Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 10 » Issue 1
COPD: balancing oxidants and antioxidants
Authors Fischer B, Voynow J, Ghio A ![]()
Received 14 June 2014
Accepted for publication 10 October 2014
Published 2 February 2015 Volume 2015:10(1) Pages 261—276
DOI https://doi.org/10.2147/COPD.S42414
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Richard Russell
Bernard M Fischer,1,* Judith A Voynow,2,* Andrew J Ghio3,*
1Department of Pediatrics, Duke University Medical Center, Durham, NC, USA; 2Department of Pediatrics, Children's Hospital of Richmond at Virginia Commonwealth University, Richmond, VA, USA; 3National Health and Environmental Effects Research Laboratory, US Environmental Protection Agency, Chapel Hill, NC, USA
*These authors contributed equally to this work
Abstract: Chronic obstructive pulmonary disease (COPD) is one of the most common chronic illnesses in the world. The disease encompasses emphysema, chronic bronchitis, and small airway obstruction and can be caused by environmental exposures, primarily cigarette smoking. Since only a small subset of smokers develop COPD, it is believed that host factors interact with the environment to increase the propensity to develop disease. The major pathogenic factors causing disease include infection and inflammation, protease and antiprotease imbalance, and oxidative stress overwhelming antioxidant defenses. In this review, we will discuss the major environmental and host sources for oxidative stress; discuss how oxidative stress regulates chronic bronchitis; review the latest information on genetic predisposition to COPD, specifically focusing on oxidant/antioxidant imbalance; and review future antioxidant therapeutic options for COPD. The complexity of COPD will necessitate a multi-target therapeutic approach. It is likely that antioxidant supplementation and dietary antioxidants will have a place in these future combination therapies.
Keywords: cigarette smoking, mucins, gene regulation, Chinese herbs, acupuncture, dietary antioxidants
Introduction
Chronic obstructive pulmonary disease (COPD) is a disease state characterized by airflow limitation that is not fully reversible;1 it can include some combination of emphysema, chronic bronchitis (CB), and small airway obstruction.2,3 The airflow limitation is usually progressive and associated with an abnormal inflammatory response of the lungs. The mechanistic basis underlying COPD is complex and can involve recurrent inflammation, oxidative stress (ie, oxidant/antioxidant imbalance), protease/antiprotease imbalance, environmental insult, and host genetics.4 Cigarette smoking serves as the major risk factor for developing COPD and is also the major source of oxidants/reactive oxygen species (ROS) to the lungs and the body in exposed individuals. It is this recurrent oxidative stress from environmental sources (eg, cigarette smoking) and persistent inflammation that can lead to extensive tissue damage and disease exacerbation susceptibility. This review focuses first on the relationship of cigarette smoking to COPD-associated oxidative stress. The second part addresses oxidant regulation of mucin gene expression and also genetic and epigenetic factors important for COPD-associated oxidative stress. Finally, we present an update about the current antioxidant nutritional and dietary supplements that have been evaluated for COPD therapy, along with alternative therapies such as acupuncture that have also been evaluated as potential COPD therapeutics.
Cigarette smoking and oxidative stress
Temperatures in the combustion zone of a burning cigarette (800°C–950°C) result in a complete pyrolysis of tobacco. However, immediately downstream, a rapid drop in temperature (200°C–600°C) and a lack of oxygen allow for an incomplete combustion. Subsequently, a complex aerosol is generated during smoking, which includes condensed liquid droplets (the particulate fraction or tar) suspended in a mixture of volatile/semivolatile compounds and combustion gases (the gas fraction). A single puff of such cigarette smoke was quantified to contain 1017 free radicals in the tar phase and 1015 in the gas phase.5,6 Numerous oxidant compounds have been identified among the 4,000 to 7,000 constituents in cigarette smoke.7,8 In the particulate fraction, phenols and semiquinones are recognized among these compounds, while superoxide (O2−), epoxides, peroxides, nitric oxide (NO; 500 to 1,000 ppm), nitrogen dioxide, peroxynitrite (ONOO−), and peroxynitrates are included in the gas phase.
Smoking produces a shift in the normal balance between oxidants and antioxidants to impact an oxidative stress both in the lungs and systemically. Oxidants included in cigarette smoke can directly injure cells and tissues, inactivate defense mechanisms, and initiate inflammation, which further elevates oxidative stress. It is difficult, if not impossible, to determine if the oxidants responsible for the stress are those originally included in cigarette smoke or those which result from the associated inflammatory response.
Specific endpoints of oxidative stress which have been quantified with smoking are numerous and diverse and have included: 1) generation of O2− , hydrogen peroxide (H2O2), and nitrite/nitrate; 2) levels of superoxide dismutase, catalase, myeloperoxidase, and cytochrome P450 (and their activities); 3) exhaled breath ethane/alkanes, thiobarbituric acid-reactive substances, and other indices of lipid peroxidation; 4) concentrations of oxidized proteins; 5) total, reduced, and oxidized glutathione; 6) activity of glutathione peroxidase, glutathione transferase, and glutathione reductase; 7) concentrations of prostanoids (F2-isoprostanes and prostaglandin F2-alpha), hydroxyeicosatetraenoic acid products, F(4)-neuroprostanes, 7-ketocholesterol, 24- and 27-hydroxycholesterol, low density lipoproteins, and other cholesterol oxidation products; 8) concentrations of uric acid and allantoin; 9) evidence of DNA damage (8-hydroxy-2′-deoxyguanosine and 8-oxo-2′-deoxyguanosine); 10) histopathology (eg, 4-hydroxy-2-nonenal and 8-hydroxydeoxyguanosine); 11) immunohistochemistry for specific proteins such as nitrotyrosine; 12) gene expression microarray analysis; 13) Trolox equivalent antioxidant capacity; 14) antioxidant reducing capacity; and 15) total radical trapping parameters. These measurements have been obtained in numerous different samples, including cells and their fractions, whole blood, serum, plasma, cord blood, sputum, exhaled breath, breath condensate, lavage fluid, lavage cells, urine, and tissues (eg, lung, vasculature, brain, muscle, testicles, and pancreas).
Oxidant generation with cigarette smoke exposure
Oxidants, including H2O2, can be directly measured in the particulate fraction of cigarette smoke.9 Metals are also included in the particulate fraction of cigarette smoke, but concentrations appear to be low.10 There is little evidence to support the assertion that iron and copper, introduced into the body by smoking, catalyze Fenton-type reactions in either the lung or any tissue.
Phagocytes (eg, macrophages and neutrophils) in smokers are elevated in number, both in the lung and systemically, and generate ROS at increased rates.11 The major cell sources of O2– are likely the nicotine adenine disphosphonucleotide (NADPH) oxidoreductases, but others may also be increased (eg, xanthine oxidase).12 Catalase, the enzyme responsible for the breakdown of H2O2, is downregulated in ex-smokers.13 In vitro studies confirm that phagocytes collected from cigarette smokers spontaneously release increased amounts of oxidants such as O2– and H2O2 compared to those from nonsmokers.14 H2O2 is elevated in bronchoalveolar lavage fluid and in exhaled breath condensate collected from cigarette smokers,15 and some portion of this was demonstrated to be attributable to the elevated number of macrophages in the lower respiratory tract of smokers and their increased release of O2– .16
Cigarette smoking is associated with lipid peroxidation and conversion of polyunsaturated fatty acids to hydroperoxides, endoperoxides, aldehydes (eg, malondialdehyde), and alkanes (eg, ethane and pentane). Levels of these end products are increased in smokers, including thiobarbituric acid-reactive products (in sputum, blood, and lung components), isoprostanes (in blood, urine, and breath condensate), 4-hydroxy-2-nonenal adducts, and breath alkanes.17,18
Cigarette smoking depletes antioxidants.19 Concentrations of ascorbate and vitamin E are decreased among smokers.20 Smokers have 15%–20% lower serum concentrations of ascorbate than do nonsmokers. These specific changes in ascorbate and vitamin E correspond to an elevated oxidative stress rather than a dietary intake. Values normalize after smoking cessation.21 Glutathione metabolism appears to be particularly provoked by smoking. Despite glutathione being acutely depleted in cells and in smokers,22,23 levels of reduced glutathione are elevated in bronchoalveolar lavage fluid of chronic smokers.24 It has been proposed that such an increase of lung glutathione in smokers may be an attempt, albeit insufficient, to counter excess oxidant production after cigarette smoke exposure.25 Exogenous antioxidants appear to have a capacity to prevent some portion of the biological effect and injury following smoking.26 Pretreatment with antioxidants can decrease lipid peroxidation following exposure to cigarette smoke.27 There are studies which suggest that vitamins C and E diminish production of oxidants by inflammatory cells and improve pulmonary function in smokers.14,28 Supplementation with N-acetylcysteine (NAC) also diminishes in vitro cytotoxicity after cigarette smoke exposure.29
Cigarette smoke is also a source of reactive nitrogen species and causes nitrosative stress. Nitric oxide, abundant in cigarette smoke and generated by inflammatory cells, has potent antioxidant and anti-inflammatory actions, but also contributes to oxidative reactions.30 NO reacts with thiols to produce nitrosothiols associated with biological effects.31 Nitrosothiol levels have been shown to be higher in breath condensate collected from smokers compared with subjects who do not smoke.32 NO in cigarette smoke can react with O2– to form peroxynitrite,33 which decreases antioxidant capacity and augments oxidative stress.34 NO and peroxynitrite can cause the nitration of tyrosine to form nitrotyrosine products of proteins measurable in body fluids and tissues.35 However, it must be pointed out that NO levels in smokers were also reported to be normal or even lower relative to nonsmokers.36 Fractional exhaled nitric oxide was reported to be decreased in smokers.37 Such reduced NO production was postulated to possibly elevate oxidative stress since this molecule can function as an antioxidant as well as being a prooxidant.38
In addition to an elevated oxidant burden in the lung, there is increased systemic oxidative stress in smokers.39 Plasma Trolox equivalent antioxidant capacity and total glutathione are decreased in cigarette smokers.40 Peripheral blood neutrophils from smokers release more oxidants than those isolated from normal subjects.41 The proposal that oxidants in cigarette smoke, whether in the particulate or gas phase, pass through the pulmonary alveolar wall into the blood to induce a disseminated systemic oxidative stress is improbable, as such radicals would rapidly react with molecules in the lung.42
Oxidative stress and disease after cigarette smoke exposure
Oxidative stress resulting from an imbalance between oxidants and antioxidants is proposed as the basis for COPD following exposure to cigarette smoking. Cigarette smoke increases the burden of oxidants in the respiratory tract, either directly included in the cigarette smoke or generated by inflammatory cells, depleting antioxidant defenses and injuring lung cells.43 Evidence of a relationship between oxidative stress and injury exists. Cell lysis and epithelial permeability are increased after exposure to cigarette smoke, and these effects can be inhibited by antioxidants (ie, glutathione).44 In addition, there are increased numbers of activated inflammatory cells in the lungs of patients with COPD relative to healthy subjects, and these cells release greater quantities of O2– and H2O2.45,46 Importantly, a correlation has been demonstrated between O2– release by peripheral white blood cells and bronchial hyperreactivity in patients with COPD, supporting a role for such oxidants in lung disease after smoking.45 As discussed later in this article, oxidants have been demonstrated to mediate mucous hypersecretion and impaired mucociliary clearance, which can contribute to injury in COPD.47 Furthermore, nitrogen-based free radicals also participate in COPD,48 and 3-nitrotyrosine, formed by nitration of tyrosine, is observed in greater concentrations in the lungs of those with COPD.49
There is systemic disease after exposure to cigarette smoke.50–52 Regarding vascular disease, cigarette smoking is associated with an oxidation of low-density lipoproteins and their deposition in the vasculature with its resultant dysfunction.53 A depletion of antioxidants has also been documented in atherosclerosis.54
Cigarette smoke-induced oxidative stress is postulated to initiate a series of cellular and molecular reactions, which include activation of kinase cascades and transcription factors, release of inflammatory mediators, initiation of inflammation, and cell injury and apoptosis (Figure 1).55 Consequently, oxidative stress is the initiating factor in the pathway by which cigarette smoke exposure leads to disease. Key among the redox-sensitive transcription factors coordinating the inflammatory response to cigarette smoke are nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) (pro-oxidative ) and nuclear factor erythroid 2-related factor 2 (Nrf2; antioxidative). The activation of these transcription factors is observed in both the lung and extrapulmonary tissues.56 An increased expression of proinflammatory mediators, including cytokines and peroxidation products of arachidonic acid (leukotrienes, prostanoids, and isoprostanes), occurs in smokers. The final product of this cascade of reactions is inflammation (pulmonary and systemic) and apoptosis; if the response is prolonged, fibrotic and neoplastic injuries can result.
