Back to Journals » Therapeutics and Clinical Risk Management » Volume 11
Continual evolution of type 2 diabetes: an update on pathophysiology and emerging treatment options
Authors Cornell S
Received 8 May 2014
Accepted for publication 21 July 2014
Published 16 April 2015 Volume 2015:11 Pages 621—632
DOI https://doi.org/10.2147/TCRM.S67387
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Susan Cornell
Chicago College of Pharmacy, Midwestern University, Downers Grove, IL, USA
Abstract: Diabetes is a complex and progressive disease that has a major societal and economic impact. The most common form of diabetes, type 2 diabetes mellitus (T2DM), is a multifactorial disease, the pathophysiology of which involves not only the pancreas but also the liver, skeletal muscle, adipose tissue, gastrointestinal tract, brain, and kidney. Novel therapies with mechanisms of action that are different from most existing drugs are emerging. One such class consists of compounds that inhibit renal sodium-glucose cotransporter 2, which is responsible for the bulk of glucose reabsorption by the kidneys. This new class of compounds improves glycemic control independently of insulin and promotes weight reduction, providing an additional tool to treat patients with T2DM. This review discusses the underlying pathophysiology of T2DM, clinical guidelines, and available and emerging treatment options, with particular emphasis on sodium-glucose cotransporter 2 inhibitors.
Keywords: diabetes, hyperglycemia, oral antidiabetic therapies, pharmacotherapy, sodium-glucose cotransporter 2
Introduction
Diabetes is a complex and potentially debilitating disease that affects an estimated 8.3% of the adult population or 382 million people worldwide.1 The region with the highest number of adults with diabetes, ie, 138 million, is the Western Pacific, which includes the People’s Republic of China.1 It is estimated that 29.1 million people in the USA (9.3% of the population) have diabetes.2 If current trends continue, it is estimated that 592 million people worldwide will have diabetes by 2035.1
Diabetes care has a major economic impact in both developed and developing countries. Estimated global health care costs to treat and prevent diabetes were at least $548 billion in 2011.1 In the USA, the total cost of diabetes was estimated to be $245 billion in 20122 and may exceed $500 billion by 2025.3
Type 2 diabetes mellitus (T2DM) accounts for 90%–95% of new cases of diabetes.2 T2DM pathophysiology involves at least seven organs and tissues, including the pancreas, liver, skeletal muscle, adipose tissue, brain, gastrointestinal tract, and kidney (Figure 1).4 Reduced sensitivity to insulin (ie, impaired insulin-mediated glucose disposal or insulin resistance) in liver, muscle, and adipose tissue, and a progressive decline in pancreatic β-cell function leading to impaired insulin secretion, eventually result in hyperglycemia, the hallmark feature of T2DM. The purpose of this review is to discuss the underlying pathophysiology of T2DM, clinical treatment guidelines, and available and emerging treatment options, with emphasis on the newest class of antihyperglycemic drugs, the sodium-glucose cotransporter 2 (SGLT2) inhibitors.
 | Figure 1 Multiorgan and tissue pathophysiology of type 2 diabetes. |
Pathophysiology
Pancreas
Impairment of insulin action and of β-cell function occurs very early in the development of T2DM.5 Insulin resistance can be detected in individuals with normal glucose tolerance who are at higher risk for development of T2DM 10–20 years before the disease is diagnosed.6 Further, individuals who are transitioning from impaired glucose tolerance to T2DM may have already lost up to 80% of their β-cell function.4 Mechanisms thought to play a role in the decline of β-cell function include the following:
Genetics
The clustering of T2DM in families has long been recognized.7 A number of genes associated with insulin and β-cell dysfunction have been identified in patients with T2DM, including genetic variants associated with pancreatic development and insulin storage and secretion.8 With insulin resistance comes an increased need for biosynthesis and release of insulin. It has been proposed that a genetic polymorphism in patients predisposed to T2DM results in failure of the β cell to adapt to the increased demand for insulin.9
Age
Numerous studies have demonstrated an age-related decline in β-cell function and insulin secretion.10 This is consistent with the increased prevalence of T2DM with aging.2
Diet and exercise
Obesity and physical inactivity are major factors in the increased prevalence of T2DM worldwide11 and are associated with insulin resistance.4 Diets high in rapidly absorbable carbohydrates result in elevated insulin and blood glucose levels,11 and the deposition of fat in liver and muscle increases insulin resistance in these tissues.4 These factors increase the demand for insulin, and in the long term may lead to progressive β-cell failure.4,11
Glucotoxicity
Chronic exposure to elevated glucose concentrations impairs β-cell function and insulin secretion. The mechanisms involved in glucotoxicity remain to be elucidated but likely involve impairment of insulin gene expression, chronic oxidative stress, and apoptosis.12
Lipotoxicity
Elevated plasma concentrations of free fatty acids (FFAs) impair insulin secretion in patients at risk for development of T2DM.13,14 Elevated FFAs in β-cells lead to increased oxidative stress and apoptosis.15
Liver
The liver is the main organ responsible for glucose production.16 Hepatic glucose production and release into the circulation comes from both gluconeogenesis and glycogenolysis.16,17 In patients with T2DM, the liver overproduces glucose because it becomes resistant to the suppressive effects of insulin.4 Other factors, such as lack of suppression of postprandial glucagon secretion from pancreatic α cells in patients with T2DM,18 increased circulating glucagon, and increased sensitivity of the liver to glucagon, also contribute to increased hepatic glucose production.4
Muscle
Insulin-stimulated transport of glucose into skeletal muscle is the primary mechanism for the disposal of an exogenous glucose load.19 The major transporter involved in the uptake of glucose into skeletal muscle, glucose transporter 4 (GLUT4),19 is also expressed in adipocytes and cardiac muscle and is responsible for insulin-stimulated glucose uptake into these tissues.20 Insulin and exercise acutely stimulate the translocation of GLUT4 to the membrane of muscle cells, resulting in increased glucose uptake.19,20 In patients with T2DM, skeletal muscle is resistant to the actions of insulin because of defects in insulin signaling and often a low level of physical activity.21 This leads to a decrease in glucose uptake that contributes to the development of hyperglycemia.
Adipose tissue
In patients with T2DM, adipocytes are resistant to the antilipolytic effect of insulin, resulting in elevation of circulating FFAs. Chronic increases in FFAs stimulate gluconeogenesis, induce hepatic and muscle insulin resistance, and impair insulin secretion.22 These lipid-induced changes in T2DM are part of a constellation of changes induced by excess FFAs, termed lipotoxicity.15 Dysfunctional adipose tissue produces excessive amounts of inflammatory and atherogenic cytokines that can induce insulin resistance and concurrently fails to adequately secrete insulin-sensitizing adipocytokines.22
Much interest has recently been focused on “brown fat”, a very metabolically active adipose tissue previously thought to be present only in small mammals and newborn humans, where it functions in cold-induced or diet-induced heat production.23 However, recent studies have shown that brown fat is also present in human adults.24–27 In contrast with white adipose tissue, which stores excess energy as triglycerides,23 brown adipose tissue uses small intracellular triglyceride vacuoles as a main energy source to produce heat.28 The mass of brown fat has been shown to be directly related to resting metabolic rate26 and inversely correlated with age, total body fat, and body mass index.24–26 In addition, exercise has been shown to induce the “browning” of white adipose tissue.29 These findings suggest that brown fat may play an important role in energy metabolism and in body weight control. However, the role of brown fat in the pathogenesis of T2DM is unclear.
