Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 13
Converging Relationships of Obesity and Hyperuricemia with Special Reference to Metabolic Disorders and Plausible Therapeutic Implications
Authors Gong M, Wen S ![]() , Nguyen T, Wang C, Jin J, Zhou L
, Nguyen T, Wang C, Jin J, Zhou L
Received 25 September 2019
Accepted for publication 9 March 2020
Published 30 March 2020 Volume 2020:13 Pages 943—962
DOI https://doi.org/10.2147/DMSO.S232377
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Muthuswamy Balasubramanyam
Min Gong, Song Wen, Thiquynhnga Nguyen, Chaoxun Wang, Jianlan Jin, Ligang Zhou
Department of Endocrinology, Shanghai Pudong Hospital, Fudan University, Shanghai 201399, People’s Republic of China
Correspondence: Ligang Zhou
Department of Endocrinology, Shanghai Pudong Hospital, Fudan University, Shanghai 201399, China
Tel +8613611927616
Email [email protected]
Background: Obesity and hyperuricemia mutually influence metabolic syndrome. This study discusses the metabolic relationships between obesity and hyperuricemia in terms of pathophysiology, complications, and treatments.
Methods: We searched for preclinical or clinical studies on the pathophysiology, complications, and therapy of obesity and hyperuricemia on the PubMed database.
Results: In this systemic review, we summarized our searching results on topics of pathophysiology, complications and therapeutic strategy. In pathophysiology, we firstly introduce genetic variations for obesity, hyperuricemia and their relationships by genetic studies. Secondly, we talk about the epigenetic influences on obesity and hyperuricemia. Thirdly, we describe the central metabolic regulation and the role of hyperuricemia. Then, we refer to the character of adipose tissue inflammation and oxidative stress in the obesity and hyperuricemia. In the last part of this topic, we reviewed the critical links of gut microbiota in the obesity and hyperuricemia. In the following part, we review the pathophysiology of major complications in obesity and hyperuricemia including insulin resistance and type 2 diabetes mellitus, chronic kidney disease, cardiovascular diseases, and cancers. Finally, we recapitulate the therapeutic strategies especially the novel pharmaceutic interventions for obesity and hyperuricemia, which concurrently show the mutual metabolic influences between two diseases.
Conclusion: The data reviewed here delineate the metabolic relationships between obesity and hyperuricemia, and provide a comprehensive overview of the therapeutic targets for the management of metabolic syndromes.
Keywords: obesity, hyperuricemia, pathophysiology, complication, pharmacotherapy
Introduction
Currently, obesogenic factors, for instance, palatable energy-condensed food, convenient transportation, attracting entertainment TV shows at home, are widespread in the modern society.1,2 These changes in lifestyle have been shown to result in disarrangement in metabolism, for example, morbid obesity and gout.3 In 2014, the age-standardized worldwide prevalence of obesity (defined as a BMI ≥30 kg/m2) was 10.8% and 15.0% and the prevalence of overweight (BMI between 25 and 30 kg/m2) was 24.4% and 27.9% in men and women, respectively. It is estimated that the global prevalence of obesity will rise to 18% in men and 21% in women by 2025.4 Obesity is frequently associated with numerous risk factors of cardiovascular disease (CVD) and cancer.5,6 Its associated complications such as diabetes, stroke, and heart attack are severe and becoming prevalent.7 Among the five metabolic syndrome components, abdominal obesity and low HDL-C level are the most prevalent (21.8% and 14.4%) in Chinese children and adolescents. Additionally, 35.9% of children and adolescents are affected by at least one of this three components.8 On the other hand, the prevalence of overweight and obesity in Chinese adults is about 70%, in which adiposity indicators and age play an important role.9
Obesity constitutes a spectrum of heterogenous diseases reflexed by the BMI definition (according to the WHO standard BMI ≥ 30 kg/m2 is defined as obesity, whereas 28 kg/m2 is the cut point in Chinese standard). However, the phenotype of obesity on the effect of metabolism significantly varies, as visceral obesity is closely linked to diseases harmful to one’s systemic well-being in Asian populations compared to subcutaneous obesity.10 Therefore, this review discusses visceral obesity. Obesity is associated with multiple complications including hyperuricemia.11 The pooled prevalence of hyperuricemia and gout in the mainland China is 13.3% (95% CI: 11.9%, 14.6%), with a range of 5.5% to 23.6% and 1.1% (95% CI: 0.7%, 1.5%), range (0.4–1.5%), respectively.12 There are numerous common factors, for instance lifestyle, that contribute to both hyperuricemia and obesity, as well as, hypertension, insulin resistance, and cardiovascular diseases. Therefore, the pathogenesis and treatment of both diseases are a major concern the scientific society and clinical practice. Regarding this, we conceptualize that the management of obesity and hyperuricemia requires comprehensive strategy concerning the metabolic similarities. Important targets in the complicated linking map of the metabolism in obesity and its complications should be studied. This review discusses these two diseases’ pathophysiology, complications and therapy.
Methods
Literature search on obesity and hyperuricemia was conducted in PubMed database. The search terms “obesity AND prevalence” and “hyperuricemia AND prevalence” were used for the epidemic studies. On the other hand, “obesity AND genetic studies” or “obesity AND GWAS” and “hyperuricemia AND genetic studies” or “hyperuricemia AND GWAS” and “obesity AND uric acid AND genetic studies” were used for genetic studies of obesity and hyperuricemia. Regarding epigenetic mechanism of obesity and hyperuricemia, the search terms “obesity AND epigenetics” and “uric acid AND epigenetics” or “uric acid AND epigenetics” were applied, while, “obesity AND brain” or “obesity AND central nervous system” or “brain AND energy homeostasis” or “central nervous system AND energy homeostasis” AND “hyperuricemia AND central nervous system” or “uric acid AND central nervous system” or “uric acid AND brain” or “hyperuricemia AND brain” were used for the role of the central nervous system in obesity and hyperuricemia. For adipose tissue inflammation, we used search terms “adipose tissue AND obesity” and “adipose tissue AND hyperuricemia” or “adipose tissue AND uric acid”. For oxidative stress, we used search terms “oxidative stress AND obesity” and “oxidative stress AND uric acid” or “oxidative stress AND hyperuricemia”. The search terms “obesity AND microbiota” and “uric acid AND microbiota” or “hyperuricemia AND microbiota” were applied to retrieve studies on microbiota. Concerning obesity and hyperuricemia associated complications, “obesity” or “hyperuricemia” combined “type 2 diabetes mellitus” or “insulin resistance” or “chronic kidney diseases” or “cardiovascular diseases” or “cancers”, respectively, were used. In addition, “obesity AND treatment” or “obesity AND pharmacotherapy” or “obesity AND management” AND “hyperuricemia AND treatment” or “hyperuricemia AND management” or “hyperuricemia AND pharmacotherapy” search terms were used to retrieve studies concerning the treatment of obesity and hyperuricemia. Recent 5 years of studies and reviews as well as review of references of the included studies were included in the study. Additionally, “insulin resistance”, “GLP-1”, “oxidative stress”, “central metabolic control”, and “adipocytokines” search terms were used to retrieve detailed content.
Etiology and Pathophysiology of Obesity and Hyperuricemia
Inherited or acquired genome alteration determines the critical components and related functions of bodily metabolism. This exhibits genetic disparity from the normal individuals and allows high-risk populations susceptible to obesogenic or hyperuricemia promoting the environment.13
The Mutant Genes Driving Obesity and Hyperuricemia
Mutant Genes Related to Obesity
Using genome-wide association studies (GWAS), increasing genetic variants associated with obesity have been identified. GWAS leads in the development of unique, personalized preventive or treatment strategies of obesity. For a comprehensive review, see.14 For instance, syndromic monogenic obesity is caused by alterations in single genes and exceptionally rare. More than 30 syndromes characterized by mental retardation notably, Prader–Willi, Bardet–Biedl and Cohen syndromes have been reported.14 In addition to obesity, this group of diseases are characterized by dysmorphic features and organ-specific abnormalities.15 Non-syndromic monogenic obesity is caused by a single gene alteration leading to a highly penetrant form of the disease. The most form mainly affects the leptin/melanocortin pathway resulting in fully penetrant obesity but are exceptionally rare in humans.16 This group of diseases includes mutations in LEP/LEPR, SH2B1, POMC, PCSK1, MC4R, NTRK2, SIM1, KSR2 and TUB genes. However, a substantially higher proportion of obesity is observed in subjects with heterozygous deleterious coding mutations in these genes, resulting in non-fully penetrant obesity referred to as oligogenic obesity. Polygenic obesity is caused by multiple gene mutations with modest effects that interact with the environment. Majorly, mutations in the FTO gene are associated with polygenetic obesity. In polygenic inheritance, each pair of genes has a small effect on traits, but different micro-genes can form an obvious phenotypic trait through additive effect accumulations.17–19 Additionally, multi-gene genetic traits are affected by environmental factors (Figure 1A). Numerous studies have shown that environmental factors such as physical activities,20 diets or sedentary lifestyle21 influence diverse polymorphisms associated with obesity. The recent studies also demonstrated that unfavorable effects of obesity variants can be compensated by behavioral changes such as improving diet and physical activity.22 Importantly, the interactions between obese genes and environmental factors seem to employ epigenetic modifications as potential mediators.23
The current results from GWAS may provide insights into the biology of obesity including BMI or other obesity-related traits. These findings show that genes near loci regulating BMI are enriched for expression in the CNS, suggesting that BMI is mainly regulated by processes such as hypothalamic control of energy intake.24 However, genes for fat distribution are analyzed to be enriched in adipose tissue, indicating fat distribution is largely regulated in local fat depots.25 Other association studies include interaction between obesity and obesogenic environment, smoking, socioeconomic status, sex, lifestyle, causality, and cardiometabolic risk factors, even cancers.26 By further investigation on related molecular mechanism underling, these studies could provide personalized obesity prevention and treatment measures.
Mutant Genes Related to Hyperuricemia and Associations with Obesity
GWAS have confirmed the importance of urate excretion in the control of serum urate levels and the risk of gout, as well as, identified the kidneys, the gut, and the liver as sites of urate regulation.27 GWAS of gout have identified loci that include genes that encode urate transporters and interacting proteins (SLC2A9, ABCG2, SLC22A11, SLC17A1–SLC17A4 and PDZK1) and proteins associated with metabolic pathways (GCKR, A1CF andIGF1R). The SLC2A9 (GLUT9) and SLC22A12 (URAT1) genes encode renal transporters responsible for the reabsorption of urate from the urine filtrate (Figure 1B). The effect of mutations in SLC2A9 was robustly associated with and explains 2–3% of the variance in serum uric acid levels in Europeans. ABCG2 acts as multifunctional transporter of ATP-binding cassette family and mediates the efflux of various compounds in an ATP-dependent manner. Variants of ABCG2 are also prominent (1%) and present both in Europeans and East-Asians. Variations in ALDH16A1 gene are rare but critical, since the ALDH16A1 protein interacts with hypoxanthine-guanine phosphoribosyl transferase (HPRT1). Its deficiency causes Lesch-Nyhan Syndrome and urate accumulation.28 Changes in serum uric acid levels resulting from common genetic variants are small, compared to the 67% caused by nongenetic factors, including obesity, high purine food or high fructose beverage. Thus, the identification of mutant genes responsible for hyperuricemia is critical for the prediction of progression from hyperuricemia to gout, especially in asymptomatic individuals. This will be essential for the development of more precise urate-lowering approaches.
Genes responsible for the relationship between gout and obesity have been extensively studied. A meta-analysis of 10 prospective studies consisting of 27,944 gout cases and 215,739 participants indicated that each 5 kg/m2 BMI increment was associated with a 55% elevated risk of gout.29 Additionally, the findings of a Mendelian randomization study showed that BMI-increasing alleles of variants in the FTO, MC4R and TMEM18 gene regions are associated with higher serum urate concentrations.30 Moreover, a mendelian genetic study posited that gene-related higher BMI is causally associated with risk of gout and higher serum urate concentrations31 (Figure 1B). Therefore, to combat hyperuricemia and gout weight management is recommended.
Epigenetic Mechanism of Obesity, Hyperuricemia, and Gout
Accumulating evidence indicates that “fetal programming of adult disease” epigenetic mechanism results in postnatal diseases including obesity.32 Recently, much attention has shifted to epigenetics as a critical mechanism through which the environment influences the human genome spatiotemporal expression that may continuously exist for decades influencing an individual’s life span or the next generation33 (Figure 1A and B).