 | Figure 1 Schematic depicting the cascade of events underlying the biological effects of cigarette smoking. |
COPD due to smoking frequently does not reverse or improve after smoking cessation, but rather can progress in ex-smokers.57 The reasons for persistence and progression of COPD despite cessation of the exposure are not fully appreciated. However, smoking cessation also does not eliminate the increased oxidative stress in the respiratory tract, suggesting that retained particles may continue to participate in oxidant generation.58 To explain this incongruity, as well as the observation that COPD in smokers is particle related, it has been proposed that the particulate fraction of cigarette smoke (ie, tar) disrupts iron homeostasis to participate in oxidative stress. Retained particles in the lung effectively complex host iron to generate free radicals. In support of a role for disrupted iron homeostasis in disease after exposure to cigarette smoke, there is increased lavage iron concentrations in smokers.59 Accumulation of iron in macrophages, proportional to the frequency and duration of cigarette smoking, has also been described among smokers.60
Oxidative stress signaling pathways culminate in COPD as well as other lung and systemic disease. Oxidants influence many of these changes by modifying the expression of specific genes (eg, MUC5AC) and epigenetic factors pivotal to the development of disease.
Investigation has failed to delineate between the oxidative stress following cigarette smoking and that associated with COPD. Indices of oxidant generation can correspond to smoking alone (with no further change observed among COPD patients) and to COPD (with further change observed among COPD patients); results appear to vary with the specific endpoint, the methodology employed, and the study population.61 Further development of biomarkers of oxidant burden may be required prior to delineating whether there are any true disparities in oxidative stress between smoking and COPD.62
Exposures other than cigarette smoking and oxidative stress
Exposures other than cigarette smoking can impact the risk for COPD. Almost all of these exposures are particle related.63,64 Such exposures can include environmental tobacco smoke, burning of biomass, and air pollution particles. All these particle exposures, environmental and occupational, can result in oxidative stress. Production of ROS results either directly from the particle supporting an inappropriate electron transfer or from an interaction of the particle with cell proteins (eg, electron transport complexes in the mitochondria). Oxidative stress is accepted as the initial step in the biological effect after particle exposures.65 In vitro exposures of cells to particles results in oxidant generation in a wide range of cell types, including phagocytic, epithelial, and endothelial cells. In vivo oxidant production following exposure to different particles has also been confirmed.66 Oxidant production by these particles initiates a pathway of cell signaling, activation of transcription factors, and release of mediators, and culminates in an inflammatory lung injury.
Oxidative stress and mucin gene expression in COPD and CB
COPD is one of the most common respiratory diseases worldwide. This disease is defined by the presence of an irreversible airflow obstruction and is characterized by the presence of both oxidative stress and airway inflammation.1 The disease encompasses several phenotypes: emphysema, small airway obstruction and obliteration,2,3 and CB. A majority of patients with COPD have CB, and this diagnosis is associated with a higher risk for respiratory-related death. CB is diagnosed in patients with a productive chronic cough for at least 3 months per year for 2 consecutive years.1 Patients with CB have a greater risk of COPD exacerbations and more respiratory symptoms due to mucus obstruction of airways and secondary bacterial and viral bronchitis. Airway mucus obstruction in CB is due to several potential factors: 1) ciliated cells are injured by proteases and by oxidative injury, resulting in failure of the mucociliary escalator;67 2) there is increased production and secretion of the major macromolecular constituent of mucus, namely, mucin glycoproteins; specifically, two respiratory tract gel-forming mucins, MUC5AC and MUC5B, are increased in the airway mucus and associated with hypertrophy and hyperplasia of mucous cells in the submucosal glands and superficial airway epithelium;68–70 3) the airway mucus is dehydrated due to tobacco smoke-triggered downregulation of the cell surface expression of the cystic fibrosis transmembrane conductance regulator;71–73 and 4) the presence of plasma proteins, glycosaminoglycans and proteoglycans,74 DNA, lipids, and inflammatory cells and pathogens in CB airway mucus that change the normal biophysical properties of mucus (reviewed in Fahy and Dickey75).
Oxidative stress, generated by tobacco smoke and augmented by ROS generated from both inflammatory cells and mitochondrial activity by cells resident in the respiratory tract, regulate mucin gene expression and mucous cell metaplasia. Tobacco smoke upregulates MUC5AC expression via epidermal growth factor receptor (EGFR)-dependent and -independent activation of activator protein 1 (AP-1),76 or via specificity protein 1 (Sp1) activation.77 Tobacco smoke also upregulates IL-8,78 which may function as an autocrine/paracrine regulator of MUC5AC.79 Tobacco smoke constituents, including aldehydes (eg, acrolein) and H2O2, also upregulate mucin gene expression. Acrolein-induced mucin production is mediated by matrix metalloproteinase (MMP)980 and by MMP14,81 and monocyte-dominant inflammation augments acrolein-induced upregulation of MUC5AC.82 Similarly, H2O2 exposure upregulates MUC5AC mRNA expression by an NADPH oxidoreductase 4 (NOX4)-dependent mechanism.83 Smoking will trigger the influx of neutrophils into the airways and release of neutrophil elastase (NE) into the airways. We have reported that NE regulates MUC5AC gene expression via a mechanism dependent on ROS84 and catalyzed by NADPH quinone oxidoreductase 1 (NQO1) activity.85 In addition, oxidant-generated hyaluronan fragments upregulate both MUC5AC and MUC5B in airway epithelial cells.86,87 Importantly, tobacco smoke also sustains goblet cell metaplasia by suppressing Bik, a proapoptotic molecule, therefore preventing apoptosis of mucous cells.88 Developmental determinants of goblet cell differentiation, SAM pointed domain ETS factor (Spdef) and FOXA3, are present in COPD airway epithelial cells,89 but the regulatory mechanisms that govern their expression are not known (reviewed in Whitsett et al90). Interestingly, several components of the Notch signaling pathway, the receptor Notch 3 and the effectors HES5 and HEY2, are decreased in healthy smokers compared to healthy nonsmokers,91 suggesting that Notch suppression sustains airway mucous cell hyperplasia/hypertrophy.
Genetic and epigenetic regulation of oxidative stress in COPD
Tobacco smoke activates lipid peroxidation and carbonyl stress (reviewed in Kirkham and Barnes92), resulting in DNA damage93 and airway epithelial cell senescence and apoptosis (reviewed in Nyunoya et al94); these are important pathways that contribute to both emphysema and airway remodeling. However, only a small portion of smokers develop COPD, emphasizing the importance of host factors in regulating tobacco smoke-induced oxidative stress and COPD. To determine the genetic factors predisposing to COPD, several approaches have been applied. Some investigators have focused on the transcriptional and epigenetic regulation of specific molecular and cellular pathways relevant to emphysema and CB phenotypes. In contrast, other investigators have leveraged consortia of large patient cohorts to interrogate unbiased genome-wide associations and discover single nucleotide polymorphisms (SNPs) that correlate with disease.
Using airway epithelial cells obtained by bronchoscopies from a large cohort of patients with and without COPD and segregated for pulmonary function test severity, signatures of gene expression have been identified by microarray in airway epithelia that are consistent with the gene expression data from lung tissues in patients with emphysema.95 The expression of at least two oxidant-regulated genes are increased in COPD and include CYP2C18, a P450 enzyme, and aryl hydrocarbon receptor nuclear translocator-like 2 (ARNTL2), a basic helix-loop-helix transcription factor that is active under conditions of low oxygen tension.95
Using a candidate gene approach, several genetic mutations in antioxidant genes are linked to COPD severity (reviewed in96). Polymorphisms in glutathione S-transferase (GST) M1, GSTP1, superoxide dismutase 3 (SOD3), and microsomal epoxide hydrolase (EPHX1) are associated with more rapid decline in lung function in COPD.
