Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Controllable drug uptake and nongenomic response through estrogen-anchored cyclodextrin drug complex
Authors Yin J, Shumyak SP, Burgess C, Zhou Z, He Z, Zhang X, Pan S, Yang T, Duan W, Qiu J, Zhou S
Received 5 February 2015
Accepted for publication 18 March 2015
Published 27 July 2015 Volume 2015:10(1) Pages 4717—4730
DOI https://doi.org/10.2147/IJN.S82255
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Thomas Webster
Juan-Juan Yin,1,2 Stepan P Shumyak,2 Christopher Burgess,2 Zhi-Wei Zhou,2 Zhi-Xu He,3 Xue-Ji Zhang,4 Shu-Ting Pan,2,5 Tian-Xin Yang,6 Wei Duan,7 Jia-Xuan Qiu,5 Shu-Feng Zhou2
1Xiaolan People’s Hospital, Southern Medical University, Zhongshan, Guangdong, People’s Republic of China; 2Department of Pharmaceutical Sciences, College of Pharmacy, University of South Florida, Tampa, FL, USA; 3Guizhou Provincial Key Laboratory for Regenerative Medicine, Stem Cell and Tissue Engineering Research Center and Sino-US Joint Laboratory for Medical Sciences, Guizhou Medical University, Guiyang, Guizhou, 4Research Center for Bioengineering and Sensing Technology, University of Science and Technology Beijing, Beijing, 5Department of Oral and Maxillofacial Surgery, the First Affiliated Hospital of Nanchang University, Nanchang, Jiangxi, People’s Republic of China; 6Department of Internal Medicine, University of Utah and Salt Lake Veterans Affairs Medical Center, Salt Lake City, UT, USA; 7School of Medicine, Deakin University, Waurn Ponds, VIC, Australia
Abstract: Breast cancer is a leading killer of women worldwide. Cyclodextrin-based estrogen receptor-targeting drug-delivery systems represent a promising direction in cancer therapy but have rarely been investigated. To seek new targeting therapies for membrane estrogen receptor-positive breast cancer, an estrogen-anchored cyclodextrin encapsulating a doxorubicin derivative Ada-DOX (CDE1-Ada-DOX) has been synthesized and evaluated in human breast cancer MCF-7 cells. First, we synthesized estrone-conjugated cyclodextrin (CDE1), which formed the complex CDE1-Ada-DOX via molecular recognition with the derivative adamantane-doxorubicin (Ada-DOX) (Kd=1,617 M-1). The structure of the targeting vector CDE1 was fully characterized using 1H- and 13C-nuclear magnetic resonance, mass spectrometry, and electron microscopy. CDE1-Ada-DOX showed two-phase drug-release kinetics with much slower release than Ada-DOX. The fluorescence polarization analysis reveals that CDE1-Ada-DOX binds to recombinant human estrogen receptor α fragments with a Kd of 0.027 µM. Competition assay of the drug complex with estrogen ligands demonstrated that estrone and tamoxifen competed with CDE1-Ada-DOX for membrane estrogen receptor binding in MCF-7 cells. Intermolecular self-assembly of CDE1 molecules were observed, showing tail-in-bucket and wire-like structures confirmed by transmission electronic microscopy. CDE1-Ada-DOX had an unexpected lower drug uptake (when the host–guest ratio was >1) than non-targeting drugs in MCF-7 cells due to ensconced ligands in cyclodextrins cavities resulting from the intermolecular self-assembly. The uptake of CDE1-Ada-DOX was significantly increased when the host–guest ratio was adjusted to be less than half at the concentration of CDE1 over 5 µM due to the release of the estrone residues. CDE1 elicited rapid activation of mitogen-activated protein kinases (p44/42 MAPK, Erk1/2) in minutes through phosphorylation of Thr202/Tyr204 in MCF-7 cells. These results demonstrate a targeted therapeutics delivery of CDE1-Ada-DOX to breast cancer cells in a controlled manner and that the drug vector CDE1 can potentially be employed as a molecular tool to differentiate nongenomic from genomic mechanism.
Keywords: breast cancer, drug vector, functionalized, membrane estrogen receptor, polysaccharide, targeted drug delivery
Introduction
The targeted drug-delivery systems formed through various types of intermolecular forces have attracted great interest because of their therapeutic potential in drug development and cancer treatment.1,2 Many elegant drug-delivery systems based on such non-bonding host–guest interactions have been developed.3–6 Cyclodextrins (CDs) are among the best nonimmunogenic vector candidates for self-assembly of targeted drug-delivery systems7,8 due to their unique hydrophobic hollow-cavity-containing structure, excellent biocompatibility, chemical modifiability of hydroxyl groups in the primary face, and strong ability to entrap drug molecules through molecular recognition.7–9 Ligand-attached CDs facilitate drug delivery to tumor cells that often express abundant target receptors on the tumor cell membrane. For example, multivalent CD-based glycoclusters specifically target clinically relevant sugar receptors;10 a recently synthesized series of drug complexes based on CDs functionalized with folic acid, arginylglycylaspartic acid (RGD tripeptide), and hyaluronic acid, in which hydrophobic therapeutics were entrapped, have been found to possess higher targeting efficiency and better drug affinity to target receptor-positive cancer cells (data from arginylglycylaspartic acid- and hyaluronic acid-conjugated CDs are unpublished).11 Ligand-attached CDs represent promising multimodal drug-delivery vectors.
However, CD-based estrogen-anchored therapeutic delivery systems have never been investigated, even though estrogens are crucial in the initiation, development, and progression of human breast cancer.12–14 Overexpression of estrogen receptors (ERs) is observed in approximately 70% of human breast cancers.12,15 In addition to residing in the nucleus (“nuclear ERs” [nERs], ie, ERα and ERβ), ERs can also reside on the membrane and endoplasmic reticulum (so-called “membrane ERs” [mERs]).16–19 Targeting the overexpressed mERs for the delivery of therapeutics represents a promising and effectual strategy for breast cancer therapy.20–22 In addition, the estrogen-conjugated CD vector is expected to preserve and elicit the estrogen response and act as a new antiestrogen for additional adjuvant endocrine treatments of ER-dependent breast cancer via targeting the ER-dependent pathways besides the targeting drug-delivery modality.21
Herein, we report an mER targeting estrone (E1)-conjugated CD vector CDE1 and prepared drug-delivery complex CDE1-Ada-DOX (Figure 1A) through molecular recognition of a doxorubicin derivative adamantane-doxorubicin (Ada-DOX) and CDE1. In this system, E1 was first conjugated to CDs (CDE1) to vectorize the encapsulated drug Ada-DOX which comprises an adamantane (Ada) molecule covalently linked to doxorubicin.11 The molecular moiety of adamantine forms stable drug inclusion with CDs with high affinity and low cytotoxicity.23,24 The drug uptake of CDE1-Ada-DOX and non-targeting CD-Ada-DOX have been examined in time course altered with host–guest ratios; notably, the drug release and uptake of Ada-DOX from CDE1-Ada-DOX in MCF-7 cells was in a controllable manner due to the unique intermolecular self-assembly of and the resulting stealth or release of the E1 residues in CDE1. Finally, we investigated whether the synthesized “estrogen-like” CDE1 could activate nonclassical rapid response through activated ER-mediated pathway in MCF-7 cells, and the potential application in differentiation of genomic and nongenomic pathways.25–27
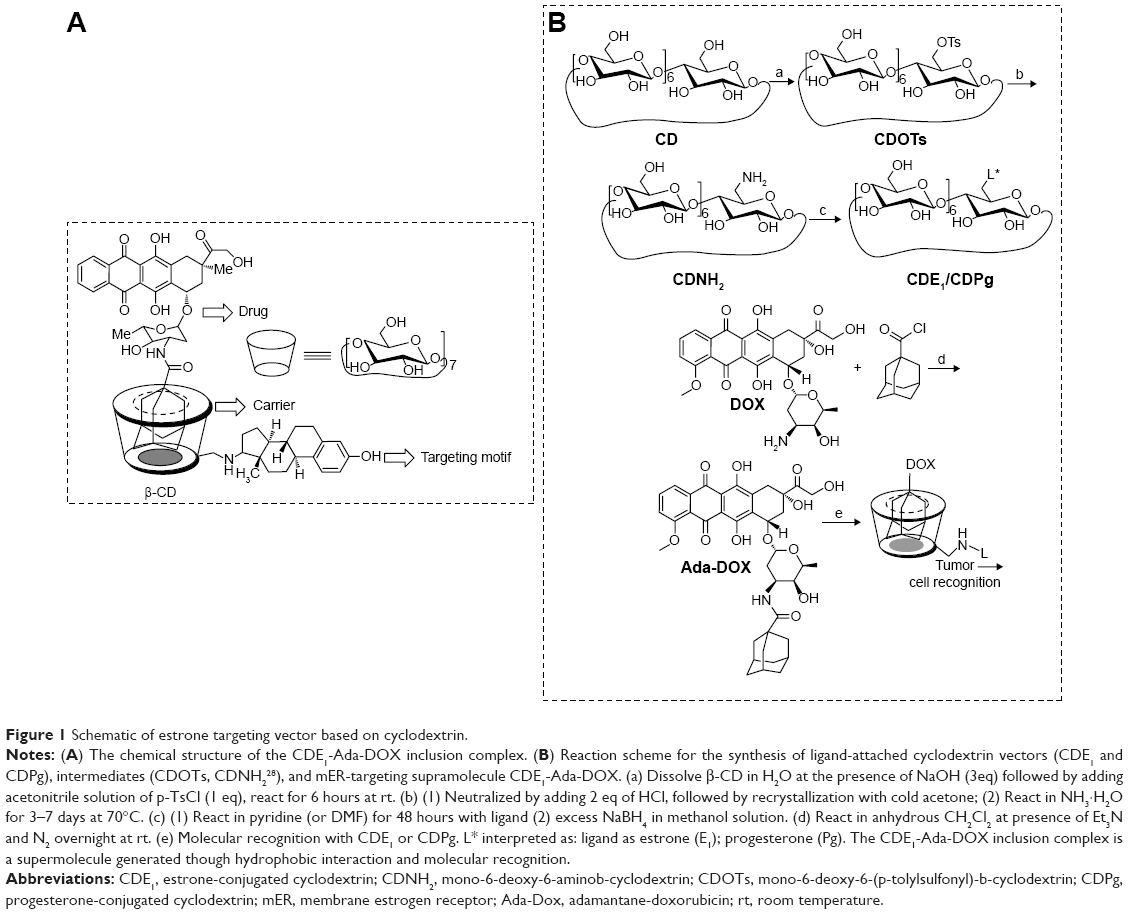 | Figure 1 Schematic of estrone targeting vector based on cyclodextrin. |
Methods
For chemical synthesis, characterization, drug release, TEM microscopy, drug uptake, and Western blot, see Supplementary materials.
