Back to Journals » International Journal of General Medicine » Volume 14
Construction and Investigation of Competing Endogenous RNA Networks and Candidate Genes Involved in SARS-CoV-2 Infection
Authors Qi M, Liu B, Li S, Ni Z, Li F
Received 20 August 2021
Accepted for publication 30 September 2021
Published 12 October 2021 Volume 2021:14 Pages 6647—6659
DOI https://doi.org/10.2147/IJGM.S335162
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Mingran Qi,1 Bin Liu,2 Shuai Li,1 Zhaohui Ni,1,3– 5 Fan Li1,3– 6
1Department of Pathogenobiology, The Key Laboratory of Zoonosis, Chinese Ministry of Education, College of Basic Medicine, Jilin University, Changchun, Jilin, People’s Republic of China; 2Cardiovascular Disease Center, The First Hospital of Jilin University, Changchun, Jilin, People’s Republic of China; 3The Key Laboratory for Bionics Engineering, Ministry of Education, Jilin University, Changchun, Jilin, People’s Republic of China; 4Engineering Research Center for Medical Biomaterials of Jilin Province, Jilin University, Changchun, Jilin, People’s Republic of China; 5Key Laboratory for Health Biomedical Materials of Jilin Province, Jilin University, Changchun, Jilin, People’s Republic of China; 6State Key Laboratory of Pathogenesis, Prevention and Treatment of High Incidence Diseases in Central Asia, Urumqi, Xinjiang, People’s Republic of China
Correspondence: Zhaohui Ni; Fan Li
Department of Pathogenobiology, The Key Laboratory of Zoonosis, Chinese Ministry of Education, College of Basic Medicine, Jilin University, No. 126 Xinmin Street, Changchun, Jilin, 130021, People’s Republic of China
Tel +86-431-85619107
Email [email protected]; [email protected]
Introduction: The current COVID-19 pandemic caused by a novel coronavirus SARS-CoV-2 is a quickly developing global health crisis, yet the mechanisms of pathogenesis in COVID-19 are not fully understood.
Methods: The RNA sequencing data of SARS-CoV-2-infected cells was obtained from the Gene Expression Omnibus (GEO). The differentially expressed mRNAs (DEmRNAs), long non-coding RNAs (DElncRNAs), and microRNAs (DEmiRNAs) were identified by edgeR, and the SARS-CoV-2-associated competing endogenous RNA (ceRNA) network was constructed based on the prediction of bioinformatic databases. The Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were conducted with the SARS-CoV-2-related DEmRNAs, and the protein–protein interaction network was also built basing on STRING database. The ROC analysis was performed for assessing the diagnostic efficiency of hub genes.
Results: The results indicated that SARS-CoV-2-related DEmRNAs were associated with the interferon signaling pathway and other antiviral processes, such as IFNL3, IFNL1 and CH25H. Our analysis suggested that lncRNA NEAT1 might regulate the host immune response through two miRNAs, hsa-miR-374-5p and hsa-miR-155-5p, which control the expression of SOCS1, IL6, IL1B, CSF1R, CD274, TLR6, and TNF. Additionally, IFI6, HRASLS2, IGFBP4 and PTN may be potential targets based on an analysis comparing the transcriptional responses of SARS-CoV-2 infection with that of other respiratory viruses.
Discussion: The unique ceRNA network identified potential non-coding RNAs and their possible targets as well as a new perspective to understand the molecular mechanisms of the host immune response to SARS-CoV-2. This study may also aid in the development of innovative diagnostic and therapeutic strategies.
Keywords: COVID-19, ceRNA network, non-coding RNA, antiviral targets
Introduction
The Coronaviruses (CoVs) are a varied group of single-stranded positive-stranded RNA viruses that include the four genera α-CoV, β-CoV, γ-CoV, and δ-CoV.1–3 Prior to 2002, coronaviruses were known to have a wide range of hosts and to be a common cause of mild upper respiratory tract illnesses in humans. In 2002, however, the severe acute respiratory syndrome coronavirus 1 (SARS-CoV-1) infected thousands of people with an 11% mortality rate.4 This was followed by the Middle East respiratory syndrome-related coronavirus (MERS-CoV) that emerged in 2012 and resulted in a 36% mortality rate.5,6 In December 2019, a new coronavirus, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) was identified and spread quickly, causing the clinical syndrome called coronavirus disease 2019 (COVID-19). By November 2020, the SARS-CoV-2 global pandemic has caused over 60 million cases and over 1.4 million deaths.7 However, there have not been any specific therapeutic drugs against SARS-CoV-2 until recently. Hence, the study of the host response to SARS-CoV-2 compared to other respiratory viruses will play an important role in discovering potential antiviral therapeutic targets and revealing the pathogenic mechanisms of SARS-CoV-2.
At present, there is abundant research on the clinical symptoms and treatment of COVID-19, but the studies of pathobiology are insufficient and the specific host responses to SARS-CoV-2 at the cellular level are unclear. Usually, the infected cells could recognize the abnormal RNA structures produced during virus replication with pattern recognition receptors (PRRs).8 These PRRs then activate downstream transcription factors to initiate the cellular antiviral processes including the defenses mediated by interferon -I/-III and the recruitment of specific leukocytes by chemokines.9,10 However, host responses to the same virus are not uniform, which causes different degrees of infection severity. In most COVID-19 cases, the primary symptoms are dry cough, fever, fatigue, and myalgia.11 More severe patients often have dyspnea and/or hypoxemia prior to developing acute respiratory distress syndrome (ARDS) and the acute lung injury that is caused by the rapid replication of SARS-CoV-2, which triggers a strong immune response and cytokine storm.12–14 The study of the cellular responses to SARS-CoV-2 is essential for understanding the pathological progression of COVID-19.