Epigenetic regulatory pathways are also activated by oxidative stress. The effect of miRNAs on signaling pathways in COPD varies with biospecimen source and the mechanism of miRNA regulation. A good example of this complexity is the biology of miR-199a-5p in COPD. One report demonstrates that increased expression of miR-199a-5p in the lung tissue of COPD patients correlates with downregulation of hypoxia inducible factor (HIF)-1α and vascular endothelial growth factor (VEGF); this function of miR-199a-p5 is demonstrated in human endothelial cells in vitro.97 Another recent report demonstrates that promoter methylation of miR-199a-5p in monocytes occurs in α-1-antitrypsin (A1AT) ZZ symptomatic patients, resulting in suppression of miR-199a-p5 expression and subsequent upregulation of the unfolded protein response.98 Both pathways engaging miR-199a-p5 result in emphysema, but by very different mechanisms.
Methylation of DNA CpG islands is broadly increased both in blood leukocytes99 and in small airway epithelia of COPD patients. The genes targeted by differential methylation in the small airways downregulate Nrf2 expression, resulting in loss of phase II metabolizing enzymes that provide antioxidant protection.100
Tobacco smoke downregulates histone deacetylases (HDACs), resulting in increased oxidation and protease activity. HDAC2 is decreased in COPD macrophages and lung tissues, and this is associated with increased H4 acetylation localized to the promoter of IL-8.101 HDAC2 downregulation impairs Nrf2 activation in the lung by decreasing the half-life of Nrf2.102 Sirtuin1 (SIRT1), a type III HDAC, is redox sensitive and can be post-translationally modified, leading to proteosomal degradation. Loss of SIRT1 correlates with tissue inhibitor of metalloproteinase (TIMP)-1 lysine acetylation and subsequent degradation of TIMP-1, the major anti-MMP protease, resulting in increased MMP-9 in human COPD lung tissue.103
Genome-wide association studies (GWAS) have identified a few SNPs associated with COPD, but the frequency of these polymorphisms in the population do not explain the much larger frequency of disease occurrence, suggesting that environmental exposures and/or epigenetic regulation have a significantly greater impact on phenotype. Other challenges for GWAS analysis include potentially missing modifying genes that interact with GWAS-identified genes, missing genes specific to ethnic backgrounds, using too broad a phenotype to test associations, and not considering the impact of expression levels of identified genes in tissues (eQTL) which would strengthen the association of genetic polymorphisms to phenotype.104
Two studies evaluated thousands of subjects phenotyped by the Global initiative for chronic Obstructive Lung Disease (GOLD) criteria for severity of COPD. The studies compared smokers with airflow obstruction to smokers with no airflow obstruction. Both studies detected significant levels of expression of SNPs on chromosome (chr) 15 spanning several genes including nicotinic acetylcholine receptors 3–5 (CHRNA3–5), and iron response element binding protein 2.105,106 One study identified family with sequence similarity 13 member A (FAM13A) on chromosome (chr) 4, a Rho A inhibitor, important for regulating oxidative stress and apoptosis in COPD lung cells. The following chromosomal localizations and gene candidates were determined to be related to forced expiratory volume in 1 second (FEV1):chr 4q24, glutathione S-transferase, C-terminal containing domain (GSTCD); chr 2q35, tensin 1 (TNS1); and chr 5q33, 5-hydroxytryptamine (serotonin) receptor 4 (HTR4). Similarly, for a decreased FEV1/forced vital capacity ratio, the following chromosomes and associated gene candidates were identified: chr 6p21, advanced glycosylation end product-specific receptor (AGER); and chr 15q23, thrombospondin, type I, domain containing 4 (THSD4).107 Finally, hedgehog interacting protein (HHIP), a developmental transcription factor, was identified as a COPD susceptibility gene at the level of significance in these studies.
Using a consortia of large COPD cohorts that included the COPDGene study (non-Hispanic white and African-American ethnic origin) and the ECLIPSE, NETT/NAS, and Norway GenKOLS studies, 6,633 individuals with moderate-to-severe COPD and 5,704 control individuals were evaluated, and three known COPD GWAS gene candidates were identified: CHRNA3, FAM13A, and HHIP. In addition, three new associations were identified: Ras interacting/interference 3 (RIN3), MMP12, and TGFB2.108
As phenotypes were characterized in detail and compared, new genome-wide associations for COPD became apparent. In one study, GWAS was conducted for a phenotype of emphysema defined by chest computed tomography scans in over 2,000 individuals with COPD. This analysis detected a near-significant association between the genomic region encompassing bicaudal D homolog 1 (BICD1) and emphysema severity.109 BICD1 is associated with dynein function and telomere shortening. Another study evaluated computed tomography scan severity of emphysema across ethnic groups and determined the following GWAS related to ethnic background: small nuclear ribonucleoprotein polypeptide F (SNRPF) and palmitoyl protein thioesterase 2 (PPT2) in non-Hispanic whites; for Hispanic subjects, α-mannosidase-related gene (MAN2B1); for Chinese subjects, DEAH box helicase 15 (DHX15) and mannosyl–glucosaminyl transferase 5B (MGAT5B), which acts on α-linked mannose; and, for African Americans, a third α-mannosidase-related gene, MAN1C1.110 The relation of GWAS to oxyhemoglobin saturations in two large cohorts, COPDGene and ECLIPSE, was tested. African Americans in COPDGene had SNP associations on chr 14 and 15. Candidate genes include forkhead box G1 (FOXG1) on chr 14, and, on chr 15, candidate genes include DNA topoisomerase-2-binding protein 1 (TOPBP1)-interacting checkpoint and replication regulator (TICRR), a regulator of cell cycle progression, and kinesin family member 7 (KIF7), a regulator of sonic hedgehog signaling. Chr 15 had no associations with COPD in non-Hispanic whites.111 GWAS for chronic mucus hypersecretion was associated with special AT-rich sequence-binding protein 1 locus (SATB1) on chr 3. This gene regulates chromatin structure. Although the original correlation was less than usually accepted for statistical correlation, the GWAS SNP was independently verified in other large COPD GWAS cohorts, and there was eQTL evidence of increased expression of SATB1 mRNA in tissues of patients with chronic mucus hypersecretion.112
Tremendous progress has been made in identifying environmental and inherited risk factors for COPD and CB. There are several target genes that increase susceptibility to COPD via oxidant-mediated pathways. Moreover, oxidant signaling pathways play a major role in airway remodeling relevant to CB. The next frontier to understanding how inheritance increases the risk of COPD disease susceptibility will be investigation of the early origins of COPD in childhood and infancy, including consideration of the role of inherited epigenetic factors. Factors that reduce ultimate lung growth113,114 or antioxidant capacity may be discovered, leading to new approaches to prevention or treatment of COPD.
Current and emerging antioxidants, diet, and alternative therapies
Studies have been undertaken to evaluate the clinical effectiveness of antioxidant therapy for COPD. Potential antioxidant therapies were reviewed in 2008,115 and again in 2013,116 with both reports suggesting that certain classes of antioxidants be evaluated for the clinical usefulness in COPD, including: thiol compounds (eg, NAC, carbocysteine), Nrf2 activators (eg, sulforaphane), antioxidant vitamins (eg, vitamin E and C), and food/diet-derived polyphenols (eg, curcumin and resveratrol) (see Table 1). Some of these antioxidants have been evaluated in rodent and/or human studies. For example, in one study, carbocysteine, a thiol antioxidant, was evaluated for its impact on COPD exacerbation rate in a randomized, placebo-controlled trial.117 In the Chinese COPD patients evaluated, the investigators reported that carbocysteine was well tolerated, and that it significantly decreased exacerbation rate over a 1-year period.117 Similarly, in a more recent randomized, placebo-controlled study with stable COPD patients in Hong Kong, 1-year treatment with high-dose NAC therapy, another thiol antioxidant, also reduced exacerbation frequency.118 This group also reported an improvement in the forced expiratory flow 25%–75% by 16 weeks of NAC therapy, which persisted for the 1-year treatment period, suggestive of decreased small airway obstruction.118 However, other, earlier studies115,116 did not demonstrate any impact of NAC therapy on lung function. The exact mechanism of action of NAC regulation of exacerbation frequency is not clear. Tse et al propose that it is the antioxidant and anti-inflammatory properties of NAC; however, one report suggests that it may also be that NAC helps restore the normal antiviral innate immune response that is suppressed by cigarette smoking.119 Thiol antioxidants show promise as one component of the COPD therapeutic “toolbox”, with their ability to attenuate exacerbation frequency, which may potentially help slow disease progression.