Binding affinity of CDE1 with Ada-DOX in aqueous solution determined using the fluorescence titration method
We next determined how strong the host molecule CDE1 interacted with the guest molecule Ada-DOX using the fluorescence titration method as described previously.29 The effect of varying CDE1 concentrations on the fluorescence intensity of Ada-DOX was examined in order to determine the association constant (Kd) between CDE1 and Ada-DOX. The concentration of Ada-DOX was set at 50 μM in the presence of CDE1 at escalating concentrations of 0, 0.17, 0.26, 0.35, 0.44, 0.53, 0.62, 0.71, 0.79, and 0.88 mM, and the fluorescence intensity was monitored using a Synergy™ H4 Hybrid Microplate Reader (BioTek Inc., Winooski, VT, USA). The solvent used was dimethylformamide:H2O (50/50, v/v). The fluorescence of Ada-DOX was measured with λex at 490 nm and λem ranging from 500 to 700 nm with an escalating step of 2 nm. The Kd value was calculated using the above-mentioned approach by nonlinear fitting for various models.
Effects of the phosphorylation of p44/42 mitogen-activated protein kinase (Erk1/2) at Thr202/Tyr204 in MCF-7 cells
To examine whether CDE1 elicited nongenomic events in MCF-7 cells, the cells were treated with CDE1 or other drugs at 1 μM for 5, 10, 15, 30, and 60 minutes to detect p44/42 mitogen-activated protein kinase (MAPK) (Erk1/2) phosphorylation at Thr202/Tyr204 using Western blotting analysis in MCF-7 cells. The experiments were repeated in triplicate.
Binding determination of the CDE1-Ada-DOX inclusion complex to recombinant human ERα fragments using the fluorescence polarization method
Fluorescence polarization (also called fluorescence anisotropy) is a versatile solution-based technique that has been widely used to investigate molecular interactions (eg, ligand–receptor binding), enzymatic activity, and nucleic acid hybridization.48 Quantitatively, fluorescence polarization (mP) is defined as the difference of the emission light intensity parallel and perpendicular to the excitation light plane normalized by the total fluorescence emission intensity. The binding of the CDE1-Ada-DOX inclusion complex to recombinant human ERα fragments consisting of amino acid residues 1–116 at the C-terminus (His tag C-terminus, Molecular Mass =12,200 Da) (catalogue number: ab153776; Abcam Plc, Cambridge, UK) was investigated using the fluorescence polarization method as described previously.31 Briefly, human ERα fragments were reconstituted in phosphate-buffered saline to the final concentration of 0.8 μM, and CDE1-Ada-DOX complex samples at concentrations from 0.04 μM to 1.26 μM were added to the protein solution. The samples were mixed well at room temperature and subject to analysis immediately. mP was measured using the Synergy H4 Hybrid Multi-Mode Microplate reader at λEx =485/20 nm and λEm =620/10 nm with xenon flash as the light source. The acquisition parameters were set as follows: 200 flashes and positioning delay 100 msec. The experiment was repeated in triplicate. The mP values were plotted against an increasing concentration of CDE1-Ada-DOX and the equilibrium binding constant (Kd) was calculated using nonlinear least squares that fit the curve data by Prism 6.03 program (GraphPad Software, Inc., La Jolla, CA, USA).
Results and discussion
In this study, E1 was successfully conjugated with β-CD to generate CDE1 as a new drug vector to target mERs of breast cancer cells. At the same time, progesterone, having a similar structure to E1, was also conjugated to CDs as the substrate extension of the conjugation reaction (Figure 1B). CDE1 accommodates the hydrophobic Ada-DOX through host–guest interactions to form the CDE1-Ada-DOX supramolecule for drug delivery since the geometry and hydrophobicity of the adamantyl group allows an excellent fit into the CD’s torus inner cavity. The structures of the synthetic compounds have been confirmed using multiple spectral methods which include 1H nuclear magnetic resonance, 13C nuclear magnetic resonance, high-resolution matrix-assisted laser desorption/ionization-time of flight mass spectroscopy, fluorescence spectroscopy, and circular dichroism spectroscopy (Figure 2). The 1:1 stoichiometric inclusion complex CDE1-Ada-DOX was readily prepared by the coprecipitation method through molecular recognition.
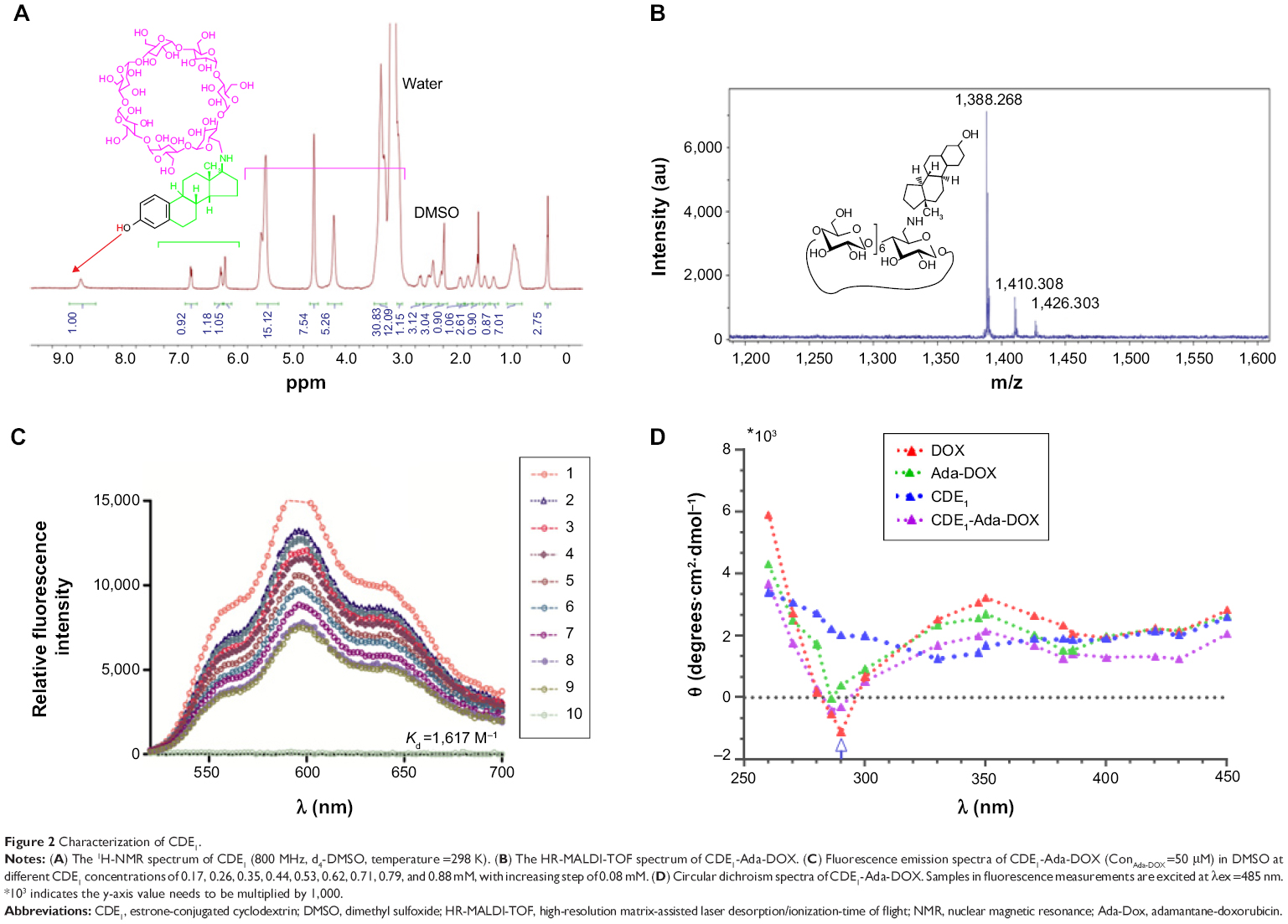 | Figure 2 Characterization of CDE1. |
The host–guest binding affinity of CDE1 with Ada-DOX (Kd) was measured using the fluorescence titration method as described previously.29 With an increasing amount of CDE1 added to the dimethylformamide and H2O (1:1) solution containing Ada-DOX, the fluorescence intensity was measured at varying emission wavelengths from 500 to 700 nm with 600 nm as the maximum peak, which decreased in a concentration-dependent manner (Figure 2C). The Kd values were calculated using the fluorescence intensity versus CDE1 concentration curve. This data showed that the one-phase and two-phase exponential decay models were the best fit. The calculated Kd was 0.93, 1.09, 0.77, and 0.73 mM when the emission wavelengths were 550, 574, 600, and 634 nm, respectively. The fluorescence quenching effect during the titration of CDE1 to Ada-DOX indicated that host–guest binding occurs.