Recently, genome-wide transcriptome studies of virus infection models have revealed that both protein-coding genes and non-coding RNAs are associated with coronavirus pathogenesis.15,16 Multiple host proteins such as BST2 and SAMHD1 were identified as modulators of innate immunity that also regulate viral replication in host cells.17,18 Long non-coding RNAs (lncRNAs) are also involved in regulating the virus life cycle and the immune response. For example, the lncRNA NEAT1 was demonstrated to play a regulatory role in increasing the viral gene expression and replication of herpes simplex virus-1 (HSV-1).19 Additionally, microRNAs (miRNAs), a type of small non-coding RNA of ~22nt, were identified to promote the degradation of genes or suppress translation by binding to the 3ʹ-untranslated regions (3ʹ-UTRs) of target genes.20 Based on the competing-endogenous RNA (ceRNA) hypothesis, some non-coding RNAs such as lncRNAs and circRNAs regulate the expression of protein-coding genes by competitively binding with miRNA response elements.21 Studies have reported that miRNAs may be antiviral targets for influenza and HIV-1 therapies.22,23 Therefore, construction of the hypothesized SARS-CoV-2-associated ceRNA network may provide a new perspective in the study of SARS-CoV-2 pathogenesis and putative diagnostic and therapeutic targets.
In this study, RNA sequencing data from SARS-CoV-2-infected cells were downloaded from the National Center of Biotechnology Information (NCBI) Gene Expression Omnibus (GEO). We sought to compare the transcriptional responses of SARS-CoV-2 with other respiratory viruses to identify differing transcriptional signatures during COVID-19. And, the flowchart of our research is shown in Figure 1. Through this analysis, we aimed to better understand the mechanisms of cellular response to SARS-CoV-2 and to identify potential antiviral targets.
 |
Figure 1 Workflow describing each step involved in the construction of the SARS-CoV-2-associated ceRNA network and identification of key targets. Abbreviation: ceRNA, competing endogenous RNA. |
Materials and Methods
Data Collection and Pre-Processing
To study the changes in the human transcriptome after SARS-CoV-2 infection, we searched for SARS-CoV-2-associated gene expression profiles in the National Center of Biotechnology Information (NCBI) Gene Expression Omnibus (GEO).24 The expression profiles generated by high throughput sequencing with the accession numbers GSE148729 and GSE156063 were chosen for further analysis. The GSE148729 dataset includes total RNA and miRNA expression profiles of SARS-CoV-1- or SARS-CoV-2-infected Calu 3 cells at different time points (4h, 12h, and 24h). The GSE156063 dataset includes mRNA expression profiles of upper airway samples from 234 patients with either COVID-19 (n=93), other viral (n=41), or non-viral (n=100) acute respiratory illnesses (ARIs). We also downloaded the gene expression profiles of other respiratory virus-infected Calu 3 cells with the accession numbers GSE28166 (avian influenza virus [H5N1]), GSE37571 (influenza A/CA/04/2009 virus), and GSE45042 (Human coronavirus EMC 2012). These expression profiles also contained multiple time points, but after considering the quality and unity of our comparative research, we selected the 24h time point of all samples (GSE148729, GSE28166, GSE37571, and GSE45042) for our analysis.For analyses of data from the public database, and this research does not involve any animal and human experiments, so approval and informed consent from the ethics committee were not required in accordance with the guidelines published by the Institutional Animal Care and Use Committee of Jilin University (Date of approval: January 1, 2016; Approval No. 2015-34).
Identification of Differentially Expressed RNAs
We used annotations of the GENECODE project (http://www.gencodegenes.org) to identify the lncRNA expression data from the total RNA expression profiles in GSE148729. The expression profiles of GSE28166, GSE37571, and GSE45042 were re-annotated according to the GPL6480 probe information, and probes without a corresponding gene symbol were removed. Next, these RNA expression data were analyzed using the edgeR package for R,25 and the p-value of differential expression was corrected for multiple comparisons by the Benjamini-Hochberg’s (BH) method for controlling the false discovery rate (FDR). Then the differentially expressed mRNAs (DEmRNAs), differentially expressed lncRNAs (DElncRNAs) and differentially expressed miRNAs (DEmiRNAs) were identified with the criteria of a |log2 fold change| > 1.5 and adjusted p < 0.05. The heat map and volcano plot showing differentially expressed RNAs were generated with the pheatmap and ggplot2 packages for R.26
Functional Enrichment Analysis
The Cluster Profiler package for R was utilized to perform a functional enrichment analysis of differentially expressed mRNAs (DEmRNAs) in GSE148729, including both Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses. The GO terms and KEGG pathways with an adjusted p < 0.05 (from the Benjamini-Hochberg method’s) were selected to represent significant enrichment, and the results were visualized using the GOplot27 and ggplot2 packages for R as well as Cytoscape Version 3.7.2.