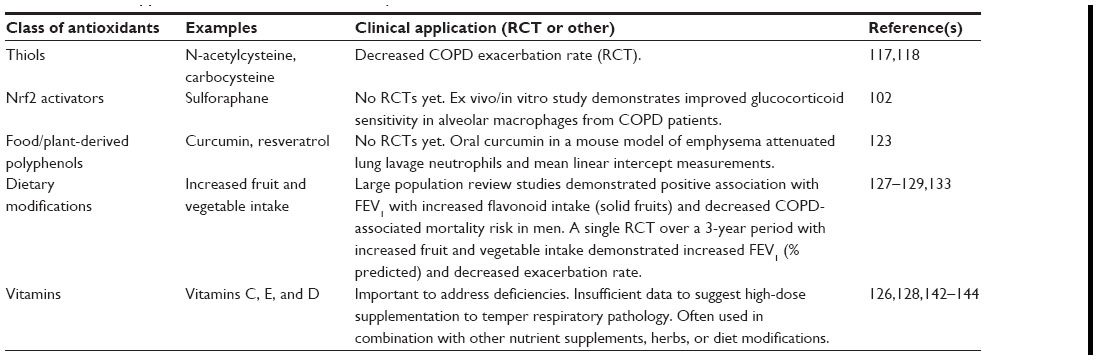 | Table 1 Clinical application of antioxidants summary table |
Nrf2 plays an important role in protection against ROS, especially those triggered by smoking. Nrf2 is a transcription factor which can bind to the antioxidant response element of important stress response genes. It regulates many of the antioxidant and phase II cytoprotective genes. Nrf2 and its importance in COPD pathophysiology has been studied in rodent models and with cells from COPD patients. Sulforaphane, derived from broccoli sprouts, is an Nrf2 activator that is being considered for potential clinical trials in COPD. Sulforaphane has been evaluated in ex vivo/in vitro studies. In one study using alveolar macrophages from COPD patients, sulforaphane treatment helped restore glucocorticoid sensitivity, through a glutathione-dependent mechanism, to increase HDAC2 activity.102 Food/plant-derived polyphenols, such as curcumin and resveratrol, have also been evaluated in a similar ex vivo/in vitro manner. Curcumin, the active constituent from turmeric, has anticancer and anti-inflammatory properties.120,121 Similar to sulforaphane, curcumin treatment of a monocytic cell line reversed cigarette smoke extract- or peroxide-induced steroid resistance by restoring HDAC2 activity and expression.122 In a mouse model of cigarette smoke-induced emphysema, oral curcumin therapy significantly attenuated lung lavage neutrophils and macrophages as well as lung mean linear intercepts as a measure of emphysema.123 Resveratrol, another polyphenolic compound that is derived from red wine, also has antioxidant and anti-inflammatory properties.115,124 Resveratrol treatment of alveolar macrophages from COPD patients significantly reduced bacterial endotoxin-induced cytokine production despite steroid insensitivity.125
Dietary modification may be another avenue of promising intervention for COPD patients.126 Two studies from the Netherlands suggest that diets rich in solid fruits may be beneficial in limiting COPD symptoms and/or mortality.127,128 The first study, by Tabak et al, focused on dietary intake of polyphenolic bioflavonoids and measured three subclasses: catechins, flavonols, and flavones. Total flavonoid intake was positively associated with FEV1 and inversely associated with cough and breathlessness.127 The second study, by Walda et al, also included patients from European countries and evaluated the 20-year mortality rate in men. Increased dietary intake of fruit and vitamin E demonstrated an inverse trend in 20-year COPD mortality with a corresponding 24% lower COPD mortality risk.128 In a follow-up to the Tabak et al study,127 a 5-year follow-up was performed and a subset of these patients with valid initial study lung function measurements were included in a study to evaluate the relationship of dietary intake to FEV1 decline over time. McKeever et al reported that people eating a more traditional diet, with intake of meat and potatoes dominating, was associated with a lower FEV1 and an increased incidence of COPD.129 These two studies focused on solid fruits, in particular apples and pears, but other fruits are rich in polyphenols and bioflavonoids. Guava is one fruit whose extract is very phenolic rich; it has been proposed as potential COPD therapy, but not yet evaluated in clinical trials.130 Blueberries are another antioxidant bioflavonoid-rich food. In healthy men, blueberry intake has been shown to improve vascular function.131 Two dietary-intervention randomized controlled trials reported differing results. One study, based in the United Kingdom, had patients increase fruit and vegetable intake to at least five servings per day, while the control group consumed two or less portions per day for 12 weeks.132 Over this short period, there were no changes in lung function nor in markers of either oxidative stress or inflammation.132 However, a study in Greece, in which the intervention group increased their fruit and vegetable consumption over a 3-year period, demonstrated a significant increase in FEV1 (% predicted) over time.133 Furthermore, the intervention group also demonstrated a significant decrease in the mean annual exacerbation rate.133 In addition, recent reports also highlight the importance of overall nutritional support, improved dietary quality, and increased dietary fiber for COPD management.134–136 Collectively, these studies suggest a diet rich in fruits providing polyphenolic bioflavonoids and other antioxidants (eg, proanthocyanidins137) along with dietary advice138 as important for the overall management of COPD.
Vitamins are generally obtained from our diets; however, is vitamin supplementation helpful for COPD management? Schols, in a recent translational paper,126 felt that there is insufficient evidence to support taking high doses of single nutrient supplements to temper respiratory pathology; however, addressing vitamin deficiencies may be considered. It is important to note that caution must be used when using high-dose vitamin supplementation. There have been reports that, for some vitamins (eg, vitamin A), high-dose supplementation can increase the risk of lung cancer and mortality in susceptible individuals.139–141 In an ex vivo/in vitro analysis, compared to placebo-treated patients, white blood cells from stable COPD patients who had been supplemented with vitamin E or C for 12 weeks had increased resistance to peroxide-induced DNA breakages.142 In a different controlled study, ascorbate intravenous infusion improved skeletal muscle fatigue resistance in COPD patients.143 The combination treatment of vitamin C, zinc, selenium, and Echinacea purpurea, a herb that has been used for treating the common cold, helped shorten COPD exacerbation episodes.144
Many people have studied the relationship of vitamin D with COPD management with varying results. A summary of the information from several reports, listed in chronological order, is shown in Table 2. Collectively, these studies suggest that COPD patients with serum 25-hydroxyvitamin D (25-OH vitamin D) below 20 ng/mL are deficient and may be at increased risk of exacerbations, worse lung function, and decline in lung function with time. This concept is supported in a review about vitamin insufficiencies and supplementation for COPD, in which Tsiligianni and van der Molen point out that the majority of COPD patients have vitamin D deficiency and that supplementation might perhaps be beneficial.145 Lehouck et al suggest that it is generally only the COPD patients with severe deficiencies of serum 25-OH vitamin D <10 ng/mL who may benefit from vitamin D supplementation.146 Vitamin D is important as an immune system regulator145 and, given that low serum 25-OH vitamin D levels have been associated with many chronic diseases (eg, autoimmune disease, cardiovascular disease, and infections),146 vitamin D supplementation may be an important therapeutic strategy for COPD that is worthy of further investigation.
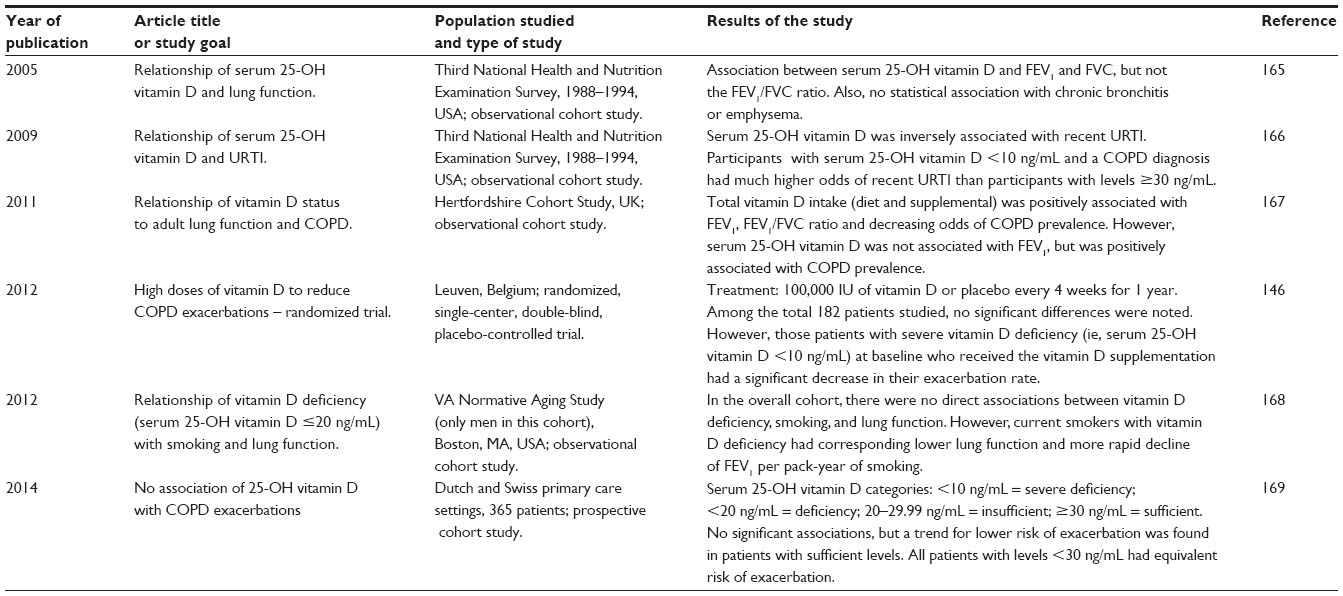 | Table 2 COPD and vitamin D reports |
Therapeutic strategies that can attenuate the inflammatory response can, in turn, attenuate oxidative stress. Alternative therapies such as herbal preparations, acupuncture, and tai chi or qigong may each help mitigate the overall inflammatory status of the COPD patient, either directly or indirectly. In the past few years, there have been multiple systematic reviews published relating to herbal therapies for COPD. One of these reports focused specifically on the combination of Tanreqing injection along with conventional therapy.147 Tanreqing is a standardized herbal formula administered by intravenous drip, which has been shown to have some antibacterial and antiviral effects along with anti-inflammatory actions.147 Tanreqing combined with conventional therapies demonstrated improvements in lung function and oxygenation, as well as decreased length of hospital stays, with no serious adverse events reported.147 Ginseng is a single herbal product that has also been evaluated for use with COPD patients. It is known for supporting qi of the lung and spleen to improve the functions of these organs and has been used for respiratory diseases.148 Studies were performed in a similar manner to that involving Tanreqing147 with ginseng given in combination with other therapies. As with Tanreqing, systematic review of the ginseng studies reported improvements in lung function and also quality of life (Qol) scores based on the St George’s Respiratory Questionnaire (SGRQ).148 Other studies have also focused on herbal therapies for improvement of Qol for COPD patients. Based on systematic review of the literature, oral Chinese herbal medicines resulted in improvement in Qol scores when compared to no treatment or routine therapies.148 A recent systematic review addressed the impact of Chinese herbal medicines on the body mass index, airflow obstruction, dyspnea, and exercise (BODE) index and the 6-minute walk test (6MWT).149 The 6MWT provides the exercise capacity score for the BODE index, while the BODE index correlates well overall with COPD mortality. Results from the systematic review suggest a benefit for the BODE index and the 6MWT by the combination of Chinese herbal medicine with routine therapies, suggesting an overall clinical benefit, too.149 The main herbs used for the studies were Astragalus membranaceus, Panax ginseng, and Cordyceps sinensis alone or, sometimes, in combination with other herbs.149 Collectively, these systematic reviews suggest a benefit of Chinese medicine herbal preparations for treatment of specific aspects of the overall COPD disease complex and that further large-scale high quality studies are warranted.