Epigenetics can reshape disease traits and phenotypes through independent genome coding majorly via chromatin remodeling, DNA methylation, histone modification, and non-coding RNAs (ncRNAs) regulation.34 Moreover, genetic polymorphisms in DNA processing enzymes such as DNA methyltransferase, methylcytosine dioxygenase, and methylene tetrahydrofolate reductase are also implicated in epigenetic modulation.33 Methylation caused by condensation of chromatin structure can result in the silence or suppression of gene expression. The ten-eleven translocation (TET) methylcytosine dioxygenase enzymes that convert methyl-C into hydroxymethyl-C also cause reversal of methylation. During early life development, abnormal DNA methylation or demethylation is implicated in the development of human obesity.35
The unstructured tails of histones are usually subject to post-translational modification, which are dynamically adjustable. Histone acetylation and deacetylation processes are mediated by histone acetyltransferase (HAT) and histone deacetylase (HDAC), respectively. The inhibition of HDAC3 activates PPARγ, which improves insulin sensitivity in diet-induced obesity.36 Epigenetic dysregulation of ncRNAs has been implicated to the onset and development of metabolic diseases.37,38 Concerning the role of ncRNA in modulating metabolism, some miRNA (Small non-coding RNAs, 18–25 nucleotides long, referred to as microRNAs (miRNAs)) levels in plasma or body fluids are considered as biomarkers of obesity and other metabolic diseases.39 Current studies have focused on the role of CircRNA in metabolism and related diseases.40 CircRNA sponges certain miRNA to suppress its function as a regulation mechanism of miRNA.26
The effects of epigenetics in urate metabolism are gradually being acknowledged by the scientific society. Recent studies have shown that the brain-signaling pathway is also modulated by epigenetic changes following exposure to high fructose intake.41 In the regulation of gouty inflammation, some class I histone deacetylases have proinflammatory roles that provoke the ability of urate crystals to initiate phagocyte activation.42 Phagocyte activation responsible for inflammation is mediated by microRNA miR-155, which is elevated in gout patients and mouse tissues from experimental acute gout compared to healthy controls.43 MiR-146a suppresses multiple urate crystal-induced inflammatory responses in vitro including IL1β expression, and has been reported to be elevated in peripheral blood mononuclear cells of inter-critical gout patients compared to individual with normal serum urate, hyperuricemia, and gout during acute flares.44
The Role of the Brain in the Control of Obesity
Appropriate energy homeostasis requires the brain as the critical metabolic regulator, which receives and concomitantly integrates peripheral signals, modulates autonomic outflow, and build up feeding behavior or patterns response to external stimuli. Peripheral organs communicate with the CNS via three pathways: humoral, metabolic, and neural. The humoral factors are mainly generated in forms of peptides by peripheral organs such as pancreas, adipose tissue, and the gastrointestinal tract.45 These metabolic factors are typically carbohydrates, lipids, ketones, amino acids, or other metabolites. Moreover, the vagal afferent relays neural signals to the nucleus of solitary tract (NTS) within the caudal brainstem for reflecting gastric motility or food content. Multiple peptides consisting of endogenous opioids, dopamine, and orexin within the CNS coordinates the effects on food intake induced by peripheral signals.46–53 The brain acts as the central command center, subsequently adjusting peripheral metabolism through autonomic neuronal pathways, or directly altering the functions of certain organs.
Two distinct populations of neurons found within the ARC mediate food intake and body weight. This constitutes the orexigenic neuropeptide Y (NPY) and agouti-related protein (AgRP) comprising NPY/AgRP neurons and neurons co-expressing anorexic neuropeptides pro-opiomelanocortin (POMC) and cocaine and amphetamine-related transcript (CART) namely POMC/CART neurons.54–56 Activities of these neurons are regulated by circulating leptin, GLP-1 or ghrelin since the hormonal signals infiltrate the blood-brain barrier anatomically near the ARC.57–62 Mutations in the Melanocortin receptor 4 (MC4R) of the melanocortin pathway are associated with the onset of monogenic obesity. Besides, mutations in POMC are associated with hyperphagia and obesity. Activation of POMC can be a potent pharmacological target in the treatment of obesity and associated complications. The administration of a 5-HT2c receptor agonist triggers the release of α-MSH, which binds to MC4R leading to the improvement in metabolism an obese mouse. This suggests the existence of a 5-HT system that interacts or regulates the melanocortin system and affects energy homeostasis.63
The rewarding center or hedonic area within the CNS also regulates appetite, satiety, motivation, and the feeding behavior. A dopaminergic reward pathway within the brain derived from midbrain to forebrain is involved in hedonic desire for hyperpalatable foods. This pathway is formed by dopamine (DA) projection primarily from the ventral tegmental area (VTA) to the nucleus accumbens (NAc) and linked to other limbic and cortical regions, including the amygdala, orbitofrontal cortex (OFC), and anterior cingulate cortex (ACC). These mesolimbic and corticolimbic links undertake the processes of reward, salience, motivation, decision making, and inhibitory control.64
Besides, DA which drives motivation, or the want of food, other molecules such as endocannabinoid and opioids are involved in pleasure or the liking of hedonic eating.65 The endocannabinoid system increases food consumption through modulating excitatory and inhibitory inputs to the VTA and NAc, which indirectly activates mesolimbic DA transmission and regulates the actions of leptin or melanocortin mediators in the hypothalamus that regulate appetite.66 Hyperpalatable foods, for instance, sugar have been shown to directly increase endogenous opioid levels and opioid agonists which in turn increases the intake of palatable foods67 (Figure 1A).
Evidence also indicates that uric acid influences metabolism via the central pathway. GLUT9 and URAT1 expressed in choroid plexus of the human brain function in transporting urate into the brain.68 High-UA diet or microinjection of UA induces the expression of pro-inflammatory cytokines, activates the NF-κB pathway resulting in hypothalamic inflammation or neuroendocrine alteration with dyslipidemia and glucose intolerance69 (Figure 1B).
Adipose Tissue Inflammation Leading to Obesity and Its Related Complications
Generally, inflammation serves as a protective mechanism required for organ remodeling, tissue repairing, wound healing, and immunity against infections. The pro-inflammatory cytokines released by the adipose tissue are released into the bloodstream to cause systemic inflammation (Figure 1A).
Great milestones in alterations in immunity associated with adipose tissue inflammation in obesity have been achieved in recent years. T lymphocytes with high distribution in inflamed adipose tissue express increased levels of interferon γ (IFN γ). IFN γ enhances the production of various inflammatory cytokines, including tumor necrosis factor-α (TNFα).70,71 IFN γ additionally, activates the interferon regulatory factors (IRFs), specifically, IRF-1, −3, −4, −7, and −9 that regulate adipogenesis and adipose tissue-associated macrophages (ATM) polarization.72–74
Macrophage infiltration is also enhanced in the adipose tissue in obesity.75 ATM acting as a pro-inflammatory cytokine releaser, is much more active than adipocytes.76 The M1 subtype of ATM promotes inflammation, whereas the M2 subtype has anti-inflammation attributes.77,78 In obese mice, increased levels of M1 and decreased levels of M2 were found in the adipose tissue.79 TZD has been shown to reduce M1 and increase M2 in obese mice, suggesting the role of M2 in improving insulin sensitivity by TZD.
Adipose Inflammation and Oxidative Stress (OS)
The adipose tissue, an endocrine and storage organ required for energy homeostasis, is primarily composed of adipocytes, containing other cells such as fibroblasts, fibroblastic pre-adipocytes, endothelial and immune cells,80 secreting hormones and cytokines (adipokines or adipocytokines) which influence the metabolism status of the whole body. However, in pathological conditions, adipokines are disturbed metabolically inducing the production of reactive oxygen species (ROS), generating oxidative stress (OS).81
Three major mechanisms influence the pathogenesis of OS.82 Firstly, the presence of excessive adipose tissue generates pro-inflammatory cytokines comprising TNF-α, interleukin (IL)-1β, and IL-6, which increase ROS and nitrogen by macrophages and monocytes.83 ROS induces further release of pro-inflammatory cytokines and expression of adhesion molecules and growth factors84 through redox-sensitive transcription factors, particularly the NF-κB and the NADPH oxidase pathway (NOX).85 On the other hand, antioxidant sources consisting of superoxide dismutase (SOD), glutathione peroxidase (GPx), and catalase (CAT), vitamin A, vitamin E, vitamin C, and β-carotene have been shown to be depleted in obese individuals.86 Similarly, the sensitivity of oxidative damage biomarkers is higher in obese individuals and correlates directly with BMI and the proportion of body fat, LDL oxidation, and triglyceride (TG) levels.87 On the other hand, the levels of antioxidant markers are lower based on the amount of body fat and central obesity.88 Secondly, excessive fat accumulation in obese patients leads to a pathological increase of serum FFA levels, which impair glucose metabolism while favoring the accumulation of metabolic substrates (fats and glucose) in peripheral consisting of the liver, muscle, and adipose tissue.89 This promotes higher mitochondrial and peroxisomal oxidation.
Thirdly, the bioactive adipokines comprising leptin, adiponectin, visfatin, and resistin, display irregulating which also contribute to OS. Because of obesity, individuals are at a higher risk of developing obesity-related diseases in the state of metabolic syndrome (MS), type 2 diabetes mellitus (T2DM), cardiovascular diseases (CVD), and even cancer. The dysregulated production of adipocytokines, presence of OS, and lack of antioxidant molecules contribute to cell structure damage, abnormal glucose, lipid metabolism, and impaired insulin signaling. This leads to vascular atherosclerosis, hypertension, β-cell damage, insulin resistance, dyslipidemia, and cell proliferation.
Hyperuricemia also contributes to the lipogenesis and inflammation of adipocytes (Figure 1B). Xanthine oxidoreductase (XO) enzyme is responsible for UA production, which increases fat deposition in the adipocytes. While extracellular UA acts as strong anti-oxidant, the intracellular UA is a pro-oxidant that stimulates NADPH oxidase enzyme, which increases intracellular oxidative stress, mitochondrial injury, and ATP depletion.90–92 Increased pro-inflammatory cytokine and decreased anti-inflammatory cytokine consisting adiponectin in gout have also been reported in mice studies. Moreover, treatment with allopurinol decreases the degree of inflammation and reduces macrophage infiltration accompanied by improved insulin resistance, hypertension, and fatty liver disease.93
The Role of Microbiota in the Pathophysiology of Obesity
Compared to conventionally raised mice and transplantation to lean mice, germ-free mice have a significantly less fat content body weight.94 Otherwise, obese and lean mice share a distinct pattern in the composition of gut microbiota.95–97 The major by-product is short-chain fatty acids (SCFAs) mainly consisting of acetate, propionate, and butyrate. These SCFAs influence whole-body metabolism.98–100 The mechanism underlying the association of obesity with microbiota has been proposed.101–107 In the human body, the change in the composition of microbiota is linked to obese and T2DM96,108,109 (Figure 1A). However, increased levels of non-digestible fibers reduce body weight, which is mediated partially by butyrate of SCFA.110,111 Additionally, there is growing evidence that microbiota modification by prebiotics or probiotics induces metabolic benefits in the host. Prebiotics lead to negative energy effect as shown by increased GLP-1 and PYY and decreased level of ghrelin.112–114
The distinct composition of microbiota is also reported between normal individuals and gout patients.115 There is similar composition of intestinal microbiota in gout and T2DM patients.101 Additionally, gout and T2DM patients share decreased levels of butyrate synthesis.101 Moreover, depletion of Faecalibacterium prausnitzii and reduced butyrate biosynthesis are shared in these two metabolic syndromes.101 The protective mechanisms of butyric acid in the human intestine include nutrition provision for intestinal mucosa, promoting growth and repair of intestinal villus, enhancing intestinal immunity, facilitating the growth of beneficial microbes, and inhibition of colonization with pathogenic bacteria.116 Therefore, decrease in butyric acid biosynthesis could cause numerous physiological disorders. Moreover, overly abundant xanthine dehydrogenase and the relative deficiency of allantoinase in the intestinal microbiota might result in accumulation of high levels of uric acid, consequently aggravating gout symptoms. Decreased immune response to joint injection of urate crystal in germ-free mice, antibiotics-treated mice, or metabolite-sensing receptor GPR43 deficiency-mice, and the administration of acetate to germ-free mice restores inflammation responses.117 However, the onset of inflammatory response is not altered in animals fed a high-fiber diet, rather the high-fiber diet induces faster resolution of the inflammatory response. Acetate plays a role in the resolution of neutrophilic inflammation by decreasing NF-κB activity and enhancing the production of anti-inflammatory mediators consisting of IL-10, TGF-β, and annexin A1.118 Additionally, probiotic-containing diet prevents hyperuricemia induced by oxonic acid, which is essential for normal blood pressure and renal function.119 Alternatively, the use of urate-lowering drugs on rats with hyperuricemia causes changes in the composition of microbiota indicating the effective treatment of hyperuricemia.120
Metabolic Alterations Related to Obesity
Obesity has impact on macronutrient metabolism, endocrinal balance, cardiovascular health, and immune normality (Figure 1). These alterations lead to exacerbation of structures or functions of organs resulting to the development of groups of metabolism-related conditions such as T2DM, hyperuricemia, CVD and even cancers.
|
Figure 1 The comparison of pathophysiology of (A) obesity and (B) hyperuricemia. Abbreviations: GFR, glomerular filtration rate, NAFLD, non-alcoholic fatty liver disease. |
Figure 1A illustrates about the pathophysiology of obesity. The genetic variants and epigenetic modification predispose the susceptibility of obesity. With the reduced satiety hormone derived from the gut, the orexigenic signal in the hypothalamus leads to the increased food intake resulting in bodyweight gain. High-fat foods also modify the composition of gut microbiota which release the LPS to the liver enhancing de novo lipogenesis. In the adipose tissue, obesity causes inflammation presented by enhanced M1 macrophage and T lymphocyte infiltration which promotes lipolysis and divert increased FFA to the liver for gluconeogenesis. The inflammation also causes adipocyte to release adipocytokine to the circulation. The consequence of the pathophysiology is likely Insulin resistance and T2DM, cardiovascular disease, chronic kidney disease.
