Back to Journals » Drug Design, Development and Therapy » Volume 11
Controlled-release nanoencapsulating microcapsules to combat inflammatory diseases
Authors Baek JS, Yeo EW, Lee YH, Tan NS ![]() , Loo SCJ
, Loo SCJ
Received 26 January 2017
Accepted for publication 11 May 2017
Published 8 June 2017 Volume 2017:11 Pages 1707—1717
DOI https://doi.org/10.2147/DDDT.S133344
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Sukesh Voruganti
Jong-Suep Baek,1 Eng Wan Yeo,1 Yin Hao Lee,2 Nguan Soon Tan,2 Say Chye Joachim Loo1,3
1School of Materials Science and Engineering, Nanyang Technological University, Singapore; 2School of Biological Sciences, Nanyang Technological University, Singapore; 3Singapore Centre on Environmental Life Sciences Engineering (SCELSE), Nanyang Technological University, Singapore
Abstract: The World Health Organization (WHO) has reported that globally 235 million people suffer from chronic and other inflammatory diseases. The short half-lives of nonsteroidal anti-inflammatory drugs (NSAIDs) and their notoriety in causing gastrointestinal discomforts, warrants these drugs to be released in a controlled and sustained manner. Although polymeric particles have been widely used for drug delivery, there are few reports that showcase their ability in encapsulating and sustaining the release of NSAIDs. In this paper, polymeric nanoencapsulating microcapsules loaded with NSAIDs were fabricated using solid/water/oil/water emulsion solvent evaporation method. Two NSAIDs, ibuprofen and naproxen, were first pre-loaded into nanoparticles and then encapsulated into a larger hollow microcapsule that contained the third NSAID, celecoxib. A high encapsulation efficiency (%) of these NSAIDs was achieved and a sustained release (up to 30 days) of these drugs in phosphate-buffered saline was observed. Then, a gastrointestinal drug – cimetidine (CIM) – was co-loaded with the NSAIDs. This floating delivery system exhibited excellent buoyancy (~88% up to 24 h) in simulated gastric fluid. It also allowed a sequential release of the drugs, whereby an immediate release of CIM followed by NSAIDs was observed. Drug release of the NSAIDs observed Fickian diffusion mechanism, whereas CIM observed non-Fickian diffusion. Therefore, this delivery system is a promising platform to control the delivery of NSAIDs to combat inflammatory diseases, thereby protecting against possible gastrointestinal side effects that may arise from the overuse of NSAIDs.
Keywords: NSAIDs, multi-drug encapsulation, sequential release, floating oral drug delivery, injectable system, oral delivery systems, diffusion, emulsion, sustained release
Introduction
Inflammation is an innate immune response to noxious stimuli, damaged cells, irritants, and microorganisms. Inflammation, as a natural biological mechanism, aids in eliminating the cause of tissue injury and healing the damaged tissues. Nonsteroidal anti-inflammatory drugs (NSAIDs) are commonly prescribed to treat inflammation and the pain associated with it.1 NSAIDs act on the cyclooxygenase (COX) family of enzymes and inhibit the conversion of arachidonic acid to prostaglandins and thromboxanes.2,3 The COX-1 enzyme is expressed in most tissues and helps to regulate hemodynamics and maintain gut integrity. However, COX-2 is found in inflamed sites and mediates fever and pain.4,5 The World Health Organization (WHO) has reported that globally 235 million people suffer from chronic and other inflammatory diseases. Therefore, the global market for anti-inflammatory therapeutics is expected to grow rapidly at a compound annual growth rate (CAGR) of 5.9% and is estimated to worth USD 106.1 billion by 2020.6
Among the NSAIDs, ibuprofen (IBU; 2-(4-(2-Methylpropyl)phenyl)propanoic acid) is one of the most commonly prescribed and is a nonselective inhibitor of both COX-1 and COX-2,7 with a prominent analgesic and antipyretic role.8 It works by inhibiting cyclooxygenases and enzymes that are involved in the synthesis of prostaglandins that affects pain, inflammation, and fever.9 However, its short elimination half-life (~2 hours) and high degree of plasma protein binding (90%–99%)10 require it to be administered regularly.11 Naproxen (NAP; 2-(6-methoxynaphthalen-2-yl)propanoic acid) is another commonly prescribed NSAID used for the alleviation of moderate to severe pains, fever, and inflammation, including stiffness caused by osteoarthritis, rheumatoid arthritis, psoriatic arthritis, and ocular inflammations.12 It is a nonselective COX inhibitor and a potent inhibitor of prostaglandin synthesis.13 However, its low water solubility often translates to poor bioavailability. Celecoxib (CEL; 4-(5-(4-methylphenyl)-3-(trifluoromethyl)pyrazol-1-yl)benzenesulfonamide), another NSAID, is a selective COX-2 inhibitor that effectively modulates inflammation and pain. CEL is used for the treatment of pain, osteoarthritis, rheumatoid arthritis, primary dysmenorrhea, and ankylosing spondylitis.14 However, it also suffers from low bioavailability due to its large volume distribution (455±166 L), fast renal clearance (14.28 L/h), and high plasma protein binding (~97%).15 Currently, regular dosing of these drugs is required to reap the therapeutic benefits of noncontrolled-release NSAIDs. However, long-term usage of these NSAIDs is not recommended as they are known to cause adverse gastrointestinal side effects. These lead to a strong demand for controlled-release anti-inflammatory therapeutics that promise fewer side effects and better efficacy.
Drug delivery systems have become an important aspect of pharmaceutical reformulation, whereby off-patent drugs can enjoy an extension of their patentability through a newly formulated controlled-release version. Although conventional drug delivery systems encapsulate and deliver only a single drug,16,17 delivery systems that encapsulate multiple drug analogues or target drugs into specific tissues18 are conceivably more potent and effective. Such multi-drug delivery systems would be best suited for chronic diseases such as cancer, Parkinson’s disease, inflammatory diseases, and HIV, which require the simultaneous use of different drugs.19–21 The encapsulation of complementary drugs, with controlled release, would, therefore, allow for reduced drug dosages, thus reducing side effects while improving patient compliance and treatment efficacy.