Acupuncture is another Chinese modality that has been evaluated as adjunctive therapy for COPD patients. Acupuncture can be administered in different ways and may be different for each patient. To control for these types of variability, Suzuki et al, in their studies, standardized the acupuncture treatment protocol and used the same acupuncture points for all patients. In a small study, all patients received conservative treatment and one-half also received acupuncture for 10 weeks. The control group was matched to the treatment group but did not receive any sham treatment. The patients receiving acupuncture showed improvement in the modified Borg dyspnea scale and in oxygen saturation during the 6MWT.150 The next study by the same group was also a small trial, which showed similar improvements after 10 weeks of acupuncture.151 Suzuki et al then performed a slightly larger trial, now placebo-controlled, that extended the acupuncture to 12 weeks and included eleven different acupuncture points similar to those used in previous studies.151,152 Similar results were reported in this controlled trial, with improvement in the Borg scale score in the acupuncture-treated patients.152 The reports from Suzuki et al were without electrical stimulation through the acupuncture needles. Studies were also performed using Acu-TENS, which is transcutaneous electrical nerve stimulation on acupuncture point sites. One study was a double-blinded, randomized controlled trial with Acu-TENS or placebo-TENS on a single acupuncture point site, once for 45 minutes. FEV1 improved in the Acu-TENS-treated patients immediately posttreatment.153 There was also a corresponding increase in blood β-endorphin levels. In a subsequent study, which was also a randomized controlled trial, the patients received 4 weeks of Acu-TENS treatment.154 This study had both placebo-TENS and sham-TENs control groups. Placebo is where the TENS stimulation electrodes are placed on the patient over the appropriate acupuncture point but without electrical stimulation applied. For the sham treatment group, electrical stimulation is applied to another part of the body that is a non-acupuncture point. Similarly to the previously mentioned study,153 Ngai et al demonstrated improvement in FEV1. They also demonstrated improvements in oxygen saturation and distance for the 6MWT and improvement in the SGRQ Qol score, with corresponding increases in blood β-endorphin levels.154 Collectively, these studies suggest that acupuncture with or without electrical stimulation promotes improved oxygenation in COPD patients.
Tai chi and qigong (TCQ) are traditional Chinese exercises with emphasis on specific focused movements, meditation, and breathing exercises. In a qigong randomized controlled study presented as a mind–body exercise intervention, the patients participating in the qigong exercises demonstrated significant improvement on the monitored functional task evaluation and the 6MWT by the 6-month follow-up.155 In a separate randomized controlled trial, TCQ were compared to other breathing and self-paced walking exercises or normal routine (ie, no extra exercise), demonstrating, by the end of the 3-month treatment period, improved lung function and 6MWT and decreased exacerbation rate in the TCQ group compared to the two other groups.156 In a systematic review of TCQ COPD trials, compared to no exercise, TCQ therapy promoted significant improvements in 6MWT, FEV1, and FEV1% predicted as well as SGRQ Qol score; however, compared to other exercise training, there was only improvement in the 6MWT.157 Similar to acupuncture, TCQ is an alternative therapy that shows potential benefit for COPD patients and is worthy of further investigation with high-quality trials. It is important to note that the placebo effect does not improve physiologic measures, but is associated with significant improvement in well-being as measured by surveys.158 Thus, the randomized, placebo-controlled trials show that complementary medicines do have a significant physiologic effect on pulmonary function outcomes in COPD.
COPD is a complex disease process and no single treatment can treat the whole disease. Clearly, from the different studies discussed, antioxidant supplementation, dietary antioxidants, Chinese herbal preparations, acupuncture, and TCQ may each have their place in COPD therapy, necessitating the use of combination and complementary therapies. One area that has not been addressed but is worth mentioning is the microbiome. The intestinal microbiome is critical to the regulation of our immune system, and alterations in the intestinal microbiome and dysbiosis may be a component of the pathogenesis of many diseases.159 Furthermore, our diets can be a major influence on the intestinal microbial composition, potentially impacting our health.160 One study in rats demonstrated that a diet rich in blueberries promoted gut health, evaluated by colonic metabolism measurements, and minimized invasive bacterial species.161 Similarly, modification to a Paleolithic hunter-gatherer-type diet might be worth considering. This type of diet promotes fruit and vegetables and eliminates cereal grains, legumes, and dairy, and has been shown to promote metabolic and physiologic improvements in sedentary adults.162 Serial analyses of gut and respiratory microbiomes in infants with cystic fibrosis have shown that nutritional factors and gut microbial colonization patterns are determinants of respiratory tract microbial development, suggesting that dietary alterations and probiotic strategies may provide early intervention opportunities.163 Similar considerations are worth investigating as potential early intervention opportunities for control of COPD progression.164
Disclosure
The authors report no conflicts of interest in this work.
References
Kim V, Criner GJ. Chronic bronchitis and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;187(3):228–237. | |
Hogg JC, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350(26):2645–2653. | |
McDonough JE, Yuan R, Suzuki M, et al. Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. N Engl J Med. 2011;365(17):1567–1575. | |
Fischer BM, Pavlisko E, Voynow JA. Pathogenic triad in COPD: oxidative stress, protease-antiprotease imbalance, and inflammation. Int J Chron Obstruct Pulmon Dis. 2011;6:413–421. | |
Pryor WA. Cigarette smoke and the involvement of free radical reactions in chemical carcinogenesis. Br J Cancer Suppl. 1987;8:19–23. | |
Church DF, Pryor WA. Free-radical chemistry of cigarette smoke and its toxicological implications. Environ Health Perspect. 1985;64:111–126. | |
Rodgman A, Perfetti TA. The Chemical Components of Tobacco and Tobacco Smoke. Boca Raton, FL: CRC Press Inc.; 2009. | |
Pryor WA. Cigarette smoke radicals and the role of free radicals in chemical carcinogenicity. Environ Health Perspect. 1997;105 Suppl 4:875–882. | |
Zang LY, Stone K, Pryor WA. Detection of free radicals in aqueous extracts of cigarette tar by electron spin resonance. Free Radic Biol Med. 1995;19(2):161–167. | |
Ghio AJ, Hilborn ED, Stonehuerner JG, et al. Particulate matter in cigarette smoke alters iron homeostasis to produce a biological effect. Am J Respir Crit Care Med. 2008;178(11):1130–1138. | |
Hempel SL, Buettner GR, O’Malley YQ, Wessels DA, Flaherty DM. Dihydrofluorescein diacetate is superior for detecting intracellular oxidants: comparison with 2′,7′-dichlorodihydrofluorescein diacetate, 5(and 6)-carboxy-2′,7′-dichlorodihydrofluorescein diacetate, and dihydrorhodamine 123. Free Radic Biol Med. 1999;27(1–2):146–159. | |
Heunks LM, Viña J, van Herwaarden CL, Folgering HT, Gimeno A, Dekhuijzen PN. Xanthine oxidase is involved in exercise-induced oxidative stress in chronic obstructive pulmonary disease. Am J Physiol. 1999;277(6 Pt 2):R1697–R1704. | |
Ho SP, Chan-Yeung M, Chow KK, Ip MS, Mak JC. Antioxidant enzyme activities in healthy Chinese adults: influence of age, gender and smoking. Respirology. 2005;10(3):305–309. | |
van Antwerpen VL, Theron AJ, Richards GA, et al. Vitamin E, pulmonary functions, and phagocyte-mediated oxidative stress in smokers and nonsmokers. Free Radic Biol Med. 1995;18(5):935–941. | |
Dekhuijzen PN, Aben KK, Dekker I, et al. Increased exhalation of hydrogen peroxide in patients with stable and unstable chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1996;154(3 Pt 1):813–816. | |
Schaberg T, Haller H, Rau M, Kaiser D, Fassbender M, Lode H. Superoxide anion release induced by platelet-activating factor is increased in human alveolar macrophages from smokers. Eur Respir J. 1992;5(4):387–393. | |
Morrow JD, Frei B, Longmire AW, et al. Increase in circulating products of lipid peroxidation (F2-isoprostanes) in smokers. Smoking as a cause of oxidative damage. N Engl J Med. 1995;332(18):1198–1203. | |
Montuschi P, Collins JV, Ciabattoni G, et al. Exhaled 8-isoprostane as an in vivo biomarker of lung oxidative stress in patients with COPD and healthy smokers. Am J Respir Crit Care Med. 2000;162(3 Pt 1):1175–1177. | |
Halliwell B. Antioxidants in human health and disease. Annu Rev Nutr. 1996;16:33–50. | |
Duthie GG, Arthur JR, James WP. Effects of smoking and vitamin E on blood antioxidant status. Am J Clin Nutr. 1991;53(Suppl 4):1061S–1063S. | |