Figure 1B illustrates the pathophysiology of hyperuricemia. The genetic variants of SLC2A9, SLC22A12 and ABCG2 and epigenetic modification predispose the susceptibility which influence urate metabolism. Increased fructose or purine results in elevated uric acid levels in circulation. Fructose can act on hypothalamus to increase food intake as well as directly increase body weight. Reduced GFR and abnormal reabsorption and secretion all contribute to elevated uric acid level, which combine the uric acid from liver may lead to hyperuricemia. The urate elimination disorder containing hyperuricosuria, nephrolithiasis, albuminuria. Excessive uric acid promotes fat storage in liver and contributes to NAFLD. Uric acid also affects adipose tissue promoting fat deposition and lipogenesis, and also cause the inflammation. Chronic hyperuricemia and metabolic syndrome may lead to the gout, insulin resistance, cardiovascular disease and chronic kidney disease.
Insulin Resistance and T2DM
The causal molecule of insulin resistance and T2DM defy explanation using a single etiological pathway, thus calling for multiple hypotheses. Recently, energy or substrate surplus rationale has been proposed and proven to be effective in the treatment of T2DM.121,122 Obesity-associated ectopic lipid storage is suggested as the critical initiator of insulin resistance (Figure 1A). Intramyocellular lipid (IMCL) accumulation in skeletal muscle leads to decreased glucose uptake,123 diverting glucose into the liver for de novo lipogenesis.124 WAT lipolysis and macrophage infiltrate increases the production of fatty acids and glycerol, which are transported into the liver leading to enhanced biogenesis of gluconeogenesis and glucose production,125–127 which requires DAG and PKCε derived from lipid procession in liver.128–130 Lipid and glucose accumulation in the liver eventually results in the selective hepatic insulin resistance. The role of exercise in the skeletal muscle is an alternative way of regulating insulin sensitivity, which activates the AMPK pathway.131 On the other hand, increased ATP level and inhibited AMPK activity have been implicated in the energy surplus state and most of the existing insulin-sensitizing medications inhibit ATP production in the mitochondria.122,132 Effective management strategies, for instance, weight loss and exercise can improve insulin resistance.133,134 Additionally, neural mechanisms for glycemic control exist, although they are compromised in type 1 diabetes mellitus (T1DM) and the late stage of T2DM which is termed as hypoglycemia associated autonomic failure (HAAF). In normal individuals, the ventromedial nucleus of hypothalamus and medial amygdala nucleus consist of the two major nucleus in the brain that respond to blood glucose levels.135 However, following long durations of T2DM, this function is attenuated leading to significant variations in blood glucose levels.
Although some studies have shown that insulin promotes urate retention at the renal tubule, and low levels of insulin might contribute to reduced levels of uric acid,136–138 a significant number of support that gout or hyperuricemia may result in insulin resistance and diabetes. Probably, the UA induced pancreatic injury,139 promotion of hepatic gluconeogenesis140,141 constitutes the mechanisms involved, which facilitate insulin resistance. In obesity patients concurrently affected by T2DM and hyperuricemia, assays of blood ROS and antioxidants show increased OS levels,142 which represses the insulin transcription factors comprising musculo-aponeurotic fibrosarcoma protein A (MafA) and pancreatic duodenal homeobox-1(PDX-1).143,144 It was shown that the XO serum levels were significantly higher in diabetic subjects and directly correlated with BMI and HbA1C.145 Additionally, the plasma level of XO activity positively correlated with BMI, insulin resistance, and inflammation and negatively correlated with adiponectin levels in healthy volunteers.146 By inhibiting XO activity, allopurinol, reduces inflammation and insulin resistance in asymptomatic patients.147 Moreover, there is a positive correlation between XO and C-reactive protein (CRP), blood glucose, insulin level, insulin resistance index, and TG/HDL-C ratio in patients with polycystic ovary syndrome.148 The plasma level of XO activity is highly correlated with BMI, left ventricular ejection fraction, and hypertrophy as well as the level of HBA1c and aminotransferases in patients with cardiac diseases.149 In conclusion, the metabolic status of hyperuricemia exacerbates insulin resistance, and plays a role in the pathogenesis of diabetes mellitus (Figure 1B).
Hyperuricemia
Hyperuricemia is caused by excessive production of purines and intake of fructose, particularly in individuals who suffer reduced renal eliminations (Figure 1B). Notably, kidney transporters such as ABC transporter G family member 2 (ABCG2), glucose transporter type 9 (GLUT9) and urate anion exchanger 1 (URAT1), are involved in urate reuptake and secretion, and their dysfunction results in underexcretion of uric acid (UA). In the gut also, a dysfunction of the ABCG2 transporter hinders the excretion of UA and contributes to its reduced excretion. The subsequent disequilibrium of resorption and excretion results in increased blood levels of UA, hyperuricemia, and risk of gout. However, environmental factors such as high dietary intake of purines are the primary triggers of hyperuricemia. Besides seafood, organ meat, and alcohol, emerging evidence suggests that fructose can be another significant source of dietary UA. Fructose is frequently used as a sweetener in beverages. The metabolism of fructose involves the transfer of a phosphate group from ATP to fructose. The reaction is catalyzed by the enzyme fructokinase and generates AMP, which is subsequently converted to UA. Previous studies also reported that high intracellular glucose could cause enhanced production of fructose.150,151 The results of epidemiological studies show that this metabolic syndrome and its associated disease conditions, including insulin resistance, obesity, hyperlipidemia, and hypertension, are strongly correlated to gout.152 However, the metabolic relationship between obesity and hyperuricemia, including the onset of gout, is obscure. The findings of a recent genetic study revealed that an increase in fat mass elevates the serum urate (SU) levels.31 Recent studies also suggest that excess fat storage could be caused by increased UA levels.153 Notably, increased SU levels are positively correlated with cardiovascular diseases (CVDs) and chronic kidney disease (CKD). These findings were confirmed using hyperuricemia (HU) rodent models, which developed features of metabolic syndrome and renal atherosclerosis. In these studies, HU was induced by inhibiting uricase,154 feeding on high fructose diets,155 and deleting the intestine glucose transporter 9 (GLUT9).156 The accumulated UA has multiple effects on metabolism.157 First, UA can influence the activity of endothelial nitric oxide synthase, which promotes insulin resistance.158,159 Alternatively, UA increases hepatic gluconeogenesis by simultaneously stimulating adenosine monophosphate dehydrogenase (AMPD) and inhibiting adenosine monophosphate kinase (AMPK).141 Second, the effects of UA on AMPK and AMPD could induce hepatic lipogenesis160 and stimulate the enzyme NADPH oxidase to promote fat deposition in the adipocytes.90 Third, UA or HU could cause hypertension and CVDs through its effect on the renin-angiotensin-aldosterone system (RAAS), Estimated Glomerular Filtration Rate (eGFR), and renal sodium excretion.161,162 Although numerous mechanisms for kidney injury or nephropathy have been proposed, the alteration of NADPH oxidase and subsequent activated reactive oxygen species (ROS) are the primary causative agents.91,92 Notably, the administration of anti-urate medication can alleviate metabolic syndrome, and weight loss is effective in the management of gout.163
The Role of Hyperuricemia on Chronic Kidney Disease (CKD)
The deposition of obesity-related lipids in the renal medulla could cause focal segmental glomerulosclerosis and reduced tubular flow rate. Excessive fructose can stimulate increased production of UA, a significant modulator of CKD progression (Figure 1B). Subsequently, CKD causes elevated UA, and hyperuricemia exacerbates renal pathology with the development of glomerular hypertrophy, sclerosis, and tubulointerstitial injury by the action of urate crystals.41,164 The excessive retention of UA could be caused by a reduced GFR. Since UA levels and lack of uricase enzyme maintain the blood pressure in humans, the use of ani-uricase enzyme to increase serum uric acid (SUA) levels should induce hypertension and nephropathy in animal models.157 In animals, nephropathy could be correlated to the activation of NADPH oxidase. Pathological analysis of untreated proteinuria patients showed focal deposition of monosodium urate crystals and secondary inflammatory response. Besides, SUA is associated with increased excretion of urine albumin as a 1mg/dL increase in SUA increases the risk of albuminuria by 80%.165 The reduced urine pH in obesity and related metabolic syndromes promotes the crystallization of UA. Hyperuricosuria, with the low pH and decreased urine synergistically, leads to the formation of urate stones, which blocks renal tubules and exacerbates the decline in renal function.
Cardiovascular Diseases (CVD)
Obesity is a major CVD risk factor. Besides, obesity adversely affects the structure and function of the cardiovascular (CV) system, by causing concentric remodeling and left ventricular (LV) hypertrophy, increasing left atrial enlargement, and inducing abnormalities in both systolic and LV diastolic function. Chronic excessive accumulation of body fat causes cardiovascular system adaption, such as an increase in cardiac output and a decrease in peripheral resistance.166–168 The expanded blood volume increases the heart preload via the Frank-Starling mechanism, induces ventricular remodeling, and eventually causes left ventricular (LV) hypertrophy.169,170 Subsequently, diastolic chamber compliance decreases, leading to a dysfunction in the LV diastolic system and decompensation of LV systolic function. Besides, some pathological changes such as atherosclerosis, muscular degeneration, increased total blood volume, and several other co-morbidities associated with obesity may exacerbate the systolic or diastolic function of the heart171 (Figure 1A).
Besides gout, which is a substantial risk factor of coronary heart diseases in men,172 HU also presents an independent risk factor with an unfavorable prognostic significance for CVDs (Figure 1B). It was associated with all-cause mortality and increased risk for ischemic stroke.173 The findings of two extensive epidemiologic studies indicate that urate-lowering therapy in patients with gout, or only HU could significantly reduce the risk for CV death.174,175 Independent of any CVD risk factor, increased UA level, even within the normal range, predisposes healthy subjects to impaired flow-mediated dilation (FMD) of the brachial artery, increased carotid intima-media thickness (IMTc), and increased stiffness of the aorta.157 Hyperuricemia could induce hypertension by up-regulating the renin/angiotensin pathway, increasing cyclooxygenase-2 (COX-2) expression, promoting the migration and proliferation of vascular myocytes, and reducing the availability of nitric oxide (NO). Uric acid and related free radicals exert pro-inflammatory actions by inducing the macrophages and cytokines and activating platelets.176 Consistently, increased UA is also associated with unstable coronary lipid-rich plaques.177 The findings of an in vitro study suggest that HU could also decrease mitochondrial DNA level, mitochondrial mass, and basal concentration of ATP in a dose-dependent manner.178
Cancers
Multiple studies provide evidence of increased risk for colon, postmenopausal breast, endometrial, kidney, esophageal, liver, and pancreatic cancer, as well as non-Hodgkin’s lymphoma and myeloma in obese individuals.179 Tumors invade stromal compartments that are rich in adipose tissue. The adipocytes function as endocrine cells that shape the tumor microenvironment and thus modulate the development and progression of tumors.180 For instance, adipose tissues secrete signaling molecules, including pro-inflammatory cytokines and are an energy reservoir for embedded cancer cells.181–183 Local alterations in adipose tissues release adipokines and proinflammatory cytokines and create chronic systemic inflammation.184 The altered production of adipokines, cytokines and hormones, such as adiponectin, leptin, TNF-α, interleukin-6 (IL-6), sex hormones and insulin then form an environment favorable for tumorigenesis.185 Elevated adipokines such as leptin stimulate the proliferation of colon, ovarian, or breast cancers among others.186 A previous study showed that leptin-deficient (ob/ob) mice are significantly less sensitive to azoxymethane-induced polyp formation than wild-type controls. Feeding the ob/ob mice on a high fructose diet (HFD), actively protected them from polyp formation compared with wild-type mice.187 However, several studies showed that the administration of adiponectin, another adipokine, decreased obesity, and the mice were found to possess anti-tumor properties.188–190 Adipokines activate transcription factors, including the hypoxia-inducible factor-1-α (HIF-1α) and NF-κB, and the peroxisome proliferator-activated receptor (PPAR) family.191 Among other factors, oxidative stress (OS) in chronic inflammation can be a critical pathological feature for tumorigenesis. Reactive oxygen species (ROS) induces pro-inflammatory cytokines such as IL-1, which could stimulate B-cell activation and trigger oxidative damage to the DNA of B-cells. Notably, variation in antioxidative genes is associated with the risk of non-Hodgkin’s lymphoma (NHL) or even pool prognosis. Consistent with this finding, a recent study evaluated the correlation between OS and diffuse large B-cell lymphoma (DLBCL). The results of the study revealed that higher concentrations of free oxygen radicals lower antioxidant substances and decrease high-density- lipoprotein cholesterol (HDL-C) levels in all stages of DLBCL. However, most of these features were alleviated following specific treatment by CHOP (Cyclophosphamide + Anthracycline + Vincristine + Prednisone) or R-CHOP (Rituximab + CHOP).192 Moreover, HDL-C could play anti-inflammatory and antioxidant roles, which suppresses the chemotactic activity of monocytes and lymphocytes, thus inhibiting cytokine-induced expression of endothelial cell adhesion molecules and protecting lymphocytes from oxidative damage. Low levels of HDL-C and elevated triglyceride (TG) and very-low-density lipoprotein (VLDL) are associated with the development of both acute leukemia and NHL. Further, several reports show that adipocyte progenitors in adipose tissues of obese individuals contribute to tumor progression by promoting angiogenesis and cancer cell proliferation in vivo.193,194 Hyperglycemic conditions induce epigenetic modifications, such as triggering the EGFR, human epidermal growth factor receptor 2 (HER2), and HER3 signaling cascades and their corresponding ligands. These changes subsequently stimulate tumor growth, even after an euglycemic environment exists.195,196
A recent meta-analysis evaluated HU and gout and showed that the two conditions are associated with higher cancer incidence and mortality.197 Hyperuricemia can increase the intracellular concentration of uric acid, which reacts with ROS and NO and causes oxidation and pro-inflammatory activity with cell transformation. The excessive intracellular concentration of uric acid down-regulates the expression of XO and thereby facilitating cell proliferation.198 The ROS, nitrogen species, and the uric acid derived from XO promote the development of both cell transformation and proliferation, as well as tumor progression and metastasis.199 Reduced expression of XO is associated with poor cell differentiation in multiple tumors but with increases in the aggressiveness and metastatic ability of breast cancer by inducing the expression of COX-2 and matrix metalloprotease.198
Therapeutic Strategies for Obesity and Hyperuricemia
The Therapeutic Approach to Obesity
The therapy of obesity encompasses lifestyle changes, anti-obesity pharmacotherapy, and bariatric surgery. Currently, treatment by bariatric surgery has been proven to be the most effective of the three therapies. Despite the restriction effect and malabsorption, bariatric surgery modulates metabolism via a mechanism that causes enhanced release of satiety hormones such as GLP-1, changed gut microbiota that alters motivation and food preferences, increased energy expenditure, and improved glucose homeostasis among others. The surgery is also associated with a reduced inflammatory response at the tissue and systemic levels, increased polarization of M2 macrophages,200 restoration of anti-inflammatory transcription factors PPAR γ, TWIST1, and KLF4,201,202 and corepressors GPS2 and SMRT.203 With improved metabolism, the uric acid level also reduces.204 Current clinical treatment guidelines place more emphasis on moderate approaches such as lifestyle modification changes and medications to achieve weight loss. Presently, the Food and Drug Administration (FDA) has approved six major types of anti-obesity drugs: Phentermine, Orlistat, Phentermine/Topiramate, Lorcaserin, Naltrexone/Bupropion, and Liraglutide205–207 (Table 1). However, there still exist multiple promising drugs in both preclinical or clinical trials which could join the list of obesity-resistant medications in the future.
