105(1):441–491. doi:10.1152/physrev.00011.2024
21. Meng D, Luo G, Liu P. Copper metabolism and cuproptosis in Alzheimer’s disease: mechanisms and therapeutic potential. Biomed Pharmacother. 2025;190:118354. doi:10.1016/j.biopha.2025.118354
22. Peng G, Huang Y, Xie G, Tang J. Exploring copper’s role in stroke: progress and treatment approaches. Front Pharmacol. 2024;15:1409317. doi:10.3389/fphar.2024.1409317
23. Gao Q, Chen Y, Hu W, et al. From cell death to neurological disease: unraveling the role of copper. Neurobiol Dis. 2025;214:107042. doi:10.1016/j.nbd.2025.107042
24. Xu M, An Y, Liu X, et al. The molecular mechanisms of cuproptosis and its role in central nervous system diseases. Cell Signal. 2026;138:112236. doi:10.1016/j.cellsig.2025.112236
25. Siddiqui N, Talib M, Tripathi PN, Kumar A. Sharma A therapeutic potential of baicalein against neurodegenerative diseases: an updated review. Health Sci Rev. 2024;11:100172. doi:10.1016/j.hsr.2024.100172
26. Siddiqui N, Saifi A, Chaudhary A, Tripathi PN, Chaudhary A, Sharma A. Multifaceted neuroprotective role of punicalagin: a review. Neurochem Res. 2024;49(6):1427–1436. doi:10.1007/s11064-023-04081-w
27. Maung MT, Carlson A, Olea-Flores M, et al. The molecular and cellular basis of copper dysregulation and its relationship with human pathologies. FASEB J. 2021;35(9):e21810. doi:10.1096/fj.202100273RR
28. Thiele DJ. Integrating trace element metabolism from the cell to the whole organism. J Nutr. 2003;133(5 Suppl 1):1579s–1580s. doi:10.1093/jn/133.5.1579S
29. Moon N, Aryan M, Westerveld D, Nathoo S, Glover S, Kamel AY. Clinical manifestations of copper deficiency: a case report and review of the literature. Nutr Clin Pract. 2021;36(5):1080–1085. doi:10.1002/ncp.10582
30. Ohgami RS, Campagna DR, McDonald A, Fleming MD. The steap proteins are metalloreductases. Blood. 2006;108(4):1388–1394. doi:10.1182/blood-2006-02-003681
31. Wyman S, Simpson RJ, McKie AT, Sharp PA. Dcytb (Cybrd1) functions as both a ferric and a cupric reductase in vitro. FEBS Lett. 2008;582(13):1901–1906. doi:10.1016/j.febslet.2008.05.010
32. Nose Y, Wood LK, Kim BE, et al. Ctr1 is an apical copper transporter in mammalian intestinal epithelial cells in vivo that is controlled at the level of protein stability. J Biol Chem. 2010;285(42):32385–32392. doi:10.1074/jbc.M110.143826
33. Ravia JJ, Stephen RM, Ghishan FK, Collins JF. Menkes copper ATPase (Atp7a) is a novel metal-responsive gene in rat duodenum, and immunoreactive protein is present on brush-border and basolateral membrane domains. J Biol Chem. 2005;280(43):36221–36227. doi:10.1074/jbc.M506727200
34. Linder MC. Copper homeostasis in mammals, with emphasis on secretion and excretion. A Review. Int J Mol Sci. 2020;21(14):4932. doi:10.3390/ijms21144932
35. Agency for Toxic Substances and Disease Registry (ATSDR) Toxicological Profiles. Toxicological Profile for Copper. Atlanta, GA: Agency for Toxic Substances and Disease Registry (US); 2024.
36. Hatori Y, Inouye S, Akagi R. Thiol-based copper handling by the copper chaperone Atox1. IUBMB Life. 2017;69(4):246–254. doi:10.1002/iub.1620
37. Polishchuk EV, Concilli M, Iacobacci S, et al. Wilson disease protein ATP7B utilizes lysosomal exocytosis to maintain copper homeostasis. Dev Cell. 2014;29(6):686–700. doi:10.1016/j.devcel.2014.04.033
38. Zhu Z, Song M, Ren J, Liang L, Mao G, Chen M. Copper homeostasis and cuproptosis in central nervous system diseases. Cell Death Dis. 2024;15(11):850. doi:10.1038/s41419-024-07206-3
39. Turnlund JR, Keyes WR, Anderson HL, Acord LL. Copper absorption and retention in young men at three levels of dietary copper by use of the stable isotope 65Cu. Am J Clin Nutr. 1989;49(5):870–878. doi:10.1093/ajcn/49.5.870
40. Arnesano F, Natile G. Interference between copper transport systems and platinum drugs. Semin Cancer Biol. 2021;76:173–188. doi:10.1016/j.semcancer.2021.05.023
41. Li SR, Tao SY, Li Q, Hu CY, Sun ZJ. Harnessing nanomaterials for copper-induced cell death. Biomaterials. 2025;313:122805. doi:10.1016/j.biomaterials.2024.122805
42. Zhang X, Tao T, Qiu Y, Guo X, Zhu X, Zhou X. Copper-mediated novel cell death pathway in tumor cells and implications for innovative cancer therapies. Biomed Pharmacother. 2023;168:115730. doi:10.1016/j.biopha.2023.115730
43. Kuo YM, Gybina AA, Pyatskowit JW, Gitschier J, Prohaska JR. Copper transport protein (Ctr1) levels in mice are tissue specific and dependent on copper status. J Nutr. 2006;136(1):21–26. doi:10.1093/jn/136.1.21
44. Song IS, Chen HH, Aiba I, et al. Transcription factor Sp1 plays an important role in the regulation of copper homeostasis in mammalian cells. Mol Pharmacol. 2008;74(3):705–713. doi:10.1124/mol.108.046771
45. Liang ZD, Tsai WB, Lee MY, Savaraj N, Kuo MT. Specificity protein 1 (sp1) oscillation is involved in copper homeostasis maintenance by regulating human high-affinity copper transporter 1 expression. Mol Pharmacol. 2012;81(3):455–464. doi:10.1124/mol.111.076422
46. Chen WJ, Wu HT, Li CL, et al. Regulatory roles of six-transmembrane epithelial antigen of the prostate family members in the occurrence and development of malignant tumors. Front Cell Dev Biol. 2021;9:752426. doi:10.3389/fcell.2021.752426
47. Gao L, Zhang A. Copper-instigated modulatory cell mortality mechanisms and progress in oncological treatment investigations. Front Immunol. 2023;14:1236063. doi:10.3389/fimmu.2023.1236063
48. Lv X, Zhao L, Song Y, Chen W, Tuo Q. Deciphering the Role of copper homeostasis in atherosclerosis: from molecular mechanisms to therapeutic targets. Int J Mol Sci. 2024;25(21):11462. doi:10.3390/ijms252111462
49. Kayaaltı Z, Akyüzlü DK, Söylemezoğlu T. Evaluation of the effect of divalent metal transporter 1 gene polymorphism on blood iron, lead and cadmium levels. Environ Res. 2015;137:8–13. doi:10.1016/j.envres.2014.11.008
50. Lin C, Zhang Z, Wang T, Chen C, James Kang Y. Copper uptake by DMT1: a compensatory mechanism for CTR1 deficiency in human umbilical vein endothelial cells. Metallomics. 2015;7(8):1285–1289. doi:10.1039/c5mt00097a
51. Waters BM, Eide DJ. Combinatorial control of yeast FET4 gene expression by iron, zinc, and oxygen. J Biol Chem. 2002;277(37):33749–33757. doi:10.1074/jbc.M206214200
52. Hassett R, Dix DR, Eide DJ, Kosman DJ. The Fe(II) permease Fet4p functions as a low affinity copper transporter and supports normal copper trafficking in Saccharomyces cerevisiae. Biochem J. 2000;351(Pt 2):477–484.
53. Cohen A, Nelson H, Nelson N. The family of SMF metal ion transporters in yeast cells. J Biol Chem. 2000;275(43):33388–33394. doi:10.1074/jbc.M004611200
54. Polishchuk R, Lutsenko S. Golgi in copper homeostasis: a view from the membrane trafficking field. Histochem Cell Biol. 2013;140(3):285–295. doi:10.1007/s00418-013-1123-8
55. Hatori Y, Lutsenko S. The role of copper chaperone Atox1 in coupling redox homeostasis to intracellular copper distribution. Antioxidants. 2016;5(3):25. doi:10.3390/antiox5030025
56. Yu CH, Lee W, Nokhrin S, Dmitriev OY. The structure of metal binding domain 1 of the copper transporter ATP7B reveals mechanism of a singular Wilson disease mutation. Sci Rep. 2018;8(1):581. doi:10.1038/s41598-017-18951-1
57. Lutsenko S, Barnes NL, Bartee MY, Dmitriev OY. Function and regulation of human copper-transporting ATPases. Physiol Rev. 2007;87(3):1011–1046. doi:10.1152/physrev.00004.2006
58. La Fontaine S, Ackland ML, Mercer JF. Mammalian copper-transporting P-type ATPases, ATP7A and ATP7B: emerging roles. Int J Biochem Cell Biol. 2010;42(2):206–209. doi:10.1016/j.biocel.2009.11.007
59. Zhu S, Shanbhag V, Hodgkinson VL, Petris MJ. Multiple di-leucines in the ATP7A copper transporter are required for retrograde trafficking to the trans-Golgi network. Metallomics. 2016;8(9):993–1001. doi:10.1039/c6mt00093b
60. Kaler SG, DiStasio AT. ATP7A-related copper transport disorders. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. Genereviews(®). Seattle: University of Washington, Seattle; 1993.