The objective of this work was, therefore, to design a drug delivery system that allows for controlled and sustained release of NSAIDs. Here, we first report on the encapsulation of three different NSAIDs (ie, IBU, NAP, and CEL – as model drugs) into nanocarriers that are housed within a polymeric microcapsule (Scheme 1). The idea of first encapsulating drugs into polymeric nanocarriers is to avoid any potential drug–drug interactions. These polymers are the US Food and Drug Administration-approved, biocompatible, and biodegradable polymers – poly(L-lactide) (PLLA) poly(caprolactone) (PCL), and poly(D,L-lactide-co-glycolide) (PLGA).22,23 Because NSAIDs are known to cause gastrointestinal problems, antacids are usually co-prescribed with these.3,24 Cimetidine (CIM), an antacid, is a histamine H2-receptor antagonist and is widely used to inhibit gastric juice secretion. Next, a modified version of the microcapsules that co-encapsulate NSAIDs (ie, IBU and NAP) and a histamine H2-receptor antagonist (ie, CIM) was developed, and its release capabilities were investigated. In essence, the development of such controlled-release delivery systems, with or without an antacid, aims at resolving the issues faced with the chronic use of NSAIDs.
 | Scheme 1 Schematic illustration of the encapsulation of NSAIDs into controlled-release microcapsules and the corresponding release of these drugs from the two drug delivery systems (PLGA shell vs PLGA/PCL shell). |
Materials and methods
Materials
PLLA (IV: 2.4) and PLGA (50:50) (IV: 1.18) were purchased from Corbion (the Netherlands). HCl solution (37% v/v Fuming), PCL (MW 10 kDa), IBU, NAP, CEL, CIM, Tween 20, Tween 80, Span 80, and polyvinyl alcohol (PVA) (MW 30–70 kDa) were purchased from Sigma-Aldrich (Steinheim, Switzerland). Extra virgin grade olive oil was purchased from Pietro Coricelli. For preparation of simulated gastric fluid (SGF), HCl solution (0.1 M HCl) was added to 0.02% (w/v) of Tween 20. The simulated intestinal fluid (SIF) consisted of phosphate-buffered saline (pH 6.8) and 0.02% (w/v) Tween 80.
Methods
PLGA and PLLA nanoparticles synthesis
Dye or NSAIDs-loaded PLGA and PLLA nanoparticles were fabricated by the solvent evaporation method. Dye (0.1% w/v) or NSAID (8% w/v) was added into PLGA or PLLA/dichloromethane (DCM) (0.2 g in 1 mL) solution. The resultant solution was mixed with 2% (w/v) PVA solution (2 mL) and ultrasonicated for a minute with ice. The primary emulsion was then transferred into distilled water (5 mL) under stirring at 400 rpm for 4 h. Next, the nanoparticle solution was washed with water followed by centrifugation (20,000 rpm, 10 min).
Preparing microcapsules encapsulating PLGA and PLLA nanoparticles
PLGA nanoparticles (0.1 g) and PLLA nanoparticles (0.1 g) were dispersed in 0.2% (w/v) Span 80 solution (1 mL) under ultrasonication. The aqueous suspension was added dropwise into PLGA or PLGA/PCL (1:1, w/w) DCM solution (5% w/v), containing CEL or CIM, under stirring (1,000 rpm, 5 min). Next, the solution was added to 0.5% (w/v) PVA solution (250 mL) and emulsified at 400 rpm using an overhead stirrer for 4 h. The solution was washed using distilled water and freeze-dried. For the microcapsules containing CIM, olive oil (10 μL) was also added to the 0.2% (w/v) Span 80 solution (1 mL) to achieve floatable microcapsules.
Encapsulation efficiency
To measure encapsulation efficiency, the microcapsules (10 mg) were solubilized in DCM (1 mL). Subsequently, deionized water (10 mL) was added to the DCM solution. The hydrophilic CIM partitions into distilled water and the supernatant was injected into high-performance liquid chromatography (HPLC) (Agilent 1100; Agilent Technologies, Santa Clara, CA, USA) with an autosampler and UV detector. The mobile phase was a mixture of acetonitrile and 0.1 M sodium acetate buffer (pH 5.0) (20:80 v/v) with the wavelength set at 230 nm. For hydrophobic IBU, NAP, and CEL, the drugs were redissolved in n-hexane to precipitate the polymer. The filtered supernatant was injected into HPLC with a mobile phase consisting of 1.8 mM phosphoric acid (pH 2.8) and acetonitrile (50:50, v/v). Encapsulation efficiency (EE) was calculated as follows:
|
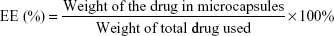
Particle characterization with confocal laser microscopy and field-emission scanning electron microscopy
To determine dye distribution within the microcapsules, a confocal laser microscope (CLMS, LSM710) was used. Few droplets of the aqueous microcapsule suspension were placed onto a glass slide and the slide was sealed with a glass cover slip. The images were taken with a 63×/1.40 oil objective lens and the AxioCam MRm camera (Carl Zeiss Microscopy GmbH, Oberkochen, Germany). Further analysis of the photos was carried out using ZEN 2012 software (Carl Zeiss, Microscopy GmbH). To prepare the microcapsules for field-emission scanning electron microscopy (FESEM), microcapsules were placed on carbon tape-attached metal stub and cut using a blade, before coating with platinum.
Drug-release and polymer degradation studies
For drug-release study, microcapsules (10 mg) were added to SGF (20 mL) in glass vials under stirring at 37°C (200 rpm). Release medium (10 mL) was withdrawn at specific time points and replenished with fresh medium (10 mL). The amount of released drug was analyzed using HPLC, as described earlier. For polymer degradation study, microcapsules (50 mg) were dispersed in SGF (20 mL) and the solution was incubated under stirring and maintained at 37°C. Microcapsules were removed at different time points. Size-exclusion chromatography (SEC) was used to measure the average molecular weight of PLGA with a reflective index detector. The flow rate was 1 mL/min. Since only PLGA is soluble in tetrahydrofuran, the polymers were separated using previously reported method.23 Subsequently, the polymers were dissolved individually into chloroform for SEC characterization.