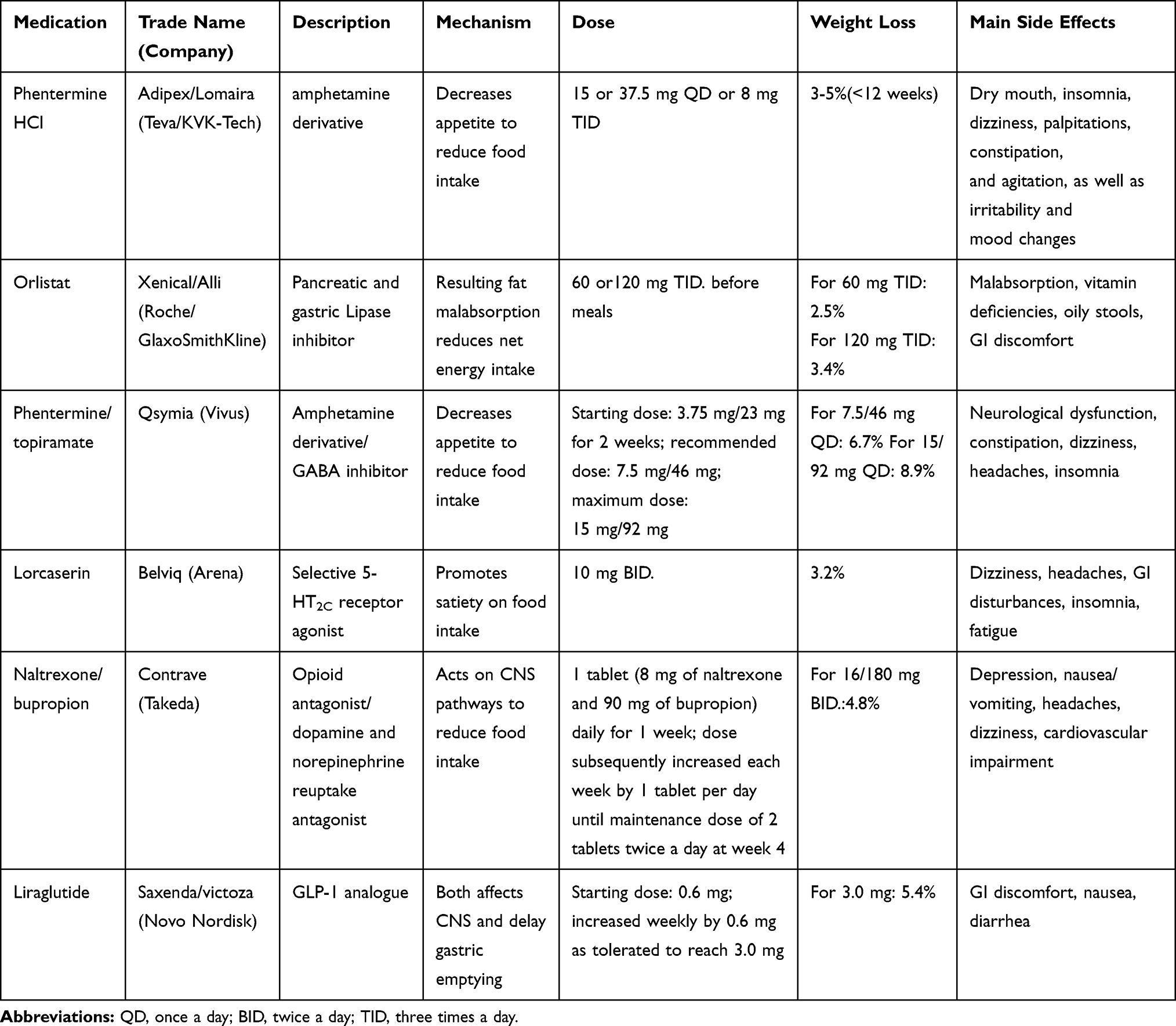 |
Table 1 Major Anti-Obesity Medications Approved by FDA |
Melanocortin Receptor-4 Agonist Setmelanotide
Setmelanotide is one of the new types of Melanocortin receptor-4 (MC4R) agonists in the development pipeline. Pre-clinical studies on non-human primates showed that Setmelanotide could reduce body weight and increase energy expenditure, and therefore improve insulin resistance and cardiac functions.208,209 Setmelanotide is currently being tested for efficacy in the treatment of a series of rare genetic obesity conditions such as Proopiomelanocortin (POMC) deficiency, Leptin receptor (LepR) deficiency, and Prader-Willi syndrome.210,211 The results of Phase 2 clinical trials on LepR deficiency have shown a substantial and durable reduction in hyperphagia and body weight over an observation period of 45–61 weeks.212 Thus, an MCR4 agonist could be a new promising choice for the treatment of melanocortin circuits related to obesity.
Glucose-Dependent Insulinotropic Polypeptide (GIP) Analogue
Glucose-Dependent Insulinotropic Polypeptide (GIP) is a 42-amino acid polypeptide hormone that belongs to the incretin and was initially isolated from intestinal K-cells. GIP can influence the pancreatic secretion of insulin and glucagon in an inverse glucose-dependent manner, which minimizes incidences of hypoglycemia and stabilizes blood glucose levels.213 Although the GIP analog improves glucose metabolism in rats and diabetic mice, it only has minimal effects on the body weight of obese mice as compared to another incretin- GLP-1.214 Nonetheless, the combination of GIP analog and GLP-1 induces more weight loss than GLP-1, suggesting dual incretin hormone-based therapy could be more efficient in the treatment of diabetic obesity rather than monotherapy.
Combination of Incretin-Based Therapy
Combining GLP-1R agonism with suitable endocrine partners for additive or synergistic metabolic benefits without compromising safety could provide an optimal incretin-based cure for obesity and its complications. Recently, this novel dual or triple incretin therapy, such as a combination of GLP-1RA and glucagon, has been shown to provide these features. Also, this combination therapy can elicit a more significant decrease in body weight than GLP-1 or glucagon monotherapy and was shown to match the results of bariatric surgery.215–218 Consequently, clinical trials based on GLP-1R and glucagon-receptor co-agonism are now ongoing.219 Besides, combining GLP-1 with incretin GIP delivers superior glycemic control relative to mono-incretin treatment.220 In rodent obesity and diabetes models, the first unimolecular GcgR/GLP-1R/GIPR triple agonist produced an unprecedented reversal of metabolic complications.221 Indeed, clinical studies are now evaluating the efficacy of the tri-agonist on obesity and diabetes, which would rival what can be achieved through bariatric surgery in animal studies.219 Also, other oral small-molecule agonists of GLP-1, such as TTP054/TTP-054 and ZYOG1 with the potential to replace injectable therapies while retaining the efficacy of GLP-1 agonist and minimizing adverse effects, are being studied.222
Fibroblast Growth Factor 21 (FGF21)
The fibroblast growth factor 21 (FGF 21) is mainly produced by the liver and regulates body weight and glucose metabolism.223 Notably, FGF 21 increases energy expenditure by enhancing the thermogenic activity of brown adipose tissue and the appearance of the beige/brite type in white fat. Another mechanism is that FGF21 can access the central nervous system (CNS) and modulate autonomic outflow to the adipose tissue. In the peripheral, FGF21 inhibits signaling by growth hormone, regulates fatty acid oxidation, and maintains lipid homeostasis in the liver of rodents.224–226 Since glucagon is a powerful FGF21 secretagogue, the preclinical success of GLP-1/glucagon co-agonism could partially be attributed to the involvement of a glucagon-FGF21 axis.227 Due to the short half-life of native FGF21, scientists have developed new stable forms of FGF21, including PEGylated FGF21,228 and FGF21-antibody conjugates as long-acting FGF21 pharmacological variants,229 Currently, multiple clinical trials are now being conducted to evaluate the efficacy of FGF 21.
The Therapeutic Strategy of Hyperuricemia
Notably, gout patients are frequently advised to lose weight. However, the findings of a recent systematic review suggest that the effects of weight loss on UA levels and achieving UA target on gout attacks are obscure and lacks sufficient data supports. Also, unfavorable effects may occur in the short term.230 Therefore, HU therapy requires comprehensive concerns far beyond only obesity management. The first concern encompasses lifestyle management, such as diet. Second, strong recommendations geared towards the active treatment of asymptomatic HU are lacking in most guidelines.231 However, gout patients should be informed that gout or HU is a chronic disease with flares, and the lack of effective treatment, especially urate-lowering therapy, frequently results in disease recurrence.232 Once a patient starts on urate-lowering treatment, the therapy should be continued for a lifelong to ensure gout flares do not recur. The typical medications, initiating therapy or titrating have been extensively reviewed in multiple literature.233,234 The present study introduces the new nontypical drug, SGLT-2 inhibitors, which has a urate-lowering property. Besides, anti-gout-based medications with immune- or anti-inflammation activities, such as biological IL-1 inhibitors are available, but do not have sufficient clinical evidence.235
Sodium-Glucose Co-Transporter Inhibitor-2 (SGLT-2i)
Sodium-glucose co-transporter-2 inhibitor (SGLT-2i), a new antidiabetic drug, has attracted immense attention by endocrinologists, cardiologists, and nephrologists. This increasing interest stems from its hypoglycemic effects that are independent of insulin action and its unique mechanisms of improving the prognosis of CVD and CKD. One of the mechanisms involves suppression of urate reabsorption, which subsequently generates a hypouricemic effect.236 Another evident feature is its ability to facilitate a reduction in body weight.237 At present, multiple clinical applications of SGLT-2i have emerged, and multiple clinical trials have reported its urate-lowering effect.238
Conclusion
In the past decades, incidences of obesity have continuously increased among all age groups, and particularly in children and teenagers.239 This pandemic has attracted immense attention from the public and has placed an enormous burden on both the family and society. Notably, genetic variations have effects on obesity primarily through influences on the central nervous system (CNS). In the peripheral system, the existence of pro-inflammatory molecules could increase the inflammatory state and contribute to a series of pathological states such as insulin resistance and hyperuricemia. Lately, the emerging role of gut microbiota in the development and progression of obesity and the use of prebiotic or probiotic agents has received massive attention. For the control of morbid obesity and its detrimental effects, bariatric surgery is frequently recommended as the most effective therapeutic strategy. However, SGLT-2i, a new non-invasive approach not only effectively lowers body weight but also acts on a specific target which ensures minimal side effects. Also, the antidiabetic medication SGLT-2i can improve the prognosis of CKD and CVD through uricosuria. As such, the prospects of comprehensively elucidating the mechanism of obesity and its complications, such as hyperuricemia, are promising, through which we can curb its adverse impacts on modern society.