61. La Fontaine S, Mercer JF. Trafficking of the copper-ATPases, ATP7A and ATP7B: role in copper homeostasis. Arch Biochem Biophys. 2007;463(2):149–167. doi:10.1016/j.abb.2007.04.021
62. Vulpe C, Levinson B, Whitney S, Packman S, Gitschier J. Isolation of a candidate gene for Menkes disease and evidence that it encodes a copper-transporting ATPase. Nat Genet. 1993;3(1):7–13. doi:10.1038/ng0193-7
63. Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat Genet. 1993;5(4):327–337. doi:10.1038/ng1293-327
64. Song W, Yue Y, Zhang Q, Wang X. Copper homeostasis dysregulation in respiratory diseases: a review of current knowledge. Front Physiol. 2024;15:1243629. doi:10.3389/fphys.2024.1243629
65. Cobine PA, Moore SA, Leary SC. Getting out what you put in: copper in mitochondria and its impacts on human disease. Biochim Biophys Acta Mol Cell Res. 2021;1868(1):118867. doi:10.1016/j.bbamcr.2020.118867
66. Cobine PA, Pierrel F, Winge DR. Copper trafficking to the mitochondrion and assembly of copper metalloenzymes. Biochim Biophys Acta. 2006;1763(7):759–772. doi:10.1016/j.bbamcr.2006.03.002
67. Horng YC, Cobine PA, Maxfield AB, Carr HS, Winge DR. Specific copper transfer from the Cox17 metallochaperone to both Sco1 and Cox11 in the assembly of yeast cytochrome C oxidase. J Biol Chem. 2004;279(34):35334–35340. doi:10.1074/jbc.M404747200
68. Jett KA, Leary SC. Building the Cu(A) site of cytochrome c oxidase: a complicated, redox-dependent process driven by a surprisingly large complement of accessory proteins. J Biol Chem. 2018;293(13):4644–4652. doi:10.1074/jbc.R117.816132
69. Banci L, Bertini I, Ciofi-Baffoni S, Hadjiloi T, Martinelli M, Palumaa P. Mitochondrial copper(I) transfer from Cox17 to Sco1 is coupled to electron transfer. Proc Natl Acad Sci U S A. 2008;105(19):6803–6808. doi:10.1073/pnas.0800019105
70. Nývltová E, Dietz JV, Seravalli J, Khalimonchuk O, Barrientos A. Coordination of metal center biogenesis in human cytochrome c oxidase. Nat Commun. 2022;13(1):3615. doi:10.1038/s41467-022-31413-1
71. Dodani SC, Leary SC, Cobine PA, Winge DR, Chang CJ. A targetable fluorescent sensor reveals that copper-deficient SCO1 and SCO2 patient cells prioritize mitochondrial copper homeostasis. J Am Chem Soc. 2011;133(22):8606–8616. doi:10.1021/ja2004158
72. Kawamata H, Manfredi G. Import, maturation, and function of SOD1 and its copper chaperone CCS in the mitochondrial intermembrane space. Antioxid Redox Signal. 2010;13(9):1375–1384. doi:10.1089/ars.2010.3212
73. Sturtz LA, Diekert K, Jensen LT, Lill R, Culotta VC. A fraction of yeast Cu,Zn-superoxide dismutase and its metallochaperone, CCS, localize to the intermembrane space of mitochondria. A physiological role for SOD1 in guarding against mitochondrial oxidative damage. J Biol Chem. 2001;276(41):38084–38089. doi:10.1074/jbc.M105296200
74. Field LS, Furukawa Y, O’Halloran TV, Culotta VC. Factors controlling the uptake of yeast copper/zinc superoxide dismutase into mitochondria. J Biol Chem. 2003;278(30):28052–28059. doi:10.1074/jbc.M304296200
75. Itoh S, Kim HW, Nakagawa O, et al. Novel role of antioxidant-1 (Atox1) as a copper-dependent transcription factor involved in cell proliferation. J Biol Chem. 2008;283(14):9157–9167. doi:10.1074/jbc.M709463200
76. Mao C, Wang M, Zhuang L, Gan B. Metabolic cell death in cancer: ferroptosis, cuproptosis, disulfidptosis, and beyond. Protein Cell. 2024;15(9):642–660. doi:10.1093/procel/pwae003
77. Gioilli BD, Kidane TZ, Fieten H, et al. Secretion and uptake of copper via a small copper carrier in blood fluid. Metallomics. 2022;14(3):mfac006. doi:10.1093/mtomcs/mfac006
78. Villalpando-Rodriguez GE, Gibson SB. Reactive oxygen species (ROS) regulates different types of cell death by acting as a rheostat. Oxid Med Cell Longev. 2021;2021:9912436. doi:10.1155/2021/9912436
79. Vo TTT, Peng TY, Nguyen TH, et al. The crosstalk between copper-induced oxidative stress and cuproptosis: a novel potential anticancer paradigm. Cell Commun Signal. 2024;22(1):353. doi:10.1186/s12964-024-01726-3
80. Gale JR, Hartnett-Scott K, Ross MM, Rosenberg PA, Aizenman E. Copper induces neuron-sparing, ferredoxin 1-independent astrocyte toxicity mediated by oxidative stress. J Neurochem. 2023;167(2):277–295. doi:10.1111/jnc.15961
81. Oh HN, Kim WK. Copper pyrithione and zinc pyrithione induce cytotoxicity and neurotoxicity in neuronal/astrocytic co-cultured cells via oxidative stress. Sci Rep. 2023;13(1):23060. doi:10.1038/s41598-023-49740-8
82. Scheiber IF, Mercer JF, Dringen R. Metabolism and functions of copper in brain. Prog Neurobiol. 2014;116:33–57. doi:10.1016/j.pneurobio.2014.01.002
83. Cruces-Sande A, Méndez-álvarez E, Soto-Otero R. Copper increases the ability of 6-hydroxydopamine to generate oxidative stress and the ability of ascorbate and glutathione to potentiate this effect: potential implications in Parkinson’s disease. J Neurochem. 2017;141(5):738–749. doi:10.1111/jnc.14019
84. Yamamoto Y, Gaynor RB. IkappaB kinases: key regulators of the NF-kappaB pathway. Trends Biochem Sci. 2004;29(2):72–79. doi:10.1016/j.tibs.2003.12.003
85. Zhou Q, Zhang Y, Lu L, et al. Copper induces microglia-mediated neuroinflammation through ROS/NF-κB pathway and mitophagy disorder. Food Chem Toxicol. 2022;168:113369. doi:10.1016/j.fct.2022.113369
86. Tang Y, Le W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol Neurobiol. 2016;53(2):1181–1194. doi:10.1007/s12035-014-9070-5
87. Caetano-Silva ME, Rund LA, Vailati-Riboni M, Pacheco MTB, Johnson RW. Copper-binding peptides attenuate microglia inflammation through suppression of NF-kB Pathway. Mol Nutr Food Res. 2021;65(22):e2100153. doi:10.1002/mnfr.202100153
88. Fu Q, Wu J, Zhou XY, et al. NLRP3/Caspase-1 pathway-induced pyroptosis mediated cognitive deficits in a mouse model of sepsis-associated encephalopathy. Inflammation. 2019;42(1):306–318. doi:10.1007/s10753-018-0894-4
89. Sun L, Ma W, Gao W, et al. Propofol directly induces caspase-1-dependent macrophage pyroptosis through the NLRP3-ASC inflammasome. Cell Death Dis. 2019;10(8):542. doi:10.1038/s41419-019-1761-4
90. Muñoz JP, Basei FL, Rojas ML, Galvis D, Zorzano A. Mechanisms of modulation of mitochondrial architecture. Biomolecules. 2023;13(8):1225. doi:10.3390/biom13081225
91. Rodrigues T, Ferraz LS. Therapeutic potential of targeting mitochondrial dynamics in cancer. Biochem Pharmacol. 2020;182:114282. doi:10.1016/j.bcp.2020.114282
92. Sheline CT, Choi DW. Cu2+ toxicity inhibition of mitochondrial dehydrogenases in vitro and in vivo. Ann Neurol. 2004;55(5):645–653. doi:10.1002/ana.20047
93. Chang H, Zhang W, Xu L, et al. Copper aggravated synaptic damage after traumatic brain injury by downregulating BNIP3-mediated mitophagy. Autophagy. 2025;21(3):548–564. doi:10.1080/15548627.2024.2409613
94. van Tol Amaral Guerra SM, Cordeiro Koppe de França L, Neto da Silva K, Scolari Grotto F, Glaser V. Copper dyshomeostasis and its relationship to AMPK activation, mitochondrial dynamics, and biogenesis of mitochondria: a systematic review of in vivo studies. J Trace Elem Med Biol. 2024;86:127549. doi:10.1016/j.jtemb.2024.127549
95. D’Ambrosi N, Rossi L. Copper at synapse: release, binding and modulation of neurotransmission. Neurochem Int. 2015;90:36–45. doi:10.1016/j.neuint.2015.07.006