Buoyancy and kinetics release profile
To determine the buoyancy of the microcapsules, the microcapsules were introduced to SGF (30 mL) under stirring for 24 h (200 rpm, 37°C). At different pre-determined time points, the numbers of microcapsules that remained afloat were visually counted. The percentage of floating microcapsules after 24 h was calculated based on the number of floating microcapsules over the total number of microcapsules initially used for this study. To analyze the rate of release, the release data were fitted to zero-order rate, first-order rate, Higuchi rate, and Korsmeyer–Peppas equations.25–27
For statistical analysis, the Student’s t-test was used, and P<0.05 was taken as statistically significant. Experimental data were obtained from three sample groups (n=3) and presented as mean ± SD.
Results and discussion
NSAIDs-loaded microcapsules
NSAIDs co-loaded microcapsules were fabricated using the solid/water/oil/water (S/W1/O/W2) emulsion solvent evaporation method. First, NSAIDs were loaded into polymeric nanoparticles and then encapsulated into larger hollow microcapsules (Scheme 1). This design allows for different drugs to be housed within different “compartments” (ie, nanoparticles) of the microcapsule thereby avoiding drug–drug interactions. For example, two different drugs can be loaded into PLGA and/or PLLA nanoparticles before encapsulating them within the microcapsule. The microcapsule, therefore, acts as a drug reservoir that controls the release kinetics of the drugs. As a proof-of-concept, this was demonstrated by first loading PLGA and PLLA nanoparticles with different dyes (ie, Nile red and fluorescein isothiocyanate (FITC), and then encapsulating them into hollow PLGA microcapsules containing a third dye (ie, Coumarin 343 [COU343]). These dyes were carefully chosen as they closely mimic the properties of the NSAIDs – Nile red, FITC, and COU343 are all highly hydrophobic (log P: 3.5, 5.03, and 3.12, respectively) and have similar molecular weights (MW: 318.4, 389.4, and 285.2 Da). Next, confocal laser scanning microscopy (CLSM) was used to determine the location of the dyes within the microcapsules (Figure 1).
 | Figure 1 (A) CLSM images of PLGA microcapsule. Z-stack comprises four confocal sections that represent Nile Red (red) encapsulated in PLGA nanoparticles, FITC (green) encapsulated in PLLA NPs, COU343 (yellow) loaded in PLGA MC shell, and merged fluorescence measured from center to top of the multiple-dye-loaded PLGA microcapsule (scale bars represent 50 μm). SEM images of (B) cross-sectional view and (C) merged fluorescence of Z-stacks (scale bar represents 50 μm). |
The dye-loaded microcapsules (Figure 1) showed simultaneous emission spectra of the dyes (COU343 – 463 nm, FITC – 530 nm, and Nile red – 590 nm), thus indicating the successful encapsulation of dye-loaded nanoparticles into the microcapsule. Merging with the optical images showed fluorescence from all the three dyes to be within the inner wall of the PLGA shell (Figure 1C) with no fluorescence observed on the outer surface of the microcapsule. This proves the feasibility of the fabrication technique in encapsulating dye-loaded nanoparticles.