Brain
Insulin can cross the blood-brain barrier and, by modulating the expression of various neuropeptides involved in food intake, suppress appetite.30 In patients with T2DM, the brain may become insulin resistant, such that the inhibitory effect of insulin on appetite is lost.30 In fact, central insulin resistance may develop in individuals at risk for T2DM who are otherwise healthy.31
Amylin, a peptide synthesized and cosecreted with insulin from β cells, decreases food intake32 by sensitizing the area postrema and nucleus of the solitary tract to other metabolic signals that reduce food intake, such as cholecystokinin and glucose.33 Amylin also slows gastric emptying, and reduces postprandial glucagon release.32 In patients with T2DM, as β-cell function progressively declines, amylin secretion is reduced and its satiety-producing effects wane.32
Leptin and ghrelin are two other hormones that act centrally to control food intake and body weight homeostasis. Leptin is mainly produced and secreted by adipocytes,34 and its blood concentration is proportional to the percentage of body fat.35 A major effect of leptin is to decrease food intake and body weight by acting on areas in the hypothalamus involved in feeding behavior and energy balance.36 Many individuals with T2DM are obese, have high circulating levels of leptin, and are resistant to exogenous leptin.34 However, a subset of obese individuals has low levels of leptin and in these individuals exogenous leptin improves obesity.37 Although exogenous leptin prevents insulin resistance in animal models of diabetes,38 treatment of obese patients with T2DM did not improve insulin sensitivity.39
Ghrelin is secreted mainly by the stomach during fasting or caloric restriction and acts in the central nervous system to promote food intake.40 Ghrelin and its receptor are also present in pancreatic islet cells.41,42 In healthy humans, exogenous ghrelin reduces insulin secretion43,44 and causes hyperglycemia.43,45 In a population-based study, low plasma ghrelin concentrations were associated with increased fasting insulin concentrations, insulin resistance, and an increased prevalence of T2DM.46
Gastrointestinal tract
Glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) are hormones released by the gastrointestinal tract (incretins) in response to nutrient ingestion.47 GLP-1 and GIP act on β cells to stimulate the release of insulin and are responsible for up to 60% of insulin secretion following a meal.48 GLP-1 also promotes satiety, slows gastric emptying, and inhibits glucagon secretion, thus reducing hepatic glucose production.49 Patients with T2DM have impaired secretion of GLP-1 and reduced responsiveness to GIP.48 This results in increased gastrointestinal motility, decreased glucose-dependent insulin secretion, increased glucagon secretion, and increased liver glucose release, all of which adversely affect glycemic control.
Kidney
Under normal conditions, more than 99% of glucose filtered by the kidneys is reabsorbed in the proximal tubule.50 The bulk of glucose is reabsorbed by the SGLT2, in concert with a facilitative glucose transporter, GLUT2.50 Once plasma glucose concentrations exceed the renal threshold for reabsorption (approximately 180 mg/dL in healthy individuals), glucose starts to appear in the urine.51 A recent study demonstrated that the capacity of the kidney to reabsorb glucose is increased in patients with T2DM compared with matched healthy individuals.52 Therefore, in patients with T2DM, the kidneys reabsorb glucose in excess and return it to the circulation, potentially worsening hyperglycemia.
The liver and kidneys are the only organs that possess the requisite enzymes for gluconeogenesis and that subsequently release newly formed glucose into the circulation.16 In the postabsorptive (fasting) state in healthy humans, renal gluconeogenesis accounts for approximately 20% of total glucose released into the circulation, with the liver contributing the remainder.16,53 It has been suggested that renal glucose synthesis is increased in patients with T2DM relative to healthy individuals.54 Therefore, in patients with T2DM, the kidneys may further exacerbate hyperglycemia by continued glucose reabsorption and increased production of glucose.
Treatment goals
T2DM is a major risk factor for the development of cardiovascular disease,55 as well as microvascular complications, including chronic kidney disease,56 end-stage renal disease requiring dialysis or transplantation,57 and blindness.2 Hyperglycemia is the key determinant of microvascular complications. Randomized controlled trials have shown that intensive glycemic control can reduce the risk of microvascular complications in patients with T2DM.58–60 Therefore, the major treatment goal for T2DM is to reduce hyperglycemia and to manage comorbidities, especially those associated with cardiovascular disease (hypertension and dyslipidemia). Lifestyle changes (diet and exercise) and intensive pharmacotherapy of hyperglycemia and of multiple risk factors substantially reduce the rates of death, cardiovascular events, and progression to end-stage renal disease and retinopathy in patients with T2DM.61
Hyperglycemia should be monitored over the long term by measurement of glycated hemoglobin (A1C) at least twice a year in all patients with diabetes and at least four times yearly in patients who are not at target.62 Management of hyperglycemia should be individualized based on life expectancy, duration of T2DM, presence of comorbidities or vascular complications, potential for hypoglycemia or other adverse events, and patients’ attitudes and access to support systems.62,63 The American Diabetes Association (ADA)/European Association for the Study of Diabetes (EASD) and the American Association of Clinical Endocrinologists (AACE) recommend general A1C target levels <7.0% and ≤6.5%, respectively, for most patients.62,63 Strict control of blood pressure (BP) and lipids is also recommended.