Prediction of lncRNA - miRNA and miRNA - mRNA Interactions
We used the differentially expressed lncRNAs (DElncRNAs), differentially expressed miRNAs (DEmiRNAs), and DEmRNAs in GSE148729 to construct a SARS-CoV-2-infection-associated ceRNA regulatory network. DIANA-LncBase Version 2 (http://www.microrna.gr/LncBase)28 was used to predict the interactions between DEmiRNAs and DElncRNAs with a cutoff score ≥ 0.4. We predicted miRNA - mRNA interactions with miRDB (http://www.mirdb.org/)29 and DIANA-TarBase Version 8 (http://www.microrna.gr/tarbase).30 The miRNA - mRNA interactions detected by both platforms were retained. The predicted targets were compared to the DElncRNAs, DEmiRNAs, and DEmRNAs, and then used to construct an lncRNA - miRNA – mRNA ceRNA network using Cytoscape Version 3.7.2. This network was based on the hypothesis that lncRNA can absorb miRNA-like sponges to regulate mRNA activity.21
Protein - Protein Interaction (PPI) Network Analysis
The Search Tool for the Retrieval of Interacting Genes (STRING) database31 was utilized to analyze the interactions of the ceRNA network-associated DEmRNAs at the protein level. A confidence score > 0.4 was the STRING threshold to establish the PPI network. We then used Cytoscape Version 3.7.2 to visualize the network and identified highly correlated gene modules with the Plug-in Molecular Complex Detection (MCODE) based on the following criteria of a degree ≥ 2, a node score ≥ 0.2, and a k-core ≥ 2.
ROC Analysis
The receiver operating characteristic (ROC) analysis of the key mRNAs in GSE156063 was performed with the pROC package for R,32 and the diagnostic efficiency of DEmRNAs between the two groups was assessed with the criteria of the area under curve (AUC) > 0.6.
Statistical Analysis
R Studio (R Version 3.6.1) and GraphPad Prism 8.0 software (GraphPad Software, San Diego, CA, USA) were used for statistical analyses, and any p < 0.05 was considered statistically significant.
Results
Identification of the SARS-CoV-2 Associated Differentially Expressed mRNAs, lncRNAs, and miRNAs
In this study, we chose the RNA expression profiles of two SARS-CoV-2-infected samples and two uninfected samples in GSE148729 for differential expression analysis. A total of 1271 mRNAs, 279 lncRNAs, and 23miRNAs were screened for |log2 fold change| > 1.5 and adjusted p < 0.05 (from the Benjamini-Hochberg’s method). There were 215 mRNAs, 70 lncRNAs, and 10 miRNAs downregulated, while 1056 mRNAs, 209 lncRNAs, and 13 miRNAs were upregulated. The expression of all significantly downregulated and upregulated mRNAs, lncRNAs, and miRNAs was visualized with heat maps of the hierarchical clustering analysis and volcano plots (Figure 2, Supplementary File 1).
 |
Figure 2 Volcano plots and heat maps for differentially expressed lncRNAs, miRNAs, and mRNAs in GSE148729. First line, volcano plots for all SARS-CoV-2-related (A) mRNAs, (B) lncRNAs, and (C) miRNAs; Second line, heat maps showing differentially-expressed (D) mRNAs, (E) lncRNAs, and (F) miRNAs with a fold change ≥ 3 (p<0.05), respectively. Abbreviations: Red, up-regulated; green, down-regulated; black, not differentially expressed; lncRNA, long non-coding RNA; miRNA, microRNA. |
Functional Analysis of the SARS-CoV-2-Associated DEmRNAs
GO and KEGG pathway analyses were performed to analyze the biological function of the SARS-CoV-2-associated DEmRNAs. The top twenty GO terms, basing on the p-values, were chosen to show the results of the enrichment analyses (Figure 3A). The top 20 GO biological process (BP) terms, molecular function (MF) terms and cellular component (CC) terms were separately shown in Figure 3B–D). The changes in the biological processes of the SARS-CoV-2-associated DEmRNAs were significantly enriched in the activation of the interferon signaling pathway, the regulation of the viral life cycle, the regulation of cytokine production, increased T cell activation, and increased mononuclear cell proliferation. In addition, the top twenty KEGG pathways that these SARS-CoV-2-associated DEmRNAs highly participated in were chosen to show the potential biological function (Figure 4A, Supplementary File 3). The results indicated that these DEmRNAs were associated with inflammation-related pathways such as the TNF, toll-like receptor, NOD-like receptor, and NFκB signaling pathways. The interactions and overlap among these essential KEGG pathways were shown by a DEmRNAs - KEGG pathway network (Figure 4B).
 |
Figure 3 GO annotations for SARS-CoV-2-related DEmRNAs. (A) The representative GO terms were visualized by GOplot R-package. (B) Top 20 GO biological process terms (p < 0.05), (C) molecular function terms (p < 0.05), and (D) cellular component terms (p < 0.05). Abbreviations: GO, Gene Ontology; DEmRNAs, differentially expressed mRNAs. |
 |
Figure 4 KEGG pathway analysis for SARS-CoV-2-related DEmRNAs. (A) Top 20 enriched KEGG pathways (p < 0.05). (B) Interactions and overlap among the essential KEGG pathways. Green hexagons represent enriched pathways; “V” shapes represent mRNAs; blue and red nodes show decreased and increased expression of mRNAs, respectively. The colors are associated with the absolute value of fold change. Abbreviations: KEGG, Kyoto Encyclopedia of Genes and Genomes; DEmRNAs, differentially expressed mRNAs. |
Construction of the SARS-CoV-2-Associated lncRNA-miRNA-mRNA ceRNA Network
To better understand the host transcriptional responses to SARS-CoV-2, we built a SARS-CoV-2-associated ceRNA network based on the interactions among the DEmRNAs, DElncRNAs, and DEmiRNAs described above. According to DIANA-LncBase, 12 of the 23 DEmiRNAs were predicted to interact with 12 of the 279 DElncRNAs. Next, using miRDB and DIANA-TarBase, these 12 DEmiRNAs were predicted to target 86 of the 1271 DEmRNAs. Finally, a SARS-CoV-2 associated-ceRNA network was constructed, which includes 110 nodes based on the interactions of 24 lncRNA - miRNA pairs and 100 miRNA - mRNA pairs (Figure 5A). Notably, we found that NEAT1 may function as a hub lncRNA with 12 interactions.
