Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 16
Construction and Identification of an NLR-Associated Prognostic Signature Revealing the Heterogeneous Immune Response in Skin Cutaneous Melanoma
Authors Geng Y ![]() , Sun YJ, Song H, Miao QJ, Wang YF, Qi JL, Xu XL
, Sun YJ, Song H, Miao QJ, Wang YF, Qi JL, Xu XL ![]() , Sun JF
, Sun JF
Received 28 March 2023
Accepted for publication 31 May 2023
Published 26 June 2023 Volume 2023:16 Pages 1623—1639
DOI https://doi.org/10.2147/CCID.S410723
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jeffrey Weinberg
Yi Geng,1 Yu-Jie Sun,1 Hao Song,1 Qiu-Ju Miao,1 Yi-Fei Wang,1 Jin-Liang Qi,2 Xiu-Lian Xu,1 Jian-Fang Sun1
1Institute of Dermatology, Peking Union Medical College and Chinese Academy of Medical Sciences, Nanjing, 210042, People’s Republic of China; 2State Key Laboratory of Pharmaceutical Biotechnology, School of Life Sciences, Nanjing University, Nanjing, 210023, People’s Republic of China
Correspondence: Xiu-Lian Xu; Jian-Fang Sun, Email [email protected]; [email protected]
Background: Skin cutaneous melanoma (SKCM) is the deadliest dermatology tumor. Ongoing researches have confirmed that the NOD-like receptors (NLRs) family are crucial in driving carcinogenesis. However, the function of NLRs signaling pathway-related genes in SKCM remains unclear.
Objective: To establish and identify an NLRs-related prognostic signature and to explore its predictive power for heterogeneous immune response in SKCM patients.
Methods: Establishment of the predictive signature using the NLRs-related genes by least absolute shrinkage and selection operator-Cox regression analysis (LASSO-COX algorithm). Through univariate and multivariate COX analyses, NLRs signature’s independent predictive effectiveness was proven. CIBERSORT examined the comparative infiltration ratios of 22 distinct types of immune cells. RT-qPCR and immunohistochemistry implemented expression validation for critical NLRs-related prognostic genes in clinical samples.
Results: The prognostic signature, including 7 genes, was obtained by the LASSO-Cox algorithm. In TCGA and validation cohorts, SKCM patients with higher risk scores had remarkably poorer overall survival. The independent predictive role of this signature was confirmed by multivariate Cox analysis. Additionally, a graphic nomogram demonstrated that the risk score of the NLRs signature has high predictive accuracy. SKCM patients in the low-risk group revealed a distinct immune microenvironment characterized by the significantly activated inflammatory response, interferon-α/γ response, and complement pathways. Indeed, several anti-tumor immune cell types were significantly accumulated in the low-risk group, including M1 macrophage, CD8 T cell, and activated NK cell. It is worth noting that our NLRs prognostic signature could serve as one of the promising biomarkers for predicting response rates to immune checkpoint blockade (ICB) therapy. Furthermore, the results of expression validation (RT-qPCR and IHC) were consistent with the previous analysis.
Conclusion: A promising NLRs signature with excellent predictive efficacy for SKCM was developed.
Keywords: cutaneous melanoma, NLR proteins, immune, survival analysis, prognosis
Introduction
Skin cutaneous melanoma (SKCM) is the most severe dermatologic malignancy, and its incidence has increased worldwide in recent years.1 SKCM accounts for 1% of all skin cancer patients, yet it is responsible for roughly 80% of all skin cancer deaths.2 Early-stage SKCM (localized or regional) can be surgically removed, and its 5-year survival rate is over 95%. The primary treatment method for advanced SKCM is combination therapy, which may include radiotherapy, chemotherapy, and targeted therapy; nevertheless, patient response rates to medication are low, and patients seldom live more than a year.3 The absence of established predictive indicators restricts the assessment of SKCM disease progression and effective clinical interventions for advanced patients.4 Hence, searching for biomarkers that could be effective in the prevention and early diagnosis is very important for SKCM.
When first discovered, nucleotide-binding and oligomerization domain-like receptors (NOD-like receptors, NLRs) were considered essential effector molecules of the innate immune response. The majority of subsequent investigations indicated that, as a class of intracellular pattern recognition receptors (PRRs), the NLRs family specifically identified the pathogen-related molecular mode (PAMPS) and damage-associated molecular patterns (DAMPs), which are involved in sensing intracellular pathogenic microbial infection and host apoptosis or damaged cells.5
The NLRs family proteins contain three main structural domains: a central oligomerization region with nucleotide binding, a leucine-rich repeat domain at the C-terminal, and an effector domain at the N-terminal.6 NLRs are mainly distributed in mucosal epithelial cells, macrophages, dendritic cells, and neutral granulocytes. Activated NLRs proteins induce the formation of inflammasomes and activate the NF-κB and MAPK signaling pathways, subsequently producing various pro-inflammatory and antiviral cytokines and chemokines, such as IL-1 and IL-18, and stimulating the initiation of acquired immunity.5,7 However, many recent studies have shown that most NLRs proteins appear to negatively regulate antiviral natural immune-related signaling pathways, including toll-like receptor (TLR), cyclic GMP-AMP synthase-stimulator of interferon (cGAS-STING), retinoic acid-inducible gene I-like receptor (RLR) signal pathways.8
Emerging evidence suggests that NLRs have a dual regulatory role in tumor immunity.9 On the one hand, immune surveillance can prevent tumorigenesis. Also, NLRs can bridge the innate immune response and subsequent acquired immune response to eliminate tumor cells and inhibit tumor progression.10 In contrast, chronic inflammation, which is predominantly mediated by NLRs, is a crucial promoter of tumorigenesis. It has been well-proven that chronic inflammation is a primary driver of tumor initiation, progression, and metastasis.11,12 The signaling pathways downstream of NLRs, such as the NF-κB and inflammasome signaling pathways, are continuously activated to generate multiple pro-inflammatory factors that induce chronic inflammation and promote tumorigenesis.13
Recent studies on the role of NLRs in tumorigenesis have primarily concentrated on colorectal, breast, prostate, and gastric cancers, whereas relatively few studies have focused on NLRs in melanoma.7 Zhai et al revealed that after the knockdown of NLRP1 expression in metastatic melanoma cell lines 1205Lu and HS294T, the activities of caspase-1, IL-1β, and NF-κB were reduced, while the activities of caspase-2, −9, and −3/7 were increased, causing increased apoptosis in melanoma cells, indicating that NLRP1 is a pro-oncogene in melanoma.14 A Swedish case-control study demonstrated that polymorphisms in the NLRP3 (rs35829419) and NLRP1 (rs12150220) significantly correlated with nodular melanoma susceptibility.15 In summary, current researches on the regulation of melanoma by NLRs mainly concern single gene and lack an overall assessment of the NLRs signaling pathway. Due to the hub function of the NLRs signaling pathway in tumor pathogenesis, this study aims to conduct a comprehensive analysis of NLRs signaling pathway-related genes, which will help discover potential therapeutic targets for SKCM.
Here, the RNA-seq profiles of SKCM and normal samples and accompanying various clinical data were systematically consolidated via the TCGA and GEO public databases. Using the obtained data, we attempt to construct and validate a robust multi-gene signature for predicting patient outcomes and investigating NLRs’ effects on the SKCM immune response and its mechanism.
Materials and Methods
Data Collection and Collation
Employing the “TCGAbiolinks” R package, the somatic mutation data, mRNA expression level from RNA-seq data (FPKM, Fragments Per Kilobase Per Million), and the accompanying various clinical data of tumor and normal samples of the TCGA Skin Cutaneous Melanoma (SKCM) cohort were collected. TCGA panimmune analysis (https://gdc.cancer.gov/about-data/publications/panimmune) was the source of tumor mutational load, neoantigen load, and TCR diversity data. Two independent SKCM patients cohorts (GSE54467, GSE133713) served as validation data sets, and their RNA-seq profiles data and patients’ clinical data were gathered from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nih.gov/geo/). When multiple microarray probes were mapped to the identical gene, the R package ‘idmap1’ calculates the median. The RNA-seq profiles and clinical materials of SKCM patients who received treatment with anti-PD1 (n=31 samples) or anti-CTLA4 (n=10 samples) were also obtained from the GEO (GSE115821).16 Through the KEGG NLRs signaling pathway in the MSigDB v7.4 database, an overall of 62 NLRs-associated genes were identified.