Pharmacotherapy
Lifestyle changes that include a healthy diet, weight loss, increased physical activity, self-monitoring of blood glucose, and diabetes self-management education have broad benefits in controlling hyperglycemia and cardiovascular risk factors in patients with T2DM.64 Moreover, early intervention with intensive lifestyle changes can prevent or delay the development of T2DM in susceptible individuals.65 However, lifestyle changes may not be adequate to control hyperglycemia in the long term, and most patients will require pharmacotherapy to achieve and maintain glycemic control.64
Patients with T2DM usually require multiple medications to adequately treat their diabetes and associated comorbidities. There is currently no single agent available that affects all seven organs and tissues involved in the pathogenesis of T2DM; most drugs target up to four of the physiologic abnormalities.64 The major classes of drugs, mechanisms of action, and major contraindications are shown in Table 1. The ADA/EASD treatment recommendations (Figure 2) stress the importance of individualization of treatment and treatment intensification with combination therapy if A1C goals are not attained. The ADA/EASD recommendations are based on the glucose-lowering effect, actions that may reduce long-term complications, tolerability, safety, ease of use, and expense of glucose-lowering medications.62 The AACE algorithm for the treatment of T2DM also emphasizes the importance of individualized glycemic goals and treatment regimens, and stratifies treatment options based on initial A1C.63
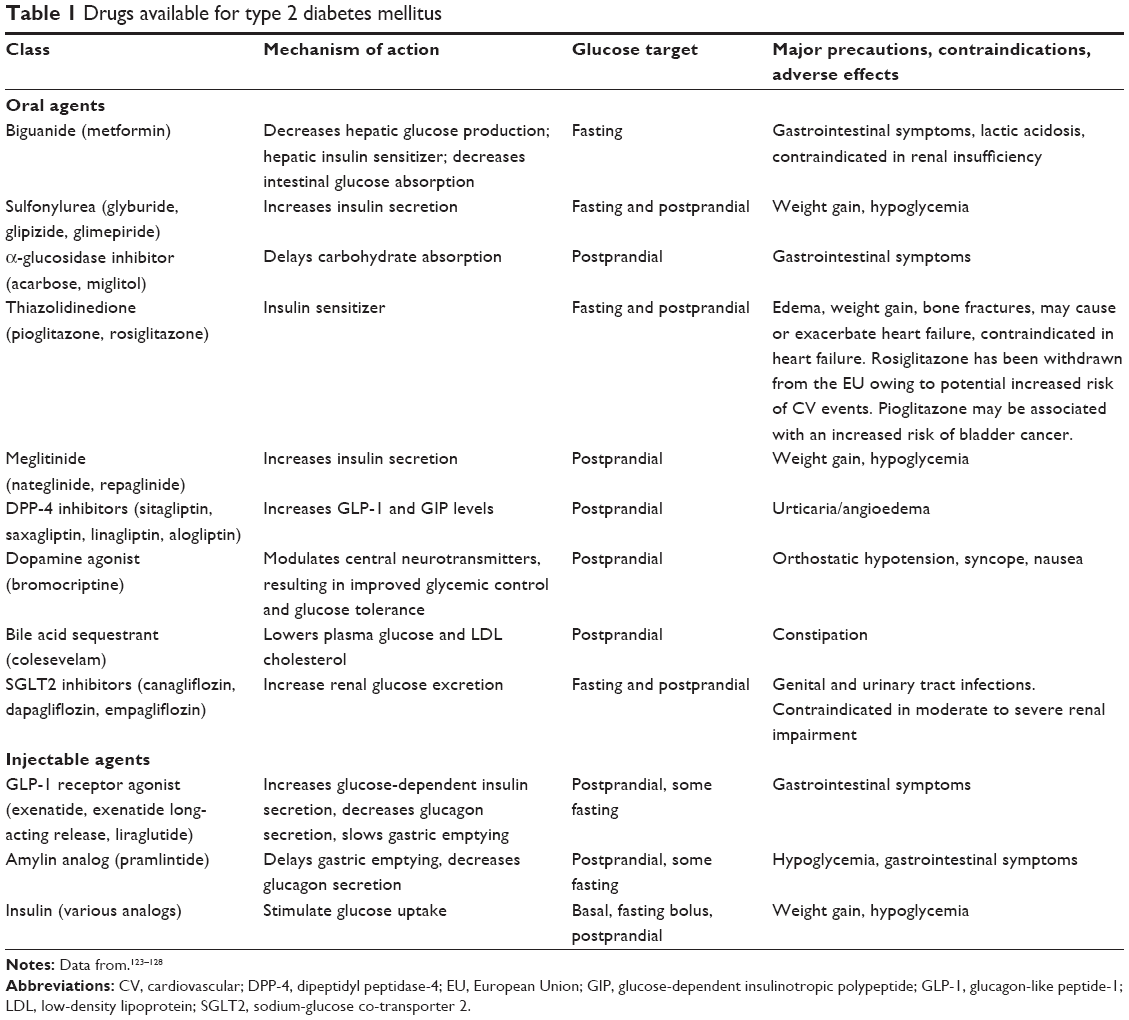 | Table 1 Drugs available for type 2 diabetes mellitus |
 | Figure 2 General recommendations for the treatment of type 2 diabetes from the American Diabetes Association/European Association for the Study of Diabetes. |
Despite the availability of several glucose-lowering drug classes, a recent survey of patients with diagnosed diabetes from 1999 to 2006 found that only 57% achieved an A1C <7%, and only 12% achieved the recommended A1C, BP, and low-density lipoprotein cholesterol goals.66 The reasons for failure to achieve recommended treatment goals may include lack of treatment initiation and intensification, patient nonadherence,67 and progressive decline in β-cell function, rendering therapies dependent on insulin secretion or action less effective over time.5 Accordingly, new pharmacologic therapies with mechanisms of action that are independent of insulin secretion or action and with acceptable safety profiles and a low risk of hypoglycemia and weight gain may enhance the ability of patients to achieve and maintain glycemic control.
New and emerging therapies
SGLT2 inhibitors
As discussed above, the kidney plays an important role in glucose homeostasis. Inhibition of the major transporter involved in glucose reabsorption by the kidney, SGLT2,68 is an attractive, insulin-independent mechanism for increasing urinary glucose excretion in the setting of hyperglycemia.69 Because the actions of SGLT2 inhibitors are independent of insulin, there is less risk of major hypoglycemic events70 compared with agents dependent on insulin action, and there is also the potential for effectiveness in advanced disease. Moreover, increased excretion of glucose and associated calories by SGLT2 inhibitors decreases body weight.71 Finally, there is a potential for combination therapy with other classes of agents with complementary mechanisms of action to optimize treatment.72 However, because the mechanism of action of SGLT2 inhibitors depends on the filtration of glucose by the kidney, as the glomerular filtration rate (GFR) declines, the amount of glucose filtered is reduced, which decreases glucosuria and the efficacy of these drugs.73 A number of selective SGLT2 inhibitors are in various stages of clinical development for the treatment of T2DM.74
Dapagliflozin
Dapagliflozin (AstraZeneca, Wilmington, DE, USA) is approved as a treatment for T2DM in the European Union, USA, and other countries. In Phase III trials, dapagliflozin reduced A1C (up to approximately −0.7%, placebo-corrected), fasting plasma glucose (FPG, up to approximately −27 mg/dL), and in most studies, body weight by 2–3 kg in patients with T2DM who were inadequately controlled with diet and exercise,75 metformin,76,77 glimepiride,78 pioglitazone,79 sitagliptin with and without metformin,80 and insulin.81 In addition, combination of dapagliflozin (5 or 10 mg/day) and metformin extended release as initial therapy in treatment-naïve patients was more effective than either drug alone in reducing A1C; dapagliflozin 10 mg alone was also noninferior to metformin.82 Finally, in patients inadequately controlled with metformin, dapagliflozin (≤10 mg/day) was noninferior to glipizide (≤20 mg/day) in reducing A1C.77
In patients with T2DM and moderate renal impairment (estimated GFR [eGFR] 30–59 mL/min/1.73 m2), mean decreases in A1C were similar for placebo (−0.32%) and for dapagliflozin 5 mg/day (−0.41%) and 10 mg/day (−0.44%) after 24 weeks of treatment.83 Although there was no significant effect on glycemic parameters in these patients, a mean reduction from baseline in body weight was observed with dapagliflozin 5 mg/day (−1.3 kg) and 10 mg/day (−1.7 kg) versus an increase (0.7 kg) with placebo.