 |
Figure 5 Construction of the SARS-Cov-2-associated lncRNA-miRNA-mRNA ceRNA network. (A) In the ceRNA network, ellipses represent mRNAs, diamonds represent lncRNAs, triangles represent miRNAs, and gray edges represent interactions among the miRNAs-lncRNAs and mRNAs. In addition, the blue and red nodes show decreased and increased expression of RNAs, respectively. The colors are associated with the absolute value of fold change. (B) The cytokine-related regulatory network. (C) The significantly enriched GO biological process terms (p < 0.05) and related genes in the ceRNA network. (D) The enriched KEGG pathways (p < 0.05) and related genes in the ceRNA network. Abbreviations: ceRNA, competing endogenous RNA; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes. |
To investigate the regulatory network of cytokine storms after SARS-COV-2 infection, we selected some cytokines which have been reported to be strongly elevated in both mild and severe COVID-19 patients, such as IL-6, IL-1β, IL-10, TNF, GM-CSF, IP-10, IL-17, MCP-3, and IL-1.47 Among these genes, IL-6 (IL6, log2 fold change = 5.678), IL-1β (IL1B, log2 fold change = 2.360) and TNF (TNF, log2 fold change = 9.306) that were overlapped with ceRNA network related mRNAs have been screened to rebuild a cytokine-related regulatory network (Figure 5B).Then we performed GO and KEGG analyses on the ceRNA network-associated DEmRNAs to speculate the function of these DElncRNAs. GO enrichment results showed that these lncRNAs were mainly involved in the regulation of immune system processes, T cell activation, and leukocyte proliferation. Furthermore, the KEGG pathway analysis indicated that these lncRNAs were also associated with the TNF, Ras, MAPK, and IL-17 signaling pathways (Figure 5C and D).
Construction of Protein - Protein Interaction (PPI) Network and Key Module Screening
To further investigate the ceRNA network-associated DEmRNAs at the protein level, we used STRING to build a protein - protein interaction (PPI) network with 64 nodes basing on the interactions among these proteins (Figure 6A). The results indicated that IL6, TNF, FOS, IL1B, SOCS1, PTGS2, GATA3, CSF1R, IL15, IL7R, FGFR3, CD274, TLR6, PRDM1, IGF2, CEBPB, and BMP2 had higher degrees of interaction and may act as hub genes. Two functionally related and interconnected key modules were then screened with the plug-in MCODE (Figure 6B and C). Among them, the hub genes IL6, TNF, FOS, IL1B, SOCS1, IL15, PTGS2, CSF1R, CD274 and TLR6 were all included in module 1 and compose a ceRNA sub-network (Figure 6D). According to this ceRNA sub-network, we found that NEAT1 could act as a key lncRNA to regulate these hub genes while SARS-CoV-2 infection.
 |
Figure 6 The protein–protein interaction (PPI) network for the ceRNA network-related genes. (A) In the network, the size of the nodes represents the degree of each node. (B and C) The two modules modularized by MCODE from the PPI network. (D) The ceRNA sub-network based on hub genes, ellipses represent mRNAs, diamonds represent lncRNAs, and triangles represent miRNAs. |
Screening of Key mRNAs to Distinguish SARS-CoV-2 Infection from Other Respiratory Virus Infections
To investigate the gene expressions that differentiate SARS-CoV-2 infections from other respiratory viruses infections, we separately performed a differential expression analysis on the mRNA expression profiles of SARS-CoV-1, avian influenza virus (H5N1), influenza virus A/CA/04/2009 I, and Human coronavirus EMC 2012 (HCoV-EMC) infected Calu 3 cells. After comparing SARS-CoV-1-, H5N1-, CA04-, and HCoV-EMC-related non-highly expressed mRNAs (log2 fold change < 1.5) to SARS-CoV-2-related highly expressed mRNAs (log2 fold change > 1.5, FDR < 0.05), 225 mRNAs were selected as potential targets (Figure 7A). Subsequently, these 225 mRNAs were validated in the GSE156063 dataset comprising of 234 mRNA expression profiles of either COVID-19 (n=93), other viral (n=41), or non-viral (n=100) ARI samples. IFI6, HRASLS2, IGFBP4 and PTN were found to be significantly highly expressed in COVID-19 samples (Figure 7B–E). All GSE156063 samples were divided into three groups including COVID-19 vs other viral and non-viral (group 1), COVID-19 vs non-viral (group 2), and COVID-19 vs other viral (group 3). The ROC analysis was conducted based on the expression of the 225 mRNAs in the three groups. The results showed that the area under curve (AUC) of IFI6 for groups 1, 2, and 3 was 0.8482, 0.9483, and 0.604, respectively (Figure 8A–C). The AUCs of HRASLS2 were 0.7927, 0.8478, and 0.6583 for groups 1, 2, and 3, respectively (Figure 8D–F). The AUCs of IGFBP4 were 0.7117, 0.7071, and 0.7071 for groups 1, 2, and 3, respectively (Figure 8G–I). And the AUCs of PTN were 0.6929, 0.6926, and 0.6913 for groups 1, 2, and 3, respectively (Figure 8J–L). The results indicated that these genes have high sensitivity and specificity. We speculated that IFI6, HRASLS2, IGFBP4 and PTN may be key genes to distinguish COVID-19 from other viral or non-viral respiratory cases.