Next, NSAIDs were loaded in place of the dyes. IBU and NAP were first encapsulated into PLGA and PLLA nanoparticles, respectively, whereas CEL was loaded directly into the shell of the microcapsule, that is, PLGA or PLGA/PCL. Particle sizes of these PLGA and PLLA nanoparticles measured by dynamic light scattering were 123.5±9.5 nm and 153.5±12.7 nm, respectively (Figure S1). Loading efficiencies of these drugs were found to be above 90%, as these hydrophobic molecules interact strongly with the hydrophobic polymers.28 Next, these drug-loaded nanoparticles were encapsulated within the microcapsule. In this study, two polymer compositions were chosen as the microcapsule shell material. This allows for an understanding of how a single polymer (PLGA) or a polymer blend (PLGA/PCL), as the microcapsule shell, would influence drug-release rates. To confirm the successful encapsulation of the nanoparticles into the hollow microcapsules, FESEM was used (Figure 2).
 | Figure 2 FESEM images of (A) IBU-loaded PLGA nanoparticles, (B) NAP-loaded PLLA nanoparticles, (C) cross-sectioned CEL-loaded PLGA microcapsules containing PLGA and PLLA nanoparticles, and (D) CEL-loaded PLGA/PCL microcapsule containing PLGA and PLLA nanoparticles. Subsequent magnifications show PLGA and PLLA nanoparticles within the hollow cavity of the microcapsule. |
Figure 2A and B show the SEM images of spherical PLGA and PLLA nanoparticles. From the SEM, these nanoparticles were confirmed to be encapsulated within the hollow microcapsules (Figure 2C and D). Similar to the dye-loaded microcapsules, these excised drug-loaded microcapsules revealed nanoparticles on the interior wall of the microcapsules. Here, Span 80 plays an important role in this nanoencapsulation in the initial water-in-oil emulsion (W1/O) system.29,30 First of all, PLGA microcapsules without Span 80 did not form any hollow cavity (Figure S2). Span 80 has a hydrophilic–lipophilic balance (HLB) of 4.3, which was suitable for stabilizing W/O emulsions. It allowed for the formation of small aqueous droplets (W1) that contain the PLGA and PLLA nanoparticles, thus protecting these nanoparticles from dissolving in the organic solvent (DCM) in the external oil (O) phase. At the end, a hollow microcapsule containing drug-loaded nanoparticles was obtained. Comparing different shell materials, that is, PLGA and PLGA/PCL microcapsules, the latter were observed to have nonuniform layer thickness (Figure 2D). This may be due to the phase separation of PCL and PLGA. As such, the blend of rubbery PCL with PLGA formed a shell that was less dense and of nonuniform thickness.31
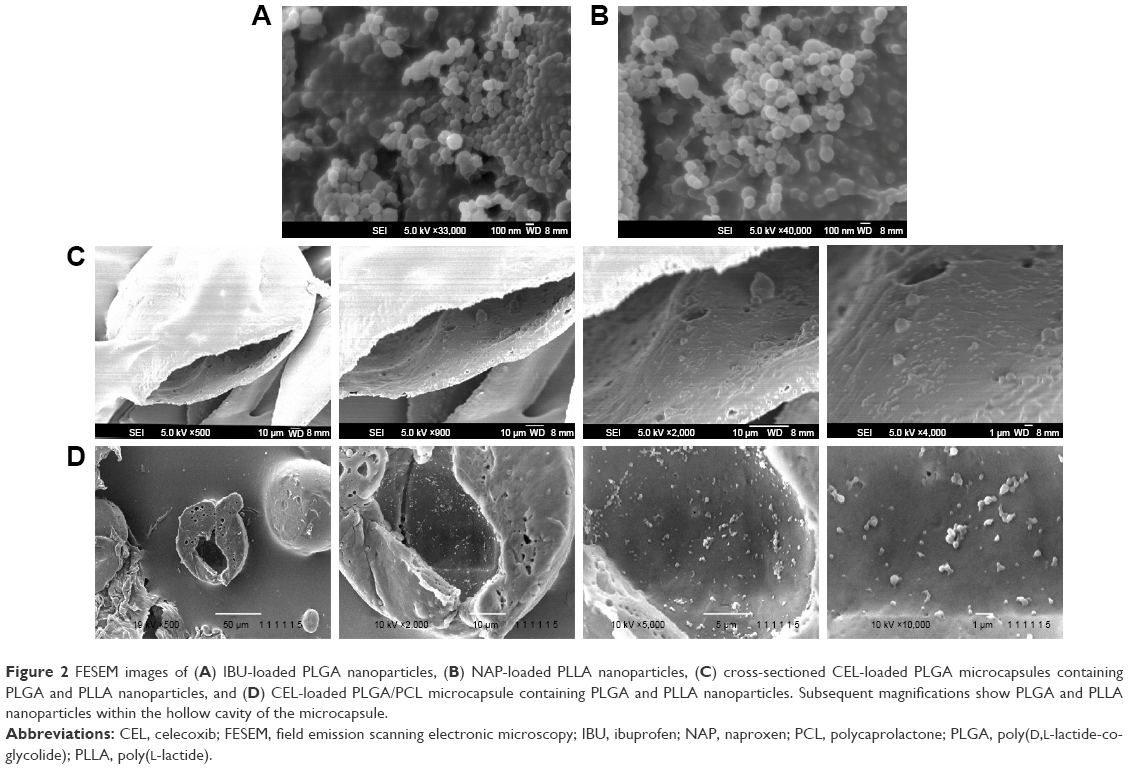
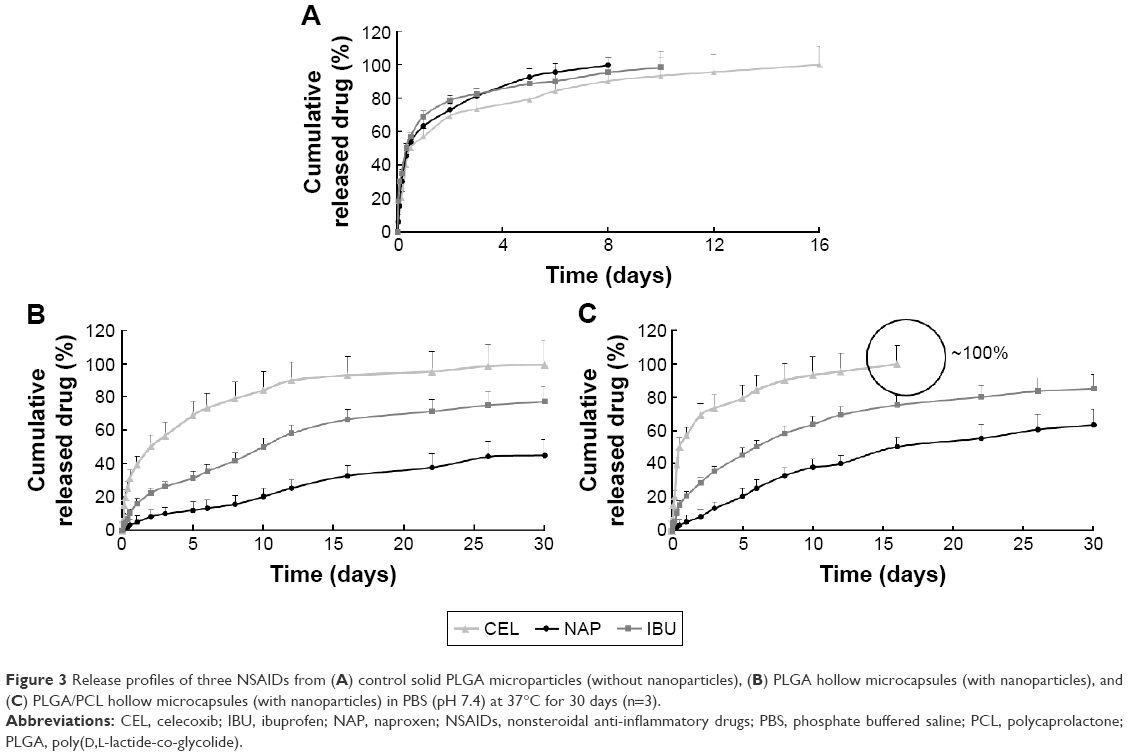
Next, drug-release kinetics of the delivery systems were investigated. Figure 3 plots the release profiles of solid microparticles and hollow microcapsules that contain all three NSAIDs. Control solid PLGA microparticles, which do not have any nanoparticles, gave similar release profiles for all three NSAIDs for up to 16 days. On the other hand, a longer sustained release was observed from both PLGA microcapsules and PLGA/PCL microcapsules (both containing nanoparticles). Therefore, encapsulating drugs first into nanoparticles allowed this sustained releasing capability. Drug-release rates were also influenced by whether the drugs were loaded into PLGA or PLLA nanoparticles. Release results show that NAP released much slower than the other drugs because of its encapsulation into PLLA nanoparticles – a more hydrophobic polymer. Comparing two different shell compositions (ie, PLGA vs PLGA/PCL), the use of PLGA/PCL as the microcapsule shell material provided faster release rates because of the associated higher water uptake of PCL that translates to a faster degradation of the polymer shell (Figure 4A and B). The faster degradation of PCL was also evident from the SEM images of the microcapsules, at 12 and 22 days of degradation, as observed from the more porous shell of PLGA/PCL (Figure 4C). A more porous PLGA/PCL microcapsule would also accelerate drug diffusion, thus leading to a more rapid drug-release rate (Figure 4C).