In a pooled analysis of 12 Phase II/III trials, dapagliflozin 10 mg/day reduced systolic BP from baseline by −4.4 mmHg and diastolic BP by −2.1 mmHg compared with changes of −0.9 mmHg in systolic BP and −0.5 mmHg in diastolic BP with placebo.84 Dapagliflozin treatment was associated with significant weight reduction of up to 3 kg over 24 weeks.76,78,79,81 The reduction in body weight with dapagliflozin is largely the result of a reduction in body fat.71 The positive effects of dapagliflozin on hyperglycemia and body weight were sustained for up to two years when added to insulin85 or metformin therapy.86,87
Dapagliflozin was generally well tolerated, and adverse events were balanced across treatment groups.75–79,81 There was a low incidence of hypoglycemia when dapagliflozin was used as monotherapy75 or as add-on to metformin,76,77 pioglitazone,79 or sitagliptin.80 The proportion of patients with hypoglycemia was higher in the dapagliflozin groups than in the placebo groups when added to glimepiride78 or insulin therapy,81 and was significantly lower compared with glipizide.77
In a 12-week randomized, double-blind study in patients with T2DM and normal renal function, dapagliflozin 10 mg/day reduced the measured GFR from baseline by −10.8% compared with −2.9% with placebo.88 Mean eGFR decreased from baseline up to week 2 and tended to return toward baseline by 12 weeks. In an analysis of 12 randomized placebo-controlled trials involving more than 4,000 patients with preserved renal function, dapagliflozin was associated with a reduction in eGFR at week 1 that slowly returned to baseline by week 24 and was maintained at that level for 2 years.89 The acute reductions in GFR may be due to reversible hemodynamic effects such as a decrease in BP or plasma volume as a result of the modest diuretic action of dapagliflozin.88 Because of the reduced efficacy in patients with renal impairment, dapagliflozin is not recommended in patients with moderate to severe renal impairment (eGFR <60 mL/min/1.73 m2).84
In data pooled from 12 randomized, placebo-controlled trials, dapagliflozin 2.5, 5, or 10 mg once daily was accompanied by an increased risk of vulvovaginitis or balanitis (4.1%–5.7% versus 0.9% with placebo).90 Diagnosed urinary tract infections occurred more frequently with dapagliflozin 5 mg (5.7%) and 10 mg (4.3%) compared with placebo (3.7%).91
Canagliflozin
Canagliflozin (Janssen Pharmaceuticals, Inc., Titusville, NJ, USA) is approved for the treatment of T2DM in the USA, European Union, and other countries. In Phase III trials, treatment with canagliflozin (100 and 300 mg/day) for 26 or 52 weeks increased glucose excretion and decreased A1C (maximum change versus placebo or comparator −1.16%) and FPG (−43 mg/dL) in patients with T2DM when used as monotherapy,92 as add-on therapy to metformin,93 or as add-on to metformin and sulfonylurea.94 In patients stabilized on metformin, canagliflozin 100 mg/day was noninferior (mean difference −0.01%; 95% confidence interval (CI) −0.11%, 0.09%) and 300 mg/day was superior (mean difference −0.12%; 95% CI −0.22%, −0.02%) to glimepiride (6 to 8 mg/day) in reducing A1C.93 Absolute decreases in body weight of up to 4.2 kg after 52 weeks of therapy with 300 mg/day have been reported.93 Canagliflozin (300 mg/day) as add-on to metformin and sulfonylurea was more effective than sitagliptin (100 mg/day) in reducing body weight, FPG, and systolic BP.95
In patients with T2DM and chronic kidney disease (eGFR ≥30 and <50 mL/min/1.73 m2), treatment with canagliflozin for 26 weeks reduced A1C from baseline by −0.33% with 100 mg/day and by −0.44% with 300 mg/day compared with −0.03% with placebo.96 Transient decreases in eGFR (−4 to −6 mL/min/1.73 m2) with canagliflozin were observed at 3 weeks and trended toward baseline at 26 weeks. As expected, based on the mechanism of action, these reductions in A1C in patients with impaired renal function were less than those reported in patients with normal renal function.92–95,97
In addition to inhibiting SGLT2, canagliflozin is a weak inhibitor of SGLT1, the transporter responsible for intestinal glucose absorption.98 In healthy individuals, canagliflozin reduced postprandial plasma glucose and insulin and delayed the appearance of oral glucose in plasma following a mixed-meal tolerance test.99 These results suggest that canagliflozin reduces postprandial glucose and insulin by increasing renal glucose excretion via SGLT2 inhibition and by delaying the intestinal absorption of glucose, possibly by inhibiting SGLT1.
An increase in symptomatic urinary tract infections92,93,96 and genital infections92–94,96 was observed with canagliflozin. In addition, canagliflozin was associated with a modest increase in bone fractures and a dose-dependent increase in low-density lipoprotein cholesterol.100
In patients with T2DM without kidney disease (eGFR >55 mL/min/1.73 m2), canagliflozin caused a small initial decrease in eGFR that was stable up to 52 weeks.93 In an analysis of pooled data from four randomized, placebo-controlled trials, canagliflozin caused early transient decreases in eGFR that remained stable or were attenuated over the course of 26 weeks.101 The changes in eGFR with canagliflozin 100 and 300 mg/day versus placebo were−1.6% and −3.0% versus −0.6% in patients <65 years of age and −2.6%, and −2.9% versus −0.4% in patients ≥65 years of age. Canagliflozin should not be used in patients with eGFR <45 mL/min/1.73 m2 and the dose is limited to 100 mg/day in patients with eGFR of 45–<60 mL/min/1.73 m2.102
Empagliflozin
Empagliflozin (Boehringer Ingelheim, Ingelheim, Germany, and Eli Lilly, Indianapolis, IN, USA) is approved in the European Union and USA. In patients with T2DM, empagliflozin increased glucose excretion and decreased FPG (up to −32 mg/dL, placebo-corrected) and A1C (maximum of −0.69%) when used as monotherapy for 12 weeks,103 as add-on to metformin for 12 weeks,104 or as add-on to pioglitazone for 24 weeks.105 As add-on therapy to metformin and sulfonylurea, placebo-corrected changes in A1C and FPG after 24 weeks were −0.65% and −0.60% for empagliflozin 10 and 25 mg/day, respectively.106 Decreases in body weight103–106 and systolic BP106 have also been reported. Improvements in glycemic control and weight reduction were sustained for up to 90 weeks.107 An increased risk of genital and urinary tract infections was observed with empagliflozin.103,104,106,107
Ipragliflozin
Ipragliflozin (Astellas, Tokyo, Japan, and Kotobuki, Hanishina, Japan) is approved in Japan for the treatment of T2DM. In a 12-week dose-ranging study in patients with T2DM, ipragliflozin (12.5, 50, 150, and 300 mg/day) dose-dependently reduced A1C by up to 0.81% (placebo-corrected) and body weight by up to 1.7 kg. A decrease in A1C of −0.72% was seen with metformin.108 In a similarly designed 12-week study in patients inadequately controlled on metformin (≥1,500 mg/day), ipragliflozin reduced A1C by a maximum of −0.48% at 300 mg.109 Urinary tract and genital infections, as well as episodes of hypoglycemia, were similar with ipragliflozin and placebo. In patients with impaired renal function ± T2DM, a single dose of ipragliflozin increased urinary glucose excretion in patients with mild (eGFR ≥60 to <90 mL/min/1.73 m2), moderate (eGFR ≥30 to <60 mL/min/1.73 m2), and severe (eGFR ≤15 to <30 mL/min/1.73 m2) renal impairment. However, absolute glucose excretion decreased as eGFR declined.110
Other SGLT2 inhibitors in earlier phases of clinical development that have been reported to have positive effects on glycemic parameters in patients with T2DM include luseogliflozin (Taisho, Tokyo, Japan),111 tofogliflozin (Chugai/Roche, Tokyo, Japan),112 ertugliflozin, (Pfizer, New York, NY, USA)113 and the SGLT2/SGLT1 inhibitor LX4211 (Lexicon Pharmaceuticals, The Woodlands, TX, USA).114
Receptor agonists
GPR119 agonists
The G-protein–coupled receptor (GPR)119 is expressed predominantly in the pancreas (β cells) and gastrointestinal tract in humans.115 Although the endogenous ligand for this receptor is unknown, stimulation with synthetic ligands results in increased release of insulin, GLP-1, and GIP.115 A number of GPR119 agonists are in development for T2DM,116,117 and published findings for JNJ-38431055 (Johnson & Johnson Research & Development, Inc., Raritan, NJ, USA) report that this agent caused an increase in post-meal GLP-1 and GIP in patients with T2DM and was not associated with hypoglycemia.118
Free fatty acid receptor 1 activators
The free fatty acid receptor 1 (FFAR1, also known as GPR40) is expressed mainly in pancreatic β cells.119 Activation of this receptor results in fatty acid–induced glucose-stimulated insulin secretion. In a 12-week Phase II clinical trial, the FFAR1 activator TAK-875 (Takeda, Osaka, Japan) significantly reduced A1C, compared with placebo, with no increase in hypoglycemia.120
11β-hydroxysteroid dehydrogenase type 1 inhibitors
Excess glucocorticoids can cause insulin resistance and, in some individuals, can lead to the development of T2DM.121 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) converts inactive cortisone to cortisol in target tissues. Therefore, inhibition of this enzyme may improve insulin sensitivity. In a randomized, placebo-controlled Phase II study of overweight or obese patients with T2DM exhibiting inadequate glycemic control with metformin, an 11β-HSD1 inhibitor, INCB13739 (Incyte, Wilmington, DE, USA), significantly reduced A1C and FPG compared with placebo.122 Body weight, insulin resistance, and total cholesterol levels were also reduced with treatment.