Moreover, we examined the releasing kinetics of Ada-DOX from the CDE1-Ada-DOX inclusion complex in phosphate-buffered saline against 50% fetal bovine serum solution using a validated fluorescence method.11 The release kinetics of Ada-DOX and CDE1-Ada-DOX from the formulations clearly demonstrated that the drug released from CDE1-Ada-DOX was significantly slower (>50%) than Ada-DOX within the incubation time period (Figure 3). Detachment of Ada-DOX from the CDE1-Ada-DOX inclusion complex showed sustained releasing kinetics. By fitting the released drug data versus time, the kinetic parameters were calculated. For Ada-DOX, the Kdα and Kdβ were 1.04 and 0.015 hours−1, with elimination half-life (t1/2)α and t1/2β of 0.67 and 45.02 hours, respectively. For CDE1-Ada-DOX, the Kdα and Kdβ were 0.02 and 0.02 hours−1, with t1/2α and t1/2β of 35.40 and 35.30 hours, respectively. The accumulative and absolute released drug for Ada-DOX and CDE1-Ada-DOX were also compared using two-way analysis of variance. At 2 hours, the cumulated drug release was 6.5% and 20.5% for CDE1-Ada-DOX and Ada-DOX, respectively (P>0.05). At 4 hours, the values were almost unchanged (6.6% versus 23.5%, P<0.05). At 6 hours, the values were increased to 11.1% and 28.4%, respectively (P<0.05). At 21 hours, drug-release values were increased to 20.9% and 49.1%, respectively (P<0.001). The measured drug released was 46.1% for CDE1-Ada-DOX and 99.0% for Ada-DOX over 75 hours of dialysis (P<0.001). These data clearly show that the release of Ada-DOX from CDE1-Ada-DOX is sustained compared with Ada-DOX over 75 hours. The sustained release of Ada-DOX from the inclusion complex would facilitate continuous drug uptake and long-term proliferation inhibition of the cancer cells.
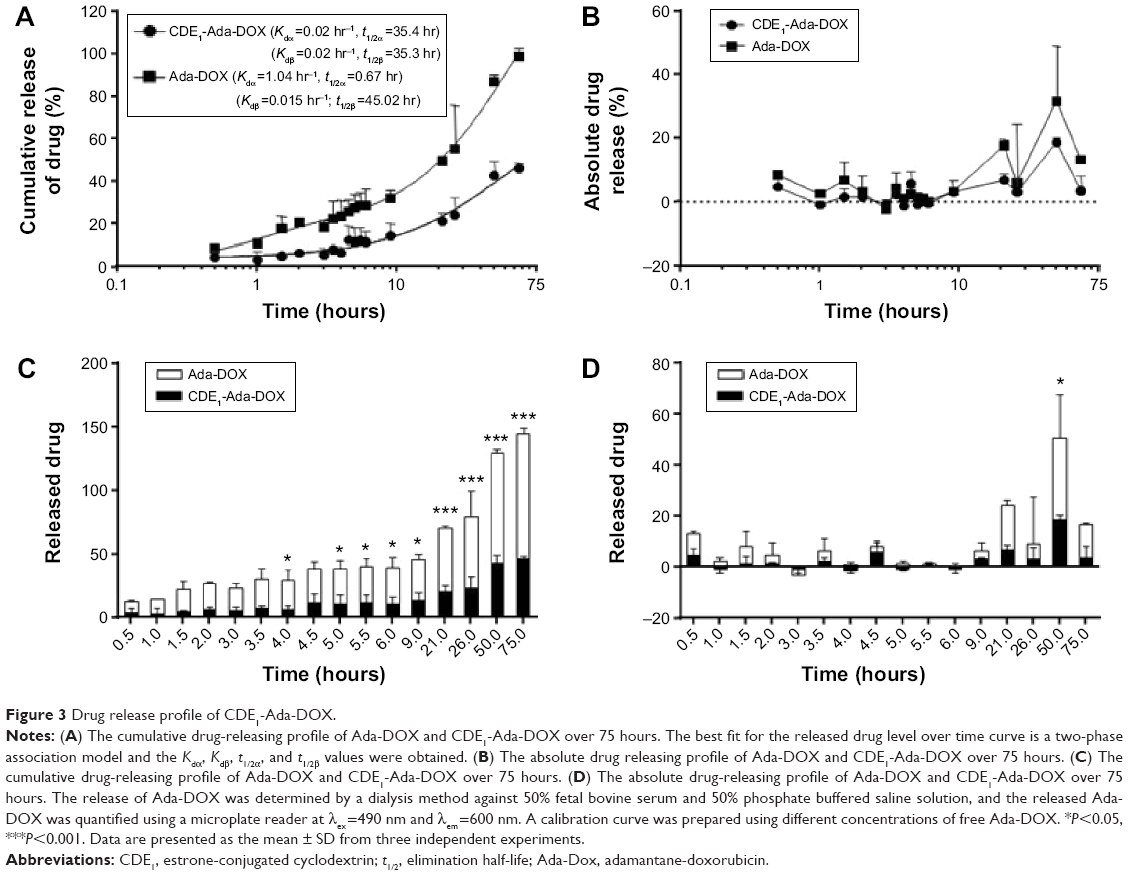 | Figure 3 Drug release profile of CDE1-Ada-DOX. |
In addition to the sustained drug release of CDE1-Ada-DOX, it has been clearly demonstrated that anchored ligands in the drug-delivery systems providing specific drug–cell surface interactions are crucial in determining the ultimate drug internalization by cancer cells.8 The cellular uptake of the mER-targeting drug complex CDE1-Ada-DOX and non-targeting compound CD-Ada-DOX was investigated to check for the targeting efficiency; flow cytometric analysis was performed in MCF-7 cells by taking advantage of intrinsic fluorescence emission from Ada-DOX. MCF-7 cells were treated with CDE1-Ada-DOX and non-targeting drug CD-Ada-DOX at different host–guest molecular molar ratios and drug concentrations in time course (Figure 4). The control (MCF-7 cells with no drug treated) and CDE1 itself did not elicit obvious fluorescence and showed very low levels of autofluorescence. The addition of CDE1 at 1, 3, or 5 μM to the MCF-7 cells slightly increased the fluorescence. Interestingly, excess CDE1 quenched the fluorescence intensity of Ada-DOX at 1 μM when formulated in ratios of 1:1, 1:3, and 1:5 (Ada-DOX:CDE1) with 12.5% and 27.2% decrease for 1:3 and 1:5, respectively, in comparison to 1:1 CDE1-Ada-DOX complex (Figure 4A). CDE1-Ada-DOX had an unexpected lower cellular uptake than non-targeting CD-Ada-DOX complex in MCF-7 cells at 1 μM with a 1:1 host–guest molar ratio (Figure 4B), while the uptake of CDE1-Ada-DOX was enhanced when the host–guest molecule molar ratio and total concentration were altered. The cells were exposed to different CD or CDE1 inclusion complexes for 2, 4, and 6 hours, respectively, with host and guest molar ratio set at 1:1. The drug uptake in MCF-7 cells treated with CD-Ada-DOX with host–guest ratio of 1:1 (CDE1 concentration =1 μM) for 2, 4, and 6 hours was 3.34-, 3.55-, and 3.61-fold of that in cells treated with CDE1-Ada-DOX with host–guest ratio of 1:1 with CDE1 concentration at 1 μM, respectively (P<0.001). The internalization efficacy of the targeting complex CDE1-Ada-DOX rebounded greatly compared with the drug without ligand attached (CD-Ada-DOX) as the host–guest molecular molar ratio increased to 1:2 with CDE1 concentration of 1 μM (Figure 4C). The drug uptake in MCF-7 cells treated with CD-Ada-DOX with host–guest ratio of 1:2 at 1 μM of CDE1 for 2, 4, and 6 hours was 1.62-, 1.36-, and 1.57-fold of that in cells treated with CDE1-Ada-DOX with host–guest ratio of 1:1 (CDE1 concentration =1 μM), respectively (P<0.001). The drug uptake of CDE1-Ada-DOX exceeded that of CD-Ada-DOX when the concentration of the guest drug was continually raised. When the host–guest molecular ratio was altered to 1:2 with a CDE1 concentration of 5 μM, the drug uptake of MCF-7 cells treated with CDE1-Ada-DOX was higher (69.9%) than cells treated with CD-Ada-DOX (P<0.01) (Figure 4D and E). These results suggest that the targeted CDE1-Ada-DOX inclusion complex improved the uptake of Ada-DOX in comparison to the non-targeted CD-Ada-DOX inclusion complex when CDE1 and Ada-DOX were formulated in appropriate ratios and drug concentrations to release ample E1 ligands from CDE1.