Lykkesfeldt J, Priemé H, Loft S, Poulsen HE. Effect of smoking cessation on plasma ascorbic acid concentration. BMJ. 1996;313(7049):91. | |
Burlakova EB, Zhizhina GP, Gurevich SM, et al. Biomarkers of oxidative stress and smoking in cancer patients. J Cancer Res Ther. 2010;6(1):47–53. | |
Wickenden JA, Clarke MC, Rossi AG, et al. Cigarette smoke prevents apoptosis through inhibition of caspase activation and induces necrosis. Am J Respir Cell Mol Biol. 2003;29(5):562–570. | |
Cantin AM, North SL, Hubbard RC, Crystal RG. Normal alveolar epithelial lining fluid contains high levels of glutathione. J Appl Physiol (1985). 1987;63(1):152–157. | |
Li XY, Donaldson K, Rahman I, MacNee W. An investigation of the role of glutathione in increased epithelial permeability induced by cigarette smoke in vivo and in vitro. Am J Respir Crit Care Med. 1994;149(6):1518–1525. | |
Cantin A, Crystal RG. Oxidants, antioxidants and the pathogenesis of emphysema. Eur J Respir Dis Suppl. 1985;139:7–17. | |
Churg A, Cherukupalli K. Cigarette smoke causes rapid lipid peroxidation of rat tracheal epithelium. Int J Exp Pathol. 1993;74(2):127–132. | |
MacNee W, Wiggs B, Belzberg AS, Hogg JC. The effect of cigarette smoking on neutrophil kinetics in human lungs. N Engl J Med. 1989;321(14):924–928. | |
Selby C, Drost E, Wraith PK, MacNee W. In vivo neutrophil sequestration within lungs of humans is determined by in vitro “filterability”. J Appl Physiol (1985). 1991;71(5):1996–2003. | |
Rahman I, MacNee W. Role of oxidants/antioxidants in smoking-induced lung diseases. Free Radic Biol Med. 1996;21(5):669–681. | |
Gaston B, Reilly J, Drazen JM, et al. Endogenous nitrogen oxides and bronchodilator S-nitrosothiols in human airways. Proc Natl Acad Sci U S A. 1993;90(23):10957–10961. | |
Corradi M, Montuschi P, Donnelly LE, Pesci A, Kharitonov SA, Barnes PJ. Increased nitrosothiols in exhaled breath condensate in inflammatory airway diseases. Am J Respir Crit Care Med. 2001;163(4):854–858. | |
Padmaja S, Huie RE. The reaction of nitric oxide with organic peroxyl radicals. Biochem Biophys Res Commun. 1993;195(2):539–544. | |
Van der Vliet A, Smith D, O’Neill CA, et al. Interactions of peroxynitrite with human plasma and its constituents: oxidative damage and antioxidant depletion. Biochem J. 1994;303(Pt 1):295–301. | |
Petruzzelli S, Puntoni R, Mimotti P, et al. Plasma 3-nitrotyrosine in cigarette smokers. Am J Respir Crit Care Med. 1997;156(6):1902–1907. | |
Corradi M, Majori M, Cacciani GC, Consigli GF, de’Munari E, Pesci A. Increased exhaled nitric oxide in patients with stable chronic obstructive pulmonary disease. Thorax. 1999;54(7):572–575. | |
Rytilä P, Rehn T, Ilumets H, et al. Increased oxidative stress in asymptomatic current chronic smokers and GOLD stage 0 COPD. Respir Res. 2006;7:69. | |
Zhang WZ, Venardos K, Chin-Dusting J, Kaye DM. Adverse effects of cigarette smoke on NO bioavailability: role of arginine metabolism and oxidative stress. Hypertension. 2006;48(2):278–285. | |
Langen RC, Korn SH, Wouters EF. ROS in the local and systemic pathogenesis of COPD. Free Radic Biol Med. 2003;35(3):226–235. | |
Bloomer RJ. Decreased blood antioxidant capacity and increased lipid peroxidation in young cigarette smokers compared to nonsmokers: impact of dietary intake. Nutr J. 2007;6:39. | |
Talukder MA, Johnson WM, Varadharaj S, et al. Chronic cigarette smoking causes hypertension, increased oxidative stress, impaired NO bioavailability, endothelial dysfunction, and cardiac remodeling in mice. Am J Physiol Heart Circ Physiol. 2011;300(1):H388–H396. | |
Yamaguchi Y, Nasu F, Harada A, Kunitomo M. Oxidants in the gas phase of cigarette smoke pass through the lung alveolar wall and raise systemic oxidative stress. J Pharmacol Sci. 2007;103(3):275–282. | |
Cano M, Thimmalappula R, Fujihara M, et al. Cigarette smoking, oxidative stress, the anti-oxidant response through Nrf2 signaling, and age-related macular degeneration. Vision Res. 2010;50(7):652–664. | |
Lannan S, Donaldson K, Brown D, MacNee W. Effect of cigarette smoke and its condensates on alveolar epithelial cell injury in vitro. Am J Physiol. 1994;266(1 Pt 1):L92–L100. | |
Rahman I, Morrison D, Donaldson K, MacNee W. Systemic oxidative stress in asthma, COPD, and smokers. Am J Respir Crit Care Med. 1996;154(4 Pt 1):1055–1060. | |
Hoidal JR, Fox RB, LeMarbe PA, Perri R, Repine JE. Altered oxidative metabolic responses in vitro of alveolar macrophages from asymptomatic cigarette smokers. Am Rev Respir Dis. 1981;123(1):85–89. | |
Rogers DF. Mucus hypersecretion in chronic obstructive pulmonary disease. In: Chadwick D, Goode JA, editors. Chronic Obstructive Pulmonary Disease: Pathogenesis to Treatment. Chichester: John Wiley & Sons; 2001:65–83. | |
Kharitonov SA, Barnes PJ. Nitric oxide, nitrotyrosine, and nitric oxide modulators in asthma and chronic obstructive pulmonary disease. Curr Allergy Asthma Rep. 2003;3(2):121–129. | |
Ricciardolo FL, Caramori G, Ito K, et al. Nitrosative stress in the bronchial mucosa of severe chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2005;116(5):1028–1035. | |
Varela-Carver A, Parker H, Kleinert C, Rimoldi O. Adverse effects of cigarette smoke and induction of oxidative stress in cardiomyocytes and vascular endothelium. Curr Pharm Des. 2010;16(23):2551–2558. | |
Müller-Schweinitzer E, Müller SE, Reineke DC, et al. Reactive oxygen species mediate functional differences in human radial and internal thoracic arteries from smokers. J Vasc Surg. 2010;51(2):438–444. | |
Burke A, Fitzgerald GA. Oxidative stress and smoking-induced vascular injury. Prog Cardiovasc Dis. 2003;46(1):79–90. | |
Harats D, Ben-Naim M, Dabach Y, Hollander G, Stein O, Stein Y. Cigarette smoking renders LDL susceptible to peroxidative modification and enhanced metabolism by macrophages. Atherosclerosis. 1989;79(2–3):245–252. | |
Cherubini A, Vigna GB, Zuliani G, Ruggiero C, Senin U, Fellin R. Role of antioxidants in atherosclerosis: epidemiological and clinical update. Curr Pharm Des. 2005;11(16):2017–2032. | |
Rahman I. Oxidative stress in pathogenesis of chronic obstructive pulmonary disease: cellular and molecular mechanisms. Cell Biochem Biophys. 2005;43(1):167–188. | |
Khanna A, Guo M, Mehra M, Royal W 3rd. Inflammation and oxidative stress induced by cigarette smoke in Lewis rat brains. J Neuroimmunol. 2013;254(1–2):69–75. | |
Rutgers SR, Postma DS, ten Hacken NH, et al. Ongoing airway inflammation in patients with COPD who do not currently smoke. Chest. 2000;117(5 Suppl 1):262S. | |
Louhelainen N, Rytila P, Haahtela T, Kinnula VL, Djukanović R. Persistence of oxidant and protease burden in the airways after smoking cessation. BMC Pulm Med. 2009;9:25. | |
Thompson AB, Bohling T, Heires A, Linder J, Rennard SI. Lower respiratory tract iron burden is increased in association with cigarette smoking. J Lab Clin Med. 1991;117(6):493–499. | |
Wesselius LJ, Nelson ME, Skikne BS. Increased release of ferritin and iron by iron-loaded alveolar macrophages in cigarette smokers. Am J Respir Crit Care Med. 1994;150(3):690–695. | |
Inonu H, Doruk S, Sahin S, et al. Oxidative stress levels in exhaled breath condensate associated with COPD and smoking. Respir Care. 2012;57(3):413–419. | |
Louhelainen N, Myllarniemi M, Rahman I, Kinnula VL. Airway biomarkers of the oxidant burden in asthma and chronic obstructive pulmonary disease: current and future perspectives. Int J Chron Obstruct Pulmon Dis. 2008;3(4):585–603. | |
Balmes J, Becklake M, Blanc P, et al; Environmental and Occupational Health Assembly, American Thoracic Society. American Thoracic Society Statement: occupational contribution to the burden of airway disease. Am J Respir Crit Care Med. 2003;167(5):787–797. | |
Sangani RG, Ghio AJ. Lung injury after cigarette smoking is particle related. Int J Chron Obstruct Pulmon Dis. 2011;6:191–198. | |
Tao F, Gonzalez-Flecha B, Kobzik L. Reactive oxygen species in pulmonary inflammation by ambient particulates. Free Radic Biol Med. 2003;35(4):327–340. | |
Kadiiska MB, Mason RP, Dreher KL, Costa DL, Ghio AJ. In vivo evidence of free radical formation in the rat lung after exposure to an emission source air pollution particle. Chem Res Toxicol. 1997;10(10):1104–1108. | |
Amitani R, Wilson R, Rutman A, et al. Effects of human neutrophil elastase and Pseudomonas aeruginosa proteinases on human respiratory epithelium. Am J Respir Cell Mol Biol. 1991;4:26–32. | |
Caramori G, Di Gregorio C, Carlstedt I, et al. Mucin expression in peripheral airways of patients with chronic obstructive pulmonary disease. Histopathology. 2004;45:477–484. | |
Caramori G, Casolari P, Di Gregorio C, et al. MUC5AC expression is increased in bronchial submucosal glands of stable COPD patients. Histopathology. 2009;55(3):321–331. | |
Kirkham S, Kolsum U, Rousseau K, Singh D, Vestbo J, Thornton DJ. MUC5B is the major mucin in the gel phase of sputum in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;178(10):1033–1039. | |
Cantin AM, Hanrahan JW, Bilodeau G, et al. Cystic fibrosis transmembrane conductance regulator function is suppressed in cigarette smokers. Am J Respir Crit Care Med. 2006;173(10):1139–1144. | |
Clunes LA, Davies CM, Coakley RD, et al. Cigarette smoke exposure induces CFTR internalization and insolubility, leading to airway surface liquid dehydration. FASEB J. 2012;26(2):533–545. | |