Summary and conclusion
The significant burden that T2DM imposes on individuals and society reinforces the need for achieving and maintaining glycemic control in these patients. The key to optimal control is early diagnosis and intensive treatment with a combination of agents that address the various pathophysiologic abnormalities in T2DM, thereby lowering fasting and postprandial glucose concentrations. Novel medications with mechanisms of action different from those of most existing drugs and with acceptable safety profiles (low rates of hypoglycemia and no weight gain) are essential for long-term glycemic control and for improving disease outcomes and comorbidities. SGLT2 inhibitors are the newest treatment option for T2DM. These agents improve glycemic control, lower fasting glucose concentrations, and promote weight loss. This new class of diabetes medication with a novel mechanism of action provides an additional option to improve glycemic control in patients with T2DM. Whether the investigational GPR119 agonists, FFAR1 activators, and 11β-HSD1 inhibitors are safe and effective treatments for T2DM will be determined by the results of longer-duration, Phase III studies.
Disclosure
Dr Cornell has received non-continuing medical education honoraria from the Johnson & Johnson Diabetes Insititute and has served on advisory boards for Sanofi, Bristol-Myers Squibb, and AstraZeneca. Editorial support was provided by Richard M Edwards and Janet E Matsuura from Complete Healthcare Communications, Inc., and was funded by Bristol–Myers Squibb and AstraZeneca.
References
International Diabetes Federation. 2013. IDF Atlas. Available from: http://www.idf.org/diabetesatlas. Accessed July 10, 2014. | ||
Centers for Disease Control and Prevention. 2014. National diabetes statistics report: estimates of diabetes and its burden in the United States, 2014. Available from: http://www.cdc.gov/diabetes/pubs/statsreport14/national-diabetes-report-web.pdf. Accessed July 16, 2014. | ||
Rowley WR, Bezold C. Creating public awareness: state 2025 diabetes forecasts. Popul Health Manag. 2012;15:194–200. | ||
DeFronzo RA. Banting Lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes. 2009;58:773–795. | ||
Fonseca VA. Defining and characterizing the progression of type 2 diabetes. Diabetes Care. 2009;32:S151–S156. | ||
Warram JH, Martin BC, Krolewski AS, Soeldner JS, Kahn CR. Slow glucose removal rate and hyperinsulinemia precede the development of type II diabetes in the offspring of diabetic parents. Ann Intern Med. 1990;113:909–915. | ||
Mills GW, Avery PJ, McCarthy MI, et al. Heritability estimates for beta cell function and features of the insulin resistance syndrome in UK families with an increased susceptibility to type 2 diabetes. Diabetologia. 2004;47:732–738. | ||
Grant RW, Moore AF, Florez JC. Genetic architecture of type 2 diabetes: recent progress and clinical implications. Diabetes Care. 2009;32:1107–1114. | ||
Kahn SE. Clinical review 135: the importance of beta-cell failure in the development and progression of type 2 diabetes. J Clin Endocrinol Metab. 2001;86:4047–4058. | ||
Chang AM, Halter JB. Aging and insulin secretion. Am J Physiol Endocrinol Metab. 2003;284:E7–E12. | ||
Hu FB. Globalization of diabetes: the role of diet, lifestyle, and genes. Diabetes Care. 2011;34:1249–1257. | ||
Poitout V, Robertson RP. Minireview: secondary beta-cell failure in type 2 diabetes – a convergence of glucotoxicity and lipotoxicity. Endocrinology. 2002;143:339–342. | ||
Carpentier A, Mittelman SD, Bergman RN, Giacca A, Lewis GF. Prolonged elevation of plasma free fatty acids impairs pancreatic beta-cell function in obese nondiabetic humans but not in individuals with type 2 diabetes. Diabetes. 2000;49:399–408. | ||
Kashyap S, Belfort R, Gastaldelli A, et al. A sustained increase in plasma free fatty acids impairs insulin secretion in nondiabetic subjects genetically predisposed to develop type 2 diabetes. Diabetes. 2003;52:2461–2474. | ||
Unger RH, Zhou YT. Lipotoxicity of beta-cells in obesity and in other causes of fatty acid spillover. Diabetes. 2001;50:S118–S121. | ||
Gerich JE, Meyer C, Woerle HJ, Stumvoll M. Renal gluconeogenesis: its importance in human glucose homeostasis. Diabetes Care. 2001;24:382–391. | ||
Heit JA, Leibson CL, Ashrani AA, Petterson TM, Bailey KR, Melton LJ 3rd. Is diabetes mellitus an independent risk factor for venous thromboembolism?: a population-based case-control study. Arterioscler Thromb Vasc Biol. 2009;29:1399–1405. | ||
Shah P, Vella A, Basu A, Basu R, Schwenk WF, Rizza RA. Lack of suppression of glucagon contributes to postprandial hyperglycemia in subjects with type 2 diabetes mellitus. J Clin Endocrinol Metab. 2000;85:4053–4059. | ||
Huang S, Czech MP. The GLUT4 glucose transporter. Cell Metab. 2007;5:237–252. | ||
Herman MA, Kahn BB. Glucose transport and sensing in the maintenance of glucose homeostasis and metabolic harmony. J Clin Invest. 2006;116:1767–1775. | ||
Cusi K, Maezono K, Osman A, et al. Insulin resistance differentially affects the PI 3-kinase- and MAP kinase-mediated signaling in human muscle. J Clin Invest. 2000;105:311–320. | ||
Bays H, Mandarino L, DeFronzo RA. Role of the adipocyte, free fatty acids, and ectopic fat in pathogenesis of type 2 diabetes mellitus: peroxisomal proliferator-activated receptor agonists provide a rational therapeutic approach. J Clin Endocrinol Metab. 2004;89:463–478. | ||
Saely CH, Geiger K, Drexel H. Brown versus white adipose tissue: a mini-review. Gerontology. 2012;58:15–23. | ||
Cypess AM, Lehman S, Williams G, et al. Identification and importance of brown adipose tissue in adult humans. N Engl J Med. 2009;360:1509–1517. | ||
Saito M, Okamatsu-Ogura Y, Matsushita M, et al. High incidence of metabolically active brown adipose tissue in healthy adult humans: effects of cold exposure and adiposity. Diabetes. 2009;58:1526–1531. | ||
van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, et al. Cold-activated brown adipose tissue in healthy men. N Engl J Med. 2009;360:1500–1508. | ||
Virtanen KA, Lidell ME, Orava J, et al. Functional brown adipose tissue in healthy adults. N Engl J Med. 2009;360:1518–1525. | ||
Ouellet V, Labbe SM, Blondin DP, et al. Brown adipose tissue oxidative metabolism contributes to energy expenditure during acute cold exposure in humans. J Clin Invest. 2012;122:545–552. | ||
Bostrom P, Wu J, Jedrychowski MP, et al. A PGC1-alpha-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature. 2012;481:463–468. | ||
Pagotto U. Where does insulin resistance start? The brain. Diabetes Care. 2009;32:S174–S177. | ||