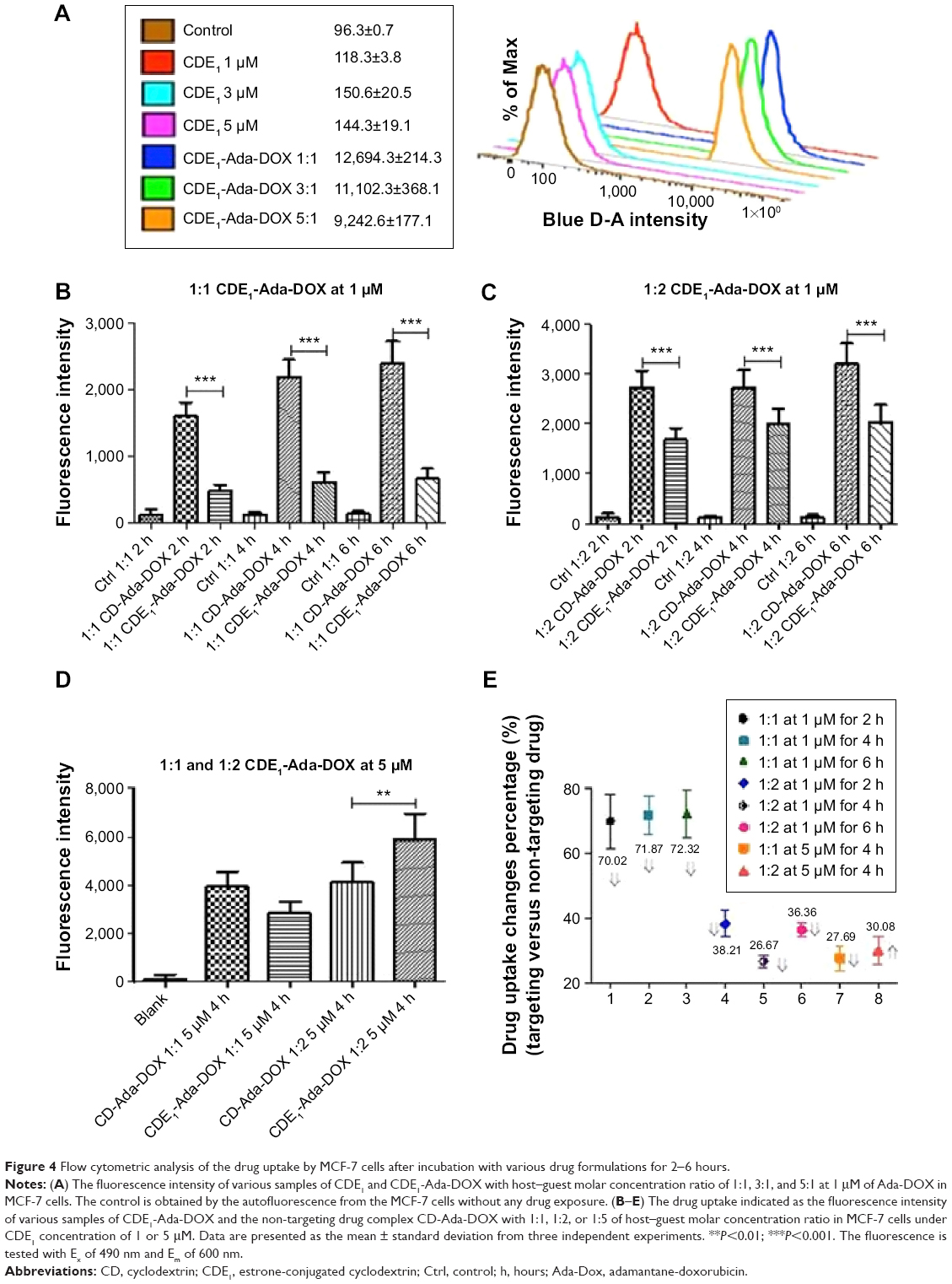 | Figure 4 Flow cytometric analysis of the drug uptake by MCF-7 cells after incubation with various drug formulations for 2-6 hours. |
With higher guest drug concentrations or altered host–guest molecular molar ratios, the release of the targeting moieties from the complex CDE1-Ada-DOX have been enhanced through binding to the mERs and have consequently facilitated the drug internalization process in a controlled manner. These interesting findings led us to propose that estrogen residues covalently bonded with the CD are stealthy under certain circumstances. In order to further substantiate the presence of stealthy ligands in CDE1 and CDE1-Ada-DOX, transmission electronic microscopy (TEM) and scanning electronic microscopy (SEM) examinations were conducted. Representative TEM and SEM images of CDE1 and CDE1-Ada-DOX are shown in Figure 5. The intermolecular assembly of CDE1 exhibited a tail-in-bucket structure and wire-like morphology for CDE1, which shows the conjugated estrogen residing inside the CD cavity of the adjacent CDE1 molecule. Figure 5A–C illustrate the long, tangled, uniform CDE1 wires. The estrogen residues in CDE1 act as linkers of CDE1 molecules due to intermolecular recognition. Figure 5D–F show the TEM images of CDE1-Ada-DOX under the same experimental conditions as CDE1. CDE1-Ada-DOX particles showed an unorganized structure under the same preparation conditions compared with the CDE1 containing no drug payload. The observation was consistent and reproducible in the TEM/SEM examinations. These findings indicate that the conjugated E1 residues of CDE1 were entrapped in the CD cavities of CDE1 in the absence of a guest molecule. Furthermore, when the guest molecule such as Ada-DOX was added into CDE1, the competition between exogenous and the intermolecular recognition with the CD cavity resulted in the consequent release of a certain amount of E1 residues, in turn disrupting intramolecular self-assembling, dissembling the wire-like and tail-in-bucket structure of CDE1. Moreover, circular dichroism analysis indicated that Ada-DOX interacted with CDE1 (Figure 2D) and caused conformational changes at the CD cavity binding site followed by chiral microenvironment changes for the whole drug complex supramolecule. The circular dichroism spectra provide supportive evidence for the morphology differences of CDE1 and CDE1-Ada-DOX complexes.