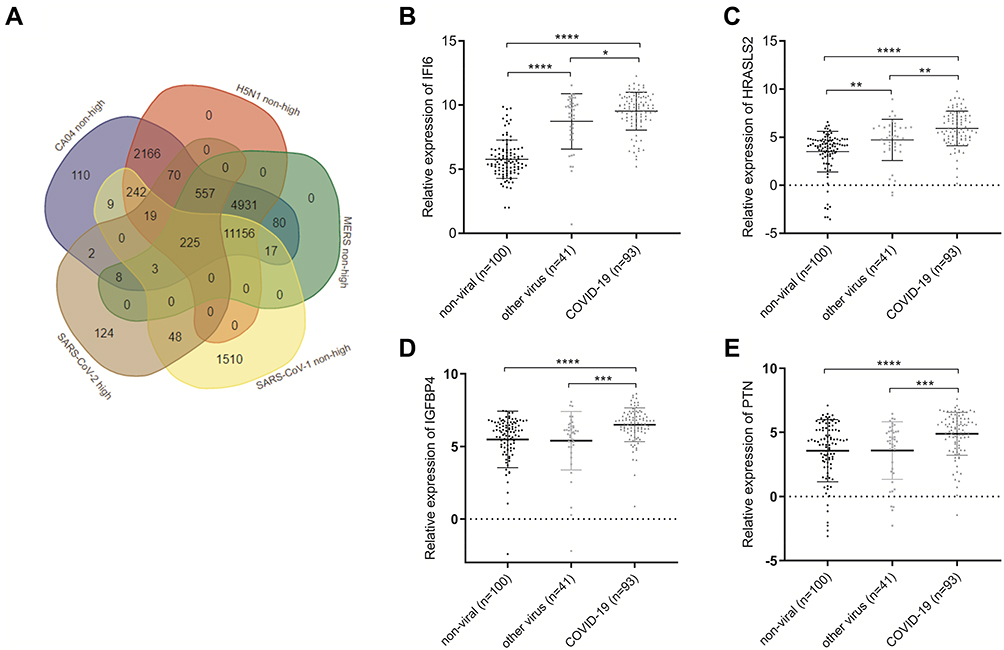 |
Figure 7 Venn diagram showing the overlap between the SARS-CoV-1-, H5N1-, CA04-, HCoV-EMC-related non-highly expressed mRNAs and the SARS-CoV-2-related highly expressed mRNAs (A). The relative expression of (B) IFI6, (C) HRASLS2, (D) IGFBP4 and (E) PTN in COVID-19 (n=93), other viral (n=41), or non-viral (n=100) acute respiratory illnesses samples, *p < 0.05, **p < 0.01,***p < 0.001, ****p < 0.0001 by unpaired t-test. Abbreviations: H5N1, avian influenza (H5N1) virus; CA04, influenza A/CA/04/2009 virus; HCoV-EMC, Human coronavirus EMC 2012. |
 |
Figure 8 ROC curves of (A–C) IFI6, (d-f) HRASLS2, (G–I) IGFBP4 and (J–L) PTN in three groups (group 1: COVID-19 vs other viral and non-viral ARIs; group 2: COVID-19 vs non-viral ARIs; group 3: COVID-19 vs other viral ARIs). Abbreviation: ARIs, acute respiratory illnesses. |
Discussion
In this study, we characterized the transcriptional responses to SARS-CoV-2 including the interactions among SARS-CoV-2-associated DEmRNAs, DElncRNAs, and DEmiRNAs to construct a SARS-CoV-2-related ceRNA network and screened for potential targets to identify COVID-19 cases from other respiratory viruses infections.
A total of 1271 mRNAs were found to be aberrantly expressed between SARS-CoV-2-infected and uninfected Calu 3 cells. Among these DEmRNAs, IFNL2 (interferon lambda 2), IFNB1 (interferon beta 1), IFNL3 (interferon lambda 3), CH25H (cholesterol 25-hydroxylase), RGAG1 (retrotransposon Gag like 9), IFNL1 (interferon lambda 1), ACTN2 (actinin alpha 2), and IFNL4 (interferon lambda 4) were up-regulated by more than 20-fold. Type I IFNs (IFN-α/β) and type III IFNs (IFN-λs) are closely associated with innate antiviral immunity,33,34 and CH25H has also been demonstrated to have broad antiviral properties.35 In addition, several inflammatory cytokines such as TNF, CXCL11, CXCL10, CXCL9, and IL6 were over-expressed in SARS-CoV-2-infected cells.
Next, functional enrichment analyses were performed on the SARS-CoV-2-associated DEmRNAs with GO and KEGG. The GO annotations showed that these DEmRNAs are mainly involved in BPs including the type I interferon signaling pathway, regulation of the viral life cycle, T cell activation, the positive regulation of cytokine production, the STAT cascade, and the JAK-STAT cascade, which are all closely associated with host immune responses. The enriched KEGG pathways included cytokine-cytokine receptor interactions, the TNF signaling pathway, the JAK-STAT signaling pathway, and the NFκB signaling pathway. These pathways were found to play critical roles in SARS-CoV-2-infections.36 Recent studies have found that some JAK/STAT inhibitors like Ruxolitinib, Baricitinib, and Tofacitinib can control cytokine storms and reduce inflammation by inhibiting JAK/STAT signaling in COVID-19 patients.37,38
Non-coding RNAs such as miRNAs, circRNAs, and lncRNAs can regulate the expression of protein-coding genes to participate in essential pathological and physiological processes, including the immune responses against viral infection.39 In this study, we built a SARS-CoV-2-associated ceRNA network consisting of 12 DElncRNAs, 12 DEmiRNAs, and 86 DEmRNAs to reveal potential regulatory mechanisms against SARS-CoV-2. In this ceRNA network, lncRNA NEAT1 may act as a hub node with 12 interactions, which has been demonstrated to play an important role during the HIV-1 life cycle and act as a host factor for HIV infection and persistence.40 Some studies have indicated that ectopic expression of NEAT1 could enhance IFN-β production and suppress Hantaan virus (HTNV) infection through modulation of the innate immune response.41 Moreover, NEAT1 has been found to be upregulated in saliva and nasopharyngeal swab from COVID-19 samples,42 but downregulated in bronchoalveolar lavage (BAL) cells from patients with mild and severe COVID-19.43 The miRNAs hsa-miR-374-5p and hsa-miR-155-5p may also act as hub miRNAs with high interactions. Hsa-miR-155-5p has been reported to modulate viral replication and viral gene expression in HSV-1 infections.44 The functional analysis showed that these ceRNA network-related mRNAs were involved in regulating the immune system, T cell activation, the TNF signaling pathway, and the IL-17 signaling pathway. We speculated that lncRNA NEAT1 may also participate in regulating the immune response, cytokine storms or antiviral processes against SARS-CoV-2 by binding with key miRNAs, such as hsa-miR-100-5p and hsa-miR-155-5p.