96. Teng X, Stefaniak E, Willison KR, Ying L. Interplay between copper, phosphatidylserine, and α-synuclein suggests a link between copper homeostasis and synaptic vesicle cycling. ACS Chem Neurosci. 2024;15(15):2884–2896. doi:10.1021/acschemneuro.4c00280
97. Nam E, Nam G, Lim MH. Synaptic copper, amyloid-β, and neurotransmitters in Alzheimer’s disease. Biochemistry. 2020;59(1):15–17. doi:10.1021/acs.biochem.9b00775
98. van den Berghe PV, Klomp LW. New developments in the regulation of intestinal copper absorption. Nutr Rev. 2009;67(11):658–672. doi:10.1111/j.1753-4887.2009.00250.x
99. Fowler L, Engqvist H, Öhman-Mägi C. Effect of copper ion concentration on bacteria and cells. Materials. 2019;12(22):3798. doi:10.3390/ma12223798
100. Tapia L, González-Agüero M, Cisternas MF, et al. Metallothionein is crucial for safe intracellular copper storage and cell survival at normal and supra-physiological exposure levels. Biochem J. 2004;378(Pt 2):617–624. doi:10.1042/bj20031174
101. Gan B. Mitochondrial regulation of ferroptosis. J Cell Biol. 2021;220(9):e202105043. doi:10.1083/jcb.202105043
102. Tian Z, Jiang S, Zhou J, Zhang W. Copper homeostasis and cuproptosis in mitochondria. Life Sci. 2023;334:122223. doi:10.1016/j.lfs.2023.122223
103. Zhang L, Deng R, Liu L, Du H, Tang D. Novel insights into cuproptosis inducers and inhibitors. Front Mol Biosci. 2024;11:1477971. doi:10.3389/fmolb.2024.1477971
104. Li Y, Du Y, Zhou Y, et al. Iron and copper: critical executioners of ferroptosis, cuproptosis and other forms of cell death. Cell Commun Signal. 2023;21(1):327. doi:10.1186/s12964-023-01267-1
105. Springer C, Humayun D, Skouta R. Cuproptosis: unraveling the mechanisms of copper-induced cell death and its implication in cancer therapy. Cancers. 2024;16(3):647. doi:10.3390/cancers16030647
106. Tsvetkov P, Detappe A, Cai K, et al. Mitochondrial metabolism promotes adaptation to proteotoxic stress. Nat Chem Biol. 2019;15(7):681–689. doi:10.1038/s41589-019-0291-9
107. Nagai M, Vo NH, Shin Ogawa L, et al. The oncology drug elesclomol selectively transports copper to the mitochondria to induce oxidative stress in cancer cells. Free Radic Biol Med. 2012;52(10):2142–2150. doi:10.1016/j.freeradbiomed.2012.03.017
108. Zulkifli M, Spelbring AN, Zhang Y, et al. FDX1-dependent and independent mechanisms of elesclomol-mediated intracellular copper delivery. Proc Natl Acad Sci U S A. 2023;120(10):e2216722120. doi:10.1073/pnas.2216722120
109. Dreishpoon MB, Bick NR, Petrova B, et al. FDX1 regulates cellular protein lipoylation through direct binding to LIAS. J Biol Chem. 2023;299(9):105046. doi:10.1016/j.jbc.2023.105046
110. Yang S, Li X, Yan J, et al. Disulfiram downregulates ferredoxin 1 to maintain copper homeostasis and inhibit inflammation in cerebral ischemia/reperfusion injury. Sci Rep. 2024;14(1):15175. doi:10.1038/s41598-024-64981-x
111. Rowland EA, Snowden CK, Cristea IM. Protein lipoylation: an evolutionarily conserved metabolic regulator of health and disease. Curr Opin Chem Biol. 2018;42:76–85. doi:10.1016/j.cbpa.2017.11.003
112. Chen H, Li D, Zhang H, et al. Mechanisms of copper metabolism and cuproptosis: implications for liver diseases. Front Immunol. 2025;16:1633711. doi:10.3389/fimmu.2025.1633711
113. Chen Z, Yu J, Fu L, et al. Unveiling the metal-driven death: ferroptosis and cuproptosis in leukemia. Eur J Med Res. 2025;30(1):1241. doi:10.1186/s40001-025-03518-y
114. Du J, Huang Z, Li Y, et al. Copper exerts cytotoxicity through inhibition of iron-sulfur cluster biogenesis on ISCA1/ISCA2/ISCU assembly proteins. Free Radic Biol Med. 2023;204:359–373. doi:10.1016/j.freeradbiomed.2023.05.017
115. Li SR, Bu LL, Cai L. Cuproptosis: lipoylated TCA cycle proteins-mediated novel cell death pathway. Signal Transduct Target Ther. 2022;7(1):158. doi:10.1038/s41392-022-01014-x
116. Graff-Radford J, Yong KXX, Apostolova LG, et al. New insights into atypical Alzheimer’s disease in the era of biomarkers. Lancet Neurol. 2021;20(3):222–234. doi:10.1016/s1474-4422(20)30440-3
117. De-paula VJ, Radanovic M, Diniz BS, Forlenza OV. Alzheimer’s disease. Subcell Biochem. 2012;65:329–352. doi:10.1007/978-94-007-5416-4_14
118. Behl C. In 2024, the amyloid-cascade-hypothesis still remains a working hypothesis, no less but certainly no more. Front Aging Neurosci. 2024;16:1459224. doi:10.3389/fnagi.2024.1459224
119. Zhang H, Wei W, Zhao M, et al. Interaction between Aβ and Tau in the pathogenesis of Alzheimer’s disease. Int J Biol Sci. 2021;17(9):2181–2192. doi:10.7150/ijbs.57078
120. Goedert M, Sisodia SS, Price DL. Neurofibrillary tangles and beta-amyloid deposits in Alzheimer’s disease. Curr Opin Neurobiol. 1991;1(3):441–447. doi:10.1016/0959-4388(91)90067-h
121. Karran E, Mercken M, De Strooper B. The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov. 2011;10(9):698–712. doi:10.1038/nrd3505
122. Brewer GJ. Alzheimer’s disease causation by copper toxicity and treatment with zinc. Front Aging Neurosci. 2014;6:92. doi:10.3389/fnagi.2014.00092
123. Squitti R, Lupoi D, Pasqualetti P, et al. Elevation of serum copper levels in Alzheimer’s disease. Neurology. 2002;59(8):1153–1161. doi:10.1212/wnl.59.8.1153
124. Li DD, Zhang W, Wang ZY, Zhao P. Serum copper, zinc, and iron levels in patients with Alzheimer’s disease: a meta-analysis of case-control studies. Front Aging Neurosci. 2017;9:300. doi:10.3389/fnagi.2017.00300
125. Huang X. A concise review on oxidative stress-mediated ferroptosis and cuproptosis in Alzheimer’s disease. Cells. 2023;12(10):1369. doi:10.3390/cells12101369
126. Gromadzka G, Tarnacka B, Flaga A, Adamczyk A. Copper dyshomeostasis in neurodegenerative diseases-therapeutic implications. Int J Mol Sci. 2020;21(23):9259. doi:10.3390/ijms21239259
127. Kitazawa M, Hsu HW, Medeiros R. Copper exposure perturbs brain inflammatory responses and impairs clearance of amyloid-beta. Toxicol Sci. 2016;152(1):194–204. doi:10.1093/toxsci/kfw081
128. Cheignon C, Tomas M, Bonnefont-Rousselot D, Faller P, Hureau C, Collin F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018;14:450–464. doi:10.1016/j.redox.2017.10.014
129. Lamtai M, Zghari O, Ouakki S, et al. Chronic copper exposure leads to hippocampus oxidative stress and impaired learning and memory in male and female rats. Toxicol Res. 2020;36(4):359–366. doi:10.1007/s43188-020-00043-4
130. Aschner M, Skalny AV, Lu R, et al. Mitochondrial pathways of copper neurotoxicity: focus on mitochondrial dynamics and mitophagy. Front Mol Neurosci. 2024;17:1504802. doi:10.3389/fnmol.2024.1504802
131. Du H, Guo L, Yan S, Sosunov AA, McKhann GM, Yan SS. Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc Natl Acad Sci U S A. 2010;107(43):18670–18675. doi:10.1073/pnas.1006586107
132. Zhang Y, Zhou Q, Lu L, et al. Copper induces cognitive impairment in mice via modulation of cuproptosis and CREB signaling. Nutrients. 2023;15(4):972. doi:10.3390/nu15040972
133. Zhu X, Wang P, Liu H, et al. Changes and significance of SYP and GAP-43 expression in the hippocampus of CIH rats. Int J Med Sci. 2019;16(3):394–402. doi:10.7150/ijms.28359
134. Marinesco S, Carew TJ. Serotonin release evoked by tail nerve stimulation in the CNS of aplysia: characterization and relationship to heterosynaptic plasticity. J Neurosci. 2002;22(6):2299–2312. doi:10.1523/jneurosci.22-06-02299.2002