Tschritter O, Preissl H, Hennige AM, et al. The cerebrocortical response to hyperinsulinemia is reduced in overweight humans: a magnetoencephalographic study. Proc Natl Acad Sci U S A. 2006;103:12103–12108. | ||
Roth JD, Maier H, Chen S, Roland BL. Implications of amylin receptor agonism: integrated neurohormonal mechanisms and therapeutic applications. Arch Neurol. 2009;66:306–310. | ||
Woods SC, Lutz TA, Geary N, Langhans W. Pancreatic signals controlling food intake; insulin, glucagon and amylin. Philos Trans R Soc Lond B Biol Sci. 2006;361:1219–1235. | ||
Friedman JM. Leptin at 14 y of age: an ongoing story. Am J Clin Nutr. 2009;89:973S–979S. | ||
Considine RV, Sinha MK, Heiman ML, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med. 1996;334:292–295. | ||
Morton GJ, Schwartz MW. Leptin and the central nervous system control of glucose metabolism. Physiol Rev. 2011;91:389–411. | ||
Farooqi IS, O’Rahilly S. Leptin: a pivotal regulator of human energy homeostasis. Am J Clin Nutr. 2009;89:980S–984S. | ||
German JP, Wisse BE, Thaler JP, et al. Leptin deficiency causes insulin resistance induced by uncontrolled diabetes. Diabetes. 2010;59:1626–1634. | ||
Mittendorfer B, Horowitz JF, DePaoli AM, McCamish MA, Patterson BW, Klein S. Recombinant human leptin treatment does not improve insulin action in obese subjects with type 2 diabetes. Diabetes. 2011;60:1474–1477. | ||
Heppner KM, Tong J. Mechanisms in endocrinology: regulation of glucose metabolism by the ghrelin system: multiple players and multiple actions. Eur J Endocrinol. 2014;171:R21–R32. | ||
Date Y, Nakazato M, Hashiguchi S, et al. Ghrelin is present in pancreatic alpha-cells of humans and rats and stimulates insulin secretion. Diabetes. 2002;51:124–129. | ||
Volante M, Allia E, Gugliotta P, et al. Expression of ghrelin and of the GH secretagogue receptor by pancreatic islet cells and related endocrine tumors. J Clin Endocrinol Metab. 2002;87:1300–1308. | ||
Broglio F, Arvat E, Benso A, et al. Ghrelin, a natural GH secretagogue produced by the stomach, induces hyperglycemia and reduces insulin secretion in humans. J Clin Endocrinol Metab. 2001;86:5083–5086. | ||
Tong J, Prigeon RL, Davis HW, et al. Ghrelin suppresses glucose-stimulated insulin secretion and deteriorates glucose tolerance in healthy humans. Diabetes. 2010;59:2145–2151. | ||
Garin MC, Burns CM, Kaul S, Cappola AR. Clinical review: the human experience with ghrelin administration. J Clin Endocrinol Metab. 2013;98:1826–1837. | ||
Pöykkö SM, Kellokoski E, Hörkkö S, Kauma H, Kesäniemi YA, Ukkola O. Low plasma ghrelin is associated with insulin resistance, hypertension, and the prevalence of type 2 diabetes. Diabetes. 2003;52:2546–2553. | ||
Freeman JS. Role of the incretin pathway in the pathogenesis of type 2 diabetes mellitus. Cleve Clin J Med. 2009;76:S12–S19. | ||
Nauck MA, Baller B, Meier JJ. Gastric inhibitory polypeptide and glucagon-like peptide-1 in the pathogenesis of type 2 diabetes. Diabetes. 2004;53:S190–S196. | ||
Dungan K, Buse JB. Glucagon-like peptide 1–based therapies for type 2 diabetes: a focus on exenatide. Clin Diabetes. 2005;23:56–62. | ||
Bakris GL, Fonseca VA, Sharma K, Wright EM. Renal sodium-glucose transport: role in diabetes mellitus and potential clinical implications. Kidney Int. 2009;75:1272–1277. | ||
Ferrannini E. Sodium-glucose transporter-2 inhibition as an antidiabetic therapy. Nephrol Dial Transplant. 2010;25:2041–2043. | ||
DeFronzo RA, Hompesch M, Kasichayanula S, et al. Characterization of renal glucose reabsorption in response to dapagliflozin in healthy subjects and subjects with type 2 diabetes. Diabetes Care. 2013;36:3169–3176. | ||
Stumvoll M, Chintalapudi U, Perriello G, Welle S, Gutierrez O, Gerich J. Uptake and release of glucose by the human kidney. Postabsorptive rates and responses to epinephrine. J Clin Invest. 1995;96:2528–2533. | ||
Meyer C, Stumvoll M, Nadkarni V, Dostou J, Mitrakou A, Gerich J. Abnormal renal and hepatic glucose metabolism in type 2 diabetes mellitus. J Clin Invest. 1998;102:619–624. | ||
Fox CS, Coady S, Sorlie PD, et al. Increasing cardiovascular disease burden due to diabetes mellitus: the Framingham Heart Study. Circulation. 2007;115:1544–1550. | ||
Coresh J, Selvin E, Stevens LA, et al. Prevalence of chronic kidney disease in the United States. JAMA. 2007;298:2038–2047. | ||
Foley RN, Collins AJ. End-stage renal disease in the United States: an update from the United States Renal Data System. J Am Soc Nephrol. 2007;18:2644–2648. | ||
Ohkubo Y, Kishikawa H, Araki E, et al. Intensive insulin therapy prevents the progression of diabetic microvascular complications in Japanese patients with non-insulin-dependent diabetes mellitus: a randomized prospective 6-year study. Diabetes Res Clin Pract. 1995;28:103–117. | ||
[No authors listed]. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998a;352:854–865. | ||
[No authors listed]. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998b;352:837–853. | ||
Gaede P, Lund-Andersen H, Parving HH, Pedersen O. Effect of a multifactorial intervention on mortality in type 2 diabetes. N Engl J Med. 2008;358:580–591. | ||
American Diabetes Association. Approaches to glycemic treatment. Sec 7. In Standards of medical care in diabetes – 2015. Diabetes Care. 2015;38(Suppl 1):S41–S48. | ||
Garber AJ, Abrahamson MJ, Barzilay JI, et al. American Association of Clinical Endocrinologists’ comprehensive diabetes management algorithm 2013 consensus statement – executive summary. Endocr Pract. 2013;19:536–557. | ||
Nathan DM, Buse JB, Davidson MB, et al; American Diabetes Association; European Association for Study of Diabetes. Medical management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy: a consensus statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care. 2009;32:193–203. | ||
Knowler WC, Barrett-Connor E, Fowler SE, et al; Diabetes Prevention Program Research Group. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346:393–403. | ||
Cheung BM, Ong KL, Cherny SS, Sham PC, Tso AW, Lam KS. Diabetes prevalence and therapeutic target achievement in the United States, 1999 to 2006. Am J Med. 2009;122:443–453. | ||
Schmittdiel JA, Uratsu CS, Karter AJ, et al. Why don’t diabetes patients achieve recommended risk factor targets? Poor adherence versus lack of treatment intensification. J Gen Intern Med. 2008;23:588–594. | ||
Vallon V, Platt KA, Cunard R, et al. SGLT2 mediates glucose reabsorption in the early proximal tubule. J Am Soc Nephrol. 2011;22:104–112. | ||
List JF, Whaley JM. Glucose dynamics and mechanistic implications of SGLT2 inhibitors in animals and humans. Kidney Int. 2011;79:S20–S27. | ||