In addition, based on the module analysis of a protein–protein interaction (PPI) network, some core mRNAs were identified. Several have been reported to modulate viral infections, such as SOCS1 (suppressor of cytokine signaling 1), which when reduced by miR-30 family members could inhibit the IFN/JAK/STAT signaling pathway to regulate immune escape by influenza viruses.45 IL15 (interleukin 15), a type of cytokine that regulates T and natural killer cell activation and proliferation, has also been demonstrated to act as an essential cytokine in both the innate and adaptive immune responses against hepatitis C virus (HCV) infection.46 IL-6 (interleukin 6), IL-1β (interleukin 1 beta) and TNF (tumor necrosis factor) have been reported to elevate both in mild and severe COVID-19 patients and closely associated with cytokine storms.47 The main hubs of the ceRNA network-related DEmRNAs were predicted to be closely involved in regulating the host immune system against viral infections. Based on our bioinformatic analyses, we hypothesized that the ceRNA network could be utilized to modulate host immune responses to SARS-CoV-2 infection such as the NEAT1 - miR-374-5p -IL15 axis and NEAT1 - miR-155-5p - IL-6/TNF/IL-1β axis. The specific mechanisms still need to be uncovered.
At present, nucleic acid detection by reverse transcription quantitative PCR (RT-qPCR) is a major diagnostic tool for COVID-19, but it also has the risk of false negatives. To evaluate the feasibility of diagnosing SARS-CoV-2 infection (COVID-19) based on host gene expression, we compared the SARS-CoV-2-related gene expression profile with those of other respiratory viruses. The mRNAs IFI6, HRASLS2, IGFBP4 and PTN were found to distinguish COVID-19 cases from other viral and non-viral acute respiratory illnesses with high sensitivity and specificity. IFI6 is one of the best studied genes in viral infection, including Hepatitis B virus, HCV, and Zika virus.48–51 IFI6 is also a type of interferon-regulated genes (IRGs), which is closely associated with antiviral innate immune response. However, HRASLS2 (phospholipase A and acyltransferase 2), a notable tumor regulatory factor, has not been studied in the context of a viral infection and needs further study. PTN (pleiotrophin) has been found to be an early indicator for the diagnosis and prognosis of non-small cell lung cancer (NSCLC),52 and PTN has been demonstrated that it could inhibit HIV-1 infection by its capacity to inhibit HIV-1 particle attachment to the surface of permissive cells.53 PTN has not been researched in respiratory viruses infection and needs more study. IGFBP4 (insulin like growth factor binding protein 4), a potential lung cancer biomarker,54 has also not been studied in viruses infection until now. We speculated that IFI6 may play a critical role in SARS-CoV-2 infection and serve as a potential diagnostic or therapeutic target for COVID-19 patients. However, this study was based on available data from a public database, which was limited by sample size, incomplete data, and the lack of a larger validation cohort. Future studies are needed to validate these findings, assess the regulation mechanisms of the integrated ceRNA network, and determine the diagnostic and antiviral value of host signatures in a larger cohort.
Conclusion
In conclusion, we constructed a SARS-CoV-2-associated ceRNA network by analyzing the RNA sequencing data of SARS-CoV-2-infected cells downloaded from the GEO platform. We speculated that lncRNA NEAT1 might regulate the host immune response to SARS-CoV-2 through acting as ceRNA, and four mRNAs were predicted to be transcriptional signatures of COVID-19 comparing to the transcriptional responses of other respiratory viruses. The potential mechanisms of these RNAs in cellular response to SARS-CoV-2 should be researched to identify their feasibility as antiviral targets.
Data Sharing Statement
All original data could be accessed from the GEO database (Accession numbers: GSE148729, GSE156063, GSE28166, GSE37571, GSE45042). The data that support the findings of this study are available on request from the corresponding author (Fan Li).
Acknowledgments
This research was supported by Science and Technology Development Plan Project of Jilin Province (Grant No. 20200901007SF). We also thank TopEdit (www.topeditsci.com) for editing and proofreading this manuscript.
Disclosure
The authors declare no competing interests.