Rab A, Rowe SM, Raju SV, Bebok Z, Matalon S, Collawn JF. Cigarette smoke and CFTR: implications in the pathogenesis of COPD. Am J Physiol Lung Cell Mol Physiol. 2013;305(8):L530–L541. | |
Girod S, Galabert C, Lecuire A, Zahm JM, Puchelle E. Phospholipid composition and surface-active properties of tracheobronchial secretions from patients with cystic fibrosis and chronic obstructive pulmonary diseases. Pediatr Pulmonol. 1992;13(1):22–27. | |
Fahy JV, Dickey BF. Airway mucus function and dysfunction. N Engl J Med. 2010;363(23):2233–2247. | |
Gensch E, Gallup M, Sucher A, et al. Tobacco smoke control of mucin production in lung cells requires oxygen radicals AP-1 and JNK. J Biol Chem. 2004;279:39085–39093. | |
Di YP, Zhao J, Harper R. Cigarette smoke induces MUC5AC protein expression through the activation of Sp1. J Biol Chem. 2012;287(33):27948–27958. | |
Moretto N, Bertolini S, Iadicicco C, et al. Cigarette smoke and its component acrolein augment IL-8/CXCL8 mRNA stability via p38 MAPK/MK2 signaling in human pulmonary cells. Am J Physiol Lung Cell Mol Physiol. 2012;303(10):L929–L938. | |
Bautista MV, Chen Y, Ivanova VS, Rahimi MK, Watson AM, Rose MC. IL-8 regulates mucin gene expression at the posttranscriptional level in lung epithelial cells. J Immunol. 2009;183(3):2159–2166. | |
Deshmukh HS, Shaver C, Case LM, et al. Acrolein-activated matrix metalloproteinase 9 contributes to persistent mucin production. Am J Respir Cell Mol Biol. 2008;38(4):446–454. | |
Deshmukh HS, McLachlan A, Atkinson JJ, et al. Matrix metalloproteinase-14 mediates a phenotypic shift in the airways to increase mucin production. Am J Respir Crit Care Med. 2009;180(9):834–845. | |
Borchers MT, Wesselkamper S, Wert SE, Shapiro SD, Leikauf GD. Monocyte inflammation augments acrolein-induced Muc5ac expression in mouse lung. Am J Physiol. 1999;277:L489–L497. | |
Kim HJ, Park YD, Moon UY, et al. The role of Nox4 in oxidative stress-induced MUC5AC overexpression in human airway epithelial cells. Am J Respir Cell Mol Biol. 2008;39(5):598–609. | |
Fischer BM, Voynow JA. Neutrophil elastase induces MUC5AC gene expression in airway epithelium via a pathway involving reactive oxygen species. Am J Respir Cell Mol Biol. 2002;26(4):447–452. | |
Zheng S, Byrd AS, Fischer BM, Grover AR, Ghio AJ, Voynow JA. Regulation of MUC5AC expression by NAD(P)H:quinone oxidoreductase 1. Free Radic Biol Med. 2007;42(9):1398–1408. | |
Casalino-Matsuda SM, Monzón ME, Forteza RM. Epidermal growth factor receptor activation by epidermal growth factor mediates oxidant-induced goblet cell metaplasia in human airway epithelium. Am J Respir Cell Mol Biol. 2006;34(5):581–591. | |
Casalino-Matsuda SM, Monzon ME, Day AJ, Forteza RM. Hyaluronan fragments/CD44 mediate oxidative stress-induced MUC5B up-regulation in airway epithelium. Am J Respir Cell Mol Biol. 2009;40(3):277–285. | |
Mebratu YA, Schwalm K, Smith KR, Schuyler M, Tesfaigzi Y. Cigarette smoke suppresses Bik to cause epithelial cell hyperplasia and mucous cell metaplasia. Am J Respir Crit Care Med. 2011;183(11):1531–1538. | |
Chen G, Korfhagen TR, Karp CL, et al. Foxa3 induces goblet cell metaplasia and inhibits innate antiviral immunity. Am J Respir Crit Care Med. 2014;189(3):301–313. | |
Whitsett JA, Haitchi HM, Maeda Y. Intersections between pulmonary development and disease. Am J Respir Crit Care Med. 2011;184(4):401–406. | |
Tilley AE, Harvey BG, Heguy A, et al. Down-regulation of the notch pathway in human airway epithelium in association with smoking and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2009;179(6):457–466. | |
Kirkham PA, Barnes PJ. Oxidative stress in COPD. Chest. 2013;144(1):266–273. | |
Aoshiba K, Zhou F, Tsuji T, Nagai A. DNA damage as a molecular link in the pathogenesis of COPD in smokers. Eur Respir J. 2012;39(6):1368–1376. | |
Nyunoya T, Mebratu Y, Contreras A, Delgado M, Chand HS, Tesfaigzi Y. Molecular processes that drive cigarette smoke-induced epithelial cell fate of the lung. Am J Respir Cell Mol Biol. 2014;50(3):471–482. | |
Steiling K, Lenburg ME, Spira A. Personalized management of chronic obstructive pulmonary disease via transcriptomic profiling of the airway and lung. Ann Am Thorac Soc. 2013;Suppl 10:S190–S196. | |
Postma DS, Kerkhof M, Boezen HM, Koppelman GH. Asthma and chronic obstructive pulmonary disease: common genes, common environments? Am J Respir Crit Care Med. 2011;183(12):1588–1594. | |
Mizuno S, Bogaard HJ, Gomez-Arroyo J, et al. MicroRNA-199a-5p is associated with hypoxia-inducible factor-1α expression in lungs from patients with COPD. Chest. 2012;142(3):663–672. | |
Hassan T, Carroll TP, Buckley PG, et al. miR-199a-5p silencing regulates the unfolded protein response in chronic obstructive pulmonary disease and α1-antitrypsin deficiency. Am J Respir Crit Care Med. 2014;189(3):263–273. | |
Qiu W, Baccarelli A, Carey VJ, et al. Variable DNA methylation is associated with chronic obstructive pulmonary disease and lung function. Am J Respir Crit Care Med. 2012;185(4):373–381. | |
Vucic EA, Chari R, Thu KL, et al. DNA methylation is globally disrupted and associated with expression changes in chronic obstructive pulmonary disease small airways. Am J Respir Cell Mol Biol. 2014;50(5):912–922. | |
Ito K, Ito M, Elliott WM, et al. Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N Engl J Med. 2005;352(19):1967–1976. | |
Malhotra D, Thimmulappa RK, Mercado N, et al. Denitrosylation of HDAC2 by targeting Nrf2 restores glucocorticosteroid sensitivity in macrophages from COPD patients. J Clin Invest. 2011;121(11):4289–4302. | |
Sundar IK, Yao H, Rahman I. Oxidative stress and chromatin remodeling in chronic obstructive pulmonary disease and smoking-related diseases. Antioxid Redox Signal. 2013;18(15):1956–1971. | |
Weiss ST. What genes tell us about the pathogenesis of asthma and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;181(11):1170–1173. | |
Pillai SG, Ge D, Zhu G, et al; ICGN Investigators. A genome-wide association study in chronic obstructive pulmonary disease (COPD):identification of two major susceptibility loci. PLoS Genet. 2009;5(3):e1000421. | |
Cho MH, Boutaoui N, Klanderman BJ, et al. Variants in FAM13A are associated with chronic obstructive pulmonary disease. Nat Genet. 2010;42(3):200–202. | |
Repapi E, Sayers I, Wain LV, et al. Genome-wide association study identifies five loci associated with lung function. Nat Genet. 2010;42(1):36–44. | |
Cho MH, McDonald ML, Zhou X, et al; NETT Genetics, ICGN, ECLIPSE and COPDGene Investigators. Risk loci for chronic obstructive pulmonary disease: a genome-wide association study and meta-analysis. Lancet Respir Med. 2014;2(3):214–225. | |
Kong X, Cho MH, Anderson W, et al; ECLIPSE Study NETT Investigators. Genome-wide association study identifies BICD1 as a susceptibility gene for emphysema. Am J Respir Crit Care Med. 2011;183(1):43–49. | |
Manichaikul A, Hoffman EA, Smolonska J, et al. Genome-wide study of percent emphysema on computed tomography in the general population. The Multi-Ethnic Study of Atherosclerosis Lung/SNP Health Association Resource Study. Am J Respir Crit Care Med. 2014;189(4):408–418. | |
McDonald ML, Cho MH, Sorheim IC, et al. Common genetic variants associated with resting oxygenation in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2014;51(5):678–687. | |
Dijkstra AE, Smolonska J, van den Berge M, et al; LifeLines Cohort study. Susceptibility to chronic mucus hypersecretion, a genome wide association study. PLoS One. 2014;9(4):e91621. | |
Gilliland FD, Berhane K, McConnell R, et al. Maternal smoking during pregnancy, environmental tobacco smoke exposure and childhood lung function. Thorax. 2000;55(4):271–276. | |
Stern DA, Morgan WJ, Wright AL, Guerra S, Martinez FD. Poor airway function in early infancy and lung function by age 22 years: a non-selective longitudinal cohort study. Lancet. 2007;370(9589):758–764. | |
Rahman I. Antioxidant therapeutic advances in COPD. Ther Adv Respir Dis. 2008;2(6):351–374. | |
Biswas S, Hwang JW, Kirkham PA, Rahman I. Pharmacological and dietary antioxidant therapies for chronic obstructive pulmonary disease. Curr Med Chem. 2013;20(12):1496–1530. | |
Zheng JP, Kang J, Huang SG, et al. Effect of carbocisteine on acute exacerbation of chronic obstructive pulmonary disease (PEACE Study):a randomised placebo-controlled study. Lancet. 2008;371(9629):2013–2018. | |
Tse HN, Raiteri L, Wong KY, et al. High-dose N-acetylcysteine in stable COPD: the 1-year, double-blind, randomized, placebo-controlled HIACE study. Chest. 2013;144(1):106–118. | |
Wu W, Patel KB, Booth JL, Zhang W, Metcalf JP. Cigarette smoke extract suppresses the RIG-I-initiated innate immune response to influenza virus in the human lung. Am J Physiol Lung Cell Mol Physiol. 2011;300(6):L821–L830. | |
Aggarwal BB, Kumar A, Bharti AC. Anticancer potential of curcumin: preclinical and clinical studies. Anticancer Res. 2003;23(1A):363–398. | |
Jagetia GC, Aggarwal BB. “Spicing up” of the immune system by curcumin. J Clin Immunol. 2007;27(1):19–35. | |
Meja KK, Rajendrasozhan S, Adenuga D, et al. Curcumin restores corticosteroid function in monocytes exposed to oxidants by maintaining HDAC2. Am J Respir Cell Mol Biol. 2008;39(3):312–323. | |
Suzuki M, Betsuyaku T, Ito Y, et al. Curcumin attenuates elastase- and cigarette smoke-induced pulmonary emphysema in mice. Am J Physiol Lung Cell Mol Physiol. 2009;296(4):L614–L623. | |