Gerich JE. Role of the kidney in normal glucose homeostasis and in the hyperglycaemia of diabetes mellitus: therapeutic implications. Diabet Med. 2010;27:136–142. | ||
Bolinder J, Ljunggren O, Kullberg J, et al. Effects of dapagliflozin on body weight, total fat mass, and regional adipose tissue distribution in patients with type 2 diabetes mellitus with inadequate glycemic control on metformin. J Clin Endocrinol Metab. 2012;97:1020–1031. | ||
Bailey CJ. SGLT2 inhibitors: glucuretic treatment for type 2 diabetes. Br J Diabetes Vasc Dis. 2010;10:193–199. | ||
Abdul-Ghani MA, Norton L, Defronzo RA. Role of sodium-glucose cotransporter 2 (SGLT 2) inhibitors in the treatment of type 2 diabetes. Endocr Rev. 2011;32:515–531. | ||
Chao EC, Henry RR. SGLT2 inhibition – a novel strategy for diabetes treatment. Nat Rev Drug Discov. 2010;9:551–559. | ||
Ferrannini E, Ramos SJ, Salsali A, Tang W, List JF. Dapagliflozin monotherapy in type 2 diabetic patients with inadequate glycemic control by diet and exercise: a randomized, double-blind, placebo-controlled, phase 3 trial. Diabetes Care. 2010;33:2217–2224. | ||
Bailey CJ, Gross JL, Pieters A, Bastien A, List JF. Effect of dapagliflozin in patients with type 2 diabetes who have inadequate glycaemic control with metformin: a randomised, double-blind, placebo-controlled trial. Lancet. 2010;375:2223–2233. | ||
Nauck MA, Del Prato S, Meier JJ, et al. Dapagliflozin versus glipizide as add-on therapy in patients with type 2 diabetes who have inadequate glycemic control with metformin: a randomized, 52-week, double-blind, active-controlled noninferiority trial. Diabetes Care. 2011;34:2015–2022. | ||
Strojek K, Yoon KH, Hruba V, Elze M, Langkilde AM, Parikh S. Effect of dapagliflozin in patients with type 2 diabetes who have inadequate glycaemic control with glimepiride: a randomized, 24-week, double-blind, placebo-controlled trial. Diabetes Obes Metab. 2011;13:928–938. | ||
Rosenstock J, Vico M, Wei L, Salsali A, List JF. Effects of dapagliflozin, an SGLT2 inhibitor, on HbA1c, body weight, and hypoglycemia risk in patients with type 2 diabetes inadequately controlled on pioglitazone monotherapy. Diabetes Care. 2012;35:1473–1478. | ||
Jabbour S, Hardy E, Sugg JE, Parikh S. Dapagliflozin as add-on therapy to sitagliptin with or without metformin: a randomized, double-blind, placebo-controlled study. Diabetes. 2012;61:A275. | ||
Wilding JPH, Woo V, Soler NG, et al; Dapagliflozin 006 Study Group. Long-term efficacy of dapagliflozin in patients with type 2 diabetes mellitus receiving high doses of insulin. Ann Intern Med. 2012b;156:405–415. | ||
Henry RR, Murray AV, Marmolejo MH, Hennicken D, Ptaszynska A, List JF. Dapagliflozin, metformin XR, or both: initial pharmacotherapy for type 2 diabetes, a randomised controlled trial. Int J Clin Pract. 2012;66:446–456. | ||
Kohan DE, Fioretto P, Tang W, List JF. Long-term study of patients with type 2 diabetes and moderate renal impairment shows that dapagliflozin reduces weight and blood pressure but does not improve glycemic control. Kidney Int. 2014;85:962–971. | ||
Farxiga® (dapagliflozin). 2014. Full Prescribing Information. Princeton, NJ, USA: Bristol-Myers Squibb, and Wilmington, DE, USA: AstraZeneca; 2014. | ||
Wilding JP, Woo V, Rohwedder K, Sugg J, Parikh S; Dapagliflozin 006 Study Group. Dapagliflozin in patients with type 2 diabetes receiving high doses of insulin: efficacy and safety over two years. Diabetes Obes Metab. 2014;16:124–136. | ||
Bailey CJ, Gross JL, Hennicken D, Iqbal N, Mansfield TA, List JF. Dapagliflozin add-on to metformin in type 2 diabetes inadequately controlled with metformin: a randomized, double-blind, placebo-controlled 102-week trial. BMC Med. 2013;11:43. | ||
Bolinder J, Ljunggren O, Johansson L, et al. Dapagliflozin maintains glycaemic control while reducing weight and body fat mass over 2 years in patients with type 2 diabetes mellitus inadequately controlled on metformin. Diabetes Obes Metab. 2014;16:159–169. | ||
Lambers Heerspink HJ, de Zeeuw D, Wie L, Leslie B, List J. Dapagliflozin a glucose-regulating drug with diuretic properties in subjects with type 2 diabetes. Diabetes Obes Metab. 2013;15:853–862. | ||
Ptaszynska A, Chalamandaris A, Sugg JE, Johnsson KM, Parikh S, List JL. Effect of dapagliflozin on renal function. Diabetes. 2012;61:A283. | ||
Johnsson KM, Ptaszynska A, Schmitz B, Sugg J, Parikh SJ, List JF. Vulvovaginitis and balanitis in patients with diabetes treated with dapagliflozin. J Diabetes Complications. 2013b;27:479–484. | ||
Johnsson KM, Ptaszynska A, Schmitz B, Sugg J, Parikh SJ, List JF. Urinary tract infections in patients with diabetes treated with dapagliflozin. J Diabetes Complications. 2013a;27:473–478. | ||
Stenlof K, Cefalu WT, Kim KA, et al. Efficacy and safety of canagliflozin monotherapy in subjects with type 2 diabetes mellitus inadequately controlled with diet and exercise. Diabetes Obes Metab. 2013;15:372–382. | ||
Cefalu WT, Leiter LA, Yoon KH, et al. Efficacy and safety of canagliflozin versus glimepiride in patients with type 2 diabetes inadequately controlled with metformin (CANTATA-SU): 52 week results from a randomised, double-blind, phase 3 non-inferiority trial. Lancet. 2013;382:941–950. | ||
Wilding JP, Mathieu C, Vercruysse F, et al. Canagliflozin (CANA), a sodium glucose co-transporter 2 inhibitor, improves glycemic control and reduces body weight in subjects with type 2 diabetes (T2D) inadequately controlled with metformin (MET) and sulfonylurea (SU). Diabetes. 2012a;61:A262. | ||
Schernthaner G, Gross JL, Rosenstock J, et al. Canagliflozin compared with sitagliptin for patients with type 2 diabetes who do not have adequate glycemic control with metformin plus sulfonylurea: a 52-week randomized trial. Diabetes Care. 2013;36:2508–2515. | ||
Yale JF, Bakris G, Cariou B, et al. Efficacy and safety of canagliflozin in subjects with type 2 diabetes and chronic kidney disease. Diabetes Obes Metab. 2013;15:463–473. | ||
Rosenstock J, Polodori D, Zhao Y, et al. Canagliflozin, an inhibitor of sodium glucose co-transporter 2, improves glycaemic control, lowers body weight, and improves beta cell function in subjects with type 2 diabetes on background metformin. Diabetologia. 2010b;53:S351. | ||
Wright EM, Loo DD, Hirayama BA. Biology of human sodium glucose transporters. Physiol Rev. 2011;91:733–794. | ||
Polidori D, Sha S, Mudaliar S, et al. Canagliflozin lowers postprandial glucose and insulin by delaying intestinal glucose absorption in addition to increasing urinary glucose excretion: results of a randomized, placebo-controlled study. Diabetes Care. 2013;36:2154–2161. | ||