References
1. Cui J, Fang L, Shi Z-L. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol. 2019;17(3):181–192. doi:10.1038/s41579-018-0118-9
2. Yang D, Leibowitz JL. The structure and functions of coronavirus genomic 3′ and 5′ ends. Virus Res. 2015;206:120–133. doi:10.1016/j.virusres.2015.02.025
3. Banerjee A, Kulcsar K, Misra V, Frieman M, Mossman K. Bats and coronaviruses. Viruses. 2019;11(1):41. doi:10.3390/v11010041
4. Zhong NS, Zheng BJ, Li YM, et al. Epidemiology and cause of severe acute respiratory syndrome (SARS) in Guangdong, People’s Republic of China, in February, 2003. Lancet. 2003;362(9393):1353–1358. doi:10.1016/s0140-6736(03)14630-2
5. Zumla A, Hui DS, Perlman S. Middle East respiratory syndrome. Lancet. 2015;386:995–1007. doi:10.1016/S0140-6736(15)60454-8
6. Hui DS, Azhar EI, Kim YJ, Memish ZA, Oh MD, Zumla A. Middle East respiratory syndrome coronavirus: risk factors and determinants of primary, household, and nosocomial transmission. Lancet Infect Dis. 2018;18:e217–e227. doi:10.1016/S1473-3099(18)30127-0
7. Dong E, Du H, Gardner L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect Dis. 2020;20(5):533–534. doi:10.1016/S1473-3099(20)30120-1
8. Charles A, Jr J, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi:10.1146/annurev.immunol.20.083001.084359
9. Lazear HM, Schoggins JW, Michael S. Diamond shared and distinct functions of Type I and Type III interferons. Immunity. 2019;50(4):907–923. doi:10.1016/j.immuni.2019.03.025
10. Sokol CL, Luster AD. The chemokine system in innate immunity. Cold Spring Harb Perspect Biol. 2015;7(5):a016303. doi:10.1101/cshperspect.a016303
11. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi:10.1016/S0140-6736(20)30183-5
12. Wölfel R, Corman VM, Guggemos W, et al. Virological assessment of hospitalized patients with COVID-2019. Nature. 2020;581(7809):465–469. doi:10.1038/s41586-020-2196-x
13. Zhe X, Shi L, Wang Y, et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med. 2020;8(4):420–422. doi:10.1016/S2213-2600(20)30076-X
14. Channappanavar R, Perlman S. Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin Immunopathol. 2017;39:529–539. doi:10.1007/s00281-017-0629-x
15. Cristinelli S, Ciuffi A. The use of single-cell RNA-Seq to understand virus-host interactions. Curr Opin Virol. 2018;29:39–50. doi:10.1016/j.coviro.2018.03.001
16. Bosinger SE, Hosiawa KA, Cameron MJ, et al. Gene expression profiling of host response in models of acute HIV infection. J Immunol. 2004;173(11):6858–6863. doi:10.4049/jimmunol.173.11.6858
17. Berry KN, Kober DL, Alvin S, Brett TJ. Limiting respiratory viral infection by targeting antiviral and immunological functions of BST-2/Tetherin: knowledge and gaps. Bioessays. 2018;40(10):e1800086. doi:10.1002/bies.201800086
18. Chen S, Bonifati S, Qin Z, et al. SAMHD1 suppresses innate immune responses to viral infections and inflammatory stimuli by inhibiting the NF-κB and interferon pathways. Proc Natl Acad Sci U S A. 2018;115(16):E3798–E3807. doi:10.1073/pnas.1801213115
19. Wang Z, Fan P, Zhao Y, et al. NEAT1 modulates herpes simplex virus-1 replication by regulating viral gene transcription. Cell Mol Life Sci. 2017;74(6):1117–1131. doi:10.1007/s00018-016-2398-4
20. Li J, Tan S, Kooger R, Zhang C, Zhang Y. MicroRNAs as novel biological targets for detection and regulation. Chem Soc Rev. 2014;43(2):506–517. doi:10.1039/c3cs60312a
21. Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146(3):353–358. doi:10.1016/j.cell.2011.07.014
22. Keshavarz M, Dianat-Moghadam H, Sofiani VH, et al. miRNA-based strategy for modulation of influenza A virus infection. Epigenomics. 2018;10(6):829–844. doi:10.2217/epi-2017-0170
23. Asahchop EL, Branton WG, Krishnan A, et al. HIV-associated sensory polyneuropathy and neuronal injury are associated with miRNA-455-3p induction. JCI Insight. 2018;3(23):e122450. doi:10.1172/jci.insight.122450
24. Edgar R, Domrachev M, Lash AE. Gene expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi:10.1093/nar/30.1.207
25. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. doi:10.1093/bioinformatics/btp616
26. Wickham H. Ggplot2: Elegant Graphics for Data Analysis. New York: Springer-Verlag; 2016. doi:10.1007/978-3-319-24277-4
27. Walter W, Sanchez-Cabo F, Ricote M. GOplot: an R package for visually combining expression data with functional analysis. Bioinformatics. 2015;31:2912–2914. doi:10.1093/bioinformatics/btv300
28. Paraskevopoulou MD, Vlachos IS, Karagkouni D, et al. DIANA-LncBase v2: indexing microRNA targets on non-coding transcripts. Nucl Acids Res. 2016;44(D1):D231–8. doi:10.1093/nar/gkv1270
29. Wong N, Wang X. miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2015;43(D1):D146–152. doi:10.1093/nar/gku1104
30. Karagkouni D, Paraskevopoulou MD, Chatzopoulos S, et al. DIANA-TarBase v8: a decade-long collection of experimentally supported miRNA-gene interactions. Nucleic Acids Res. 2018;46(Database issue):D239–D245. doi:10.1093/nar/gkx1141