Construction and Identification of the Gene Signature
The TCGA SKCM cohort was subjected to a univariate Cox regression analysis to discover NLRs-related prognostic genes. The optimal candidate NLRs-related genes with the most suitable parameters were subsequently identified using LASSO arithmetic. The risk score was developed by integrating these optimal candidate genes’ expression levels and weighted them according to the LASSO coefficient as follows:
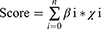
Where βi represents each gene’s coefficient, and χi indicates the relative expression quantity for every single gene (FPKM). SKCM patients with high risk can be distinguished from those with low risk via the median risk score.
Assessment of Immune Cell Infiltration
SKCM patients’ immune score, stromal score, and tumor purity have been determined utilizing the ESTIMATE algorithm (Estimation of STromal and Immune cells in MAlignant Tumour tissues using Expression data) from the “estimate” R package.17 By applying the CIBERSORT algorithm (Cell type Identification By Estimating Relative Subsets Of RNA Transcripts) with default parameters, we further investigated relative infiltration ratios for 22 distinct immune cell types. The TCGA-SKCM patients’ leukocyte fraction was obtained via the TCGA panimmune data sets (https://gdc.cancer.gov/about-data/publications/panimmune).
Comparative Analysis of Cancer Hallmark Profiles
In order to compare the biological differences between low-risk and high-risk SKCM patients, the assessment for each sample’s cancer hallmark features was implemented by ssGSEA (single sample gene set enrichment analysis) utilizing the R package “GSVA”. The reference gene dataset “h.all.v7.2.symbols.gmt” for cancer hallmarks was selected from the public resource “MSigDB”.
Clinical Specimens Collection
We collected 9 samples of SKCM and paracancerous fresh tissues exclusively from patients who underwent surgery. In addition, paraffin-embedded sections were provided by 10 SKCM patients and 8 pigmented nevus patients who underwent a biopsy procedure.
RNA Extraction and RT-qPCR
Trizol (Invitrogen, USA) was utilized to separate total RNA from SKCM and paraneoplastic tissues. The EVO M-MLV Mix kit (Accurate, China) was applied for reverse transcription and cDNA synthesis. SYBR Green Premix Pro Taq HS qPCR kit (Accurate, China) and the Roche Lightcycler 480 (Roche) were the kit and data analysis platform employed during the RT-qPCR reaction procedure. The reaction volume was 10µL per well, which included 5µL 2X SYBR Green mixture, 1µL cDNA, 0.5µM each primer, and sterile distilled water. The 2−ΔΔCq algorithm served to determine specific mRNAs’ relative expression amounts. Primers involved in the RT-qPCR experiments are given in Table 1.
 |
Table 1 Primers Used in the Real-Time qPCR Assay |
Immunohistochemistry (IHC)
The embedded paraffin tissue sections were first deparaffinized, hydrated, and blocked. The tissue sections were subsequently exposed to primary anti-MAPK10 antibody (1:400, Abcam, USA) overnight at 4°C. After incubating the sections with HRP-conjugated antibody (DAKO, USA), a DAB color development kit (Beyotime, China) was applied to staining. Two pathologists microscopically observed and recorded scores for all sections. Positive cell fraction was as follows: 0:0–5%; 1:6–25%; 2:26–50%; 3:51%-75%; 4: >75%. The scoring criteria for staining intensity were: score 0: unstained; 1: yellowish; 2: yellow to brown; 3: brown. Microscopically, the result of the staining intensity score multiplied by the fraction of positive cells was the immunohistochemical score. The immunohistochemical scores were finally classified into 4 grades: negative (-):0; mildly positive (+): score 1–4; intermediate positive (++): score 5–8; extremely positive (+++): score 9–12.
Statistical Analysis
R software (version 4.1.2) was employed to analyze all statistical data. NLRs signature’s prognostic significance was estimated by Kaplan-Meier curves and Log rank test using the R package “survminer”. The predictive presentation of NLRs signature scores was calculated using the R package ‘tROC’. R package “corrplot” was adopted to analyze Pearson’s correlation between the mRNA expression levels of 7 genes included in the NLRs signature and risk scores. Wilcoxon-rank sum test was utilized to assess differences in continuous variables between two distinct groups. NS: not significant; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Results
Critical NLRs Signaling Pathway-Related Genes in SKCM
Multiple investigations have demonstrated that the mutations in genes related to the NLRs signaling pathway are responsible for many diseases, including inflammation, autoimmune disorders, and multiple tumors.18,19 Among the SKCM cohort from TCGA, several NLRs family members were found recurrently mutated. As shown in Figure 1A, we depicted the genomic profile of the top 20 NLRs signaling pathway-related genes with higher somatic mutation frequency. Most of the mutations were missense mutations. NLRP1 and NLRP3 were the leading genes recurrently mutated in over 10% of samples. They belong to the NLRP subfamily, which primarily mediates “inflammasome” formation.20 NOD1 and NOD2 encode cytosolic proteins, triggering signal transduction by activating NF-κB and MAPK.21 It has been reported that both could be involved in tumorigenesis and tumor progression of breast, lung, and gastric tumors.22,23 Mutations of NOD1 and NOD2 were spread in 3% and 5% samples, respectively, suggesting their potential role in the development of SKCM.
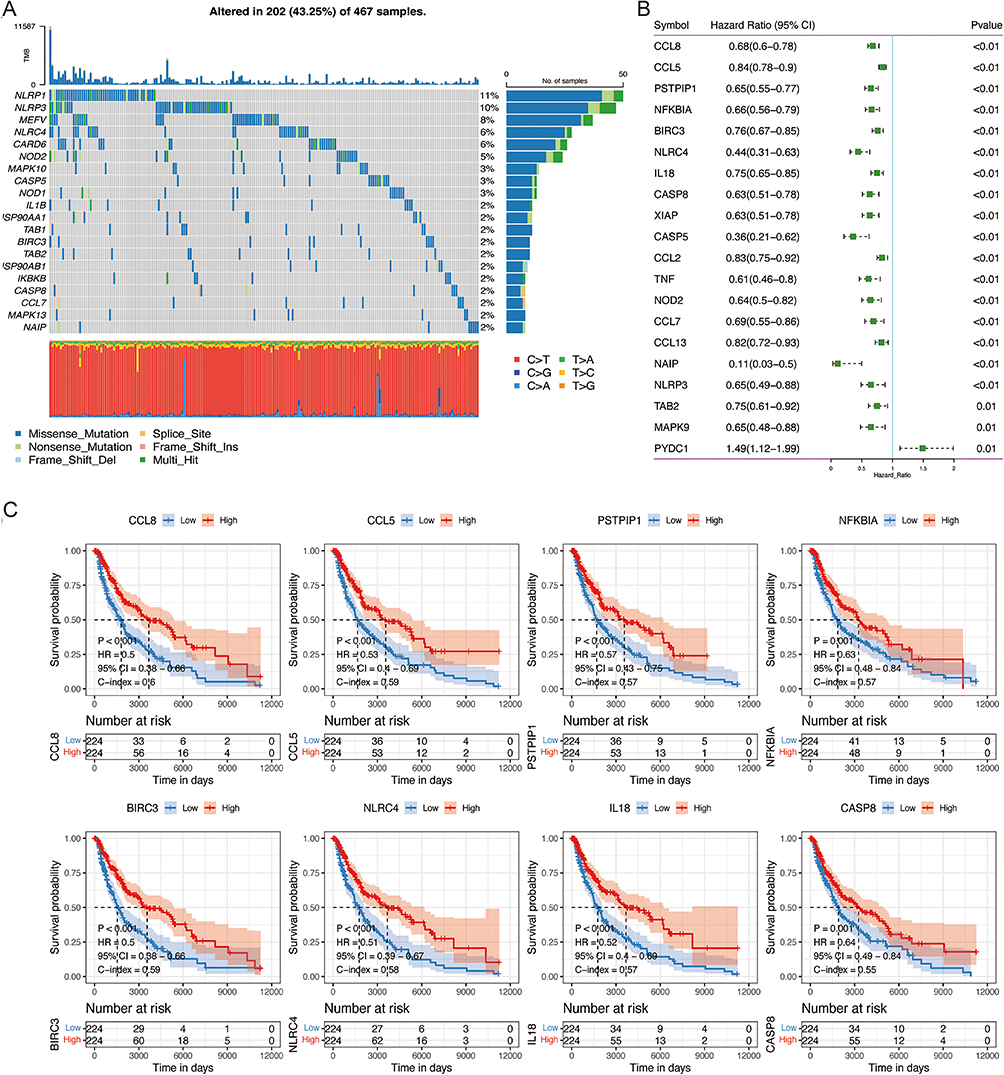 |
Figure 1 Critical NLRs signaling pathway-related genes in TCGA SKCM cohort. (A) Genomic profile of top 20 NLRs signaling pathway-related genes with higher somatic mutation frequency. (B) Forest plot showing the hazard ratio and p-value of the NLRs signaling pathway-related genes from univariate Cox regression analysis in the TCGA cohort. (C) Kaplan-Meier curves for OS by expression of CCL8, CCL5, PSTPIP1, NFKBIA, BIRC3, NLRC4, IL18, and CASP8, where the median expression was cut-off. |
Then we screened the candidate informative NLRs signaling pathway-related genes in SKCM through their prognostic efficacy. Univariate Cox analysis revealed that 35 NLRs-associated genes were notably related to SKCM patients’ overall survival (OS). The top 19 significant genes (sorted by P-value) exhibited a protective role in SKCM (Figure 1B, P< 0.01), including NLRs Family CARD Domain Containing 4 (NLRC4), chemokines (CCL5, CCL8), and apoptosis regulator CASP8 (P< 0.05, Figure 1C).