Wood LG, Wark PA, Garg ML. Antioxidant and anti-inflammatory effects of resveratrol in airway disease. Antioxid Redox Signal. 2010;13(10):1535–1548. | |
Knobloch J, Hag H, Jungck D, Urban K, Koch A. Resveratrol impairs the release of steroid-resistant cytokines from bacterial endotoxin-exposed alveolar macrophages in chronic obstructive pulmonary disease. Basic Clin Pharmacol Toxicol. 2011;109(2):138–143. | |
Schols AM. Nutrition as a metabolic modulator in COPD. Chest. 2013;144(4):1340–1345. | |
Tabak C, Arts IC, Smit HA, Heederik D, Kromhout D. Chronic obstructive pulmonary disease and intake of catechins, flavonols, and flavones: the MORGEN Study. Am J Respir Crit Care Med. 2001;164(1):61–64. | |
Walda IC, Tabak C, Smit HA, et al. Diet and 20-year chronic obstructive pulmonary disease mortality in middle-aged men from three European countries. Eur J Clin Nutr. 2002;56(7):638–643. | |
McKeever TM, Lewis SA, Cassano PA, et al. Patterns of dietary intake and relation to respiratory disease, forced expiratory volume in 1 s, and decline in 5-y forced expiratory volume. Am J Clin Nutr. 2010;92(2):408–415. | |
Flores G, Dastmalchi K, Wu SB, et al. Phenolic-rich extract from the Costa Rican guava (Psidium friedrichsthalianum) pulp with antioxidant and anti-inflammatory activity. Potential for COPD therapy. Food Chem. 2013;141(2):889–895. | |
Rodriguez-Mateos A, Rendeiro C, Bergillos-Meca T, et al. Intake and time dependence of blueberry flavonoid-induced improvements in vascular function: a randomized, controlled, double-blind, crossover intervention study with mechanistic insights into biological activity. Am J Clin Nutr. 2013;98(5):1179–1191. | |
Baldrick FR, Elborn JS, Woodside JV, et al. Effect of fruit and vegetable intake on oxidative stress and inflammation in COPD: a randomised controlled trial. Eur Respir J. 2012;39(6):1377–1384. | |
Keranis E, Makris D, Rodopoulou P, et al. Impact of dietary shift to higher-antioxidant foods in COPD: a randomised trial. Eur Respir J. 2010;36(4):774–780. | |
Collins PF, Elia M, Stratton RJ. Nutritional support and functional capacity in chronic obstructive pulmonary disease: a systematic review and meta-analysis. Respirology. 2013;18(4):616–629. | |
Fonseca Wald EL, van den Borst B, Gosker HR, Schols AM. Dietary fibre and fatty acids in chronic obstructive pulmonary disease risk and progression: a systematic review. Respirology. 2014;19(2):176–184. | |
van de Bool C, Mattijssen-Verdonschot C, van Melick PP, et al. Quality of dietary intake in relation to body composition in patients with chronic obstructive pulmonary disease eligible for pulmonary rehabilitation. Eur J Clin Nutr. 2014;68(2):159–165. | |
Bagchi D, Swaroop A, Preuss HG, Bagchi M. Free radical scavenging, antioxidant and cancer chemoprevention by grape seed proanthocyanidin: an overview. Mutat Res Fundam Mol Mech Mutagen. Epub April 19, 2014. | |
Farooqi N, Nordström L, Lundgren R, Sandström T, Håglin L. Changes in body weight and physical performance after receiving dietary advice in patients with chronic obstructive pulmonary disease (COPD):1-year follow-up. Arch Gerontol Geriatr. 2011;53(1):70–75. | |
Omenn GS, Goodman GE, Thornquist MD, et al. Effects of a combination of beta carotene and vitamin A on lung cancer and cardiovascular disease. N Engl J Med. 1996;334(18):1150–1155. | |
Omenn GS, Goodman GE, Thornquist MD, et al. Risk factors for lung cancer and for intervention effects in CARET, the Beta-Carotene and Retinol Efficacy Trial. J Natl Cancer Inst. 1996;88(21):1550–1559. | |
Goodman GE, Thornquist MD, Balmes J, et al. The Beta-Carotene and Retinol Efficacy Trial: incidence of lung cancer and cardiovascular disease mortality during 6-year follow-up after stopping beta-carotene and retinol supplements. J Natl Cancer Inst. 2004;96(23):1743–1750. | |
Wu TC, Huang YC, Hsu SY, Wang YC, Yeh SL. Vitamin E and vitamin C supplementation in patients with chronic obstructive pulmonary disease. Int J Vitam Nutr Res. 2007;77(4):272–279. | |
Rossman MJ, Garten RS, Groot HJ, et al. Ascorbate infusion increases skeletal muscle fatigue resistance in patients with chronic obstructive pulmonary disease. Am J Physiol Regul Integr Comp Physiol. 2013;305(10):R1163–R1170. | |
Isbaniah F, Wiyono WH, Yunus F, Setiawati A, Totzke U, Verbruggen MA. Echinacea purpurea along with zinc, selenium and vitamin C to alleviate exacerbations of chronic obstructive pulmonary disease: results from a randomized controlled trial. J Clin Pharm Ther. 2011;36(5):568–576. | |
Tsiligianni IG, van der Molen T. A systematic review of the role of vitamin insufficiencies and supplementation in COPD. Respir Res. 2010;11:171. | |
Lehouck A, Mathieu C, Carremans C, et al. High doses of vitamin D to reduce exacerbations in chronic obstructive pulmonary disease: a randomized trial. Ann Intern Med. 2012;156(2):105–114. | |
Zhong Y, Mao B, Wang G, et al. Tanreqing injection combined with conventional Western medicine for acute exacerbations of chronic obstructive pulmonary disease: a systematic review. J Altern Complement Med. 2010;16(12):1309–1319. | |
An X, Zhang AL, Yang AW, et al. Oral ginseng formulae for stable chronic obstructive pulmonary disease: a systematic review. Respir Med. 2011;105(2):165–176. | |
Chen X, May B, Di YM, et al. Oral Chinese herbal medicine combined with pharmacotherapy for stable COPD: a systematic review of effect on BODE index and six minute walk test. PLoS One. 2014;9(3):e91830. | |
Suzuki M, Namura K, Ohno Y, et al. The effect of acupuncture in the treatment of chronic obstructive pulmonary disease. J Altern Complement Med. 2008;14(9):1097–1105. | |
Suzuki M, Namura K, Ohno Y, et al. Combined standard medication and acupuncture for COPD: a case series. Acupunct Med. 2012;30(2):96–102. | |
Suzuki M, Muro S, Ando Y, et al. A randomized, placebo-controlled trial of acupuncture in patients with chronic obstructive pulmonary disease (COPD):the COPD-acupuncture trial (CAT). Arch Intern Med. 2012;172(11):878–886. | |
Jones AY, Ngai SP, Hui-Chan CW, Yu HP. Acute effects of Acu-TENS on FEV1 and blood β-endorphin level in chronic obstructive pulmonary disease. Altern Ther Health Med. 2011;17(5):8–13. | |
Ngai SP, Jones AY, Hui-Chan CW, Ko FW, Hui DS. Effect of 4 weeks of Acu-TENS on functional capacity and beta-endorphin level in subjects with chronic obstructive pulmonary disease: a randomized controlled trial. Respir Physiol Neurobiol. 2010;173(1):29–36. | |
Ng BH, Tsang HW, Jones AY, So CT, Mok TY. Functional and psychosocial effects of health qigong in patients with COPD: a randomized controlled trial. J Altern Complement Med. 2011;17(3):243–251. | |
Chan AW, Lee A, Suen LK, Tam WW. Tai chi Qigong improves lung functions and activity tolerance in COPD clients: a single blind, randomized controlled trial. Complement Ther Med. 2011;19(1):3–11. | |
Ding M, Zhang W, Li K, Chen X. Effectiveness of t’ai chi and qigong on chronic obstructive pulmonary disease: a systematic review and meta-analysis. J Altern Complement Med. 2014;20(2):79–86. | |
Wechsler ME, Kelley JM, Boyd IO, et al. Active albuterol or placebo, sham acupuncture, or no intervention in asthma. N Engl J Med. 2011; 365(2):119–126. | |
Hill DA, Artis D. Intestinal bacteria and the regulation of immune cell homeostasis. Annu Rev Immunol. 2010;28:623–667. | |
Flint HJ, Scott KP, Louis P, Duncan SH. The role of the gut microbiota in nutrition and health. Nat Rev Gastroenterol Hepatol. 2012;9(10):577–589. | |
Lacombe A, Li RW, Klimis-Zacas D, et al. Lowbush wild blueberries have the potential to modify gut microbiota and xenobiotic metabolism in the rat colon. PLoS One. 2013;8(6):e67497. | |
Frassetto LA, Schloetter M, Mietus-Synder M, Morris RC Jr, Sebastian A. Metabolic and physiologic improvements from consuming a paleolithic, hunter-gatherer type diet. Eur J Clin Nutr. 2009;63(8):947–955. | |
Madan JC, Koestler DC, Stanton BA, et al. Serial analysis of the gut and respiratory microbiome in cystic fibrosis in infancy: interaction between intestinal and respiratory tracts and impact of nutritional exposures. MBio. 2012;3(4):e00251-12. | |
Han MK, Huang YJ, Lipuma JJ, et al. Significance of the microbiome in obstructive lung disease. Thorax. 2012;67(5):456–463. | |
Black PN, Scragg R. Relationship between serum 25-hydroxyvitamin D and pulmonary function in the third national health and nutrition examination survey. Chest. 2005;128(6):3792–3798. | |
Ginde AA, Mansbach JM, Camargo CA Jr. Association between serum 25-hydroxyvitamin D level and upper respiratory tract infection in the Third National Health and Nutrition Examination Survey. Arch Intern Med. 2009;169(4):384–390. | |
Shaheen SO, Jameson KA, Robinson SM, et al. Relationship of vitamin D status to adult lung function and COPD. Thorax. 2011;66(8):692–698. | |
Lange NE, Sparrow D, Vokonas P, Litonjua AA. Vitamin D deficiency, smoking, and lung function in the Normative Aging Study. Am J Respir Crit Care Med. 2012;186(7):616–621. | |
Puhan MA, Siebeling L, Frei A, Zoller M, Bischoff-Ferrari H, Ter Riet G. No association of 25-hydroxyvitamin D with exacerbations in primary care patients with COPD. Chest. 2014;145(1):37–43. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.