 | Figure 3 Release profiles of three NSAIDs from (A) control solid PLGA microparticles (without nanoparticles), (B) PLGA hollow microcapsules (with nanoparticles), and (C) PLGA/PCL hollow microcapsules (with nanoparticles) in PBS (pH 7.4) at 37°C for 30 days (n=3). |
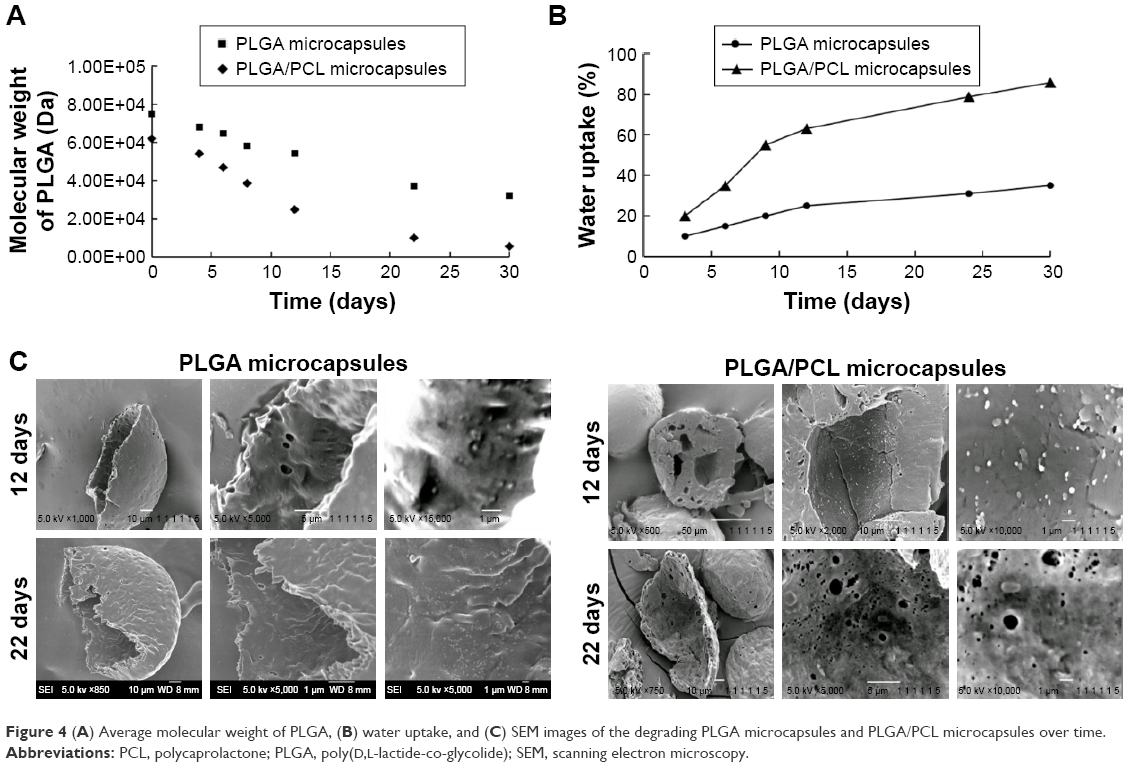 | Figure 4 (A) Average molecular weight of PLGA, (B) water uptake, and (C) SEM images of the degrading PLGA microcapsules and PLGA/PCL microcapsules over time. |
The oral route is the most convenient and economical route for drug administration. In view of the possible gastrointestinal problems with the use of NSAIDs, antacids are usually prescribed together with NSAIDs.32 Notably, IBU and NAP, both nonselective COX inhibitors, can cause gastric side effects. To address these issues, we designed the delivery system to provide a rapid release of an antacid (ie, CIM), followed by a controlled and sustained release of NSAIDs (ie, IBU and NAP). Here, IBU and NAP were encapsulated within PLGA and PLLA nanoparticles, respectively, whereas CIM (in the place of CEL) was encapsulated in the shell of either PLGA or PLGA/PCL microcapsules. Loading efficiency of IBN and NAP in their respective nanoparticles was again >90%. However, CIM being hydrophilic in nature (log P −0.11) had a lower encapsulation efficiency (~47%) in the shell of the microcapsules.
To increase oral bioavailability of drugs, these microcapsules should possess good buoyancy property, which allows for a prolonged gastric residence time (GRT) and for drugs to be released in the upper gastrointestinal tract.32,33 For example, floating lipid beads showed better oral bioavailability (162.7%) of Cinnarizine as compared to non-floatable Stugeron®.34 In addition, Ofloxacin-loaded floating hollow microcapsules exhibited a higher oral bioavailability (124.1%) as compared to commercial Ofloxacin tablets.35 The buoyancy of the PLGA microcapsules and PLGA/PCL microcapsules is plotted in Figure 5. The results show that PLGA microcapsules possessed better floating capabilities compared to PLGA/PCL microcapsules. The excellent buoyancy of the former was due to its low density, attributed to the oil-filled hollow cavity, along with the hydrophobic nature of PLGA that restricted water influx into the microcapsules. PLGA microcapsules exhibited 88% of the microcapsules floating at 24 h, whereas PLGA/PCL microcapsules had only 42% of microcapsules floating at 24 h. For this reason, the PLGA microcapsules were chosen for further studies.