Establishment and Identification of the Prognostic NLRs Signature
The LASSO-Cox regression analysis was applied to remove redundant factors to establish a prognostic signature comprised of critical genes connected with NLRs signaling pathways (Figure 2A and 2B). Based on the optimal value of λ, 7 genes were filtered out and constructed the prognostic signature. These 7 NLRs signaling pathway-related genes were the most predictive factors for OS of SKCM patients, including CCL8, XIAP, PSTPIP1, NFKBIA, CASP8, MAPK10, and CARD18 (Figure 2C). The risk score of each SKCM patient was computed by utilizing the expression levels of the seven genes mentioned above. According to the median risk score, all SKCM patients were separated into two distinct groups. Evidently, SKCM patients who had elevated risk scores exhibited a considerably reduced OS compared to those with low-risk scores (Figure 2D and E). Kaplan-Meier survival analysis further compared the noteworthy differences in OS between the two SKCM patient groups. (Figure 2G, P< 0.001). The expression patterns of the 7 NLRs signaling pathway-related genes, constituting the prognostic signature, differed in the two groups. CARD18 and MAPK10 were up-regulated in SKCM patients of the high-risk group, whereas CCL8, XIAP, PSTPIP1, NFKBIA, and CASP8 expression levels were elevated in the low-risk group (Figure 2F). NLRs signature’s predictive efficiency was estimated through the time-dependent ROC curve. In years 1, 3, and 5, the AUC (area under the ROC curve) for SKCM patients in the TCGA cohort reached 0.657, 0.666, and 0.678, respectively (Figure 2H).
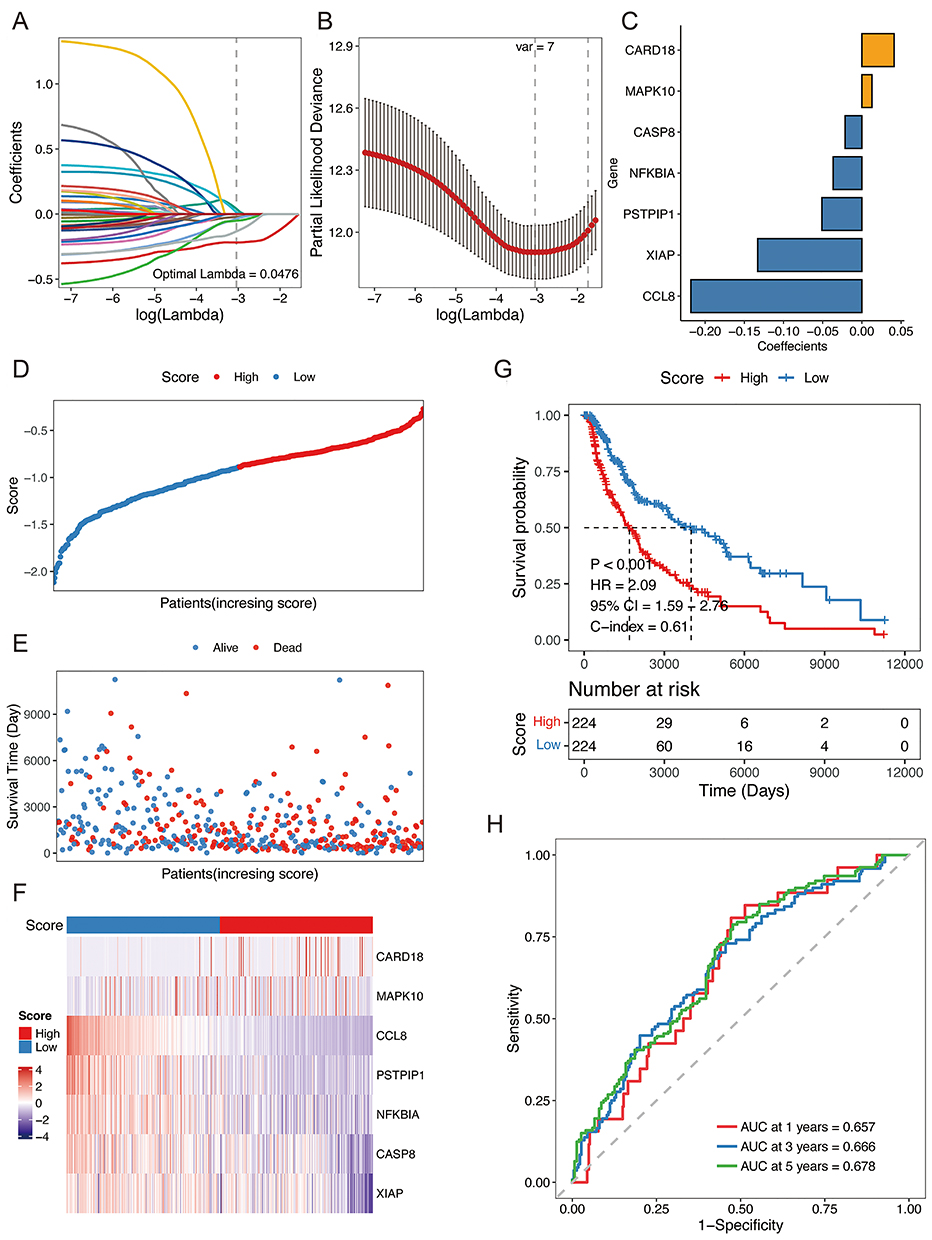 |
Figure 2 Construction of the prognostic NLRs-related gene signature in the TCGA cohort. (A) LASSO coefficient profiles of the prognostic genes. (B) Cross-validation for turning parameter selection in the LASSO regression model. Two vertical dashed lines indicated the optimal values using the minimum criteria. Optimal RNAs with the best discriminative capability were selected for developing the risk score. (C) LASSO coefficients of the 7 optimized genes for constructing the prognostic model. (D) The distribution of the risk scores. (E) The distributions of OS status, OS time, and risk score in TCGA SKCM cohort. (F) Heatmap indicated mRNA expression distribution of the 7 genes between the high-risk and low-risk groups. (G) Kaplan-Meier curves for the OS by risk groups. (H) The ROC curves of the risk score in predicting 1-, 3-, and 5-year OS. |
In addition, we assessed the robustness and generalizability of the NLRs signature in two independent validation sets. We calculated each patient’s risk score regarding the coefficients and expression levels of NLRs signaling pathway-associated genes in the signature. We separated them into two groups as the TCGA SKCM cohort did. As expected, SKCM patients in the high-risk group demonstrated considerably decreased OS versus the low-risk group. (log-rank P-value = 0.021, 0.025 for GSE133713 and GSE54467, respectively, Figure 3A and B). It is worth noting that the prognostic NLRs signature also exhibited outstanding predictive accuracy in the two validation sets (Figure 3C and D). In years 1, 3, and 5, the AUC was 0.987, 0.780, and 0.820 for patients from GSE133713, respectively.
 |
Figure 3 Validation of the prognostic NLRs-related gene signature in the two independent validation cohorts. (A and B) Kaplan-Meier curves for the OS by risk groups for GSE133713 and GSE54467 datasets, respectively. (C and D) The ROC curves of the risk score in predicting 1-, 3-, and 5-year OS in GSE133713 and GSE54467 datasets, respectively. |
Independent Predictive Efficacy of the NLRs Signature and Its Association with Clinicopathological Features
We also investigated the relationship between the NLRs signature and clinicopathologic features. SKCM patients older than 60 years in the TCGA SKCM cohort had exceedingly higher risk scores as opposed to relatively younger patients (P-value=0.022, Figure 4A). However, neither gender nor tumor stage was remarkably linked with the risk score (Figure 4B and C).