135. Ben-Ari Y, Gaiarsa JL, Tyzio R, Khazipov R. GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol Rev. 2007;87(4):1215–1284. doi:10.1152/physrev.00017.2006
136. Zhang Y, Smolen P, Alberini CM, Baxter DA, Byrne JH. Computational model of a positive BDNF feedback loop in hippocampal neurons following inhibitory avoidance training. Learn Mem. 2016;23(12):714–722. doi:10.1101/lm.042044.116
137. Palasz E, Wysocka A, Gasiorowska A, Chalimoniuk M, Niewiadomski W, Niewiadomska G. BDNF as a promising therapeutic agent in Parkinson’s disease. Int J Mol Sci. 2020;21(3):1170. doi:10.3390/ijms21031170
138. Wang Z, Zhang YH, Guo C, et al. Tetrathiomolybdate treatment leads to the suppression of inflammatory responses through the TRAF6/NFκB Pathway in LPS-Stimulated BV-2 Microglia. Front Aging Neurosci. 2018;10:9. doi:10.3389/fnagi.2018.00009
139. Jäntti H, Sitnikova V, Ishchenko Y, et al. Microglial amyloid beta clearance is driven by PIEZO1 channels. J Neuroinflammation. 2022;19(1):147. doi:10.1186/s12974-022-02486-y
140. Zheng Z, White C, Lee J, et al. Altered microglial copper homeostasis in a mouse model of Alzheimer’s disease. J Neurochem. 2010;114(6):1630–1638. doi:10.1111/j.1471-4159.2010.06888.x
141. Sehar U, Rawat P, Reddy AP, Kopel J, Reddy PH. Amyloid Beta in Aging and Alzheimer’s Disease. Int J Mol Sci. 2022;23(21):12924. doi:10.3390/ijms232112924
142. Strope TA, Wilkins HM. Amyloid precursor protein and mitochondria. Curr Opin Neurobiol. 2023;78:102651. doi:10.1016/j.conb.2022.102651
143. Kim SB, Mun BR, Kim SY, et al. Therapeutic effects of a novel synthetic α-secretase. Front Aging Neurosci. 2024;16:1383905. doi:10.3389/fnagi.2024.1383905
144. Barnham KJ, McKinstry WJ, Multhaup G, et al. Structure of the Alzheimer’s disease amyloid precursor protein copper binding domain. A regulator of neuronal copper homeostasis. J Biol Chem. 2003;278(19):17401–17407. doi:10.1074/jbc.M300629200
145. Barbier P, Zejneli O, Martinho M, et al. Role of tau as a microtubule-associated protein: structural and functional aspects. Front Aging Neurosci. 2019;11:204. doi:10.3389/fnagi.2019.00204
146. Voss K, Harris C, Ralle M, Duffy M, Murchison C, Quinn JF. Modulation of tau phosphorylation by environmental copper. Transl Neurodegener. 2014;3(1):24. doi:10.1186/2047-9158-3-24
147. Crouch PJ, Hung LW, Adlard PA, et al. Increasing Cu bioavailability inhibits Abeta oligomers and tau phosphorylation. Proc Natl Acad Sci U S A. 2009;106(2):381–386. doi:10.1073/pnas.0809057106
148. Ou Z, Pan J, Tang S, et al. Global trends in the incidence, prevalence, and years lived with disability of Parkinson’s disease in 204 countries/territories from 1990 to 2019. Front Public Health. 2021;9:776847. doi:10.3389/fpubh.2021.776847
149. Bloem BR, Okun MS, Klein C. Parkinson’s disease. Lancet. 2021;397(10291):2284–2303. doi:10.1016/s0140-6736(21)00218-x
150. Lo Bianco C, Ridet JL, Schneider BL, Deglon N, Aebischer P. alpha -Synucleinopathy and selective dopaminergic neuron loss in a rat lentiviral-based model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2002;99(16):10813–10818. doi:10.1073/pnas.152339799
151. Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388(6645):839–840. doi:10.1038/42166
152. Huang M, Zhang Y, Liu X. The mechanism of cuproptosis in Parkinson’s disease. Ageing Res Rev. 2024;95:102214. doi:10.1016/j.arr.2024.102214
153. Carboni E, Lingor P. Insights on the interaction of alpha-synuclein and metals in the pathophysiology of Parkinson’s disease. Metallomics. 2015;7(3):395–404. doi:10.1039/c4mt00339j
154. Davies KM, Bohic S, Carmona A, et al. Copper pathology in vulnerable brain regions in Parkinson’s disease. Neurobiol Aging. 2014;35(4):858–866. doi:10.1016/j.neurobiolaging.2013.09.034
155. Lorentzon E, Kumar R, Horvath I, Wittung-Stafshede P. Differential effects of Cu(2+) and Fe(3+) ions on in vitro amyloid formation of biologically-relevant α-synuclein variants. Biometals. 2020;33(2–3):97–106. doi:10.1007/s10534-020-00234-4
156. Rasia RM, Bertoncini CW, Marsh D, et al. Structural characterization of copper(II) binding to alpha-synuclein: insights into the bioinorganic chemistry of Parkinson’s disease. Proc Natl Acad Sci U S A. 2005;102(12):4294–4299. doi:10.1073/pnas.0407881102
157. Mason RJ, Paskins AR, Dalton CF, Smith DP. Copper binding and subsequent aggregation of α-synuclein are modulated by N-Terminal acetylation and ablated by the H50Q missense mutation. Biochemistry. 2016;55(34):4737–4741. doi:10.1021/acs.biochem.6b00708
158. Calvo JS, Mulpuri NV, Dao A, Qazi NK, Meloni G. Membrane insertion exacerbates the α-Synuclein-Cu(II) dopamine oxidase activity: metallothionein-3 targets and silences all α-synuclein-Cu(II) complexes. Free Radic Biol Med. 2020;158:149–161. doi:10.1016/j.freeradbiomed.2020.07.006
159. Wimalasena K, Adetuyi O, Eldani M. Metabolic energy decline coupled dysregulation of catecholamine metabolism in physiologically highly active neurons: implications for selective neuronal death in Parkinson’s disease. Front Aging Neurosci. 2024;16:1339295. doi:10.3389/fnagi.2024.1339295
160. Gou DH, Huang TT, Li W, et al. Inhibition of copper transporter 1 prevents α-synuclein pathology and alleviates nigrostriatal degeneration in AAV-based mouse model of Parkinson’s disease. Redox Biol. 2021;38:101795. doi:10.1016/j.redox.2020.101795
161. Gonzalez-Alcocer A, Gopar-Cuevas Y, Soto-Dominguez A, et al. Combined chronic copper exposure and aging lead to neurotoxicity in vivo. Neurotoxicology. 2023;95:181–192. doi:10.1016/j.neuro.2023.02.002
162. Zhang F, Chen F, Shen R, et al. Naphthalimide fluorescent skeleton for facile and accurate quantification of glutathione. Anal Chem. 2023;95(9):4301–4309. doi:10.1021/acs.analchem.2c04098
163. Sian J, Dexter DT, Lees AJ, et al. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann Neurol. 1994;36(3):348–355. doi:10.1002/ana.410360305
164. Rebai A, Chbili C, Ben Amor S, Hassine A, Ben Ammou S, Saguem S. Effects of glutathione S-transferase M1 and T1 deletions on Parkinson’s disease risk among a North African population. Rev Neurol. 2021;177(3):290–295. doi:10.1016/j.neurol.2020.03.013
165. Pieńkowska N, Bartosz G, Sadowska-Bartosz I. Effect of 6-hydroxydopamine increase the glutathione level in SH-SY5Y human neuroblastoma cells. Acta Biochim Pol. 2023;70(2):457–464. doi:10.18388/abp.2020_6847
166. Matsumura N, Kinoshita C, Bhadhprasit W, Nakaki T, Aoyama K. A purine derivative, paraxanthine, promotes cysteine uptake for glutathione synthesis. J Pharmacol Sci. 2023;151(1):37–45. doi:10.1016/j.jphs.2022.11.001
167. Trist BG, Davies KM, Cottam V, et al. Amyotrophic lateral sclerosis-like superoxide dismutase 1 proteinopathy is associated with neuronal loss in Parkinson’s disease brain. Acta Neuropathol. 2017;134(1):113–127. doi:10.1007/s00401-017-1726-6
168. Abdeen AH, Trist BG, Nikseresht S, et al. Parkinson-like wild-type superoxide dismutase 1 pathology induces nigral dopamine neuron degeneration in a novel murine model. Acta Neuropathol. 2025;149(1):22. doi:10.1007/s00401-025-02859-6
169. Craufurd D, Thompson JC, Snowden JS. Behavioral changes in Huntington Disease. Neuropsychiatry Neuropsychol Behav Neurol. 2001;14(4):219–226.