Acknowledgment
This work was supported by National Natural Science Foundation of China (81370932), the Key Studies (Special) Department Fund of the Pudong, New Area Health Planning Commission (PWZzk2017-03), and Integrative Medicine special fund of Shanghai Municipal Health Planning Committee (ZHYY-ZXYJHZX-2-201712), Outstanding Leaders Training Program of Pudong Health Bureau of Shanghai (PWR12014-06), the Outstanding Clinical Discipline Project of Shanghai Pudong (PWYgy-2018-08), the Natural Science Foundation of China (Project no. 21675034), Shanghai Natural Science Foundation (19ZR1447500).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Nicolaidis S. Environment and obesity. Metabolism. 2019;100:153942. doi:10.1016/j.metabol.2019.07.006
2. Gregory JW. Prevention of obesity and metabolic syndrome in children. Front Endocrinol (Lausanne). 2019;10:669. doi:10.3389/fendo.2019.00669
3. Hruby A, Manson JE, Qi L, et al. Determinants and consequences of obesity. Am J Public Health. 2016;106(9):1656–1662. doi:10.2105/AJPH.2016.303326
4. NCD Risk Factor Collaboration. Trends in adult body-mass index in 200 countries from 1975 to 2014: a pooled analysis of 1698 population-based measurement studies with 19·2 million participants. Lancet. 2016;387(10026):1377–1396. doi:10.1016/S0140-6736(16)30054-X
5. Lavie CJ, Milani RV, Ventura HO. Obesity and cardiovascular disease: risk factor, paradox, and impact of weight loss. J Am Coll Cardiol. 2009;53(21):1925–1932. doi:10.1016/j.jacc.2008.12.068
6. Koene RJ, Prizment AE, Blaes A, et al. Shared Risk factors in cardiovascular disease and cancer. Circulation. 2016;133(11):1104–1114. doi:10.1161/CIRCULATIONAHA.115.020406
7. Yu YH. Making sense of metabolic obesity and hedonic obesity. J Diabetes. 2017;9(7):656–666. doi:10.1111/1753-0407.12529
8. Zhu Y, Zheng H, Zou Z, et al. Metabolic syndrome and related factors in Chinese children and adolescents: analysis from a Chinese national study. J Atheroscler Thromb. 2019. doi:10.5551/jat.50591
9. Xu W, Zhang H, Paillard-Borg S, et al. Prevalence of overweight and obesity among Chinese Adults: role of adiposity indicators and age. Obes Facts. 2016;9(1):17–28. doi:10.1159/000443003
10. Lv X, Zhou W, Sun J, et al. Visceral adiposity is significantly associated with type 2 diabetes in middle-aged and elderly Chinese women: a cross-sectional study. J Diabetes. 2017;9(10):920–928. doi:10.1111/1753-0407.12499
11. Thottam GE, Krasnokutsky S, Pillinger MH. Gout and metabolic syndrome: a tangled web. Curr Rheumatol Rep. 2017;19(10):60. doi:10.1007/s11926-017-0688-y
12. Liu R, Han C, Wu D, et al. Prevalence of Hyperuricemia and Gout in Mainland China from 2000 to 2014: a systematic review and meta-analysis. Biomed Res Int. 2015;2015:762820. doi:10.1155/2015/762820
13. Qasim A, Turcotte M, de Souza RJ, et al. On the origin of obesity: identifying the biological, environmental and cultural drivers of genetic risk among human populations. Obesity Rev. 2018;19(2):121–149. doi:10.1111/obr.v19.2
14. Pigeyre M, Yazdi FT, Kaur Y, Meyre D. Recent progress in genetics, epigenetics and metagenomics unveils the pathophysiology of human obesity. Clin Sci (Lond). 2016;130(12):943–986.
15. Waalen J. The genetics of human obesity. Transl Res. 2014;164(4):293–301. doi:10.1016/j.trsl.2014.05.010
16. Choquet H, Meyre D. Molecular basis of obesity: current status and future prospects. Curr Genomics. 2011;12(3):154–168. doi:10.2174/138920211795677921
17. Yu Z, Han S, Cao X, et al. Genetic polymorphisms in adipokine genes and the risk of obesity: a systematic review and meta-analysis. Obesity (Silver Spring). 2012;20(2):396–406. doi:10.1038/oby.2011.148
18. Cecil JE, Palmer CN, Fischer B, et al. Variants of the peroxisome proliferator-activated receptor gamma- and beta-adrenergic receptor genes are associated with measures of compensatory eating behaviors in young children. Am J Clin Nutr. 2007;86(1):167–173. doi:10.1093/ajcn/86.1.167
19. Ariza M, Garolera M, Jurado MA, et al. Dopamine genes (DRD2/ANKK1-TaqA1 and DRD4-7R) and executive function: their interaction with obesity. PLoS One. 2012;7(7):e41482. doi:10.1371/journal.pone.0041482
20. Andreasen CH, Stender-Petersen KL, Mogensen MS, et al. Low physical activity accentuates the effect of the FTO rs9939609 polymorphism on body fat accumulation. Diabetes. 2008;57(1):95–101. doi:10.2337/db07-0910
21. Qi Q, Chu AY, Kang JH, et al. Sugar-sweetened beverages and genetic risk of obesity. N Engl J Med. 2012;367(15):1387–1396. doi:10.1056/NEJMoa1203039
22. Rohde K, Keller M, la Cour Poulsen L, et al. Genetics and epigenetics in obesity. Metabolism. 2019;92:37–50. doi:10.1016/j.metabol.2018.10.007
23. Landrier J-F, Derghal A, Mounien L. MicroRNAs in obesity and related metabolic disorders. Cells. 2019;8(8):859. doi:10.3390/cells8080859
24. Locke AE, Kahali B, Berndt SI, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518(7538):197–206. doi:10.1038/nature14177
25. Shungin D, Winkler TW, Croteau-Chonka DC, et al. New genetic loci link adipose and insulin biology to body fat distribution. Nature. 2015;518(7538):187–196. doi:10.1038/nature14132
26. Goodarzi MO. Genetics of obesity: what genetic association studies have taught us about the biology of obesity and its complications. lancet Diabetes Endocrinol. 2018;6(3):223–236. doi:10.1016/S2213-8587(17)30200-0
27. Major TJ, Dalbeth N, Stahl EA, et al. An update on the genetics of hyperuricaemia and gout. Nat Rev Rheumatol. 2018;14(6):341–353. doi:10.1038/s41584-018-0004-x
28. Lesch M, Nyhan WL. A familial disorder of uric acid metabolism and central nervous system function. Am J Med. 1964;36(4):561–570. doi:10.1016/0002-9343(64)90104-4
29. Aune D, Norat T, Vatten LJ. Body mass index and the risk of gout: a systematic review and dose-response meta-analysis of prospective studies. Eur J Nutr. 2014;53(8):1591–1601. doi:10.1007/s00394-014-0766-0
30. Lyngdoh T, Vuistiner P, Marques-Vidal P, et al. Serum uric acid and adiposity: deciphering causality using a bidirectional Mendelian randomization approach. PLoS One. 2012;7(6):e39321. doi:10.1371/journal.pone.0039321
31. Larsson SC, Burgess S, Michaelsson K. Genetic association between adiposity and gout: a Mendelian randomization study. Rheumatology (Oxford). 2018;57(12):2145–2148. doi:10.1093/rheumatology/key229
32. Gali Ramamoorthy T, Begum G, Harno E, et al. Developmental programming of hypothalamic neuronal circuits: impact on energy balance control. Front Neurosci. 2015;9:126. doi:10.3389/fnins.2015.00126
33. Haggarty P. Genetic and metabolic determinants of human epigenetic variation. Curr Opin Clin Nutr Metab Care. 2015;18(4):334–338. doi:10.1097/MCO.0000000000000194
34. Zhang Y, Ren J. Epigenetics and obesity cardiomyopathy: from pathophysiology to prevention and management. Pharmacol Ther. 2016;161:52–66. doi:10.1016/j.pharmthera.2016.03.005
35. Yara S, Lavoie JC, Levy E. Oxidative stress and DNA methylation regulation in the metabolic syndrome. Epigenomics. 2015;7(2):283–300. doi:10.2217/epi.14.84
36. Jiang X, Ye X, Guo W, et al. Inhibition of HDAC3 promotes ligand-independent PPARgamma activation by protein acetylation. J Mol Endocrinol. 2014;53(2):191–200. doi:10.1530/JME-14-0066
37. Arner P, Kulyte A. MicroRNA regulatory networks in human adipose tissue and obesity. Nat Rev Endocrinol. 2015;11(5):276–288. doi:10.1038/nrendo.2015.25
38. Marques-Rocha JL, Samblas M, Milagro FI, et al. Noncoding RNAs, cytokines, and inflammation-related diseases. FASEB J. 2015;29(9):3595–3611. doi:10.1096/fj.14-260323
39. Deiuliis JA. MicroRNAs as regulators of metabolic disease: pathophysiologic significance and emerging role as biomarkers and therapeutics. Int J Obes (Lond). 2016;40(1):88–101. doi:10.1038/ijo.2015.170
40. Kulcheski FR, Christoff AP, Margis R. Circular RNAs are miRNA sponges and can be used as a new class of biomarker. J Biotechnol. 2016;238:42–51. doi:10.1016/j.jbiotec.2016.09.011
41. Yerlikaya A, Dagel T, King C, et al. Dietary and commercialized fructose: sweet or sour? Int Urol Nephrol. 2017;49(9):1611–1620. doi:10.1007/s11255-017-1544-8
42. Cleophas MC, Crişan TO, Lemmers H, et al. Suppression of monosodium urate crystal-induced cytokine production by butyrate is mediated by the inhibition of class I histone deacetylases. Ann Rheum Dis. 2016;75(3):593–600. doi:10.1136/annrheumdis-2014-206258
43. Jin HM, Kim T-J, Choi J-H, et al. MicroRNA-155 as a proinflammatory regulator via SHIP-1 down-regulation in acute gouty arthritis. Arthritis Res Ther. 2014;16(2):R88. doi:10.1186/ar4531
44. Dalbeth N, Pool B, Shaw OM, et al. Role of miR-146a in regulation of the acute inflammatory response to monosodium urate crystals. Ann Rheum Dis. 2015;74(4):786–790. doi:10.1136/annrheumdis-2014-205409
45. Park HK, Ahima RS. Physiology of leptin: energy homeostasis, neuroendocrine function and metabolism. Metabolism. 2015;64(1):24–34. doi:10.1016/j.metabol.2014.08.004
46. Kukkonen JP, Leonard CS. Orexin/hypocretin receptor signalling cascades. Br J Pharmacol. 2014;171(2):314–331. doi:10.1111/bph.12324
47. Macneil DJ. The role of melanin-concentrating hormone and its receptors in energy homeostasis. Front Endocrinol (Lausanne). 2013;4:49. doi:10.3389/fendo.2013.00049
48. Peterson SM, Pack TF, Wilkins AD, et al. Elucidation of G-protein and beta-arrestin functional selectivity at the dopamine D2 receptor. Proc Natl Acad Sci U S A. 2015;112(22):7097–7102. doi:10.1073/pnas.1502742112
49. Heyman E, Gamelin F-X, Aucouturier J, et al. The role of the endocannabinoid system in skeletal muscle and metabolic adaptations to exercise: potential implications for the treatment of obesity. Obes Rev. 2012;13(12):1110–1124. doi:10.1111/obr.2012.13.issue-12
50. Nathan PJ, Bullmore ET. From taste hedonics to motivational drive: central mu-opioid receptors and binge-eating behaviour. Int J Neuropsychopharmacol. 2009;12(7):995–1008. doi:10.1017/S146114570900039X
51. Fang PH, Yu M, Ma Y-P, et al. Central nervous system regulation of food intake and energy expenditure: role of galanin-mediated feeding behavior. Neurosci Bull. 2011;27(6):407–412. doi:10.1007/s12264-011-1841-7
52. Hayes DJ, Greenshaw AJ. 5-HT receptors and reward-related behaviour: a review. Neurosci Biobehav Rev. 2011;35(6):1419–1449. doi:10.1016/j.neubiorev.2011.03.005
53. Kovacs KJ. CRH: the link between hormonal-, metabolic- and behavioral responses to stress. J Chem Neuroanat. 2013;54:25–33. doi:10.1016/j.jchemneu.2013.05.003
54. Sohn JW. Network of hypothalamic neurons that control appetite. BMB Rep. 2015;48(4):229–233. doi:10.5483/BMBRep.2015.48.4.272
55. Lin Y, Hall RA, Kuhar MJ. CART peptide stimulation of G protein-mediated signaling in differentiated PC12 cells: identification of PACAP 6-38 as a CART receptor antagonist. Neuropeptides. 2011;45(5):351–358. doi:10.1016/j.npep.2011.07.006
56. Loh K, Herzog H, Shi YC. Regulation of energy homeostasis by the NPY system. Trends Endocrinol Metab. 2015;26(3):125–135. doi:10.1016/j.tem.2015.01.003
57. Adamska E, Ostrowska L, Górska M, et al. The role of gastrointestinal hormones in the pathogenesis of obesity and type 2 diabetes. Prz Gastroenterol. 2014;9(2):69–76. doi:10.5114/pg.2014.42498
58. Simpson K, Parker J, Plumer J, Bloom S. CCK, PYY and PP: the control of energy balance. In: Handb Exp Pharmacol. Berlin, Heidelberg: Springer; 2012:209–230.
59. Potes CS, Lutz TA. Brainstem mechanisms of amylin-induced anorexia. Physiol Behav. 2010;100(5):511–518. doi:10.1016/j.physbeh.2010.03.001
60. Begg DP, Woods SC. The central insulin system and energy balance. In: Handb Exp Pharmacol. Berlin, Heidelberg: Springer; 2012:111–129.