US Food and Drug Administration. Endocrinologic and Metabolic Drugs Advisory Committee, January 10, 2013. Available from: http://www.fda.gov/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/EndocrinologicandMetabolicDrugsAdvisoryCommittee/ucm331504.htm. Accessed July 16, 2014. | ||
Sinclair A, Bode B, Harris S, et al. Efficacy and safety of canagliflozin compared with placebo in older patients with type 2 diabetes mellitus: a pooled analysis of clinical studies. BMC Endocr Disord. 2014;14:37. | ||
Invokana® (canagliflozin). Full Prescribing Information. Titusville, NJ, USA: Janssen Pharmaceuticals; 2013. | ||
Ferrannini E, Seman L, Seewaldt-Becker E, Hantel S, Pinnetti S, Woerle HJ. A Phase IIb, randomized, placebo-controlled study of the SGLT2 inhibitor empagliflozin in patients with type 2 diabetes. Diabetes Obes Metab. 2013a;15:721–728. | ||
Rosenstock J, Seman LJ, Jelaska A, et al. Efficacy and safety of empagliflozin, a sodium glucose cotransporter 2 (SGLT2) inhibitor, as add-on to metformin in type 2 diabetes with mild hyperglycaemia. Diabetes Obes Metab. 2013;15:1154–1160. | ||
Kovacs CS, Seshiah V, Swallow R, et al; EMPA-REG PIO™ trial investigators. Empagliflozin improves glycaemic and weight control as add-on therapy to pioglitazone or pioglitazone plus metformin in patients with type 2 diabetes: a 24-week, randomized, placebo-controlled trial. Diabetes Obes Metab. 2014;16:147–158. | ||
Haring HU, Merker L, Seewaldt-Becker E, et al; EMPA-REG METSU Trial Investigators. Empagliflozin as add-on to metformin plus sulfonylurea in patients with type 2 diabetes: a 24-week, randomized, double-blind, placebo-controlled trial. Diabetes Care. 2013;36:3396–3404. | ||
Woerle HJ, Ferrannini E, Berk A, et al. Safety and efficacy of empagliflozin as monotherapy or add-on to metformin in a 78-week open-label extension study in patients with type 2 diabetes. Diabetes. 2012;61:LB13. | ||
Fonseca VA, Ferrannini E, Wilding JP, et al. Active- and placebo-controlled dose-finding study to assess the efficacy, safety, and tolerability of multiple doses of ipragliflozin in patients with type 2 diabetes mellitus. J Diabetes Complications. 2013;27:268–273. | ||
Wilding JP, Ferrannini E, Fonseca VA, Wilpshaar W, Dhanjal P, Houzer A. Efficacy and safety of ipragliflozin in patients with type 2 diabetes inadequately controlled on metformin: a dose-finding study. Diabetes Obes Metab. 2013a;15:403–409. | ||
Ferrannini E, Veltkamp SA, Smulders RA, Kadokura T. Renal glucose handling: impact of chronic kidney disease and sodium-glucose cotransporter 2 inhibition in patients with type 2 diabetes. Diabetes Care. 2013b;36:1260–1265. | ||
Seino Y, Sasaki T, Fukatsu A, Samukawa Y, Sakai S, Watanabe T. Luseogliflozin (TS-071), a selective SGLT2 inhibitor, improves glycemic control and lowers body weight in Japanese patients with type 2 diabetes mellitus. Diabetes. 2012;61:A266. | ||
Kadowaki T, Ikeda S, Takano Y, et al. Tofogliflozin, a novel and selective SGLT2 inhibitor improves glycemic control and lowers body weight in patients with type 2 diabetes mellitus inadequately controlled on stable metformin or diet and excercise alone. Diabetes. 2012;61:A22. | ||
Mandema J, Sweeney K, Terra S, Sahasrabudhe V. Model-based meta-analysis of the HbA1c lowering effect of PF-04971729, a sodium glucose co-transporter-2 inhibitor (SGLT2i), in comparison with other SGLT2i and anti-diabetic agents (ADA). Diabetes. 2012;61:A260. | ||
Zambrowicz B, Freiman J, Brown PM, et al. LX4211, a dual SGLT1/SGLT2 inhibitor, improved glycemic control in patients with type 2 diabetes in a randomized, placebo-controlled trial. Clin Pharmacol Ther. 2012;92:158–169. | ||
Overton HA, Fyfe MC, Reynet C. GPR119, a novel G protein-coupled receptor target for the treatment of type 2 diabetes and obesity. Br J Pharmacol. 2008;153:S76–S81. | ||
GlaxoSmithKline. A study in type 2 diabetics of single and multiple doses of orally administered GSK1292263 to investigate the safety, tolerability, pharmacokinetics and pharmacodynamics. Available from: http://clinicaltrials.gov/ct2/show/NCT01119846?term=gsk1292263&rank=1. Accessed July 16, 2014. | ||
CymaBay Therapeutics, Inc. Study to evaluate the efficacy, safety, tolerability, and pharmacokinetics of MBX-2982 administered daily for 4 weeks as monotherapy in patients with type 2 diabetes. Available from: http://clinicaltrials.gov/ct2/show/NCT01035879?term=mbx2982&rank=1. Accessed July 16, 2014. | ||
Katz LB, Gambale JJ, Rothenberg PL, et al. Effects of JNJ-38431055, a novel GPR119 receptor agonist, in randomized, double-blind, placebo-controlled studies in subjects with type 2 diabetes. Diabetes Obes Metab. 2012;14:709–716. | ||
Kebede MA, Alquier T, Latour MG, Poitout V. Lipid receptors and islet function: therapeutic implications? Diabetes Obes Metab. 2009;11:10–20. | ||
Burant CF, Viswanathan P, Marcinak J, et al. TAK-875 versus placebo or glimepiride in type 2 diabetes mellitus: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet. 2012;379:1403–1411. | ||
Cooper MS, Stewart PM. 11Beta-hydroxysteroid dehydrogenase type 1 and its role in the hypothalamus-pituitary-adrenal axis, metabolic syndrome, and inflammation. J Clin Endocrinol Metab. 2009;94:4645–4654. | ||
Rosenstock J, Banarer S, Fonseca VA, et al; INCB13739-202 Principal Investigators. The 11-beta-hydroxysteroid dehydrogenase type 1 inhibitor INCB13739 improves hyperglycemia in patients with type 2 diabetes inadequately controlled by metformin monotherapy. Diabetes Care. 2010a;33:1516–1522. | ||
Inzucchi SE, Bergenstal RM, Buse JB, et al; American Diabetes Association (ADA); European Association for the Study of Diabetes (EASD). Management of hyperglycemia in type 2 diabetes: a patient-centered approach: position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care. 2012;35:1364–1379. | ||
Inzucchi SE, Bergenstal RM, Buse JB, et al; Management of hyperglycemia in type 2 diabetes, 2015: a patient-centered approach: update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care. 2015;38:140–149. | ||
Blonde L. Current antihyperglycemic treatment strategies for patients with type 2 diabetes mellitus. Cleve Clin J Med. 2009;76 Suppl 5:S4–S11. | ||
Zangeneh F, Kudva YC, Basu A. Insulin sensitizers. Mayo Clin Proc. 2003;78:471–479. | ||
Welchol® (colesevelam HCl). Full Prescribing Information. Parsippany, NJ, USA: Daiichi Sankyo, Inc.; 2010. | ||
Cycloset® (bromocriptine mesylate). Full Prescribing Information. Tiverton, RI: VeroScience, LLC; 2010. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.