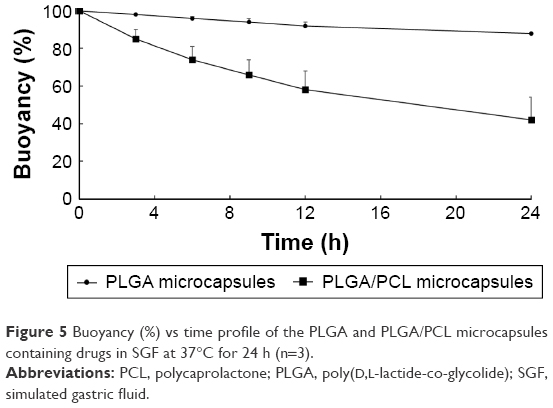 | Figure 5 Buoyancy (%) vs time profile of the PLGA and PLGA/PCL microcapsules containing drugs in SGF at 37°C for 24 h (n=3). |
As floating microspheres had been reported to achieve a prolonged GRT (5 h) in humans,36,37 the release study was conducted under the same conditions that would mimic drug dissolution in the upper gastrointestinal tract (in SGF for 5 h), followed by release in the intestinal tract (in SIF for remaining 19 h).38 Hence, the release studies were conducted in SGF (5 h) and SIF (24 h). Figure 6 plots the release profiles of the three drugs from the PLGA microcapsules. Since gastric discomfort is the main side effect of NSAIDs,39 the release of over 75% of CIM in the stomach within 5–7 h to combat this would be advantageous. As shown in Figure 6, the microcapsules exhibited sequential release of the drugs (first CIM, followed by NSAIDs) in SGF and SIF over 24 h. Notably, the release profile of the microcapsules indicated that small amounts of NSAIDs would be released into the stomach according to the gastric residence period, and the rest would be released into the intestine.40 CIM release was observed to be the fastest, while the encapsulation of two NSAIDs within PLGA and PLLA nanoparticles achieved a delayed and more sustained release profile.
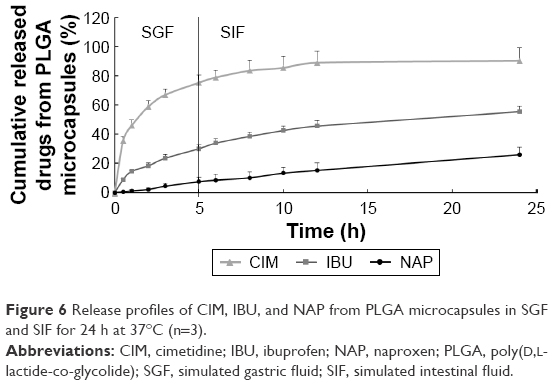 | Figure 6 Release profiles of CIM, IBU, and NAP from PLGA microcapsules in SGF and SIF for 24 h at 37°C (n=3). |
This formulation could now achieve sequential release of two NSAIDs and an antacid for oral delivery. This delivery system could modulate release kinetics of drugs regardless of their hydrophobic nature. To explain the mechanism of release profiles of these drugs from the formulations, the release results were fitted to the following models: Higuchi’s equation and Korsmeyer–Peppas equation.25–27 Table 1 summarizes the r2 coefficient values of all the three drugs and their respective rate constants from these models.
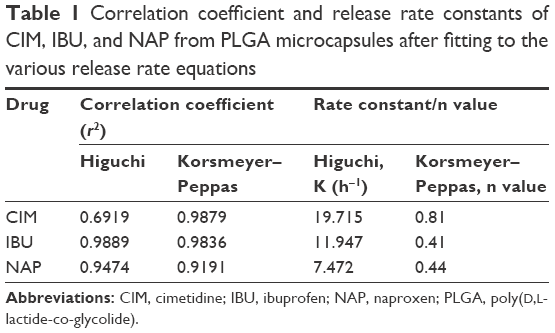 | Table 1 Correlation coefficient and release rate constants of CIM, IBU, and NAP from PLGA microcapsules after fitting to the various release rate equations |
Higuchi’s equation fits well for IBU and NAP, indicating the drug-release mechanism was diffusion controlled. In addition, the n values of Korsmeyer–Peppas for IBU and NAP were <0.5, indicating the Fickian diffusion mechanism. The release of these two drugs from nanoparticles through the microcapsule likely mimics that of a drug-loaded matrix. However, the Korsmeyer–Peppas equation predicted an n value of 0.81 for hydrophilic CIM that is housed within the PLGA shell of these microcapsules. This n value indicates a non-Fickian diffusion that refers to combination of both diffusion and erosion controlled rate release. It was reported that the decrease in average molecular weight of PLGA microparticles followed first-order degradation kinetics for both acidic and basic conditions. Specifically, at acidic pH (ie, SGF), faster degradation and release of drug would occur due to the crystallization of oligomeric degradation products at acidic pH.41 Hence, CIM can be released from the microcapsule shell through both diffusion and extensive degradation of the capsule shell (Figure 4). This oral formulation for controlled and sustained release of an antacid with NSAIDs would provide benefits of the latter while protecting the stomach from gastrointestinal side effects.
The usage of complementary drugs for the treatment of diseases is now gaining traction in the pharmaceutical industry.42,43 For instance, for Parkinson’s disease, patients would require administration of three drugs to manage this neurological disease.18 At the same time, there have been recent advances in complementary therapy of drugs with nutraceuticals in combating gastrointestinal illnesses.44–46 Complementary delivery, therefore, synergizes drugs of different mechanisms of action by co-encapsulating them into delivery systems. This study, therefore, proves the ability of delivering complementary drugs through the careful design of an oral delivery system. Although this study provides knowledge on the feasibility of delivering drugs, that is, an antacid with NSAIDs, from a single delivery system, its limitation lies in the need to further evaluate its effectiveness through more realistic animal models. In addition, large-scale manufacturing of the microcapsules has not been explored, although this simple, economical, and single-step fabrication process may be an attractive approach. Therefore, part of the future plan for this delivery system is to scale up this fabrication process and to evaluate its efficacy through relevant in vivo tests.