61. Gahete MD, Rincón-Fernández D, Villa-Osaba A, et al. Ghrelin gene products, receptors, and GOAT enzyme: biological and pathophysiological insight. J Endocrinol. 2014;220(1):R1–R24. doi:10.1530/JOE-13-0391
62. Holst JJ. The physiology of glucagon-like peptide 1. Physiol Rev. 2007;87(4):1409–1439. doi:10.1152/physrev.00034.2006
63. Zhou L, Sutton GM, Rochford JJ, et al. Serotonin 2C receptor agonists improve type 2 diabetes via melanocortin-4 receptor signaling pathways. Cell Metab. 2007;6(5):398–405. doi:10.1016/j.cmet.2007.10.008
64. Volkow ND, Wang G-J, Tomasi D, et al. Obesity and addiction: neurobiological overlaps. Obes Rev. 2013;14(1):2–18. doi:10.1111/obr.2013.14.issue-1
65. Volkow ND, Wang GJ, Baler RD. Reward, dopamine and the control of food intake: implications for obesity. Trends Cogn Sci. 2011;15(1):37–46. doi:10.1016/j.tics.2010.11.001
66. D’Addario C, Micioni Di Bonaventura MV, Pucci M, et al. Endocannabinoid signaling and food addiction. Neurosci Biobehav Rev. 2014;47:203–224. doi:10.1016/j.neubiorev.2014.08.008
67. Stice E, Figlewicz DP, Gosnell BA, et al. The contribution of brain reward circuits to the obesity epidemic. Neurosci Biobehav Rev. 2013;37(9):2047–2058. doi:10.1016/j.neubiorev.2012.12.001
68. Uemura N, Murakami R, Chiba Y, et al. Immunoreactivity of urate transporters, GLUT9 and URAT1, is located in epithelial cells of the choroid plexus of human brains. Neurosci Lett. 2017;659:99–103. doi:10.1016/j.neulet.2017.09.001
69. Lu W, Xu Y, Shao X, et al. Uric acid produces an inflammatory response through activation of NF-kappaB in the hypothalamus: implications for the pathogenesis of metabolic disorders. Sci Rep. 2015;5(1):12144. doi:10.1038/srep12144
70. Rocha VZ, Folco EJ, Sukhova G, et al. Interferon-gamma, a Th1 cytokine, regulates fat inflammation: a role for adaptive immunity in obesity. Circ Res. 2008;103(5):467–476. doi:10.1161/CIRCRESAHA.108.177105
71. Wong N, Fam BC, Cempako GR, et al. Deficiency in interferon-gamma results in reduced body weight and better glucose tolerance in mice. Endocrinology. 2011;152(10):3690–3699. doi:10.1210/en.2011-0288
72. Abe M, Matsuda M, Kobayashi H, et al. Effects of statins on adipose tissue inflammation: their inhibitory effect on MyD88-independent IRF3/IFN-beta pathway in macrophages. Arterioscler Thromb Vasc Biol. 2008;28(5):871–877. doi:10.1161/ATVBAHA.107.160663
73. Wang XA, Zhang R, Jiang D, et al. Interferon regulatory factor 9 protects against hepatic insulin resistance and steatosis in male mice. Hepatology. 2013;58(2):603–616. doi:10.1002/hep.26368
74. Eguchi J, Kong X, Tenta M, et al. Interferon regulatory factor 4 regulates obesity-induced inflammation through regulation of adipose tissue macrophage polarization. Diabetes. 2013;62(10):3394–3403. doi:10.2337/db12-1327
75. Weisberg SP, McCann D, Desai M, et al. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112(12):1796–1808. doi:10.1172/JCI200319246
76. Di Gregorio GB, Yao-Borengasser A, Rasouli N, et al. Expression of CD68 and macrophage chemoattractant protein-1 genes in human adipose and muscle tissues: association with cytokine expression, insulin resistance, and reduction by pioglitazone. Diabetes. 2005;54(8):2305–2313. doi:10.2337/diabetes.54.8.2305
77. Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3(1):23–35. doi:10.1038/nri978
78. Hume DA. The many alternative faces of macrophage activation. Front Immunol. 2015;6:370. doi:10.3389/fimmu.2015.00370
79. Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117(1):175–184. doi:10.1172/JCI29881
80. Cristancho AG, Lazar MA. Forming functional fat: a growing understanding of adipocyte differentiation. Nat Rev Mol Cell Biol. 2011;12(11):722–734. doi:10.1038/nrm3198
81. Fernandez-Sanchez A, Madrigal-Santillán E, Bautista M, et al. Inflammation, oxidative stress, and obesity. Int J Mol Sci. 2011;12(5):3117–3132. doi:10.3390/ijms12053117
82. Marseglia L, Manti S, D’Angelo G, et al. Oxidative stress in obesity: a critical component in human diseases. Int J Mol Sci. 2014;16(1):378–400. doi:10.3390/ijms16010378
83. Fonseca-Alaniz MH, Takada J, Alonso-Vale MIC, et al. Adipose tissue as an endocrine organ: from theory to practice. J Pediatr (Rio J). 2007;83(5):S192–S203. doi:10.1590/S0021-75572007000700011
84. Lavrovsky Y, Chatterjee B, Clark RA, Roy AK. Role of redox-regulated transcription factors in inflammation, aging and age-related diseases. Exp Gerontol. 2000;35(5):521–532. doi:10.1016/S0531-5565(00)00118-2
85. Shoelson SE, Herrero L, Naaz A. Obesity, inflammation, and insulin resistance. Gastroenterology. 2007;132(6):2169–2180. doi:10.1053/j.gastro.2007.03.059
86. Amirkhizi F, Siassi F, Minaie S, Djalali M, Rahimi A, Chamari M. Is obesity associated with increased plasma lipid peroxidation and oxidative stress in women? ARYA Atheroscler. 2007;2.
87. Pihl E, Zilmer K, Kullisaar T, et al. Atherogenic inflammatory and oxidative stress markers in relation to overweight values in male former athletes. Int J Obes (Lond). 2006;30(1):141–146. doi:10.1038/sj.ijo.0803068
88. Chrysohoou C, Panagiotakos DB, Pitsavos C, et al. The implication of obesity on total antioxidant capacity in apparently healthy men and women: the ATTICA study. Nutr Metab Cardiovasc Dis. 2007;17(8):590–597. doi:10.1016/j.numecd.2006.05.007
89. Tereshin EV. [A role of fatty acids in the development of oxidative stress in aging. A hypothesis]. Adv Gerontol. 2007;20(1):59–65. Russian.
90. Sautin YY, Nakagawa T, Zharikov S, et al. Adverse effects of the classic antioxidant uric acid in adipocytes: NADPH oxidase-mediated oxidative/nitrosative stress. Am J Physiol Cell Physiol. 2007;293(2):C584–C596. doi:10.1152/ajpcell.00600.2006
91. Sanchez-Lozada LG, Lanaspa MA, Cristóbal-García M, et al. Uric acid-induced endothelial dysfunction is associated with mitochondrial alterations and decreased intracellular ATP concentrations. Nephron Exp Nephrol. 2012;121(3–4):e71–e78. doi:10.1159/000345509
92. Cristobal-Garcia M, García-Arroyo FE, Tapia E, et al. Renal oxidative stress induced by long-term hyperuricemia alters mitochondrial function and maintains systemic hypertension. Oxid Med Cell Longev. 2015;2015:535686. doi:10.1155/2015/535686
93. Baldwin W, McRae S, Marek G, et al. Hyperuricemia as a mediator of the proinflammatory endocrine imbalance in the adipose tissue in a murine model of the metabolic syndrome. Diabetes. 2011;60(4):1258–1269. doi:10.2337/db10-0916
94. Turnbaugh PJ, Ley RE, Mahowald MA, et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444(7122):1027–1031. doi:10.1038/nature05414
95. Ley RE, Backhed F, Turnbaugh P, et al. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A. 2005;102(31):11070–11075. doi:10.1073/pnas.0504978102
96. Ley RE, Turnbaugh PJ, Klein S, et al. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444(7122):1022–1023. doi:10.1038/4441022a
97. Cani PD, Amar J, Iglesias MA, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56(7):1761–1772. doi:10.2337/db06-1491
98. Lewis K, Lutgendorff F, Phan V, et al. Enhanced translocation of bacteria across metabolically stressed epithelia is reduced by butyrate†. Inflamm Bowel Dis. 2010;16(7):1138–1148. doi:10.1002/ibd.21177
99. Usami M, Kishimoto K, Ohata A, et al. Butyrate and trichostatin A attenuate nuclear factor kappaB activation and tumor necrosis factor alpha secretion and increase prostaglandin E2 secretion in human peripheral blood mononuclear cells. Nutr Res. 2008;28(5):321–328. doi:10.1016/j.nutres.2008.02.012
100. Freeland KR, Wolever TM. Acute effects of intravenous and rectal acetate on glucagon-like peptide-1, peptide YY, ghrelin, adiponectin and tumour necrosis factor-alpha. Br J Nutr. 2010;103(3):460–466. doi:10.1017/S0007114509991863
101. Louis P, Flint HJ. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol Lett. 2009;294(1):1–8. doi:10.1111/fml.2009.294.issue-1
102. Backhed F, Ding H, Wang T, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A. 2004;101(44):15718–15723. doi:10.1073/pnas.0407076101
103. Rabot S, Membrez M, Bruneau A, et al. Germ-free C57BL/6J mice are resistant to high-fat-diet-induced insulin resistance and have altered cholesterol metabolism. FASEB J. 2010;24(12):4948–4959. doi:10.1096/fj.10-164921
104. Musso G, Gambino R, Cassader M. Interactions between gut microbiota and host metabolism predisposing to obesity and diabetes. Annu Rev Med. 2011;62(1):361–380. doi:10.1146/annurev-med-012510-175505
105. Aronsson L, Huang Y, Parini P, et al. Decreased fat storage by Lactobacillus paracasei is associated with increased levels of angiopoietin-like 4 protein (ANGPTL4). PLoS One. 2010;5(9):e13087. doi:10.1371/journal.pone.0013087
106. Backhed F, Manchester JK, Semenkovich CF, et al. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Natl Acad Sci U S A. 2007;104(3):979–984. doi:10.1073/pnas.0605374104
107. Vrieze A, Holleman F, Zoetendal EG, et al. The environment within: how gut microbiota may influence metabolism and body composition. Diabetologia. 2010;53(4):606–613. doi:10.1007/s00125-010-1662-7
108. Armougom F, Henry M, Vialettes B, et al. Monitoring bacterial community of human gut microbiota reveals an increase in Lactobacillus in obese patients and Methanogens in anorexic patients. PLoS One. 2009;4(9):e7125. doi:10.1371/journal.pone.0007125
109. Santacruz A, Collado MC, García-Valdés L, et al. Gut microbiota composition is associated with body weight, weight gain and biochemical parameters in pregnant women. Br J Nutr. 2010;104(1):83–92. doi:10.1017/S0007114510000176
110. Cani PD, Joly E, Horsmans Y, et al. Oligofructose promotes satiety in healthy human: a pilot study. Eur J Clin Nutr. 2006;60(5):567–572. doi:10.1038/sj.ejcn.1602350
111. Priebe MG, Wang H, Weening D, et al. Factors related to colonic fermentation of nondigestible carbohydrates of a previous evening meal increase tissue glucose uptake and moderate glucose-associated inflammation. Am J Clin Nutr. 2010;91(1):90–97. doi:10.3945/ajcn.2009.28521
112. Cani PD, Lecourt E, Dewulf EM, et al. Gut microbiota fermentation of prebiotics increases satietogenic and incretin gut peptide production with consequences for appetite sensation and glucose response after a meal. Am J Clin Nutr. 2009;90(5):1236–1243. doi:10.3945/ajcn.2009.28095
113. Parnell JA, Reimer RA. Weight loss during oligofructose supplementation is associated with decreased ghrelin and increased peptide YY in overweight and obese adults. Am J Clin Nutr. 2009;89(6):1751–1759. doi:10.3945/ajcn.2009.27465
114. van Can JG, IJzerman TH, van Loon LJC, et al. Reduced glycaemic and insulinaemic responses following isomaltulose ingestion: implications for postprandial substrate use. Br J Nutr. 2009;102(10):1408–1413. doi:10.1017/S0007114509990687
115. Guo Z, Zhang J, Wang Z, et al. Intestinal microbiota distinguish gout patients from healthy humans. Sci Rep. 2016;6(1):20602. doi:10.1038/srep20602
116. Li G, Yao W, Jiang H. Short-chain fatty acids enhance adipocyte differentiation in the stromal vascular fraction of porcine adipose tissue. J Nutr. 2014;144(12):1887–1895. doi:10.3945/jn.114.198531
117. Vieira AT, Macia L, Galvão I, et al. A Role for gut microbiota and the metabolite-sensing receptor GPR43 in a murine model of gout. Arthritis Rheumatol. 2015;67(6):1646–1656. doi:10.1002/art.39107
118. Vieira AT, Galvão I, Macia LM, et al. Dietary fiber and the short-chain fatty acid acetate promote resolution of neutrophilic inflammation in a model of gout in mice. J Leukoc Biol. 2017;101(1):275–284. doi:10.1189/jlb.3A1015-453RRR
119. Garcia-Arroyo FE, Gonzaga G, Muñoz-Jiménez I, et al. Probiotic supplements prevented oxonic acid-induced hyperuricemia and renal damage. PLoS One. 2018;13(8):e0202901. doi:10.1371/journal.pone.0202901
120. Yu Y, Liu Q, Li H, et al. Alterations of the gut microbiome associated with the treatment of hyperuricaemia in male rats. Front Microbiol. 2018;9:2233. doi:10.3389/fmicb.2018.02233
121. Samuel VT, Shulman GI. The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J Clin Invest. 2016;126(1):12–22. doi:10.1172/JCI77812
122. Ye J. Mechanisms of insulin resistance in obesity. Front Med. 2013;7(1):14–24. doi:10.1007/s11684-013-0262-6
123. Dresner A, Laurent D, Marcucci M, et al. Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J Clin Invest. 1999;103(2):253–259. doi:10.1172/JCI5001
124. Petersen KF, Dufour S, Savage DB, et al. The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc Natl Acad Sci U S A. 2007;104(31):12587–12594. doi:10.1073/pnas.0705408104
125. Perry RJ, Camporez J-P, Kursawe R, et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell. 2015;160(4):745–758. doi:10.1016/j.cell.2015.01.012
126. Perry RJ, Zhang X-M, Zhang D, et al. Leptin reverses diabetes by suppression of the hypothalamic-pituitary-adrenal axis. Nat Med. 2014;20(7):759–763. doi:10.1038/nm.3579
127. Previs SF, Cline GW, Shulman GI. A critical evaluation of mass isotopomer distribution analysis of gluconeogenesis in vivo. Am J Physiol. 1999;277(1):E154–E160. doi:10.1152/ajpendo.1999.277.1.E154
128. Samuel VT, Liu Z-X, Qu X, et al. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem. 2004;279(31):32345–32353. doi:10.1074/jbc.M313478200
129. Qu X, Seale JP, Donnelly R. Tissue and isoform-selective activation of protein kinase C in insulin-resistant obese Zucker rats - effects of feeding. J Endocrinol. 1999;162(2):207–214. doi:10.1677/joe.0.1620207
130. Magkos F, Su X, Bradley D, et al. Intrahepatic diacylglycerol content is associated with hepatic insulin resistance in obese subjects. Gastroenterology. 2012;142(7):1444–6 e2. doi:10.1053/j.gastro.2012.03.003
131. Stanford KI, Goodyear LJ. Exercise and type 2 diabetes: molecular mechanisms regulating glucose uptake in skeletal muscle. Adv Physiol Educ. 2014;38(4):308–314. doi:10.1152/advan.00080.2014
132. Zhang Y, Ye J. Mitochondrial inhibitor as a new class of insulin sensitizer. Acta Pharm Sin B. 2012;2(4):341–349. doi:10.1016/j.apsb.2012.06.010
133. Petersen KF, Dufour S, Befroy D, et al. Reversal of nonalcoholic hepatic steatosis, hepatic insulin resistance, and hyperglycemia by moderate weight reduction in patients with type 2 diabetes. Diabetes. 2005;54(3):603–608. doi:10.2337/diabetes.54.3.603
134. Lim EL, Hollingsworth KG, Aribisala BS, et al. Reversal of type 2 diabetes: normalisation of beta cell function in association with decreased pancreas and liver triacylglycerol. Diabetologia. 2011;54(10):2506–2514. doi:10.1007/s00125-011-2204-7
135. Zhou L, Podolsky N, Sang Z, et al. The medial amygdalar nucleus: a novel glucose-sensing region that modulates the counterregulatory response to hypoglycemia. Diabetes. 2010;59(10):2646–2652. doi:10.2337/db09-0995
136. Choi HK, Mount DB, Reginato AM, et al. Pathogenesis of gout. Ann Intern Med. 2005;143(7):499–516. doi:10.7326/0003-4819-143-7-200510040-00009
137. Yoo TW, Sung KC, Shin HS, et al. Relationship between serum uric acid concentration and insulin resistance and metabolic syndrome. Circ J. 2005;69(8):928–933. doi:10.1253/circj.69.928