170. Hui BSM, Zhi LR, Retinasamy T, et al. The role of interferon-α in neurodegenerative diseases: a systematic review. J Alzheimers Dis. 2023;94(s1):S45–s66. doi:10.3233/jad-221081
171. Daldin M, Fodale V, Cariulo C, et al. Polyglutamine expansion affects huntingtin conformation in multiple Huntington’s disease models. Sci Rep. 2017;7(1):5070. doi:10.1038/s41598-017-05336-7
172. Takeuchi T, Nagai Y. Protein Misfolding and aggregation as a therapeutic target for polyglutamine diseases. Brain Sci. 2017;7(10):128. doi:10.3390/brainsci7100128
173. McColgan P, Tabrizi SJ. Huntington’s disease: a clinical review. Eur J Neurol. 2018;25(1):24–34. doi:10.1111/ene.13413
174. Ross CA, Tabrizi SJ. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 2011;10(1):83–98. doi:10.1016/s1474-4422(10)70245-3
175. Stoker TB, Mason SL, Greenland JC, Holden ST, Santini H, Barker RA. Huntington’s disease: diagnosis and management. Pract Neurol. 2022;22(1):32–41. doi:10.1136/practneurol-2021-003074
176. Xiao G, Fan Q, Wang X, Zhou B. Huntington disease arises from a combinatory toxicity of polyglutamine and copper binding. Proc Natl Acad Sci U S A. 2013;110(37):14995–15000. doi:10.1073/pnas.1308535110
177. Lobato AG, Ortiz-Vega N, Zhu Y, Neupane D, Meier KK, Zhai RG. Copper enhances aggregational toxicity of mutant huntingtin in a Drosophila model of Huntington’s Disease. Biochim Biophys Acta Mol Basis Dis. 2024;1870(1):166928. doi:10.1016/j.bbadis.2023.166928
178. Fox JH, Kama JA, Lieberman G, et al. Mechanisms of copper ion mediated Huntington’s disease progression. PLoS One. 2007;2(3):e334. doi:10.1371/journal.pone.0000334
179. Pamp K, Bramey T, Kirsch M, De Groot H, Petrat F. NAD(H) enhances the Cu(II)-mediated inactivation of lactate dehydrogenase by increasing the accessibility of sulfhydryl groups. Free Radic Res. 2005;39(1):31–40. doi:10.1080/10715760400023671
180. Petrat F, Bramey T, Kirsch M, De Groot H. Initiation of a superoxide-dependent chain oxidation of lactate dehydrogenase-bound NADH by oxidants of low and high reactivity. Free Radic Res. 2005;39(10):1043–1057. doi:10.1080/10715760500231786
181. Grima JC, Daigle JG, Arbez N, et al. Mutant huntingtin disrupts the nuclear pore complex. Neuron. 2017;94(1):93–107.e106. doi:10.1016/j.neuron.2017.03.023
182. Shirendeb UP, Calkins MJ, Manczak M, et al. Mutant huntingtin’s interaction with mitochondrial protein Drp1 impairs mitochondrial biogenesis and causes defective axonal transport and synaptic degeneration in Huntington’s disease. Hum Mol Genet. 2012;21(2):406–420. doi:10.1093/hmg/ddr475
183. Sap KA, Geijtenbeek KW, Schipper-Krom S, Guler AT, Reits EA. Ubiquitin-modifying enzymes in Huntington’s disease. Front Mol Biosci. 2023;10:1107323. doi:10.3389/fmolb.2023.1107323
184. Zhang B, Binks T, Burke R. The E3 ubiquitin ligase Slimb/β-TrCP is required for normal copper homeostasis in Drosophila. Biochim Biophys Acta Mol Cell Res. 2020;1867(10):118768. doi:10.1016/j.bbamcr.2020.118768
185. Zhang B, Kirn LA, Burke R. The Vhl E3 ubiquitin ligase complex regulates melanisation via sima, cnc and the copper import protein Ctr1A. Biochim Biophys Acta Mol Cell Res. 2021;1868(7):119022. doi:10.1016/j.bbamcr.2021.119022
186. Qin X, Wu P, Wen T, et al. Comparative assessment of blood Metal/metalloid levels, clinical heterogeneity, and disease severity in amyotrophic lateral sclerosis patients. Neurotoxicology. 2022;89:12–19. doi:10.1016/j.neuro.2022.01.003
187. Barberio J, Lally C, Kupelian V, Hardiman O, Flanders WD. Estimated familial amyotrophic lateral sclerosis proportion: a literature review and meta-analysis. Neurol Genet. 2023;9(6):e200109. doi:10.1212/nxg.0000000000200109
188. Al-Chalabi A, Calvo A, Chio A, et al. Analysis of amyotrophic lateral sclerosis as a multistep process: a population-based modelling study. Lancet Neurol. 2014;13(11):1108–1113. doi:10.1016/s1474-4422(14)70219-4
189. Gong YH, Elliott JL. Metallothionein expression is altered in a transgenic murine model of familial amyotrophic lateral sclerosis. Exp Neurol. 2000;162(1):27–36. doi:10.1006/exnr.2000.7323
190. Hilton JB, White AR, Crouch PJ. Metal-deficient SOD1 in amyotrophic lateral sclerosis. J Mol Med. 2015;93(5):481–487. doi:10.1007/s00109-015-1273-3
191. Yamazaki K, Tahara S, Ohyama T, Kuroi K, Nakabayashi T. SOD1 gains pro-oxidant activity upon aberrant oligomerization: change in enzymatic activity by intramolecular disulfide bond cleavage. Sci Rep. 2022;12(1):11750. doi:10.1038/s41598-022-15701-w
192. Tajiri M, Aoki H, Shintani A, Sue K, Akashi S, Furukawa Y. Metal distribution in Cu/Zn-superoxide dismutase revealed by native mass spectrometry. Free Radic Biol Med. 2022;183:60–68. doi:10.1016/j.freeradbiomed.2022.03.014
193. Tokuda E, Furukawa Y. Copper homeostasis as a therapeutic target in amyotrophic lateral sclerosis with SOD1 mutations. Int J Mol Sci. 2016;17(5):636. doi:10.3390/ijms17050636
194. Min JH, Sarlus H, Harris RA. Copper toxicity and deficiency: the vicious cycle at the core of protein aggregation in ALS. Front Mol Neurosci. 2024;17:1408159. doi:10.3389/fnmol.2024.1408159
195. Tokuda E, Nomura T, Ohara S, et al. A copper-deficient form of mutant Cu/Zn-superoxide dismutase as an early pathological species in amyotrophic lateral sclerosis. Biochim Biophys Acta Mol Basis Dis. 2018;1864(6 Pt A):2119–2130. doi:10.1016/j.bbadis.2018.03.015
196. Bourassa MW, Brown HH, Borchelt DR, Vogt S, Miller LM. Metal-deficient aggregates and diminished copper found in cells expressing SOD1 mutations that cause ALS. Front Aging Neurosci. 2014;6:110. doi:10.3389/fnagi.2014.00110
197. Chen QY, Wu P, Wen T, et al. Association of cerebral spinal fluid copper imbalance in amyotrophic lateral sclerosis. Front Aging Neurosci. 2022;14:970711. doi:10.3389/fnagi.2022.970711
198. Kysenius K, Hilton JB, Paul B, Hare DJ, Crouch PJ. Anatomical redistribution of endogenous copper in embryonic mice overexpressing SOD1. Metallomics. 2019;11(1):141–150. doi:10.1039/c8mt00242h
199. Tiwari A, Liba A, Sohn SH, et al. Metal deficiency increases aberrant hydrophobicity of mutant superoxide dismutases that cause amyotrophic lateral sclerosis. J Biol Chem. 2009;284(40):27746–27758. doi:10.1074/jbc.M109.043729
200. Lynch SM, Colón W. Dominant role of copper in the kinetic stability of Cu/Zn superoxide dismutase. Biochem Biophys Res Commun. 2006;340(2):457–461. doi:10.1016/j.bbrc.2005.12.024
201. Boyd SD, Ullrich MS, Skopp A, Winkler DD. Copper sources for Sod1 activation. Antioxidants. 2020;9(6):500. doi:10.3390/antiox9060500
202. Carrì MT, D’Ambrosi N, Cozzolino M. Pathways to mitochondrial dysfunction in ALS pathogenesis. Biochem Biophys Res Commun. 2017;483(4):1187–1193. doi:10.1016/j.bbrc.2016.07.055
203. Lu P, Yan HJ, Yang C, et al. High fat suppresses SOD1 activity by reducing copper chaperone for SOD1 associated with neurodegeneration and memory decline. Life Sci. 2021;272:119243. doi:10.1016/j.lfs.2021.119243
204. Son M, Elliott JL. Mitochondrial defects in transgenic mice expressing Cu,Zn superoxide dismutase mutations: the role of copper chaperone for SOD1. J Neurol Sci. 2014;336(1–2):1–7. doi:10.1016/j.jns.2013.11.004
205. Zetterström P, Stewart HG, Bergemalm D, et al. Soluble misfolded subfractions of mutant superoxide dismutase-1s are enriched in spinal cords throughout life in murine ALS models. Proc Natl Acad Sci U S A. 2007;104(35):14157–14162. doi:10.1073/pnas.0700477104
206. Bar-Or A, Li R. Cellular immunology of relapsing multiple sclerosis: interactions, checks, and balances. Lancet Neurol. 2021;20(6):470–483. doi:10.1016/s1474-4422(21)00063-6
207. Bierhansl L, Hartung HP, Aktas O, Ruck T, Roden M, Meuth SG. Thinking outside the box: non-canonical targets in multiple sclerosis. Nat Rev Drug Discov. 2022;21(8):578–600. doi:10.1038/s41573-022-00477-5
208. Hametner S, Wimmer I, Haider L, Pfeifenbring S, Brück W, Lassmann H. Iron and neurodegeneration in the multiple sclerosis brain. Ann Neurol. 2013;74(6):848–861. doi:10.1002/ana.23974
209. Wergeland S, Torkildsen Ø, Myhr KM, Mørk SJ, Bø L. The cuprizone model: regional heterogeneity of pathology. APMIS. 2012;120(8):648–657. doi:10.1111/j.1600-0463.2012.02882.x