31. Szklarczyk D, Franceschini A, Wyder S, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43(Database issue):D447–52. doi:10.1093/nar/gku1003
32. Robin X, Turck N, Hainard A, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011;12:77. doi:10.1186/1471-2105-12-77
33. Takaoka A, Yanai H. Interferon signalling network in innate defence. Cell Microbiol. 2006;8(6):907–922. doi:10.1111/j.1462-5822.2006.00716.x
34. Kotenko SV. IFN-λs. Curr Opin Immunol. 2011;23(5):583–590. doi:10.1016/j.coi.2011.07.007
35. Magoro T, Dandekar A, Jennelle LT, et al. IL-1β/TNF-α/IL-6 inflammatory cytokines promote STAT1-dependent induction of CH25H in Zika virus-infected human macrophages. J Biol Chem. 2019;294(40):14591–14602. doi:10.1074/jbc.RA119.007555
36. Totura AL, Baric RS. SARS coronavirus pathogenesis: host innate immune responses and viral antagonism of interferon. Curr Opin Virol. 2012;2(3):264–275. doi:10.1016/j.coviro.2012.04.004
37. Zhang X, Zhang Y, Qiao W, Zhang J, Qi Z. Baricitinib, a drug with potential effect to prevent SARS-COV-2 from entering target cells and control cytokine storm induced by COVID-19. Int Immunopharmacol. 2020;86:106749. doi:10.1016/j.intimp.2020.106749
38. Satarker S, Tom AA, Shaji RA, Alosious A, Luvis M, Nampoothiri M. JAK-STAT pathway inhibition and their implications in COVID-19 therapy. Postgrad Med. 2021;133(5):489–507. doi:10.1080/00325481.2020.1855921
39. Ghosal S, Das S, Sen R, Chakrabarti J. HumanViCe: host ceRNA network in virus infected cells in human. Front Genet. 2014;5:249. doi:10.3389/fgene.2014.00249
40. Zhang Q, Chen CY, Yedavalli VS, Jeang KT. NEAT1 long noncoding RNA and paraspeckle bodies modulate HIV-1 posttranscriptional expression. mBio. 2013;4(1):e00596–12. doi:10.1128/mBio.00596-12
41. Hongwei M, Han P, Wei Y, et al. The long noncoding RNA NEAT1 exerts antihantaviral effects by acting as positive feedback for RIG-I signaling. J Virol. 2017;91(9):e02250–16. doi:10.1128/JVI.02250-16
42. Rodrigues AC, Adamoski D, Genelhould G, et al. NEAT1 and MALAT1 are highly expressed in saliva and nasopharyngeal swab samples of COVID-19 patients. Mol Oral Microbiol. 2021. doi:10.1111/omi.12351
43. Shaath H, Vishnubalaji R, Elkord E, Alajez NM. Single-cell transcriptome analysis highlights a role for neutrophils and inflammatory macrophages in the pathogenesis of severe COVID-19. Cells. 2020;9(11):2374. doi:10.3390/cells9112374
44. Wang Z, Kun L, Wang X, Huang W. MiR-155-5p modulates HSV-1 replication via the epigenetic regulation of SRSF2 gene expression. Epigenetics. 2019;14(5):494–503. doi:10.1080/15592294.2019.1600388
45. Lin X, Shiman Y, Ren P, Sun X, Jin M. Human microRNA-30 inhibits influenza virus infection by suppressing the expression of SOCS1, SOCS3, and NEDD4. Cell Microbiol. 2020;22(5):e13150. doi:10.1111/cmi.13150
46. Jiménez-Sousa MA, Berenguer J, Rallón N, et al. IL15 polymorphism is associated with advanced fibrosis, inflammation-related biomarkers and virological response in human immunodeficiency virus/hepatitis C virus coinfection. Liver Int. 2016;36(9):1258–1266. doi:10.1111/liv.13079
47. Wang J, Jiang M, Chen X, Montaner LJ. Cytokine storm and leukocyte changes in mild versus severe SARS-CoV-2 infection: review of 3939 COVID-19 patients in China and emerging pathogenesis and therapy concepts. J Leukoc Biol. 2020;108(1):17–41. doi:10.1002/JLB.3COVR0520-272R
48. Prabhu N, Ho AW, Wong KHS, et al. Gamma interferon regulates contraction of the influenza virus-specific CD8 T cell response and limits the size of the memory population. J Virol. 2013;87(23):12510–12522. doi:10.1128/JVI.01776-13
49. Park G-H, Kim K-Y, Cho SW, et al. Association between Interferon-Inducible Protein 6 (IFI6) Polymorphisms and Hepatitis B virus clearance. Genomics Inform. 2013;11(1):15–23. doi:10.5808/GI.2013.11.1.15
50. Meyer K, Kwon Y-C, Shuanghu Liu CH, Hagedorn RB, Ray RR. Interferon-α inducible protein 6 impairs EGFR activation by CD81 and inhibits hepatitis C virus infection. Sci Rep. 2015;5:9012. doi:10.1038/srep09012
51. Dukhovny A, Lamkiewicz K, Chen Q, et al. A CRISPR activation screen identifies genes that protect against zika virus infection. J Virol. 2019;93(16):e00211–19. doi:10.1128/JVI.00211-19
52. Du ZY, Shi MH, Ji CH, Yu Y. Serum pleiotrophin could be an early indicator for diagnosis and prognosis of non-small cell lung cancer. Asian Pac J Cancer Prev. 2015;16(4):1421–1425. doi:10.7314/apjcp.2015.16.4.1421
53. Said EA, Courty J, Svab J, Delbé J, Krust B, Hovanessian AG. Pleiotrophin inhibits HIV infection by binding the cell surface-expressed nucleolin. FEBS J. 2005;272(18):4646–4659. doi:10.1111/j.1742-4658.2005.04870.x
54. Nur SI, Ozturk A, Kavas M, et al. IGFBP-4: a promising biomarker for lung cancer. J Med Biochem. 2021;40(3):237–244. doi:10.5937/jomb0-25629
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.