138. Quinones-Galvan A, Ferrannini E. Renal effects of insulin in man. J Nephrol. 1997;10(4):188–191.
139. Scott FW, Trick KD, Stavric B, et al. Uric acid-induced decrease in rat insulin secretion. Proc Soc Exp Biol Med. 1981;166(1):123–128. doi:10.3181/00379727-166-41033
140. Wan X, Xu C, Lin Y, et al. Uric acid regulates hepatic steatosis and insulin resistance through the NLRP3 inflammasome-dependent mechanism. J Hepatol. 2016;64(4):925–932. doi:10.1016/j.jhep.2015.11.022
141. Cicerchi C, Li N, Kratzer J, et al. Uric acid-dependent inhibition of AMP kinase induces hepatic glucose production in diabetes and starvation: evolutionary implications of the uricase loss in hominids. FASEB J. 2014;28(8):3339–3350. doi:10.1096/fsb2.v28.8
142. Epingeac M, Gaman MA, Diaconu CC, Gad M, Gaman AM. The evaluation of oxidative stress levels in obesity. Revista Chimie Bucharest Original Ed. 2019;70:2241–2244.
143. Matsuoka TA, Kaneto H, Miyatsuka T, et al. Regulation of MafA expression in pancreatic beta-cells in db/db mice with diabetes. Diabetes. 2010;59(7):1709–1720. doi:10.2337/db08-0693
144. Kawamori D, Kajimoto Y, Kaneto H, et al. Oxidative stress induces nucleo-cytoplasmic translocation of pancreatic transcription factor PDX-1 through activation of c-Jun NH(2)-terminal kinase. Diabetes. 2003;52(12):2896–2904. doi:10.2337/diabetes.52.12.2896
145. Miric DJ, Kisic BM, Filipovic-Danic S, et al. Xanthine oxidase activity in type 2 diabetes mellitus patients with and without diabetic peripheral neuropathy. J Diabetes Res. 2016;2016:4370490. doi:10.1155/2016/4370490
146. Washio KW, Kusunoki Y, Murase T, et al. Xanthine oxidoreductase activity is correlated with insulin resistance and subclinical inflammation in young humans. Metabolism. 2017;70:51–56. doi:10.1016/j.metabol.2017.01.031
147. Takir M, Kostek O, Ozkok A, et al. Lowering uric acid with allopurinol improves insulin resistance and systemic inflammation in asymptomatic hyperuricemia. J Investig Med. 2015;63(8):924–929. doi:10.1097/JIM.0000000000000242
148. Isık H, Aynıoglu O, Tımur H, et al. Is Xanthine oxidase activity in polycystic ovary syndrome associated with inflammatory and cardiovascular risk factors? J Reprod Immunol. 2016;116:98–103. doi:10.1016/j.jri.2016.06.002
149. Fujimura Y, Yamauchi Y, Murase T, et al. Relationship between plasma xanthine oxidoreductase activity and left ventricular ejection fraction and hypertrophy among cardiac patients. PLoS One. 2017;12(8):e0182699. doi:10.1371/journal.pone.0182699
150. Nakagawa T, Hu H, Zharikov S, et al. A causal role for uric acid in fructose-induced metabolic syndrome. Am J Physiol Renal Physiol. 2006;290(3):F625–F631. doi:10.1152/ajprenal.00140.2005
151. Bjornstad P, Lanaspa MA, Ishimoto T, et al. Fructose and uric acid in diabetic nephropathy. Diabetologia. 2015;58(9):1993–2002. doi:10.1007/s00125-015-3650-4
152. Roddy E, Doherty M. Epidemiology of gout. Arthritis Res Ther. 2010;12(6):223. doi:10.1186/ar3199
153. Choi YJ, Shin H-S, Choi HS, et al. Uric acid induces fat accumulation via generation of endoplasmic reticulum stress and SREBP-1c activation in hepatocytes. Lab Invest. 2014;94(10):1114–1125. doi:10.1038/labinvest.2014.98
154. Mazzali M, Hughes J, Kim Y-G, et al. Elevated uric acid increases blood pressure in the rat by a novel crystal-independent mechanism. Hypertension. 2001;38(5):1101–1106. doi:10.1161/hy1101.092839
155. Perez-Pozo SE, Schold J, Nakagawa T, et al. Excessive fructose intake induces the features of metabolic syndrome in healthy adult men: role of uric acid in the hypertensive response. Int J Obes (Lond). 2010;34(3):454–461. doi:10.1038/ijo.2009.259
156. DeBosch BJ, Kluth O, Fujiwara H, et al. Early-onset metabolic syndrome in mice lacking the intestinal uric acid transporter SLC2A9. Nat Commun. 2014;5(1):4642. doi:10.1038/ncomms5642
157. El Din UAS, Salem MM, Abdulazim DO. Uric acid in the pathogenesis of metabolic, renal, and cardiovascular diseases: a review. J Adv Res. 2017;8(5):537–548. doi:10.1016/j.jare.2016.11.004
158. Roy D, Perreault M, Marette A. Insulin stimulation of glucose uptake in skeletal muscles and adipose tissues in vivo is NO dependent. Am J Physiol. 1998;274(4):E692–E699. doi:10.1152/ajpendo.1998.274.4.E692
159. Bjornstad P, Snell-Bergeon JK, McFann K, et al. Serum uric acid and insulin sensitivity in adolescents and adults with and without type 1 diabetes. J Diabetes Complications. 2014;28(3):298–304. doi:10.1016/j.jdiacomp.2013.12.007
160. Lanaspa MA, Epperson LE, Li N, et al. Opposing activity changes in AMP deaminase and AMP-activated protein kinase in the hibernating ground squirrel. PLoS One. 2015;10(4):e0123509. doi:10.1371/journal.pone.0123509
161. Xu W, Huang Y, Li L, et al. Hyperuricemia induces hypertension through activation of renal epithelial sodium channel (ENaC). Metabolism. 2016;65(3):73–83. doi:10.1016/j.metabol.2015.10.026
162. Tani S, Nagao K, Hirayama A. Effect of febuxostat, a xanthine oxidase inhibitor, on cardiovascular risk in hyperuricemic patients with hypertension: a prospective, open-label, pilot study. Clin Drug Investig. 2015;35(12):823–831. doi:10.1007/s40261-015-0349-8
163. Roddy E, Mallen CD, Doherty M. Gout. BMJ. 2013;347(oct01 3):f5648. doi:10.1136/bmj.f5648
164. Cirillo P, Sato W, Reungjui S, et al. Uric acid, the metabolic syndrome, and renal disease. J Am Soc Nephrol. 2006;17(12 Suppl 3):S165–S168. doi:10.1681/ASN.2006080909
165. Jalal DI, Rivard CJ, Johnson RJ, et al. Serum uric acid levels predict the development of albuminuria over 6 years in patients with type 1 diabetes: findings from the Coronary Artery Calcification in Type 1 Diabetes study. Nephrol Dial Transplant. 2010;25(6):1865–1869. doi:10.1093/ndt/gfp740
166. Bastien M, Poirier P, Lemieux I, et al. Overview of epidemiology and contribution of obesity to cardiovascular disease. Prog Cardiovasc Dis. 2014;56(4):369–381. doi:10.1016/j.pcad.2013.10.016
167. Kaltman AJ, Goldring RM. Role of circulatory congestion in the cardiorespiratory failure of obesity. Am J Med. 1976;60(5):645–653. doi:10.1016/0002-9343(76)90499-X
168. Messerli FH, Nunez BD, Ventura HO, Snyder DW. Overweight and sudden death. Increased ventricular ectopy in cardiopathy of obesity. Arch Intern Med. 1987;147(10):1725–1728. doi:10.1001/archinte.1987.00370100039008
169. Messerli FH. Cardiopathy of obesity–a not-so-Victorian disease. N Engl J Med. 1986;314(6):378–380. doi:10.1056/NEJM198602063140608
170. Ku CS, Lin S-L, Wang D-J, et al. Left ventricular filling in young normotensive obese adults. Am J Cardiol. 1994;73(8):613–615. doi:10.1016/0002-9149(94)90347-6
171. Poirier P, Giles TD, Bray GA, et al. Obesity and cardiovascular disease: pathophysiology, evaluation, and effect of weight loss. Arterioscler Thromb Vasc Biol. 2006;26(5):968–976. doi:10.1161/01.ATV.0000216787.85457.f3
172. Abbott RD, Brand FN, Kannel WB, et al. Gout and coronary heart disease: the Framingham Study. J Clin Epidemiol. 1988;41(3):237–242. doi:10.1016/0895-4356(88)90127-8
173. Storhaug HM, Norvik JV, Toft I, et al. Uric acid is a risk factor for ischemic stroke and all-cause mortality in the general population: a gender specific analysis from The Tromso Study. BMC Cardiovasc Disord. 2013;13(1):115. doi:10.1186/1471-2261-13-115
174. Chen JH, Lan J-L, Cheng C-F, et al. Effect of Urate-lowering therapy on all-cause and cardiovascular mortality in hyperuricemic patients without gout: a case-matched cohort study. PLoS One. 2015;10(12):e0145193. doi:10.1371/journal.pone.0145193
175. Chen JH, Lan J-L, Cheng C-F, et al. Effect of urate-lowering therapy on the risk of cardiovascular disease and all-cause mortality in patients with gout: a case-matched cohort study. J Rheumatol. 2015;42(9):1694–1701. doi:10.3899/jrheum.141542
176. Battelli MG, Bortolotti M, Polito L, et al. The role of xanthine oxidoreductase and uric acid in metabolic syndrome. Biochim Biophys Acta Mol Basis Dis. 2018;1864(8):2557–2565. doi:10.1016/j.bbadis.2018.05.003
177. Ando K, Takahashi H, Watanabe T, et al. Impact of serum uric acid levels on coronary plaque stability evaluated using integrated backscatter intravascular ultrasound in patients with coronary artery disease. J Atheroscler Thromb. 2016;23(8):932–939. doi:10.5551/jat.33951
178. Sánchez-Lozada LG, Lanaspa MA, Cristóbal-García M, et al. Uric acid-induced endothelial dysfunction is associated with mitochondrial alterations and decreased intracellular ATP concentrations. Nephron Exp Nephrol. 2012;121(3–4):e71–8. doi:10.1159/000345509
179. Basen-Engquist K, Chang M. Obesity and cancer risk: recent review and evidence. Curr Oncol Rep. 2011;13(1):71–76. doi:10.1007/s11912-010-0139-7
180. Park J, Euhus DM, Scherer PE. Paracrine and endocrine effects of adipose tissue on cancer development and progression. Endocr Rev. 2011;32(4):550–570. doi:10.1210/er.2010-0030
181. Nieman KM, Kenny HA, Penicka CV, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med. 2011;17(11):1498–1503. doi:10.1038/nm.2492
182. Tessitore L, Vizio B, Pesola D, et al. Adipocyte expression and circulating levels of leptin increase in both gynaecological and breast cancer patients. Int J Oncol. 2004;24(6):1529–1535.