210. Colombo E, Triolo D, Bassani C, et al. Dysregulated copper transport in multiple sclerosis may cause demyelination via astrocytes. Proc Natl Acad Sci U S A. 2021;118(27):e2025804118. doi:10.1073/pnas.2025804118
211. Shao Y, Chen C, Zhu T, et al. TRPM2 contributes to neuroinflammation and cognitive deficits in a cuprizone-induced multiple sclerosis model via NLRP3 inflammasome. Neurobiol Dis. 2021;160:105534. doi:10.1016/j.nbd.2021.105534
212. Yu W, Jiang LH, Zheng Y, Hu X, Luo J, Yang W. Inactivation of TRPM2 channels by extracellular divalent copper. PLoS One. 2014;9(11):e112071. doi:10.1371/journal.pone.0112071
213. Mezzaroba L, Alfieri DF, Colado Simão AN, Vissoci Reiche EM. The role of zinc, copper, manganese and iron in neurodegenerative diseases. Neurotoxicology. 2019;74:230–241. doi:10.1016/j.neuro.2019.07.007
214. Arakawa Y, Itoh S, Fukazawa Y, et al. Association between oxidative stress and microRNA expression pattern of ALS patients in the high-incidence area of the Kii Peninsula. Brain Res. 2020;1746:147035. doi:10.1016/j.brainres.2020.147035
215. Rasoul AA, Khudhur ZO, Hamad MS, et al. The role of oxidative stress and haematological parameters in relapsing-remitting multiple sclerosis in Kurdish population. Mult Scler Relat Disord. 2021;56:103228. doi:10.1016/j.msard.2021.103228
216. Tapiero H, Townsend DM, Tew KD. Trace elements in human physiology and pathology. Copper Biomed Pharmacother. 2003;57(9):386–398. doi:10.1016/s0753-3322(03)00012-x
217. Boga S, Jain D, Schilsky ML. Trientine induced colitis during therapy for Wilson disease: a case report and review of the literature. BMC Pharmacol Toxicol. 2015;16:30. doi:10.1186/s40360-015-0031-z
218. Akil M, Schwartz JA, Dutchak D, Yuzbasiyan-Gurkan V, Brewer GJ. The psychiatric presentations of Wilson’s disease. J Neuropsychiatry Clin Neurosci. 1991;3(4):377–382. doi:10.1176/jnp.3.4.377
219. Członkowska A, Litwin T, Chabik G. Wilson disease: neurologic features. Handb Clin Neurol. 2017;142:101–119. doi:10.1016/b978-0-444-63625-6.00010-0
220. Chang IJ, Hahn SH. The genetics of Wilson disease. Handb Clin Neurol. 2017;142:19–34. doi:10.1016/b978-0-444-63625-6.00003-3
221. Wu F, Wang J, Pu C, Qiao L, Jiang C. Wilson’s disease: a comprehensive review of the molecular mechanisms. Int J Mol Sci. 2015;16(3):6419–6431. doi:10.3390/ijms16036419
222. Członkowska A, Litwin T, Dusek P, et al. Wilson disease. Nat Rev Dis Primers. 2018;4(1):21. doi:10.1038/s41572-018-0018-3
223. Gromadzka G, Antos A, Sorysz Z, Litwin T. Psychiatric symptoms in Wilson’s disease-consequence of ATP7B gene mutations or just coincidence?-possible causal cascades and molecular pathways. Int J Mol Sci. 2024;25(22):12354. doi:10.3390/ijms252212354
224. Stremmel W, Merle U, Weiskirchen R. Clinical features of Wilson disease. Ann Transl Med. 2019;7(Suppl 2):S61. doi:10.21037/atm.2019.01.20
225. Shribman S, Poujois A, Bandmann O, Czlonkowska A, Warner TT. Wilson’s disease: update on pathogenesis, biomarkers and treatments. J Neurol Neurosurg Psychiatry. 2021;92(10):1053–1061. doi:10.1136/jnnp-2021-326123
226. Borchard S, Bork F, Rieder T, et al. The exceptional sensitivity of brain mitochondria to copper. Toxicol In Vitro. 2018;51:11–22. doi:10.1016/j.tiv.2018.04.012
227. Litwin T, Gromadzka G, Szpak GM, Jabłonka-Salach K, Bulska E, Członkowska A. Brain metal accumulation in Wilson’s disease. J Neurol Sci. 2013;329(1–2):55–58. doi:10.1016/j.jns.2013.03.021
228. Zhong W, Huang Z, Tang X. A study of brain MRI characteristics and clinical features in 76 cases of Wilson’s disease. J Clin Neurosci. 2019;59:167–174. doi:10.1016/j.jocn.2018.10.096
229. Dusek P, Litwin T, Członkowska A. Neurologic impairment in Wilson disease. Ann Transl Med. 2019;7(Suppl 2):S64. doi:10.21037/atm.2019.02.43
230. Terwel D, Löschmann YN, Schmidt HH, Schöler HR, Cantz T, Heneka MT. Neuroinflammatory and behavioural changes in the Atp7B mutant mouse model of Wilson’s disease. J Neurochem. 2011;118(1):105–112. doi:10.1111/j.1471-4159.2011.07278.x
231. Bulcke F, Dringen R, Scheiber IF. Neurotoxicity of Copper. Adv Neurobiol. 2017;18:313–343. doi:10.1007/978-3-319-60189-2_16
232. Kaler SG. Neurodevelopment and brain growth in classic Menkes disease is influenced by age and symptomatology at initiation of copper treatment. J Trace Elem Med Biol. 2014;28(4):427–430. doi:10.1016/j.jtemb.2014.08.008
233. Petris MJ, Mercer JF, Culvenor JG, Lockhart P, Gleeson PA, Camakaris J. Ligand-regulated transport of the Menkes copper P-type ATPase efflux pump from the Golgi apparatus to the plasma membrane: a novel mechanism of regulated trafficking. EMBO J. 1996;15(22):6084–6095.
234. Jaksch M, Paret C, Stucka R, et al. Cytochrome c oxidase deficiency due to mutations in SCO2, encoding a mitochondrial copper-binding protein, is rescued by copper in human myoblasts. Hum Mol Genet. 2001;10(26):3025–3035. doi:10.1093/hmg/10.26.3025
235. Goldstein DS, Holmes CS, Kaler SG. Relative efficiencies of plasma catechol levels and ratios for neonatal diagnosis of Menkes disease. Neurochem Res. 2009;34(8):1464–1468. doi:10.1007/s11064-009-9933-8
236. Kaler SG. ATP7A-related copper transport diseases-emerging concepts and future trends. Nat Rev Neurol. 2011;7(1):15–29. doi:10.1038/nrneurol.2010.180
237. Yi L, Kaler SG. Interaction between the AAA ATPase p97/VCP and a concealed UBX domain in the copper transporter ATP7A is associated with motor neuron degeneration. J Biol Chem. 2018;293(20):7606–7617. doi:10.1074/jbc.RA117.000686
238. Ojha R, Prasad AN. Menkes disease: what a multidisciplinary approach can do. J Multidiscip Healthc. 2016;9:371–385. doi:10.2147/jmdh.S93454
239. Kaler SG, Goldstein DS, Holmes C, Salerno JA, Gahl WA. Plasma and cerebrospinal fluid neurochemical pattern in Menkes disease. Ann Neurol. 1993;33(2):171–175. doi:10.1002/ana.410330206
240. Chen J, Jiang Y, Shi H, Peng Y, Fan X, Li C. The molecular mechanisms of copper metabolism and its roles in human diseases. Pflugers Arch. 2020;472(10):1415–1429. doi:10.1007/s00424-020-02412-2
241. Horn N, Wittung-Stafshede P. ATP7A-regulated enzyme metalation and trafficking in the Menkes disease puzzle. Biomedicines. 2021;9(4):391. doi:10.3390/biomedicines9040391
242. Gacheru S, McGee C, Uriu-Hare JY, et al. Expression and accumulation of lysyl oxidase, elastin, and type I procollagen in human Menkes and mottled mouse fibroblasts. Arch Biochem Biophys. 1993;301(2):325–329. doi:10.1006/abbi.1993.1151
243. Cosimo QC, Daniela L, Elsa B, Carlo DV, Giuseppe F. Kinky hair, kinky vessels, and bladder diverticula in Menkes disease. J Neuroimaging. 2011;21(2):e114–116. doi:10.1111/j.1552-6569.2010.00476.x
244. Kaler SG, Gallo LK, Proud VK, et al. Occipital horn syndrome and a mild Menkes phenotype associated with splice site mutations at the MNK locus. Nat Genet. 1994;8(2):195–202. doi:10.1038/ng1094-195
245. Beyens A, Van Meensel K, Pottie L, et al. Defining the Clinical, molecular and ultrastructural characteristics in occipital horn syndrome: two new cases and review of the literature. Genes. 2019;10(7):528. doi:10.3390/genes10070528
246. Tang J, Robertson S, Lem KE, Godwin SC, Kaler SG. Functional copper transport explains neurologic sparing in occipital horn syndrome. Genet Med. 2006;8(11):711–718. doi:10.1097/01.gim.0000245578.94312.1e
247. Campbell BCV, Khatri P. Stroke. Lancet. 2020;396(10244):129–142. doi:10.1016/s0140-6736(20)31179-x
248. Alsbrook DL, Di Napoli M, Bhatia K, et al. Neuroinflammation in acute ischemic and hemorrhagic stroke. Curr Neurol Neurosci Rep. 2023;23(8):407–431. doi:10.1007/s11910-023-01282-2
249. Atkin MA, Gasper A, Ullegaddi R, Powers HJ. Oxidative susceptibility of unfractionated serum or plasma: response to antioxidants in vitro and to antioxidant supplementation. Clin Chem. 2005;51(11):2138–2144. doi:10.1373/clinchem.2005.051078
250. Xiao Y, Yuan Y, Liu Y, et al. Circulating multiple metals and incident stroke in Chinese adults. Stroke. 2019;50(7):1661–1668. doi:10.1161/strokeaha.119.025060