Conclusion
In this article, nanoencapsulated microcapsules were designed to deliver NSAIDs. The drug-release results showed that a more sustained release of IBU and NAP were observed when they were pre-loaded into PLGA and PLLA nanoparticles, before encapsulating them into larger hollow PLGA and PLGA/PCL microcapsules. These microcapsules exhibited sustained release of NSAIDs in phosphate-buffered saline beyond 30 days. Next, CIM (an antacid) was co-loaded with the NSAIDs, and a sequential release of CIM followed by NSAIDs in SGF was observed. CIM, which was loaded into the shell of the microcapsules, had a more rapid and immediate release, followed by a delayed and sustained release of the NSAIDs. This floating delivery system could, therefore, be a promising platform to control the delivery of NSAIDs to combat inflammatory diseases while protecting against possible gastrointestinal side effects that may arise from the overuse of NSAIDs.
Acknowledgments
This work was made possible through the financial support of the School of Materials Science and Engineering (M020070110), the Nanyang Technological University-National Healthcare Group (ARG/14012), the Ministry of Education (RG11/16), and the Singapore Centre on Environmental Life Sciences Engineering (M4330001.C70.703012).
Disclosure
The authors report no conflicts of interest in this work.
References
Voilley N, de Weille J, Mamet J, Lazdunsk M. Nonsteroid anti-inflammatory drugs inhibit both the activity and the inflammation-induced expression of acid-sensing ion channels in nociceptors. J Neurosci. 2001;21(20):8026–8033. | ||
Smith WL, DeWitt DK, Garavito RN. Cyclooxygenases: structural, cellular, and molecular biology. Annu Rev Biochem. 2000;69:145–182. | ||
Lanas A, Boers M, Nuevo J. Gastrointestinal events in at-risk patients starting non-steroidal anti-inflammatory drugs (NSAIDs) for rheumatic diseases: the EVIDENCE study of European routine practice. Ann Rheum Dis. 2015;74(4):675–681. | ||
Dubois RN, Abramson SB, Crofford L, et al. Cyclooxygenase in biology and disease. FASEB J. 1988;12(12):1063–1073. | ||
Seibert K, Masferrer J, Zhang Y, et al. Mediation of inflammation by cyclooxygenase-2. Agents Actions Suppl. 1995;46:41–50. | ||
Koutroubakis IE, Ramos-Rivers C, Regueiro M, et al. Five-year period prevalence and characteristics of anemia in a large US inflammatory bowel disease cohort. J Clin Gastroenterol. 2015;50(8):638–643. | ||
Chavez ML, DeKorte CJ. Valdecoxib: a review. Clin Ther. 2003;25(3):817–851. | ||
Bushra R, Nousheen A. An overview of clinical pharmacology of Ibuprofen. Oman Med J. 2010;25(3):155–1661. | ||
Wahbi AA, Hassan E, Hamdy D, Khamis E, Baray M. Spectrophotometric methods for the determination of Ibuprofen in tablets. Pak J Pharm Sci. 2005;18(4):1–6. | ||
Antal EJ, Wright CE 3rd, Brown BL, Albert KS, Aman LC, Levin NW. The influence of hemodialysis on the pharmacokinetics of ibuprofen and its major metabolites. J Clin Pharmacol. 1986;26(3):184–190. | ||
Kantor TG. Ibuprofen. Ann Intern Med. 1979;91(6):877–882. | ||
Goldstein JL, Hochberg MC, Fort JG, Zhang Y, Hwang C, Sostek M. Clinical trial: the incidence of NSAID-associated endoscopic gastric ulcers in patients treated with PN 400 (naproxen plus esomeprazole magnesium) vs. enteric-coated naproxen alone. Aliment Pharmacol Ther. 2010;32(3):401–413. | ||
Dhir A, Padi SS, Naidu PS, Kulkarni SK. Protective effect of naproxen (non-selective COX-inhibitor) or rofecoxib (selective COX-2 inhibitor) on immobilization stress-induced behavioral and biochemical alterations in mice. Eur J Pharmacol. 2006;535(1–3):192–198. | ||
Davies NM, McLachlan AJ, Day RO, Williams KM. Clinical pharmacokinetics and pharmacodynamics of celecoxib: a selective cyclo-oxygenase-2 inhibitor. Clin Pharmacokinet. 2000;38(3):225–242. | ||
Paulson SK, Kaprak TA, Gresk CJ, et al. Plasma protein binding of celecoxib in mice, rat, rabbit, dog and human. Biopharm Drug Dispos. 2009;20(6):293–299. | ||
Kankala RK, Kuthati Y, Sie HW, et al. Multi-laminated metal hydroxide nanocontainers for oral-specific delivery for bioavailability improvement and treatment of inflammatory paw edema in mice. J Colloid Interface Sci. 2015;458:217–228. | ||
Joseph KA and Yan Z. Polymeric Nanoparticle Receptors as Synthetic Antibodies for Nonsteroidal Anti-Inflammatory Drugs (NSAIDs). ACS Biomater Sci Eng. 2015:1;425–43016. | ||
Baek JS, Cho CW. A multifunctional lipid nanoparticle for co-delivery of paclitaxel and curcumin for targeted delivery and enhanced cytotoxicity in multidrug resistant breast cancer cells. Oncotarget. 2017;8(18):30369–30382. | ||
Baek JS, Cho CW. Controlled release and reversal of multidrug resistance by co-encapsulation of paclitaxel and verapamil in solid lipid nanoparticles. Int J Pharm. 2015;478(2):617–624. | ||
Baek JS, Choo CC, Qian C, Tan NS, Shen Z, Loo SC. Multi-drug-loaded microcapsules with controlled release for management of Parkinson’s disease. Small. 2016;12(27):3712–3722. | ||
Coradini K, Friedrich RB, Fonseca FN, et al. A novel approach to arthritis treatment based on resveratrol and curcumin co-encapsulated in lipid-core nanocapsules: In vivo studies. Eur J Pharm Sci. 2015;78:163–170. | ||