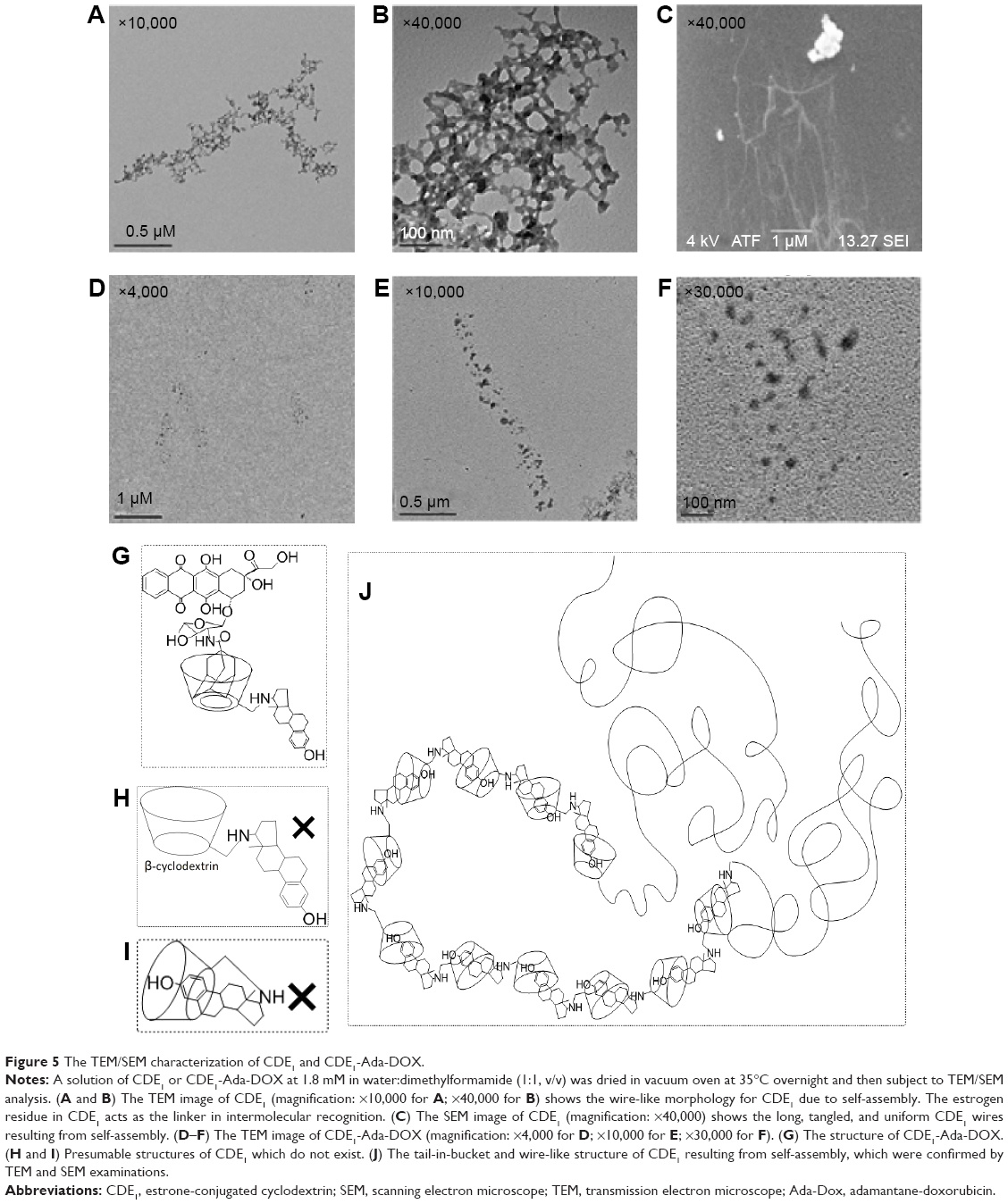 | Figure 5 The TEM/SEM characterization of CDE1 and CDE1-Ada-DOX. |
Herein, we consider that targeting mERs expressed on MCF-7 cells is an effective means of affecting the uptake of the drug complex into these cells since the drug complex is taken up by ER-mediated endocytosis. Our results showed that CDE1 preserved the binding ability after E1 conjugated to CD. More importantly, drug uptake can be augmented significantly if the estrogen molecules in the complex are released to ensure the estrogen residue tailed outside the CD cavity. The intermolecular recognition between the covalent attached estrogen residue of one CDE1 and the CD cavity of another CDE1 molecule results in the host–guest molar ratio-dependent difference in drug uptake since the targeting moiety has been entrapped, and in turn reduces or loses its affinity to mERs. When an appropriate guest molecule approaches the CDE1, the estrogen residues are pushed out and released. The competition between complexation of the drug and intermolecular inclusion of the estrogen in the CD cavity leads to altered drug uptake. In other words, the equilibrium of the host–guest molecules and the complex is the critical factor for controlling and optimizing the release kinetics of the targeting and drug moieties, mER binding, drug uptake efficiency, dissimilarity, and biological responses.
Furthermore, the cholesterol levels have been monitored in both cancerous cells (MCF-7 and A549 cells) and normal cells (MCF-10A and T80 cells) to investigate cholesterol depletion of lipid rafts on the cell membrane after drug exposure of CDE1 and the drug complexes, since CD derivatives have been reported to be able to extract cholesterol from bilayer membrane by the CDs cavity, and modulate the activity of multiple signaling pathways.30 Cholesterol and estrogens have structural similarity. It was shown that the cholesterol level was not significantly affected after CDE1 treatment due to the preoccupancy of the CD cavity by E1 residues from the intramolecular self-assembly (Figure 6).
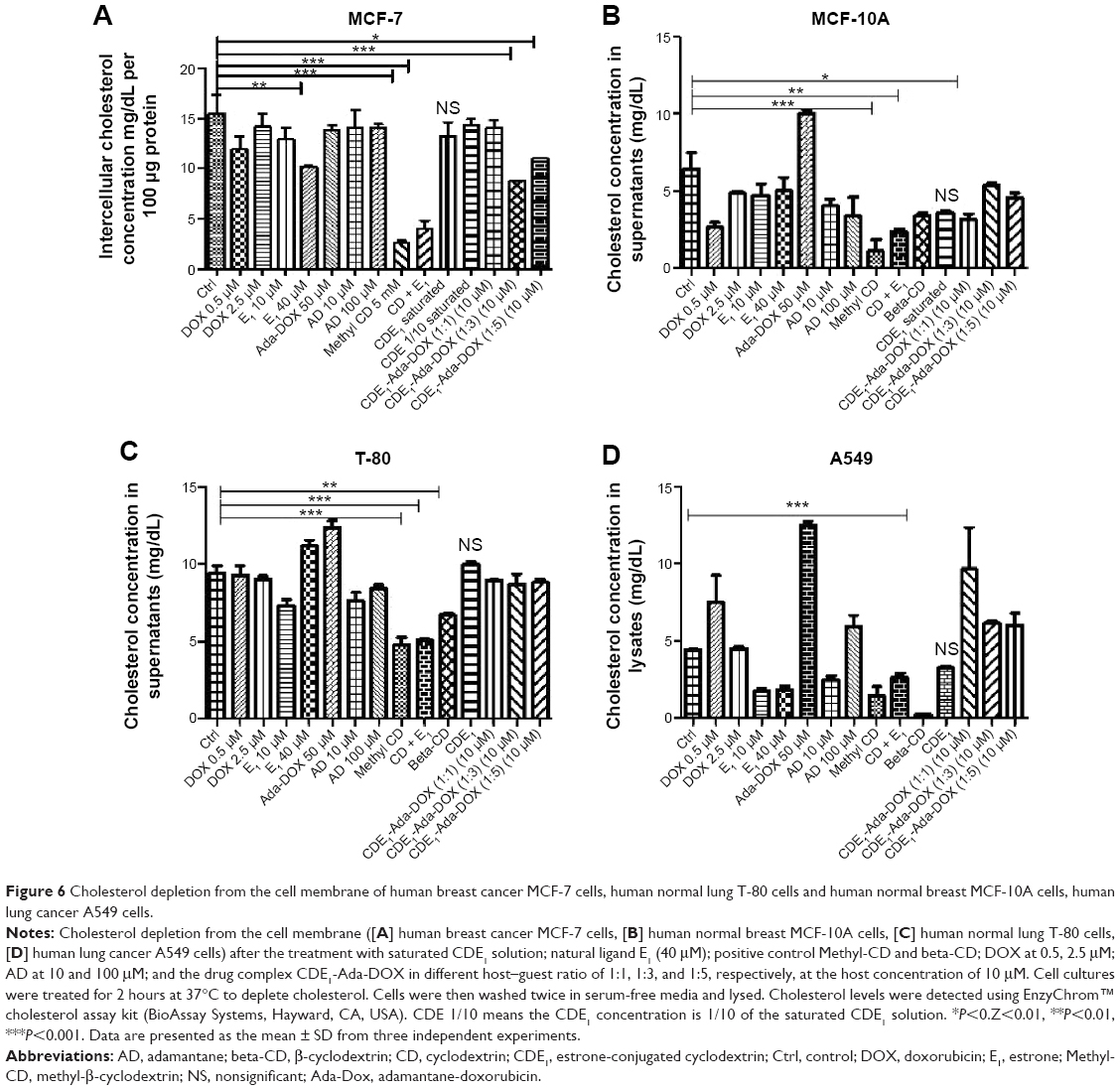 | Figure 6 Cholesterol depletion from the cell membrane of human breast cancer MCF-7 cells, human normal lung T-80 cells and human normal breast MCF-10A cells, human lung cancer A549 cells. |
Therefore, the magnitude of drug binding and internalization in cancer cells could be modulated by disrupting the intramolecular self-assembly of CDE1, and changing the drug exposure levels and composition (eg, host–guest molecular molar ratio) in a controllable manner based on our findings.