251. Zhang M, Li W, Wang Y, Wang T, Ma M, Tian C. Association between the change of serum copper and ischemic stroke: a systematic review and meta-analysis. J Mol Neurosci. 2020;70(3):475–480. doi:10.1007/s12031-019-01441-6
252. Takizawa S, Nagata E, Nakayama T, Masuda H, Asahara T. Recent progress in endothelial progenitor cell culture systems: potential for stroke therapy. Neurol Med Chir. 2016;56(6):302–309. doi:10.2176/nmc.ra.2016-0027
253. Chen X, Cai Q, Liang R, et al. Copper homeostasis and copper-induced cell death in the pathogenesis of cardiovascular disease and therapeutic strategies. Cell Death Dis. 2023;14(2):105. doi:10.1038/s41419-023-05639-w
254. Jiang Y, Wang LP, Dong XH, et al. Trace amounts of copper in drinking water aggravate cerebral ischemic injury via impairing endothelial progenitor cells in mice. CNS Neurosci Ther. 2015;21(8):677–680. doi:10.1111/cns.12427
255. Xu J, Xu G, Fang J. Association between serum copper and stroke risk factors in adults: evidence from the national health and nutrition examination survey, 2011-2016. Biol Trace Elem Res. 2022;200(3):1089–1094. doi:10.1007/s12011-021-02742-x
256. Sakata H, Niizuma K, Wakai T, Narasimhan P, Maier CM, Chan PH. Neural stem cells genetically modified to overexpress Cu/Zn-superoxide dismutase enhance amelioration of ischemic stroke in mice. Stroke. 2012;43(9):2423–2429. doi:10.1161/strokeaha.112.656900
257. An Y, Li S, Huang X, Chen X, Shan H, Zhang M. The role of copper homeostasis in brain disease. Int J Mol Sci. 2022;23(22):13850. doi:10.3390/ijms232213850
258. Ye H, Li H, Gao Z. Copper binding induces nitration of NPY under nitrative stress: complicating the role of NPY in Alzheimer’s disease. Chem Res Toxicol. 2018;31(9):904–913. doi:10.1021/acs.chemrestox.8b00128
259. Domin H, Przykaza Ł, Jantas D, Kozniewska E, Boguszewski PM, Śmiałowska M. Neuropeptide Y Y2 and Y5 receptors as promising targets for neuroprotection in primary neurons exposed to oxygen-glucose deprivation and in transient focal cerebral ischemia in rats. Neuroscience. 2017;344:305–325. doi:10.1016/j.neuroscience.2016.12.040
260. Pain S, Brot S, Gaillard A. Neuroprotective effects of neuropeptide Y against neurodegenerative disease. Curr Neuropharmacol. 2022;20(9):1717–1725. doi:10.2174/1570159x19666210906120302
261. Stevens SL, Shaw TE, Dykhuizen E, et al. Reduced cerebral injury in CRH-R1 deficient mice after focal ischemia: a potential link to microglia and atrocytes that express CRH-R1. J Cereb Blood Flow Metab. 2003;23(10):1151–1159. doi:10.1097/01.Wcb.0000086957.72078.D4
262. Litin RB, Goldstein NP, Randall RV, Power MH, Diessner GR. Effect of D,L-penicillamine on the urinary excretion of copper and calcium in hepatolenticular degeneration (Wilson’s disease). Neurology. 1960;10:123–126. doi:10.1212/wnl.10.2.123
263. Hedera P. Clinical management of Wilson disease. Ann Transl Med. 2019;7(Suppl 2):S66. doi:10.21037/atm.2019.03.18
264. Walshe JM. Penicillamine, a new oral therapy for Wilson’s disease. Am J Med. 1956;21(4):487–495. doi:10.1016/0002-9343(56)90066-3
265. Squitti R, Rossini PM, Cassetta E, et al. d-penicillamine reduces serum oxidative stress in Alzheimer’s disease patients. Eur J Clin Invest. 2002;32(1):51–59. doi:10.1046/j.1365-2362.2002.00933.x
266. Zhong M, Kou H, Zhao P, et al. Nasal delivery of D-penicillamine hydrogel upregulates a disintegrin and metalloprotease 10 expression via melatonin receptor 1 in Alzheimer’s disease models. Front Aging Neurosci. 2021;13:660249. doi:10.3389/fnagi.2021.660249
267. Yarze JC. The mechanisms of penicillamine, trientine, and zinc in the treatment of Wilson’s disease. Am J Gastroenterol. 1995;90(6):1026.
268. Tang S, Bai L, Hou W, et al. Comparison of the effectiveness and safety of d-penicillamine and zinc salt treatment for symptomatic Wilson disease: a systematic review and meta-analysis. Front Pharmacol. 2022;13:847436. doi:10.3389/fphar.2022.847436
269. Kamlin COF, Jenkins TM, Heise JL, Amin NS. Trientine tetrahydrochloride, from bench to bedside: a narrative review. Drugs. 2024;84(12):1509–1518. doi:10.1007/s40265-024-02099-0
270. Kirk FT, Munk DE, Swenson ES, et al. Effects of trientine and penicillamine on intestinal copper uptake: a mechanistic 64 Cu PET/CT study in healthy humans. Hepatology. 2024;79(5):1065–1074. doi:10.1097/hep.0000000000000708
271. Brewer GJ, Askari F, Dick RB, et al. Treatment of Wilson’s disease with tetrathiomolybdate: v. Control of free copper by tetrathiomolybdate and a comparison with trientine. Transl Res. 2009;154(2):70–77. doi:10.1016/j.trsl.2009.05.002
272. Brewer GJ, Askari F, Lorincz MT, et al. Treatment of Wilson disease with ammonium tetrathiomolybdate: IV. Comparison of tetrathiomolybdate and trientine in a double-blind study of treatment of the neurologic presentation of Wilson disease. Arch Neurol. 2006;63(4):521–527. doi:10.1001/archneur.63.4.521
273. Brewer GJ, Hedera P, Kluin KJ, et al. Treatment of Wilson disease with ammonium tetrathiomolybdate: III. Initial therapy in a total of 55 neurologically affected patients and follow-up with zinc therapy. Arch Neurol. 2003;60(3):379–385. doi:10.1001/archneur.60.3.379
274. Tokuda E, Okawa E, Watanabe S, Ono S, Marklund SL. Dysregulation of intracellular copper homeostasis is common to transgenic mice expressing human mutant superoxide dismutase-1s regardless of their copper-binding abilities. Neurobiol Dis. 2013;54:308–319. doi:10.1016/j.nbd.2013.01.001
275. Tokuda E, Ono S, Ishige K, et al. Ammonium tetrathiomolybdate delays onset, prolongs survival, and slows progression of disease in a mouse model for amyotrophic lateral sclerosis. Exp Neurol. 2008;213(1):122–128. doi:10.1016/j.expneurol.2008.05.011
276. Choi BY, Jang BG, Kim JH, et al. Copper/zinc chelation by clioquinol reduces spinal cord white matter damage and behavioral deficits in a murine MOG-induced multiple sclerosis model. Neurobiol Dis. 2013;54:382–391. doi:10.1016/j.nbd.2013.01.012
277. Zheng Q, Zhu H, Lv C, et al. Clioquinol rescues yeast cells from Aβ42 toxicity via the inhibition of oxidative damage. Biotechnol J. 2024;19(6):e2300662. doi:10.1002/biot.202300662
278. Cherny RA, Atwood CS, Xilinas ME, et al. Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron. 2001;30(3):665–676. doi:10.1016/s0896-6273(01)00317-8
279. Lin G, Zhu F, Kanaan NM, et al. Clioquinol decreases levels of phosphorylated, truncated, and oligomerized tau protein. Int J Mol Sci. 2021;22(21):12063. doi:10.3390/ijms222112063
280. Roberts BR, Lim NK, McAllum EJ, et al. Oral treatment with Cu(II)(atsm) increases mutant SOD1 in vivo but protects motor neurons and improves the phenotype of a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci. 2014;34(23):8021–8031. doi:10.1523/jneurosci.4196-13.2014
281. McAllum EJ, Lim NK, Hickey JL, et al. Therapeutic effects of CuII(atsm) in the SOD1-G37R mouse model of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14(7–8):586–590. doi:10.3109/21678421.2013.824000
282. Soon CPW, Donnelly PS, Turner BJ, et al. Diacetylbis(N(4)-methylthiosemicarbazonato) copper(II) (CuII(atsm)) protects against peroxynitrite-induced nitrosative damage and prolongs survival in amyotrophic lateral sclerosis mouse model. J Biol Chem. 2011;286(51):44035–44044. doi:10.1074/jbc.M111.274407
283. Southon A, Szostak K, Acevedo KM, et al. Cu(II) (atsm) inhibits ferroptosis: implications for treatment of neurodegenerative disease. Br J Pharmacol. 2020;177(3):656–667. doi:10.1111/bph.14881
284. Weiss KH, Członkowska A, Hedera P, Ferenci P. WTX101 - an investigational drug for the treatment of Wilson disease. Expert Opin Investig Drugs. 2018;27(6):561–567. doi:10.1080/13543784.2018.1482274
285. Weiss KH, Askari FK, Czlonkowska A, et al. Bis-choline tetrathiomolybdate in patients with Wilson’s disease: an open-label, multicentre, phase 2 study. Lancet Gastroenterol Hepatol. 2017;2(12):869–876. doi:10.1016/s2468-1253(17)30293-5
286. Sun F, Zhao J, Zhang H, et al. Proteomics evidence of the role of TDMQ20 in the cholinergic system and synaptic transmission in a mouse model of Alzheimer’s disease. ACS Chem Neurosci. 2022;13(21):3093–3107. doi:10.1021/acschemneuro.2c00455