Lee WL, Guo WM, Ho VH, et al. Delivery of doxorubicin and paclitaxel from double-layered microparticles: The effects of layer thickness and dual-drug vs. single-drug loading. Acta Biomater. 2015;27:53–65. | ||
Lee WL, Tan JW, Tan CN, Loo SC. Modulating drug release from gastric-floating microcapsules through spray-coating layers. PLos One. 2014;9(12):e114284. | ||
Lim AW, Löbmann K, Grohganz H, Rades T, Chieng N. Investigation of physical properties and stability of indomethacin-cimetidine and naproxen-cimetidine co-amorphous systems prepared by quench cooling, coprecipitation and ball milling. J Pharm Pharmacol. 2016;68(1):36–45. | ||
Cooper J, Gunn C. Tutorial Pharmacy. 6th ed. New Delhi, India: CBS Publishers and Distributors; 1986:225. | ||
Hadjiioannou TP, Christian GD, Koupparis MA. Quantitative Calculations in Pharmaceutical Practice and Research. New York, NY: VCH Publishers Inc; 1993:345–348. | ||
Higuchi T. Mechanism of sustained-action medication. Theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J Pharm Sci. 1963;52:1145–1149. | ||
Satoh T, Higuchi Y, Kawakami S, Hashida M, Kagechika H, Shudo K, et al. Encapsulation of the synthetic retinoids Am80 and LE540 into polymeric micelles and the retinoids’ release control. J Control Release. 2009;136(3):187–195. | ||
Dinarvand R, Mahmoodi S, Farboud E, Salehi M, Atyabi F. Preparation of gelatin microspheres containing lactic acid – effect of cross-linking on drug release. Acta Pharm. 2005;55(1):57–67. | ||
Lee YS, Johnson PJ, Robbins PT, Bridson RH. Production of nanoparticles-in-microparticles by a double emulsion method: a comprehensive study. Eur J Pharm Biopharm. 2013;83(2):168–173. | ||
Lee WL, Loei C, Widjaja E, Loo SC. Altering the drug release profiles of double-layered ternary-phase microparticles. J Control Release. 2011;151(3):229–238. | ||
Cong HH, Khaziakhmetova VN, Zigashina LE. Rat paw oedema modeling and NSAIDs: Timing of effects. Int J Risk Saf Med. 2015;27(Suppl 1):S76–S77. | ||
Allesø M, Chieng N, Rehder S, Rantanen J, Rades T, Aaltonen J. Enhanced dissolution rate and synchronized release of drugs in binary systems through formulation: Amorphous naproxen-cimetidine mixtures prepared by mechanical activation. J Control Release. 2009;136(1):45–53. | ||
Abouelatta SM, Aboelwafa AA, Khalil RM, ElGazayerly ON. Floating lipid beads for the improvement of bioavailability of poorly soluble basic drugs: in-vitro optimization and in-vivo performance in humans. Eur J Pharm Biopharm. 2015;89:82–92. | ||
Zhang C, Tang J, Liu D, Li X, Cheng L, Tang X. Design and evaluation of an innovative floating and bioadhesive multiparticulate drug delivery system based on hollow structure. Int J Pharm. 2016;503(1–2):41–55. | ||
Kortejarvi H, Urtti A, Yliperttula M. Pharmacokinetic simulation of biowaiver criteria: the effects of gastric emptying, dissolution, absorption and elimination rates. Eur J Pharm Sci. 2007;30(2):155–166. | ||
Joseph NJ, Lakshmi S, Jayakrishnan A. A floating-type oral dosage form for piroxicam based on hollow polycarbonate microspheres: in vitro and in vivo evaluation in rabbits. J Control Release. 2002;79(1–3):71–79. | ||
Sato Y, Kawashima Y, Takeuchi H, Yamamoto H, Fujibayashi Y. Pharmacoscintigraphic evaluation of riboflavin-containing microballoons for a floating controlled drug delivery system in healthy humans. J Control Release. 2004;98(1):75–85. | ||
Gabriel SE, Jaakkimainen L, Bombardier C. Risk for serious gastrointestinal complications related to use of nonsteroidal anti-inflammatory drugs. A meta-analysis. Ann Intern Med. 1991;115(10):787–796. | ||
Sato Y, Kawashima Y, Takeuchi H, Yamamoto H. In vitro evaluation of floating and drug releasing behaviors of hollow microspheres (microballoons) prepared by the emulsion solvent diffusion method. Eur J Pharm Biopharm. 2004;57(2):235–243. | ||
Zolnik BS, Burgess DJ. Effect of acidic pH on PLGA microsphere degradation and release. J Control Release. 2007;122(3):338–344. | ||
Yadav V, Shinto L, Bourdette D. Complementary and alternative medicine for the treatment of multiple sclerosis. Expert Rev Clin Immunol. 2010;6(3):381–395. | ||
Taylor D, editor. The Pharmaceutical Industry and the Future of Drug Development. In: Pharmaceuticals in the Environment. Cambridge: Royal Society of Chemistry; 2015:1–33. | ||
Yang N, Sampathkumar K, Loo SC. Recent advances in complementary and replacement therapy with nutraceuticals in combating gastrointestinal illnesses. Clin Nutr. 2016. Epub August 30, 2016. | ||
Byrav DSP, Medhi B, Prakash A, Chakrabarti A, Vaiphei K, Khanduja KL. Comparative evaluation of different doses of PPAR-γ agonist alone and in combination with sulfasalazine in experimentally induced inflammatory bowel disease in rats. Pharmacol Rep. 2013;65(4):951–959. | ||
Rossi G, Pengo G, Caldin M, et al. Comparison of microbiological, histological, and immunomodulatory parameters in response to treatment with either combination therapy with prednisone and metronidazole or probiotic VSL#3 strains in dogs with idiopathic inflammatory bowel disease. PLoS One. 2014;9(4):e94699. |
Supplementary materials
 | Figure S1 DLS results of (A) PLGA nanoparticles and (B) PLLA nanoparticles. |
 | Figure S2 Cross-sectioned PLGA microcapsules without Span 80 loaded PLGA and PLLA nanoparticles. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.