To confirm the binding ability of CDE1-Ada-DOX to mERs in breast cancer cells, we performed the competition assay with CDE1-Ada-DOX using E1 and a selective ER modulator, tamoxifen, as the inhibitors in MCF-7 cells. The results are shown in Figure 7A. The flow cytometric analysis showed that E1 at 5-50 μM inhibited CDE1-Ada-DOX uptake in a concentration-dependent manner in MCF-7 cells. E1 at 5, 10, and 50 μM diminished the uptake of CDE1-Ada-DOX by 5.1%, 4.3%, and 8.3%, respectively, in MCF-7 cells (P<0.001). Furthermore, tamoxifen also concentration-dependently reduced the uptake of CDE1-Ada-DOX in MCF-7 cells. Tamoxifen at 1, 5, and 10 μM inhibited the uptake of CDE1-Ada-DOX by 7.0%, 20.5%, and 19.2%, respectively, in MCF-7 cells (P<0.001). The results demonstrate that both E1 and tamoxifen competed with CDE1-Ada-DOX for mER binding in MCF-7 cells in a concentration-dependent manner with higher competing potency for tamoxifen compared to E1.
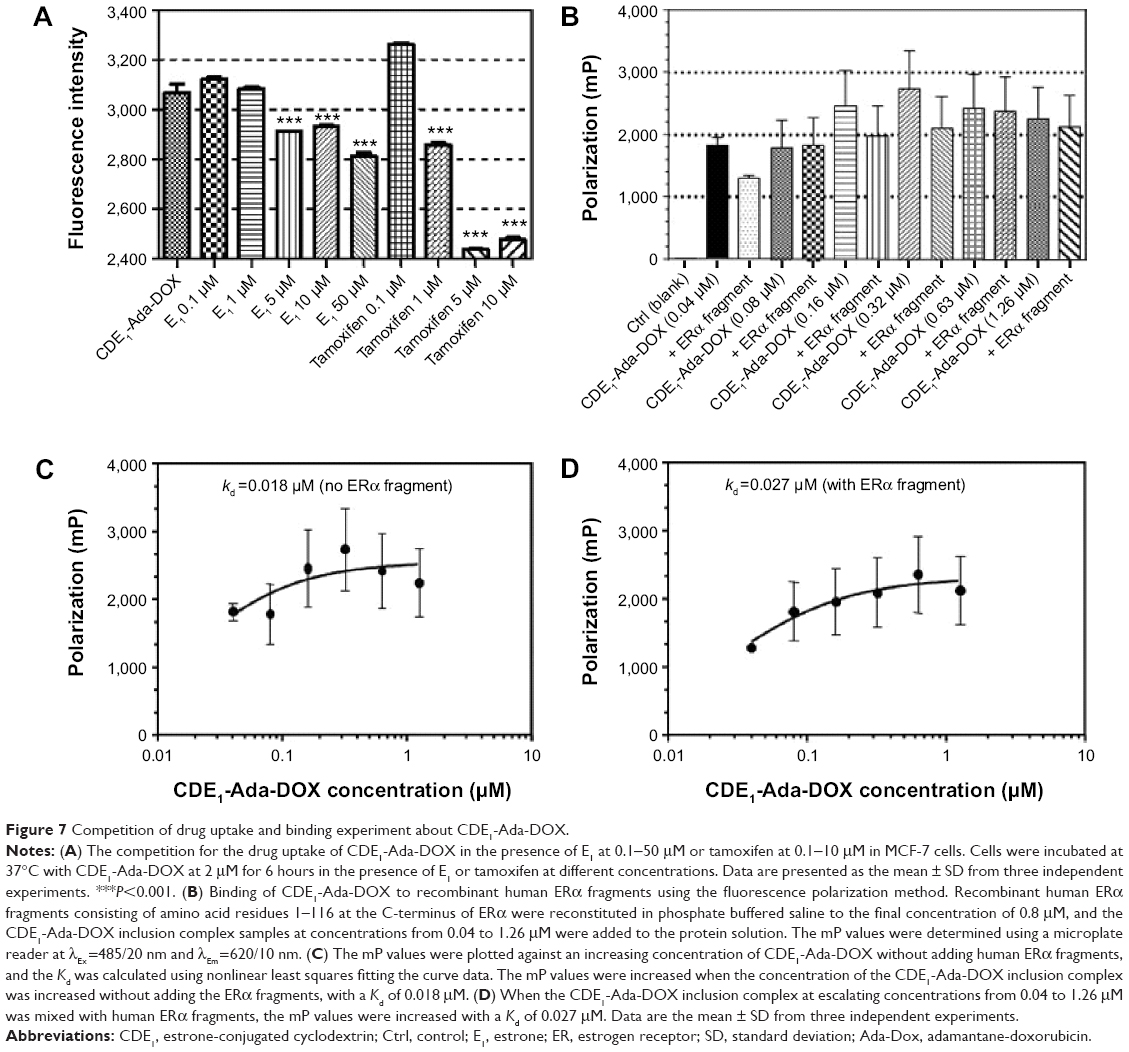 | Figure 7 Competition of drug uptake and binding experiment about CDE1-Ada-DOX. |
Binding to mERs on tumor cells by tamoxifen or E1 prevented the entry of other ER ligands such as CDE1-Ada-DOX targeting complex to the binding pocket. These findings provide further evidence that estrogen residues from CDE1-Ada-DOX complex recognize and bind to mER/mERs on MCF-7 cells.
Additionally, we used a fragment of human ERα containing the ligand binding domain (recombinant human ERα fragments consisting of amino acid residues 1–116 at the C-terminus, His tag C-terminus, Mr =12,200 Da; Abcam Plc) to examine whether CDE1-Ada-DOX could bind to it using the fluorescence polarization approach.31 The polarization (mP) data over the concentration of the CDE1-Ada-DOX inclusion complex in the absence or presence of human ERα fragments at a fixed concentration of 0.08 μM are shown in Figure 7B–D. The mP values were increased when the concentration of the CDE1-Ada-DOX inclusion complex was increased without adding the ERα fragments, with a Kd of 0.018 μM.
When the CDE1-Ada-DOX inclusion complex at escalating concentrations from 0.04 to 1.26 μM was mixed with human ERα fragments, the mP values were increased with a Kd of 0.027 μM. These results demonstrate the interaction of CDE1-Ada-DOX with human ERα fragments containing the ligand-binding domain.
In addition to the targeting drug-delivery modality of the novel “estrogen-like” molecule CDE1, the cellular response triggered by CDE1 in a manner different from the classical nER-mediated pathway was investigated by Western blotting assay (Figure 8). Estrogens bind to nuclear ERα and ERβ, triggering the classical pathway of estrogen-dependent action and finally eliciting remarkable genomic responses. The action of nuclear ERs includes binding lipophilic hormone molecule in cytoplasm, translocation of the ligand–ER complex to the nucleus, dimerization, interaction with estrogen-specific response elements in the promoter areas of target genes, and finally initiating gene transcription.18,19,32 The effects of steroid hormone action dependent on ER occur within hours or even days. On the other hand, some ligands can elicit rapid nongenomic signaling cascades in a much shorter time (from seconds to minutes) upon estrogen binding.18,19,33,34 These rapid nongenomic effects of estrogens result in calcium mobilization, cyclic adenosine monophosphate stimulation, phospholipase C activation, inositol phosphate generation, and activation of membrane-associated signaling pathways, including protein kinase A, phosphotidylinositol-3 kinase, and MAPK (p44/42 MAPK, also called extracellular-signal-regulated kinase [Erk1/2]) signaling pathways.18,19 Importantly, crosstalk via second messengers between mER- and nER-initiated signaling responses can regulate transcriptional activation of multiple target genes in a coordinated manner.18,19 The estrogens E1 and E2, the selective ER modulator tamoxifen, and the synthetic estrogen 17α-ethinyl estradiol were used as positive control to trigger the rapid response of Erk1/2; adamantane and 1-adamantanol were used as structural fraction controls to the drug complex CDE1-Ada-DOX. It was demonstrated that CDE1 resulted in rapid phosphorylation response of p44/42 MAPK (p-Erk1/2) within 30 minutes in ER-positive cancerous cells such as MCF-7 cells, A549 cells, OVCAR3 cells, and even in the normal lung T80 cells, while no significant response was observed in normal breast MCF-10A cells. Cells treated with CDE1 at 1 μM showed a maximum p44/p42 MAPK phosphorylation at Thr202/Tyr204 at 15 minutes drug exposure and increases by 22.2%, 25.5%, and 59.2% at 30 minutes for MCF-7, A549 and OVCAR3 cells respectively, compared with controls (P<0.05). These results show that treatment of MCF-7 cells with CDE1 activated the mER-mediated signaling pathway as indicated by the significantly increased phosphorylation of p44/42 MAPK (Erk1/2) rapidly within 5-30 minutes at Thr202/Tyr204 in MCF-7 cancer cells. Estrogens such as E2 induce a number of rapid signaling events in cells that express mERs such as GPR30,19 except in classical pathways.18,19,32 Different stimuli including mitogens, growth factors, cytokines, virus infection, ligands for heterotrimeric G protein-coupled receptors, transforming agents, and carcinogens can activate the p44/42 MAPK (Erk1/2) pathway.35–37 There is evidence that E2-induced