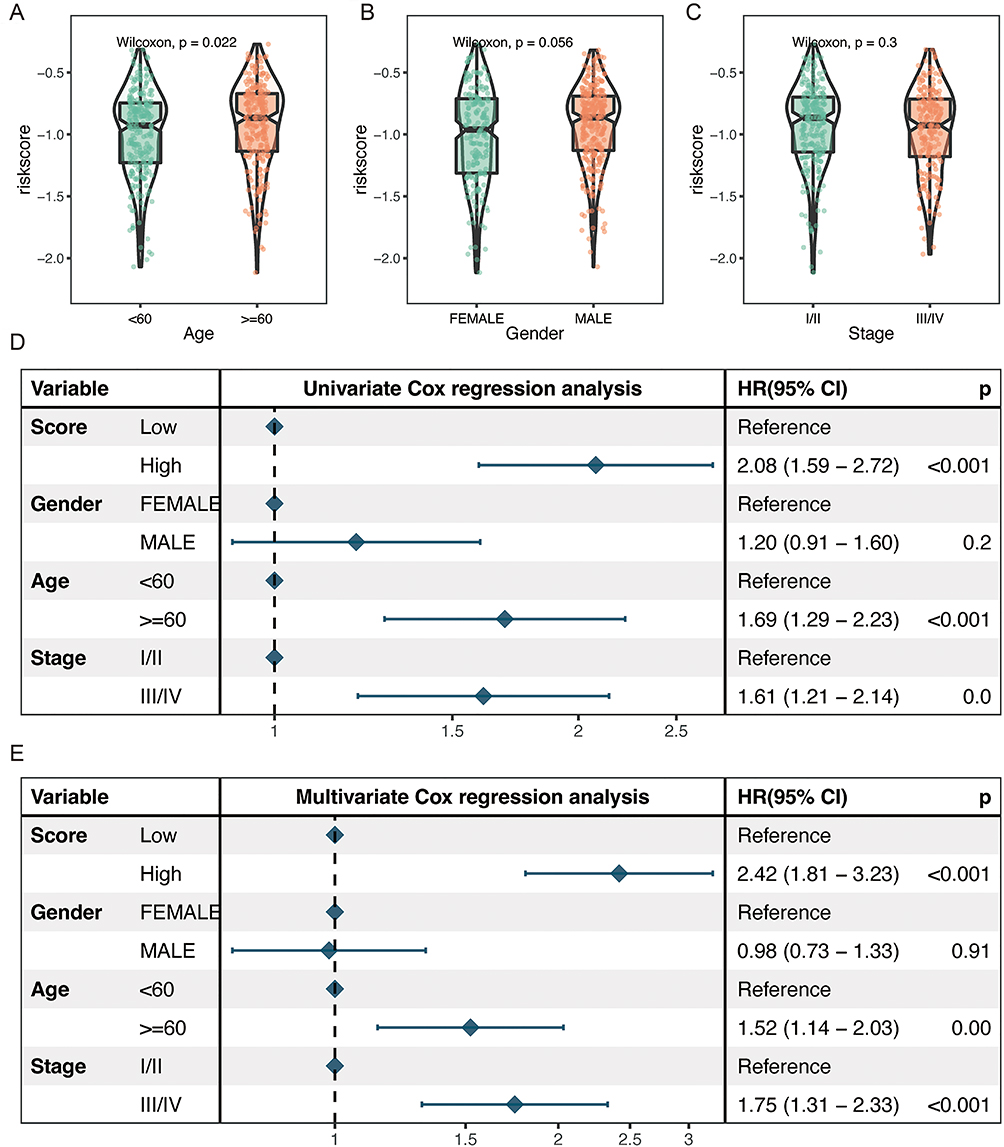 |
Figure 4 Independent predictive efficacy of the NLRs signature and its association with clinicopathological features. Distribution of the risk score by different age (A), gender (B), and tumor stage (C) groups in TCGA SKCM cohort. Results of the univariate (D) and multivariate (E) Cox regression analyses regarding OS of risk score and clinicopathological features in the TCGA cohort. |
Then, we carried out univariate and multivariate Cox analyses to further assess the NLRs signature’s unique predictive ability. Univariate Cox analysis disclosed that the risk score (high or low), age (older than 60 or not), and TNM stage (I/II or III/IV) in the TCGA SKCM cohort were notably associated with OS (P<0.001, Figure 4D). Multivariate Cox analysis further determined the independent function of the NLRs signature as an indicator of prognosis. (P< 0.001, Figure 4E).
Moreover, a graphic nomogram was developed for the potential clinical application of the NLRs signature. Based on the tumor stage, age, and risk score, the 1-, 3-, and 5-year survival rates for SKCM patients may be forecasted (Figure 5A). The calibration chart demonstrated that the nomogram’s predictive accuracy is in high agreement with the actual observed results, underscoring the significance of this integrated nomogram in clinical applications (Figure 5B). Furthermore, decision curve analysis (DCA) supported the predictive effectiveness of the NLRs signature in clinical decision-making at 1, 3, and 5 years (Figure 5C). These results highlighted that the NLRs signature could serve as a favorable predictive model for the long-term outcome of SKCM patients.
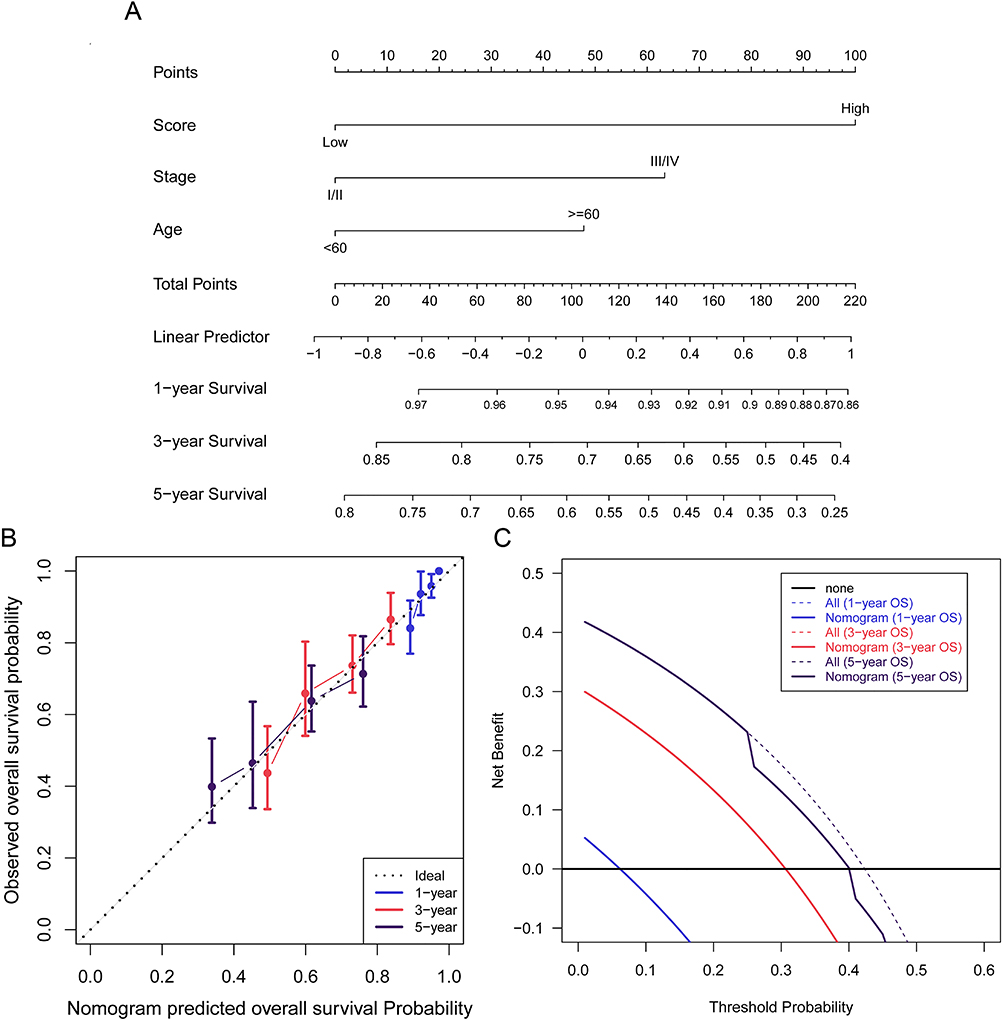 |
Figure 5 The construction and verification of the nomogram in the TCGA cohort. (A) A nomogram based on the NLRs prognostic signature risk score, tumor stage, and age for 1-, 3- and 5-year OS prediction. (B) The calibration plot showed the agreement between 1-, 3- and 5-year OS prediction and actual observation. (C) Results of the decision curve analysis (DCA). |
The NLRs Signature Characterized Differential Tumor Molecular Features
To further delve into the underlying pathogenesis of the established NLRs signature, we first checked somatic mutations in different risk groups of the TCGA SKCM patients. Mutations of specific genes were commonly mutated in both high-risk and low-risk tumors, including TTN, MUC16, BRAF, DNAH5, and PCLO, with mutation frequency over 40% (Figure 6A and B). Among them, MUC16 mutations were comparatively prevalent in the low-risk group (69% vs 64%), which corresponded to the previous findings that MUC16 mutation could affect the prognosis of SKCM patients by participating in the regulation of the distribution of tumor-infiltrating immune cell species and immune-associated signal transduction pathways.24
 |
Figure 6 The NLRs signature characterized differential tumor molecular features in the TCGA cohort. (A and B) Oncoprint depicts the recurrent somatic mutations with maximum mutation frequency in the different risk groups of the TCGA SKCM cohort. (C) The ssGSEA activity score of the 50 cancer hallmark pathways between the high-and low-risk groups in the TCGA cohort. |
In order to investigate cancer hallmarks connected to the NLRs signature, the ssGSEA activity scores of 50 cancer hallmark pathways were computed for TCGA SKCM patients. Comparing SKCM patients with elevated scores, we discovered that the low-risk group’s inflammatory response, interferon-α/γ response, and complement pathways were remarkably activated (Figure 6C).