183. Park J, Scherer PE. Adipocyte-derived endotrophin promotes malignant tumor progression. J Clin Invest. 2012;122(11):4243–4256. doi:10.1172/JCI63930
184. Berg AH, Scherer PE. Adipose tissue, inflammation, and cardiovascular disease. Circ Res. 2005;96(9):939–949. doi:10.1161/01.RES.0000163635.62927.34
185. Doyle SL, Donohoe CL, Lysaght J, et al. Visceral obesity, metabolic syndrome, insulin resistance and cancer. Proc Nutr Soc. 2012;71(1):181–189. doi:10.1017/S002966511100320X
186. Park J, Morley TS, Kim M, et al. Obesity and cancer–mechanisms underlying tumour progression and recurrence. Nat Rev Endocrinol. 2014;10(8):455–465. doi:10.1038/nrendo.2014.94
187. Endo H, Hosono K, Uchiyama T, et al. Leptin acts as a growth factor for colorectal tumours at stages subsequent to tumour initiation in murine colon carcinogenesis. Gut. 2011;60(10):1363–1371. doi:10.1136/gut.2010.235754
188. Saxena NK, Fu PP, Nagalingam A, et al. Adiponectin modulates C-jun N-terminal kinase and mammalian target of rapamycin and inhibits hepatocellular carcinoma. Gastroenterology. 2010;139(5):1762–1765. doi:10.1053/j.gastro.2010.07.001
189. Kim AY, Lee YS, Kim KH, et al. Adiponectin represses colon cancer cell proliferation via AdipoR1- and -R2-mediated AMPK activation. Mol Endocrinol. 2010;24(7):1441–1452. doi:10.1210/me.2009-0498
190. Denzel MS, Hebbard LW, Shostak G, et al. Adiponectin deficiency limits tumor vascularization in the MMTV-PyV-mT mouse model of mammary cancer. Clin Cancer Res. 2009;15(10):3256–3264. doi:10.1158/1078-0432.CCR-08-2661
191. Mendonca FM, de Sousa FR, Barbosa AL, et al. Metabolic syndrome and risk of cancer: which link? Metabolism. 2015;64(2):182–189. doi:10.1016/j.metabol.2014.10.008
192. Găman M, Epingeac ME, Găman A. The evaluation of oxidative stress and high-density lipoprotein cholesterol levels in diffuse large B-cell lymphoma. Revista Chimie. 2019;70(3):977–980. doi:10.37358/RC.19.3.7043
193. Brown JM, McIntosh MK. Conjugated linoleic acid in humans: regulation of adiposity and insulin sensitivity. J Nutr. 2003;133(10):3041–3046. doi:10.1093/jn/133.10.3041
194. Naugler WE, Karin M. The wolf in sheep’s clothing: the role of interleukin-6 in immunity, inflammation and cancer. Trends Mol Med. 2008;14(3):109–119. doi:10.1016/j.molmed.2007.12.007
195. Park J, Sarode VR, Euhus D, et al. Neuregulin 1-HER axis as a key mediator of hyperglycemic memory effects in breast cancer. Proc Natl Acad Sci U S A. 2012;109(51):21058–21063. doi:10.1073/pnas.1214400109
196. Wang ZV, Mu J, Schraw TD, et al. PANIC-ATTAC: a mouse model for inducible and reversible beta-cell ablation. Diabetes. 2008;57(8):2137–2148. doi:10.2337/db07-1631
197. Xie Y, Xu P, Liu K, et al. Hyperuricemia and gout are associated with cancer incidence and mortality: a meta-analysis based on cohort studies. J Cell Physiol. 2019;234(8):14364–14376. doi:10.1002/jcp.v234.8
198. Fini MA, Elias A, Johnson RJ, et al. Contribution of uric acid to cancer risk, recurrence, and mortality. Clin Transl Med. 2012;1(1):16. doi:10.1186/2001-1326-1-16
199. Battelli MG, Bortolotti M, Polito L, et al. Metabolic syndrome and cancer risk: the role of xanthine oxidoreductase. Redox Biol. 2019;21:101070. doi:10.1016/j.redox.2018.101070
200. Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol. 2010;72(1):219–246. doi:10.1146/annurev-physiol-021909-135846
201. Liao X, Sharma N, Kapadia F, et al. Kruppel-like factor 4 regulates macrophage polarization. J Clin Invest. 2011;121(7):2736–2749. doi:10.1172/JCI45444
202. Chawla A, Nguyen KD, Goh YP. Macrophage-mediated inflammation in metabolic disease. Nat Rev Immunol. 2011;11(11):738–749. doi:10.1038/nri3071
203. Toubal A, Clément K, Fan R, et al. SMRT-GPS2 corepressor pathway dysregulation coincides with obesity-linked adipocyte inflammation. J Clin Invest. 2013;123(1):362–379. doi:10.1172/JCI64052
204. Dalbeth N, Chen P, White M, et al. Impact of bariatric surgery on serum urate targets in people with morbid obesity and diabetes: a prospective longitudinal study. Ann Rheum Dis. 2014;73(5):797–802. doi:10.1136/annrheumdis-2013-203970
205. Heymsfield SB, Wadden TA, Longo DL. Mechanisms, pathophysiology, and management of obesity. N Engl J Med. 2017;376(3):254–266. doi:10.1056/NEJMra1514009
206. Velazquez A, Apovian CM. Updates on obesity pharmacotherapy. Ann N Y Acad Sci. 2018;1411(1):106–119. doi:10.1111/nyas.13542
207. Solas M, Milagro FI, Martínez-Urbistondo D, et al. Precision obesity treatments including pharmacogenetic and nutrigenetic approaches. Trends Pharmacol Sci. 2016;37(7):575–593. doi:10.1016/j.tips.2016.04.008
208. Kievit P, Halem H, Marks DL, et al. Chronic treatment with a melanocortin-4 receptor agonist causes weight loss, reduces insulin resistance, and improves cardiovascular function in diet-induced obese rhesus macaques. Diabetes. 2013;62(2):490–497. doi:10.2337/db12-0598
209. Fani L, Bak S, Delhanty P, et al. The melanocortin-4 receptor as target for obesity treatment: a systematic review of emerging pharmacological therapeutic options. Int J Obes (Lond). 2014;38(2):163–169. doi:10.1038/ijo.2013.80
210. Low MJ. Neuroendocrinology: new hormone treatment for obesity caused by POMC-deficiency. Nat Rev Endocrinol. 2016;12(11):627–628. doi:10.1038/nrendo.2016.156
211. Kuhnen P, Clément K, Wiegand S, et al. Proopiomelanocortin deficiency treated with a melanocortin-4 receptor agonist. N Engl J Med. 2016;375(3):240–246. doi:10.1056/NEJMoa1512693
212. Clement K, Biebermann H, Farooqi IS, et al. MC4R agonism promotes durable weight loss in patients with leptin receptor deficiency. Nat Med. 2018;24(5):551–555. doi:10.1038/s41591-018-0015-9
213. Christensen M, Vedtofte L, Holst JJ, et al. Glucose-dependent insulinotropic polypeptide: a bifunctional glucose-dependent regulator of glucagon and insulin secretion in humans. Diabetes. 2011;60(12):3103–3109. doi:10.2337/db11-0979
214. Norregaard PK, Deryabina MA, Tofteng Shelton P, et al. A novel GIP analogue, ZP4165, enhances glucagon-like peptide-1-induced body weight loss and improves glycaemic control in rodents. Diabetes Obes Metab. 2018;20(1):60–68. doi:10.1111/dom.2018.20.issue-1
215. Day JW, Ottaway N, Patterson JT, et al. A new glucagon and GLP-1 co-agonist eliminates obesity in rodents. Nat Chem Biol. 2009;5(10):749–757. doi:10.1038/nchembio.209
216. Pocai A, Carrington PE, Adams JR, et al. Glucagon-like peptide 1/glucagon receptor dual agonism reverses obesity in mice. Diabetes. 2009;58(10):2258–2266. doi:10.2337/db09-0278
217. Cegla J, Troke RC, Jones B, et al. Coinfusion of low-dose GLP-1 and glucagon in man results in a reduction in food intake. Diabetes. 2014;63(11):3711–3720. doi:10.2337/db14-0242
218. Tan TM, Field BCT, McCullough KA, et al. Coadministration of glucagon-like peptide-1 during glucagon infusion in humans results in increased energy expenditure and amelioration of hyperglycemia. Diabetes. 2013;62(4):1131–1138. doi:10.2337/db12-0797
219. Tschop MH, Finan B, Clemmensen C, et al. Unimolecular polypharmacy for treatment of diabetes and obesity. Cell Metab. 2016;24(1):51–62. doi:10.1016/j.cmet.2016.06.021
220. Finan B, Müller TD, Clemmensen C, et al. Reappraisal of GIP pharmacology for metabolic diseases. Trends Mol Med. 2016;22(5):359–376. doi:10.1016/j.molmed.2016.03.005
221. Finan B, Yang B, Ottaway N, et al. A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nat Med. 2015;21(1):27–36. doi:10.1038/nm.3761
222. Fosgerau K, Hoffmann T. Peptide therapeutics: current status and future directions. Drug Discov Today. 2015;20(1):122–128. doi:10.1016/j.drudis.2014.10.003
223. Giralt M, Gavalda-Navarro A, Villarroya F. Fibroblast growth factor-21, energy balance and obesity. Mol Cell Endocrinol. 2015;418(Pt 1):66–73. doi:10.1016/j.mce.2015.09.018
224. Fisher FM, Maratos-Flier E. Understanding the Physiology of FGF21. Annu Rev Physiol. 2016;78(1):223–241. doi:10.1146/annurev-physiol-021115-105339
225. Badman MK, Koester A, Flier JS, et al. Fibroblast growth factor 21-deficient mice demonstrate impaired adaptation to ketosis. Endocrinology. 2009;150(11):4931–4940. doi:10.1210/en.2009-0532
226. Cuevas-Ramos D, Aguilar-Salinas CA. Modulation of energy balance by fibroblast growth factor 21. Horm Mol Biol Clin Investig. 2016;30(1).
227. Habegger KM, Stemmer K, Cheng C, et al. Fibroblast growth factor 21 mediates specific glucagon actions. Diabetes. 2013;62(5):1453–1463. doi:10.2337/db12-1116
228. Ye X, Qi J, Ren G, et al. Long-lasting anti-diabetic efficacy of PEGylated FGF-21 and liraglutide in treatment of type 2 diabetic mice. Endocrine. 2015;49(3):683–692. doi:10.1007/s12020-014-0503-8
229. Weng Y, Chabot JR, Bernardo B, et al. Pharmacokinetics (PK), pharmacodynamics (PD) and integrated PK/PD modeling of a novel long acting FGF21 clinical candidate PF-05231023 in diet-induced obese and leptin-deficient obese mice. PLoS One. 2015;10(3):e0119104. doi:10.1371/journal.pone.0119104
230. Nielsen SM, Bartels EM, Henriksen M, et al. Weight loss for overweight and obese individuals with gout: a systematic review of longitudinal studies. Ann Rheum Dis. 2017;76(11):1870–1882. doi:10.1136/annrheumdis-2017-211472
231. Paul BJ, Anoopkumar K, Krishnan V. Asymptomatic hyperuricemia: is it time to intervene? Clin Rheumatol. 2017;36(12):2637–2644. doi:10.1007/s10067-017-3851-y
232. Shoji A, Yamanaka H, Kamatani N. A retrospective study of the relationship between serum urate level and recurrent attacks of gouty arthritis: evidence for reduction of recurrent gouty arthritis with antihyperuricemic therapy. Arthritis Rheum. 2004;51(3):321–325. doi:10.1002/art.20405
233. Robinson PC. Gout - An update of aetiology, genetics, co-morbidities and management. Maturitas. 2018;118:67–73. doi:10.1016/j.maturitas.2018.10.012
234. Pascart T, Richette P. Current and future therapies for gout. Expert Opin Pharmacother. 2017;18(12):1201–1211. doi:10.1080/14656566.2017.1351945
235. Soskind R, Abazia DT, Bridgeman MB. Updates on the treatment of gout, including a review of updated treatment guidelines and use of small molecule therapies for difficult-to-treat gout and gout flares. Expert Opin Pharmacother. 2017;18(11):1115–1125. doi:10.1080/14656566.2017.1349099
236. Heerspink HJ, Perkins BA, Fitchett DH, et al. Sodium glucose cotransporter 2 inhibitors in the treatment of diabetes mellitus: cardiovascular and kidney effects, potential mechanisms, and clinical applications. Circulation. 2016;134(10):752–772. doi:10.1161/CIRCULATIONAHA.116.021887
237. Pereira MJ, Eriksson JW. Emerging role of SGLT-2 inhibitors for the treatment of obesity. Drugs. 2019;79(3):219–230. doi:10.1007/s40265-019-1057-0
238. Borghi C, Palazzuoli A, Landolfo M, Cosentino E. Hyperuricemia: a novel old disorder-relationship and potential mechanisms in heart failure. Heart Fail Rev. 2019;25(1):43–51.
239. Bussler S, Penke M, Flemming G, et al. Novel insights in the metabolic syndrome in childhood and adolescence. Horm Res Paediatr. 2017;88(3–4):181–193. doi:10.1159/000479510
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.