p44/42 MAPK (Erk1/2) activation requires GPR30, and occurs via transactivation of the epidermal growth factor receptor.38,39 In the Raf–MEK–MAPK/Erk pathway, receptor tyrosine kinases and G protein-coupled receptors activate Ras, which in turn activates c-Raf.35,37 Activation of c-Raf involves phosphorylation at multiple residues including Ser338, Tyr341, Thr491, Ser494, Ser497, and Ser499. p21-activated protein kinase can phosphorylate c-Raf at Ser338 and the Src family phosphorylates Tyr341 of c-Raf.35,37 Activated c-Raf activates MAPK kinase (called MKK, MEK, or MAP2K) at Ser217/221 located in the activation loop of subdomain VIII, and MEK1/2 activate p44 and p42 through phosphorylation of activation loop residues Thr202/Tyr204 and Thr185/Tyr187, respectively.37 p44/42 MAPK (Erk1/2) are negatively regulated by a family of dual-specificity (Thr/Tyr) MAPK phosphatases, along with MEK inhibitors such as U0126 and PD98059.40
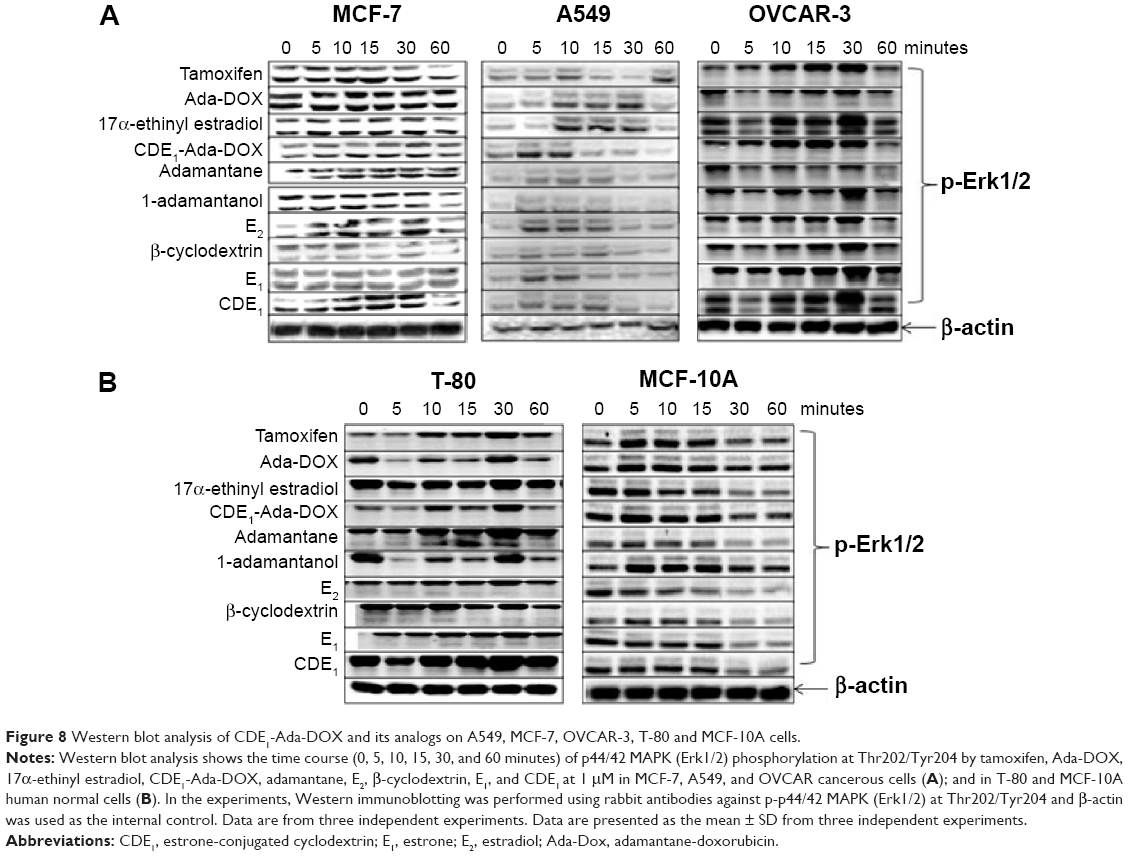 | Figure 8 Western blot analysis of CDE1-Ada-DOX and its analogs on A549, MCF-7, OVCAR-3, T-80 and MCF-10A cells. |
In this study, treatment of MCF-7 cells with the new synthetic CDE1 resulted in rapid phosphorylation of p44/42 MAPK (Erk1/2) in minutes. These findings suggest that CDE1 can interact with and activate mERs and might be used as a probe for studying mER-mediated nongenomic events in comparison with nER-mediated genomic responses that involve DNA binding and gene expression initiation; it also needs a much longer time. It was demonstrated that adamantane and 1-adamantanol molecules do not affect p44/42 MAPK phosphorylation in tumor cells significantly, while upregulation of p-Erk1/2 from the drug complex CDE1-Ada-DOX results from CDE1 and partially from the Ada-DOX since Ada-DOX treatment also cause slight pErk1/2 over-expression.
It is well known that estrogens activate the rapid, nonclassical signaling cascades via mERs, and there is crosstalk between mER-mediated nonclassical and nER-mediated classical pathways.39,41,42 Deregulation of both pathways plays important roles in the pathogenesis of cancer and other diseases.43 It is recognized that selective ER ligands are needed to delineate the role of extranuclear and nuclear ERs in disease development and therapeutics where ERs represent important therapeutic targets. Several estrogen conjugates, including CD–E2, E2–BSA, E2–peroxidase, and E2–dendrimers, have been reported and used to probe the nonclassical and classical cascades.44–47 CD derivatives are generally considered cell-membrane impermeable. Therefore, CDE1 can be employed as a molecular tool to differentiate nongenomic response from genomic response based on the findings.
Conclusion
We synthesized the novel estrogen-anchored conjugate CDE1 and the corresponding drug inclusion with the doxorubicin derivative Ada-DOX (CDE1-Ada-DOX). The structures of these new compounds were confirmed with rigorous spectral methods. A tail-in-bucket and wire-like structure of CDE1 via intermolecular self-assembly was observed by TEM and SEM examination; in contrast, CDE1-Ada-DOX exhibited unorganized structure due to disruption of self-assembly in the presence of guest molecules. The binding of Kd between CDE1 and Ada-DOX through hydrophobic interactions was determined to be 0.77 mM by fluorescence titration. CDE1-Ada-DOX showed sustained and two-phase exponential drug-release kinetics over 75 hours. Notably, for the mER-targeted CDE1-Ada-DOX inclusion complex, the critical factor for drug uptake efficiency in MCF-7 cells relied on the equilibrium between the host–guest and drug complex. By altering the ratios between the host and the guest molecules and the breakdown of the self-assembly nanostructure, CDE1-Ada-DOX delivered the anticancer drug into MCF-7 cells in a controlled manner. E2 and tamoxifen suppressed the drug uptake in MCF-7 cells treated with CDE1-Ada-DOX through competition for mER binding. Moreover, CDE1-Ada-DOX binds to recombinant human ERα fragments with a Kd of 0.027 μM determined by fluorescence polarization. The treatment of MCF-7 cells with CDE1-Ada-DOX elicited rapid activation of MAPKs (p44/42 MAPK, Erk1/2) in minutes through phosphorylation of Thr202/Tyr204. These results demonstrate a targeted delivery of the DOX derivative Ada-DOX to mER-positive breast cancer cells using CDE1 as the drug carrier vectors in a controlled manner. The estrogen conjugates elicit nongenomic (but not genomic) events in MCF-7 cells. CDE1 can be used as a powerful probe to explore the classical and nonclassical steroid-mediated pathways that are critical in the initiation, development, and progression of certain type of cancer (breast and ovarian cancer).
Acknowledgments
The authors are grateful for the support from the National Natural Science Foundation of China (Grant No 81372383), and the startup fund from College of Pharmacy, University of South Florida. This work has been supported in part by the Florida Center of Excellence for Drug Discovery and Innovation at the University of South Florida. The authors thank Dr S Karoly and Dr A Garces at the Lisa Muma Weitz Advanced Microscopy Core Laboratory for their support in data acquisition of flow cytometry and electronic microscopy. We also thank Dr R Sprunt at Moffitt Cancer Center for the MALDI-TOF mass spectroscopy.
Disclosure
The authors report no conflicts of interest in this work.