The NLRs Signature Indicated the Heterogeneous Tumor Immune Microenvironment
To continue searching for the connection between the NLRs signature and immune response, we first assessed SKCM patients’ general immune status. Leukocyte fraction was inversely related to the risk score (P<0.05, Figure 7A). Outcomes of the ESTIMATE algorithm confirmed the negative association between the NLRs signature score and the immune and stromal score of the tumor microenvironment (Figure 7B and C). Unsurprisingly, the tumor purity was positively correlated with the NLRs signature score (Figure 7D). These results might explain the superior survival in the low-risk group, as it showed an activated immune response. Then we measured the relative infiltration ratios of 22 distinct types of immune cells by CIBERSORT. Significant accumulations of several anti-cancer immune cells, which comprises CD8 T cell, activated NK cell, CD4 memory activated T cell, γδ T cell, and M1 macrophage had been observed in the low-risk group (cut-off criteria was the median value) (P<0.05, Figure 7E). In contrast, the majority of immune cells infiltrating the high-risk group were in a resting state, such as the M0 macrophage, resting NK cell, resting mast cell, and CD4 memory resting T cell. Notably, SKCM patients in the higher-risk group possessed higher levels of M2 macrophage infiltration, which is consistent with its predominant immunosuppressive role in the tumor immune microenvironment of SKCM.25 Also, previous researches have demonstrated that a higher ratio of M1/M2 tumor-associated macrophage may improve the prognosis for SKCM patients.26 7 NLRs signature genes were connected with 22 immune cell infiltration distributions, verifying the correlation between risk scores and tumor immune microenvironment (Figure 7F).
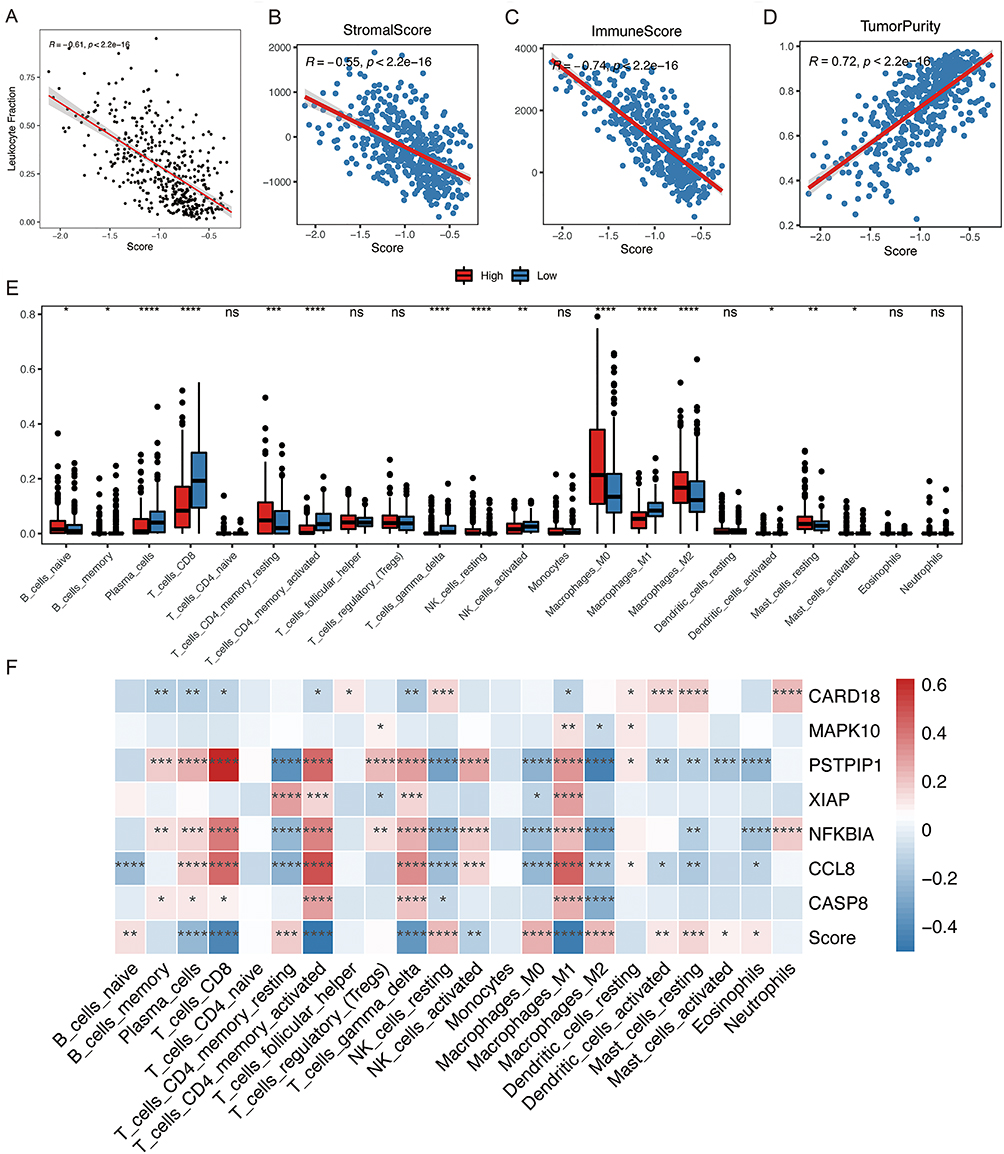 |
Figure 7 The NLRs signature indicated the heterogeneous tumor immune microenvironment. Scatter plots depict the correlations between the risk score and leukocyte fraction (A), stromal score (B), immune score (C), and tumor purity (D). (E) Comparison of 22 immune cell subtypes infiltration between high- and low-risk groups in TCGA SKCM cohort. (F) Heatmap depicting the correlation between the 7 signature genes expressions, risk score, and the infiltration scores of 22 immune cells; *P < 0.05; **P < 0.01; ***P < 0.001; ****P <0.0001. Abbreviation: ns, not significant. |
Potential Clinical Application of the NLRs Signature in Immunotherapy
ICB therapy has achieved obvious clinical advantages in multiple tumor types, especially SKCM.27 In the TCGA SKCM cohort, we observed a negative association between the NLRs signature score and several inhibitory checkpoints, including CD274, PDCD1, and CTLA4, which were the targets of current ICB strategies (Figure 8A). Furthermore, the NLRs signature score also showed a slightly positive association with CD276 (Figure 8A). Higher mutation load and neoantigen load were considered the biomarkers of clinical response for ICB,28 but they did not show significant dissimilarity in the different risk groups (Figure 8B). On the contrary, there was a difference in T cell receptor (TCR) clonal diversity between the two groups, with significantly higher TCR clonal diversity in the low-risk group, suggesting T cell clonal expansion within the low-risk group (Figure 8B).