287. Zhao J, Shi Q, Tian H, et al. TDMQ20, a specific copper chelator, reduces memory impairments in Alzheimer’s disease mouse models. ACS Chem Neurosci. 2021;12(1):140–149. doi:10.1021/acschemneuro.0c00621
288. Huang L, Zeng Y, Li Y, et al. Distribution in rat blood and brain of TDMQ20, a copper chelator designed as a drug-candidate for Alzheimer’s disease. Pharmaceutics. 2022;14(12):2691. doi:10.3390/pharmaceutics14122691
289. Guthrie LM, Soma S, Yuan S, et al. Elesclomol alleviates Menkes pathology and mortality by escorting Cu to cuproenzymes in mice. Science. 2020;368(6491):620–625. doi:10.1126/science.aaz8899
290. Adlard PA, Cherny RA, Finkelstein DI, et al. Rapid restoration of cognition in Alzheimer’s transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Abeta. Neuron. 2008;59(1):43–55. doi:10.1016/j.neuron.2008.06.018
291. Bush AI. Drug development based on the metals hypothesis of Alzheimer’s disease. J Alzheimers Dis. 2008;15(2):223–240. doi:10.3233/jad-2008-15208
292. Faux NG, Ritchie CW, Gunn A, et al. PBT2 rapidly improves cognition in Alzheimer’s Disease: additional phase II analyses. J Alzheimers Dis. 2010;20(2):509–516. doi:10.3233/jad-2010-1390
293. Huntington Study Group Reach2HD Investigators. Safety, tolerability, and efficacy of PBT2 in Huntington’s disease: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2015;14(1):39–47. doi:10.1016/s1474-4422(14)70262-5
294. Finkelstein DI, Billings JL, Adlard PA, et al. The novel compound PBT434 prevents iron mediated neurodegeneration and alpha-synuclein toxicity in multiple models of Parkinson’s disease. Acta Neuropathol Commun. 2017;5(1):53. doi:10.1186/s40478-017-0456-2
295. Cendrowska-Pinkosz M, Krauze M, Juśkiewicz J, Fotschki B, Ognik K. The influence of copper nanoparticles on neurometabolism marker levels in the brain and intestine in a rat model. Int J Mol Sci. 2023;24(14):11321. doi:10.3390/ijms241411321
296. Kempuraj D, Thangavel R, Kempuraj DD, et al. Neuroprotective effects of flavone luteolin in neuroinflammation and neurotrauma. Biofactors. 2021;47(2):190–197. doi:10.1002/biof.1687
297. Wiciński M, Domanowska A, Wódkiewicz E, Malinowski B. Neuroprotective properties of resveratrol and its derivatives-influence on potential mechanisms leading to the development of Alzheimer’s disease. Int J Mol Sci. 2020;21(8):2749. doi:10.3390/ijms21082749
298. Tosatti JAG, Fontes A, Caramelli P, Gomes KB. Effects of resveratrol supplementation on the cognitive function of patients with Alzheimer’s disease: a systematic review of randomized controlled trials. Drugs Aging. 2022;39(4):285–295. doi:10.1007/s40266-022-00923-4
299. Zhao L, Wang JL, Wang YR, Fa XZ. Apigenin attenuates copper-mediated β-amyloid neurotoxicity through antioxidation, mitochondrion protection and MAPK signal inactivation in an AD cell model. Brain Res. 2013;1492:33–45. doi:10.1016/j.brainres.2012.11.019
300. Arowoogun J, Akanni OO, Adefisan AO, Owumi SE, Tijani AS, Adaramoye OA. Rutin ameliorates copper sulfate-induced brain damage via antioxidative and anti-inflammatory activities in rats. J Biochem Mol Toxicol. 2021;35(1):e22623. doi:10.1002/jbt.22623
301. Rakshit A, Khatua K, Shanbhag V, Comba P, Datta A. Cu(2+) selective chelators relieve copper-induced oxidative stress in vivo. Chem Sci. 2018;9(41):7916–7930. doi:10.1039/c8sc04041a
302. Roy S, Lutsenko S. Mechanism of Cu entry into the brain: many unanswered questions. Neural Regen Res. 2024;19(11):2421–2429. doi:10.4103/1673-5374.393107
303. Bonda DJ, Liu G, Men P, Perry G, Smith MA, Zhu X. Nanoparticle delivery of transition-metal chelators to the brain: oxidative stress will never see it coming! CNS Neurol Disord Drug Targets. 2012;11(1):81–85. doi:10.2174/187152712799960709
304. Tosato M, Di Marco V. Metal chelation therapy and Parkinson’s disease: a critical review on the thermodynamics of complex formation between relevant metal ions and promising or established drugs. Biomolecules. 2019;9(7):269. doi:10.3390/biom9070269
305. Lannfelt L, Blennow K, Zetterberg H, et al. Safety, efficacy, and biomarker findings of PBT2 in targeting Abeta as a modifying therapy for Alzheimer’s disease: a phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2008;7(9):779–786. doi:10.1016/s1474-4422(08)70167-4
306. Abolaji AO, Fasae KD, Iwezor CE, Farombi EO. D-Penicillamine prolongs survival and lessens copper-induced toxicity in Drosophila melanogaster. Toxicol Res. 2020;9(4):346–352. doi:10.1093/toxres/tfaa032
307. Ala A, Walker AP, Ashkan K, Dooley JS, Schilsky ML. Wilson’s disease. Lancet. 2007;369(9559):397–408. doi:10.1016/s0140-6736(07)60196-2
308. Li X, Hu F, Xu G. Membranous nephropathy caused by dimercaptosuccinic acid in a patient with Wilson’s disease: a case report and literature review. BMC Nephrol. 2023;24(1):147. doi:10.1186/s12882-023-03201-6
309. Yin JM, Sun LB, Zheng JS, Wang XX, Chen DX, Li N. Copper chelation by trientine dihydrochloride inhibits liver RFA-induced inflammatory responses in vivo. Inflamm Res. 2016;65(12):1009–1020. doi:10.1007/s00011-016-0986-2
310. Huuskonen MT, Tuo QZ, Loppi S, et al. The copper bis(thiosemicarbazone) complex Cu(II)(atsm) Is Protective Against Cerebral Ischemia Through Modulation Of The Inflammatory Milieu. Neurotherapeutics. 2017;14(2):519–532. doi:10.1007/s13311-016-0504-9
311. Li YQ, Tan SS, Wu D, Zhang Q, Wang T, Zheng G. The role of intracellular and extracellular copper compartmentalization in Alzheimer’s disease pathology and its implications for diagnosis and therapy. Front Neurosci. 2025;19:1553064. doi:10.3389/fnins.2025.1553064
312. Hung LW, Villemagne VL, Cheng L, et al. The hypoxia imaging agent CuII(atsm) is neuroprotective and improves motor and cognitive functions in multiple animal models of Parkinson’s disease. J Exp Med. 2012;209(4):837–854. doi:10.1084/jem.20112285
313. Tarin M, Babaie M, Eshghi H, Matin MM, Saljooghi AS. Elesclomol, a copper-transporting therapeutic agent targeting mitochondria: from discovery to its novel applications. J Transl Med. 2023;21(1):745. doi:10.1186/s12967-023-04533-5
314. Davies KM, Mercer JF, Chen N, Double KL. Copper dyshomoeostasis in Parkinson’s disease: implications for pathogenesis and indications for novel therapeutics. Clin Sci. 2016;130(8):565–574. doi:10.1042/cs20150153
315. Cui Z, Lockman PR, Atwood CS, et al. Novel D-penicillamine carrying nanoparticles for metal chelation therapy in Alzheimer’s and other CNS diseases. Eur J Pharm Biopharm. 2005;59(2):263–272. doi:10.1016/j.ejpb.2004.07.009
316. Tang H, Xu M, Shi F, et al. Effects and mechanism of nano-copper exposure on hepatic cytochrome P450 enzymes in rats. Int J Mol Sci. 2018;19(7):2140. doi:10.3390/ijms19072140
317. Saputra F, Uapipatanakul B, Lee JS, et al. Co-treatment of copper oxide nanoparticle and carbofuran enhances cardiotoxicity in zebrafish embryos. Int J Mol Sci. 2021;22(15):8259. doi:10.3390/ijms22158259
318. Liu R, Meng F, Zhang L, et al. Luteolin isolated from the medicinal plant Elsholtzia rugulosa (Labiatae) prevents copper-mediated toxicity in β-amyloid precursor protein Swedish mutation overexpressing SH-SY5Y cells. Molecules. 2011;16(3):2084–2096. doi:10.3390/molecules16032084
319. Xu Y, Yang J, Lu Y, et al. Copper(II) coordination and translocation in luteolin and effect on radical scavenging. J Phys Chem B. 2020;124(2):380–388. doi:10.1021/acs.jpcb.9b10531
320. Majewski M, Ognik K, Thoene M, Rawicka A, Juśkiewicz J. Resveratrol modulates the blood plasma levels of Cu and Zn, the antioxidant status and the vascular response of thoracic arteries in copper deficient Wistar rats. Toxicol Appl Pharmacol. 2020;390:114877. doi:10.1016/j.taap.2020.114877
321. Khalid S, Afzal N, Khan JA, et al. Antioxidant resveratrol protects against copper oxide nanoparticle toxicity in vivo. Naunyn Schmiedebergs Arch Pharmacol. 2018;391(10):1053–1062. doi:10.1007/s00210-018-1526-0
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Cuproptosis and Orthopedic Diseases: Molecular Mechanisms of Copper Metabolic Imbalance in the Skeletal System and Clinical Translation Prospects
Huang F, Wang J, Pan X
Orthopedic Research and Reviews 2026, 18:597482
Published Date: 19 May 2026