References
Schneider HJ. Binding mechanisms in supramolecular complexes. Angew Chem Int Ed Engl. 2009;48(22):3924–3977. | ||
Kubik S. Molecular cages and capsules with functionalized inner surfaces. Top Curr Chem. 2012;319:1–34. | ||
Del Valle EMM. Cyclodextrins and their uses: a review. Process Biochem. 2004;39(9):1033–1046. | ||
Zhang X, Wang C. Supramolecular amphiphiles. Chem Soc Rev. 2011;40(1):94–101. | ||
Benito JM, Gómez-García M, Ortiz Mellet C, Baussanne I, Defaye J, García Fernández JM. Optimizing saccharide-directed molecular delivery to biological receptors: design, synthesis, and biological evaluation of glycodendrimer-cyclodextrin conjugates. J Am Chem Soc. 2004;126(33):10355–10363. | ||
Paliwal SR, Paliwal R, Pal HC, et al. Estrogen-anchored pH-sensitive liposomes as nanomodule designed for site-specific delivery of doxorubicin in breast cancer therapy. Mol Pharm. 2012;9(1):176–186. | ||
Zhang J, Ma PX. Cyclodextrin-based supramolecular systems for drug delivery: recent progress and future perspective. Adv Drug Deliv Rev. 2013;65(9):1215–1233. | ||
Davis ME, Brewster ME. Cyclodextrin-based pharmaceutics: past, present and future. Nat Rev Drug Discov. 2004;3(12):1023–1035. | ||
Yin JJ, Zhou ZW, Zhou SF. Cyclodextrin-based targeting strategies for tumor treatment. Drug Deliv Transl Res. 2013;3(4):364–374. | ||
André S, Kaltner H, Furuike T, Nishimura SI, Gabius HJ. Persubstituted cyclodextrin-based glycoclusters as inhibitors of protein-carbohydrate recognition using purified plant and mammalian lectins and wild-type and lectin-gene-transfected tumor cells as targets. Bioconjug Chem. 2004;15(1):87–98. | ||
Yin JJ, Sharma S, Shumyak SP, et al. Synthesis and biological evaluation of novel folic acid receptor-targeted, β-cyclodextrin-based drug complexes for cancer treatment. PloS One. 2013;8(5):e62289. | ||
Deblois G, Giguère V. Oestrogen-related receptors in breast cancer: control of cellular metabolism and beyond. Nat Rev Cancer. 2013;13(1):27–36. | ||
Manavathi B, Dey O, Gajulapalli VN, Bhatia RS, Bugide S, Kumar R. Derailed estrogen signaling and breast cancer: an authentic couple. Endocr Rev. 2013;34(1):1–32. | ||
Yue W, Yager JD, Wang JP, Jupe ER, Santen RJ. Estrogen receptor-dependent and independent mechanisms of breast cancer carcinogenesis. Steroids. 2013;78(2):161–170. | ||
Eroles P, Bosch A, Pérez-Fidalgo JA, Lluch A. Molecular biology in breast cancer: intrinsic subtypes and signaling pathways. Cancer Treat Rev. 2012;38(6):698–707. | ||
Liang J, Shang Y. Estrogen and cancer. Annu Rev Physiol. 2013;75:225–240. | ||
Dahlman-Wright K, Cavailles V, Fuqua SA, et al. International Union of Pharmacology. LXIV. Estrogen receptors. Pharmacol Rev. 2006;58(4):773–781. | ||
Kampa M, Pelekanou V, Notas G, Stathopoulos EN, Castanas E. The estrogen receptor: two or more molecules, multiple variants, diverse localizations, signaling and functions. Are we undergoing a paradigm-shift as regards their significance in breast cancer? Hormones (Athens). 2013;12(1):69–85. | ||
Soltysik K, Czekaj P. Membrane estrogen receptors – is it an alternative way of estrogen action? J Physiol Pharmacol. 2013;64(2):129–142. | ||
Renoir JM, Marsaud V, Lazennec G. Estrogen receptor signaling as a target for novel breast cancer therapeutics. Biochem Pharmacol. 2013;85(4):449–465. | ||
Ali S, Buluwela L, Coombes RC. Antiestrogens and their therapeutic applications in breast cancer and other diseases. Annu Rev Med. 2011;62:217–232. | ||
Nilsson S, Koehler KF, Gustafsson JA. Development of subtype-selective oestrogen receptor-based therapeutics. Nat Rev Drug Discov. 2011;10(10):778–792. | ||
Grillaud M, Russier J, Bianco A. Polycationic adamantane-based dendrons of different generations display high cellular uptake without triggering cytotoxicity. J Am Chem Soc. 2014;136(2):810–819. | ||
Bohm I, Isenbügel K, Ritter H, Branscheid R, Kolb U. Cyclodextrin and adamantane host-guest interactions of modified hyperbranched poly(ethylene imine) as mimetics for biological membranes. Angew Chem Int Ed Engl. 2011;50(34):7896–7899. | ||
Silva E, Kabil A, Kortenkamp A. Cross-talk between non-genomic and genomic signalling pathways – distinct effect profiles of environmental estrogens. Toxicol Appl Pharmacol. 2010;245(2):160–170. | ||
Marino M, Galluzzo P, Ascenzi P. Estrogen signaling multiple pathways to impact gene transcription. Curr Genomics. 2006;7(8):497–508. | ||
Parveen S, Sahoo SK. Long circulating chitosan/PEG blended PLGA nanoparticle for tumor drug delivery. Eur J Pharmacol. 2011;670(2–3):372–383. | ||
Tang W, Ng SC. Facile synthesis of mono-6-amino-6-deoxy-alpha-, beta-, gamma-cyclodextrin hydrochlorides for molecular recognition, chiral separation and drug delivery. Nat Protoc. 2008;3(4):691–697. | ||
Anand R, Malanga M, Manet I, et al. Citric acid-γ-cyclodextrin crosslinked oligomers as carriers for doxorubicin delivery. Photochem Photobiol Sci. 2013;12(10):1841–1854. | ||
Kabouridis PS, Janzen J, Magee AL, Ley SC. Cholesterol depletion disrupts lipid rafts and modulates the activity of multiple signaling pathways in T lymphocytes. Eur J Immunol. 2000;30(3):954–963. | ||
Parker GJ, Law TL, Lenoch FJ, Bolger RE. Development of high throughput screening assays using fluorescence polarization: nuclear receptor-ligand-binding and kinase/phosphatase assays. J Biomol Screen. 2000;5(2):77–88. | ||
Nilsson S, Mäkelä S, Treuter E, et al. Mechanisms of estrogen action. Physiol Rev. 2001;81(4):1535–1565. | ||
Chaudhri RA, Olivares-Navarrete R, Cuenca N, Hadadi A, Boyan BD, Schwartz Z. Membrane estrogen signaling enhances tumorigenesis and metastatic potential of breast cancer cells via estrogen receptor-α36 (ERα36). J Biol Chem. 2012;287(10):7169–7181. | ||
Albanito L, Madeo A, Lappano R, et al. G protein-coupled receptor 30 (GPR30) mediates gene expression changes and growth response to 17beta-estradiol and selective GPR30 ligand G-1 in ovarian cancer cells. Cancer Res. 2007;67(4):1859–1866. | ||
Meloche S, Pouysségur J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene. 2007;26(22):3227–3239. | ||
Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298(5600):1911–1912. | ||
Dent P. Crosstalk between ERK, AKT, and cell survival. Cancer Biol Ther. 2014;15(3):245–246. | ||
Filardo EJ, Quinn JA, Bland KI, Frackelton AR Jr. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol. 2000;14(10):1649–1660. | ||
Banerjee S, Chambliss KL, Mineo C, Shaul PW. Recent insights into non-nuclear actions of estrogen receptor-alpha. Steroids. 2014;81:64–69. | ||
Böttner M, Thelen P, Jarry H. Estrogen receptor-beta: tissue distribution and the still largely enigmatic physiological function. J Steroid Biochem Mol Biol. 2014;139:245–251. | ||
Wehling M, Lösel R. Non-genomic steroid hormone effects: membrane or intracellular receptors? J Steroid Biochem Mol Biol. 2006;102(1–5):180–183. | ||
Watson CS, Alyea RA, Jeng YJ, Kochukov MY. Nongenomic actions of low concentration estrogens and xenoestrogens on multiple tissues. Mol Cell Endocrinol. 2007;274(1–2):1–7. | ||
Deroo BJ, Korach KS. Estrogen receptors and human disease. J Clin Invest. 2006;116(3):561–570. | ||
Kim HY, Sohn J, Wijewickrama GT, et al. Click synthesis of estradiol-cyclodextrin conjugates as cell compartment selective estrogens. Bioorg Med Chem. 2010;18(2):809–821. | ||
Clark S, Rainville J, Zhao X, Katzenellenbogen BS, Pfaff D, Vasudevan N. Estrogen receptor-mediated transcription involves the activation of multiple kinase pathways in neuroblastoma cells. J Steroid Biochem Mol Biol. 2014;139:45–53. | ||
Bulayeva NN, Gametchu B, Watson CS. Quantitative measurement of estrogen-induced ERK 1 and 2 activation via multiple membrane-initiated signaling pathways. Steroids. 2004;69(3):181–192. | ||
Adlanmerini M, Solinhac R, Abot A, et al. Mutation of the palmitoylation site of estrogen receptor-α in vivo reveals tissue-specific roles for membrane versus nuclear actions. Proc Natl Acad Sci U S A. 2014;111(2):E283–E290. | ||
Lea WA, Simeonov A. Fluorescence polarization assays in small molecule screening. Expert Opin Drug Discov. 2011;6(1):17–32. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.