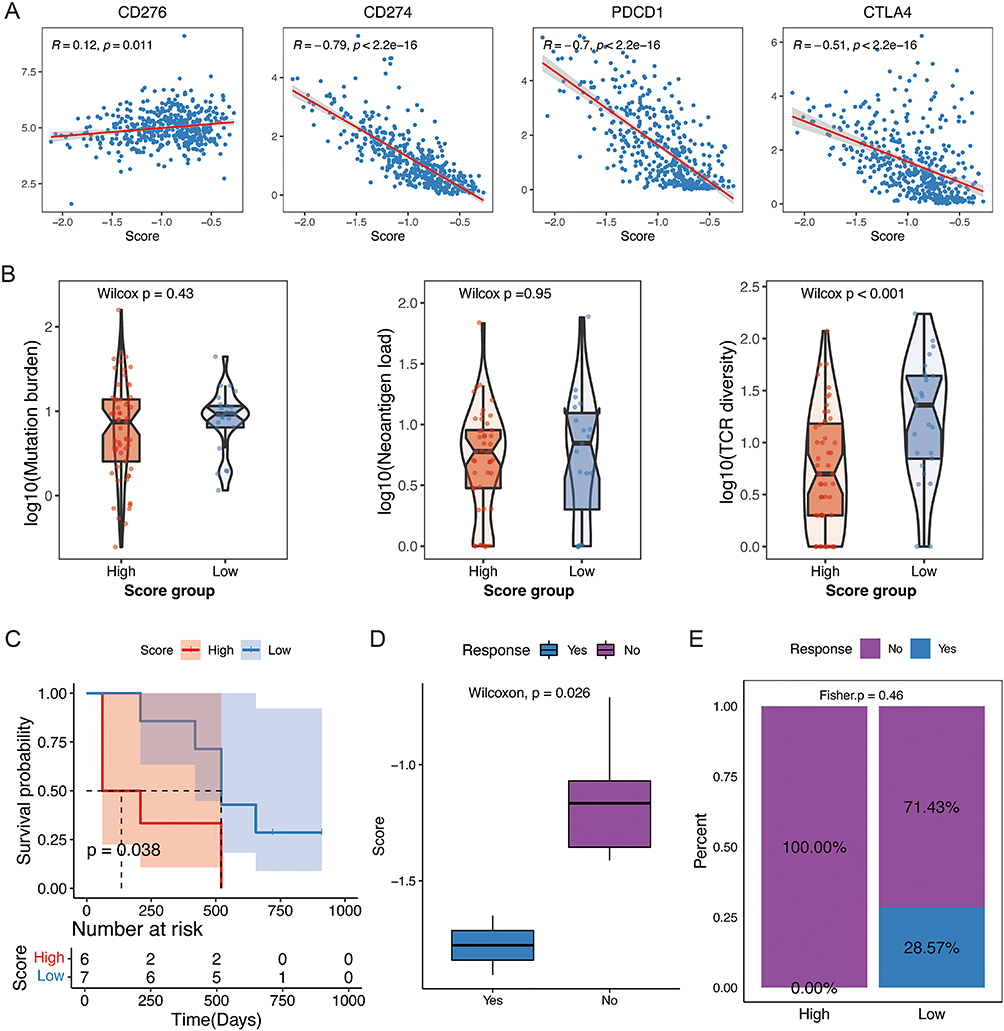 |
Figure 8 Potential clinical application of NLR signature in immunotherapy. (A) Scatter plots depicting correlations between the risk score and the expression of four immune checkpoint genes (CD276, CD274, PDCD1, CTLA4). (B) Comparison of the mutation burden, neoantigen load, and TCR diversity by different risk groups. (C) Kaplan-Meier curves for the OS of patients treated with ICB by different risk groups. (D) Distribution of the risk score between responders and non-responders. (E) The proportion of responders and non-responders between the high- and low-risk groups. |
To explore the potential application of NLRs signature in ICB therapy exhaustively, we computed the NLRs signature scores in SKCM patients following the management of anti-CTLA4 and/or anti-PD1 antibodies (GSE115821). SKCM patients with a lower score exhibited significantly prolonged OS (Figure 8C). Meanwhile, the NLRs signature score differed between the response and the non-response group to ICB (Figure 8D). Patients with lower scores exhibited a greater clinical response to ICB (Figure 8E). In summary, the prognostic NLRs signature for SKCM could also be considered a promising biomarker for ICB therapy and probably supplement the effect of mutation burden.
Expression Validation of the NLRs Signature in Clinical Samples
SKCM tumor tissues exhibited elevated MAPK10, XIAP, and CCL8 expression levels, as determined by RT-qPCR. PSTPIP1, NFKBIA, and CASP8 were notably overexpressed in paraneoplastic tissues, whereas there was no dramatic difference in CARD18 expression between the two tissues (Figure 9A). According to the coefficient in the signature, MAPK10 tends to be a risk factor for SKCM, and the RT-qPCR result of MAPK10 was also compatible with the signature. Therefore, immunohistochemistry was utilized to compare the expression of MAPK10 protein in 10 patients with SKCM and 8 patients with benign pigmented nevi. Immunohistochemistry findings suggested that MAPK10 protein was predominantly localized in the cytoplasm (Figure 9C); the expression level of MAPK10 was stronger in SKCM tissues versus benign pigmented nevus tissues (Figure 9B).
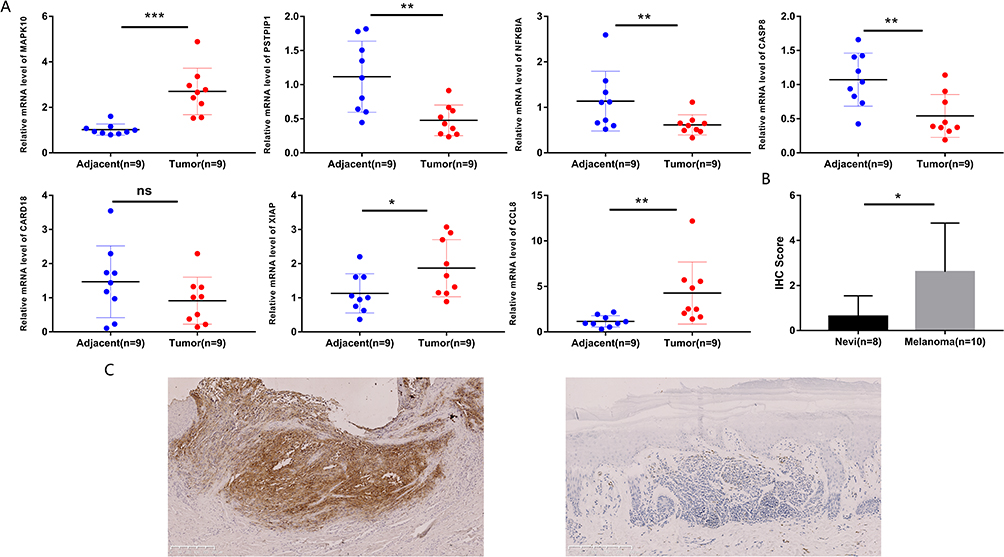 |
Figure 9 Expression Validation of the NLR-related prognostic gene signature in Clinical Samples. (A) Expressions of 7 critical NLR signature genes in tumor and para-tumor samples by RT-qPCR. (B) The immunostaining scores of SKCM tissues (n = 10) and nevus tissues (n = 8) were displayed using t-test. (C) Immunohistochemistry staining for MAPK10 in clinical SKCM (Left) and nevus tissue (Right) (Scale bar, 250 μm); *P < 0.05; **P < 0.01; ***P < 0.001. Abbreviation: ns, not significant. |
Discussion
Here, we established and verified a necessary prognostic NLRs signature in the TCGA SKCM cohort and two external GEO datasets. High-risk SKCM patients tended to experience a consistently poorer prognosis across all three cohorts. It was evidenced that the NLRs signature was a separate risk indicator for SKCM prognosis. Additionally, the prognostic ability of the nomogram derived from the constructed signature and clinicopathological factors for SKCM patients was highly accurate. To date, a few gene signatures for assessing the prognosis of SKCM patients have been constructed.29,30 These multigene signatures originating from RNA-seq or RT-qPCR data exhibited strong prediction performance. At the same time, it is still challenging for them to utilize effectively in clinical practice.
Recent studies underscore that several members of the NLRs family, which includes NOD1, NOD2, NLRP3, and NLRP12, have an essential function in modulating intestinal inflammation and tumor development.31,32 Among seven genes used to construct the risk signature, CARD18 (caspase recruitment domain family, member 18) was mildly up-regulated in the asbestos-related lung squamous cell carcinomas tissue.33 MAPK10 (mitogen-activated protein kinase 10 gene) is an essential MAP kinase (MAPK) gene family member. MAPK10 is a member of the c-Jun N-terminal kinase family, which belongs to a subfamily of mitogen-activated protein kinases (MAPK). C-Jun N-terminal kinases are involved in tumor cells’ proliferation, differentiation, and apoptosis.34 Several studies have suggested that MAPK10 is highly expressed in gastric and ovarian cancers and is a prognostic risk factor for tumors.35–37 Our investigation verified the elevated expression of MAPK10 in SKCM tumor tissues via RT-qPCR and IHC. CCL8 (chemotactic cytokines 8) expression correlated positively with M1 macrophages, and higher CCL8 expression had significantly longer overall survival in SKCM, which may be attributed to the positive role played by M1 macrophages in anti-tumor immune activity.38 PSTPIP1 (Proline-serine-threonine phosphatase interacting protein 1 gene) has been linked to acne syndrome, pyoderma gangrenosum, and pyogenic arthritis, with implications for inflammasome activation and overexpression of IL-1 and IL-18.39 Recent research has revealed that NFKBIA is one of the inhibitors of NF-κB and that the SNP rs1957106 CT and TT genotypes of NFKBIA are related to decreased NFKBIA expression in patients with malignant glioma and may serve as a biomarker for poor prognosis.40 CASP8 (caspase protease 8 gene) mediated cell apoptosis and inhibited tumor progression by inducing PD-L1 degradation via upregulating TNFAIP3 expression in SKCM.41 XIAP (X-linked inhibitor of the apoptosis protein gene) has been evidenced to be overexpressed in aggressive and metastatic melanoma cell lines. XIAP performed a multifunctional pro-metastasis role and may be one of the candidate therapeutic targets for SKCM. Currently, several compounds targeting XIAP have been developed and demonstrated promising anti-tumor activities in preclinical studies, including Embelin, BV6, Smac mimetics, and LCL161.42–45 These conclusions supported their potential as a promising biomarker for SKCM.
In 2011, the USA FDA approved the anti-CTLA4 antibody for SKCM medication, marking a new stage of development in tumor immunotherapy. Previous studies have shown numerous malignancies, especially SKCM and non-small cell lung cancer, to be responsive to immunotherapy. Our investigation revealed the connection between the NLRs signature genes and the SKCM immune environment for the first time. For the SKCM patients with lower scores, the inflammatory response, interferon-α/γ response, and complement pathways related to tumor immune response were dramatically more active than in the high-risk group. Various anti-cancer immune cell types, consisting of CD8 T cell, M1 macrophage, activated NK cell, γδ T cell, and CD4 memory-activated T cell, were also highly accumulated in the low-risk group. Thus, the tumor type of the low-risk group patients can be defined as “hot tumors”.46 However, the majority of extensively infiltrated immune cells in the high-risk SKCM patients were in a resting state. Also, the pro-tumorigenic M2-polarized macrophages were accumulated in the high-risk patients. Past research highlighted its contribution to hypoxia induction, lymphangiogenic regulation, angiogenic, tumor cell proliferation, immune suppression, and metastasis.47 These findings suggested that the 7 genes comprising the NLRs signature may influence SKCM patients’ prognosis by modulating the tumor immune microenvironment. Not surprisingly, this research also revealed that low-risk SKCM patients responded more positively to ICB therapy.
Finally, external validation of the critical NLRs prognostic genes was implemented via RT-qPCR and IHC. Compared with SKCM tumor tissues, PSTPIP1, NFKBIA, and CASP8 expression levels were more elevated in paraneoplastic tissues by RT-qPCR. On the contrary, the expression level of MAPK10 was higher in SKCM tumor tissues, as confirmed by RT-qPCR and IHC. These results were consistent with the previous data analyses, indicating the validity of the signature.
Conclusion
In summary, a promising NLRs signature with excellent predictive efficacy for SKCM was developed. Further analysis supported its potential effects in characterizing the tumor microenvironment of SKCM. Our research identified a new possible prognostic and therapeutic biomarker for SKCM patients. For a comprehensive knowledge of the diagnosis, prognosis, and mechanisms involved in SKCM, future studies should conduct more external validation of the key genes in this study, such as IHC, and analyze the biological function of the 7 critical NLRs signature genes.
Institutional Review Board Statement
The study complied with the latest Declaration of Helsinki and was approved by the Ethics committee of the Institute of Dermatology, Chinese Academy of Medical Sciences (2021-KY-044).
Informed Consent Statement
Informed consent was received from each patient participating in the study.
Acknowledgments
The authors would like to express their appreciation to Beijing GAP Biotechnology Co., Ltd. for providing their assistance with the technical consultation.
Funding
This work was supported by the National Natural Science Foundation of China (81772916, 82103470) and the Jiangsu Natural Science Foundation (BK20171132).
Disclosure
The authors of this study declare that no conflicts of interest exist.
References
1. Lv H, Liu X, Zeng X, et al. Comprehensive analysis of cuproptosis-related genes in immune infiltration and prognosis in melanoma. Front Pharmacol. 2022;13:930041. doi:10.3389/fphar.2022.930041
2. Banach K, Kowalska J, Rzepka Z, Beberok A, Rok J, Wrzesniok D. The role of uva radiation in ketoprofen-mediated braf-mutant amelanotic melanoma cells death - a study at the cellular and molecular level. Toxicol in Vitro. 2021;72:105108. doi:10.1016/j.tiv.2021.105108
3. Zhang W, Xie X, Huang Z, et al. The integration of single-cell sequencing, TCGA, and geo data analysis revealed that prrt3-as1 is a biomarker and therapeutic target of skcm. Front Immunol. 2022;13:919145. doi:10.3389/fimmu.2022.919145
4. Singh M, Vianden C, Cantwell MJ, et al. Intratumoral cd40 activation and checkpoint blockade induces t cell-mediated eradication of melanoma in the brain. Nat Commun. 2017;8(1):1447. doi:10.1038/s41467-017-01572-7
5. Wilmanski JM, Petnicki-Ocwieja T, Kobayashi KS. Nlr proteins: integral members of innate immunity and mediators of inflammatory diseases. J Leukoc Biol. 2008;83(1):13–30. doi:10.1189/jlb.0607402
6. Kim YK, Shin JS, Nahm MH. Nod-like receptors in infection, immunity, and diseases. Yonsei Med J. 2016;57(1):5–14. doi:10.3349/ymj.2016.57.1.5
7. Velloso FJ, Trombetta-Lima M, Anschau V, Sogayar MC, Correa RG. Nod-like receptors: major players (and targets) in the interface between innate immunity and cancer. Biosci Rep. 2019;39:4. doi:10.1042/BSR20181709
8. Zheng C. The emerging roles of nod-like receptors in antiviral innate immune signaling pathways. Int J Biol Macromol. 2021;169:407–413. doi:10.1016/j.ijbiomac.2020.12.127
9. Cui J, Chen Y, Wang HY, Wang RF. Mechanisms and pathways of innate immune activation and regulation in health and cancer. Hum Vaccin Immunother. 2014;10(11):3270–3285. doi:10.4161/21645515.2014.979640
10. Bai L, Li W, Zheng W, Xu D, Chen N, Cui J. Promising targets based on pattern recognition receptors for cancer immunotherapy. Pharmacol Res. 2020;159:105017. doi:10.1016/j.phrs.2020.105017
11. Saxena M, Yeretssian G. Nod-like receptors: master regulators of inflammation and cancer. Front Immunol. 2014;5:327. doi:10.3389/fimmu.2014.00327
12. Goutagny N, Estornes Y, Hasan U, Lebecque S, Caux C. Targeting pattern recognition receptors in cancer immunotherapy. Target Oncol. 2012;7(1):29–54. doi:10.1007/s11523-012-0213-1
13. Zhong Y, Kinio A, Saleh M. Functions of nod-like receptors in human diseases. Front Immunol. 2013;4:333. doi:10.3389/fimmu.2013.00333
14. Zhai Z, Liu W, Kaur M, et al. Nlrp1 promotes tumor growth by enhancing inflammasome activation and suppressing apoptosis in metastatic melanoma. Oncogene. 2017;36(27):3820–3830. doi:10.1038/onc.2017.26
15. Verma D, Bivik C, Farahani E, et al. Inflammasome polymorphisms confer susceptibility to sporadic malignant melanoma. Pigment Cell Melanoma Res. 2012;25(4):506–513. doi:10.1111/j.1755-148X.2012.01008.x
16. Auslander N, Zhang G, Lee JS, et al. Robust prediction of response to immune checkpoint blockade therapy in metastatic melanoma. Nat Med. 2018;24(10):1545–1549. doi:10.1038/s41591-018-0157-9
17. Yoshihara K, Shahmoradgoli M, Martinez E, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013;4:2612. doi:10.1038/ncomms3612
18. Wang H, Xia L, Yao CC, et al. Nlrp4 negatively regulates type i interferon response and influences the outcome in anti-programmed cell death protein (pd)-1/pd-ligand 1 therapy. Cancer Sci. 2022;113(3):838–851. doi:10.1111/cas.15243
19. Ozbayer C, Kurt H, Bayramoglu A, et al. The role of nod1/card4 and nod2/card15 genetic variations in lung cancer risk. Inflamm Res. 2015;64(10):775–779. doi:10.1007/s00011-015-0859-0
20. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proil-beta. Mol Cell. 2002;10(2):417–426. doi:10.1016/s1097-2765(02)00599-3
21. Chen G, Shaw MH, Kim YG, Nunez G. Nod-like receptors: role in innate immunity and inflammatory disease. Annu Rev Pathol. 2009;4:365–398. doi:10.1146/annurev.pathol.4.110807.092239
22. Da SCJ, Miranda Y, Austin-Brown N, et al. Nod1-dependent control of tumor growth. Proc Natl Acad Sci USA. 2006;103(6):1840–1845. doi:10.1073/pnas.0509228103
23. Lener MR, Oszutowska D, Castaneda J, et al. Prevalence of the nod2 3020insc mutation in aggregations of breast and lung cancer. Breast Cancer Res Treat. 2006;95(2):141–145. doi:10.1007/s10549-005-9057-z
24. Wang Z, Hou H, Zhang H, Duan X, Li L, Meng L. Effect of muc16 mutations on tumor mutation burden and its potential prognostic significance for cutaneous melanoma. Am J Transl Res. 2022;14(2):849–862.
25. Komohara Y, Fujiwara Y, Ohnishi K, Takeya M. Tumor-associated macrophages: potential therapeutic targets for anti-cancer therapy. Adv Drug Deliv Rev. 2016;99(Pt B):180–185. doi:10.1016/j.addr.2015.11.009
26. Partecke LI, Gunther C, Hagemann S, et al. Induction of m2-macrophages by tumour cells and tumour growth promotion by m2-macrophages: a quid pro quo in pancreatic cancer. Pancreatology. 2013;13(5):508–516. doi:10.1016/j.pan.2013.06.010
27. He X, Xu C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 2020;30(8):660–669. doi:10.1038/s41422-020-0343-4
28. Samstein RM, Lee CH, Shoushtari AN, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. 2019;51(2):202–206. doi:10.1038/s41588-018-0312-8
29. Song LB, Luan JC, Zhang QJ, et al. The identification and validation of a robust immune-associated gene signature in cutaneous melanoma. J Immunol Res. 2021;2021:6686284. doi:10.1155/2021/6686284
30. Mei Y, Chen MM, Liang H, Ma L. A four-gene signature predicts survival and anti-ctla4 immunotherapeutic responses based on immune classification of melanoma. Commun Biol. 2021;4(1):383. doi:10.1038/s42003-021-01911-x
31. Udden S, Peng L, Gan JL, et al. Nod2 suppresses colorectal tumorigenesis via downregulation of the tlr pathways. Cell Rep. 2017;19(13):2756–2770. doi:10.1016/j.celrep.2017.05.084
32. Couturier-Maillard A, Secher T, Rehman A, et al. Nod2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J Clin Invest. 2013;123(2):700–711. doi:10.1172/JCI62236
33. Wright CM, Savarimuthu FS, Tan ME, et al. Ms4a1 dysregulation in asbestos-related lung squamous cell carcinoma is due to cd20 stromal lymphocyte expression. PLoS One. 2012;7(4):e34943. doi:10.1371/journal.pone.0034943
34. Hammouda MB, Ford AE, Liu Y, Zhang JY. The jnk signaling pathway in inflammatory skin disorders and cancer. Cells. 2020;9(4). doi:10.3390/cells9040857
35. Gao Y, Wang Y, Wang X, et al. Mir-335-5p suppresses gastric cancer progression by targeting mapk10. Cancer Cell Int. 2021;21(1):71. doi:10.1186/s12935-020-01684-z
36. Qiao B, Wang Q, Zhao Y, Wu J. Mir-205-3p functions as a tumor suppressor in ovarian carcinoma. Reprod Sci. 2020;27(1):380–388. doi:10.1007/s43032-019-00047-y
37. Nie S, Ni N, Chen N, et al. Development of a necroptosis-related gene signature and the immune landscape in ovarian cancer. J Ovarian Res. 2023;16(1):82. doi:10.1186/s13048-023-01155-9
38. Lian W, Zheng X. Identification and validation of tme-related signatures to predict prognosis and response to anti-tumor therapies in skin cutaneous melanoma. Funct Integr Genomics. 2023;23(2):153. doi:10.1007/s10142-023-01051-x
39. Marzano AV, Trevisan V, Gattorno M, Ceccherini I, De Simone C, Crosti C. Pyogenic arthritis, pyoderma gangrenosum, acne, and hidradenitis suppurativa (papash): a new autoinflammatory syndrome associated with a novel mutation of the pstpip1 gene. JAMA Dermatol. 2013;149(6):762–764. doi:10.1001/jamadermatol.2013.2907
40. Zhao Z, Zhong X, Wu T, et al. Identification of a nfkbia polymorphism associated with lower nfkbia protein levels and poor survival outcomes in patients with glioblastoma multiforme. Int J Mol Med. 2014;34(5):1233–1240. doi:10.3892/ijmm.2014.1932
41. Zou J, Xia H, Zhang C, et al. Casp8 acts through a20 to inhibit pd-l1 expression: the mechanism and its implication in immunotherapy. Cancer Sci. 2021;112(7):2664–2678. doi:10.1111/cas.14932
42. Danquah M. Embelin and its derivatives: design, synthesis, and potential delivery systems for cancer therapy. Pharmaceuticals. 2022;15(9). doi:10.3390/ph15091131
43. Ahmad I, Dera AA, Irfan S, et al. Bv6 enhances apoptosis in lung cancer cells by ameliorating caspase expressions through attenuation of xiap, ciap-1, and ciap-2 proteins. J Cancer Res Ther. 2022;18(6):1651–1657. doi:10.4103/jcrt.JCRT_1281_20
44. Miles MA, Caruso S, Baxter AA, Poon I, Hawkins CJ. Smac mimetics can provoke lytic cell death that is neither apoptotic nor necroptotic. Apoptosis. 2020;25(7–8):500–518. doi:10.1007/s10495-020-01610-8
45. Cremona M, Vandenberg CJ, Farrelly AM, et al. BRCA mutations lead to xiap overexpression and sensitise ovarian cancer to inhibitor of apoptosis (iap) family inhibitors. Br J Cancer. 2022;127(3):488–499. doi:10.1038/s41416-022-01823-5
46. Ye F, Cai Z, Wang B, et al. Tgf-beta antagonizes ifn-gamma-mediated adaptive immune evasion via activation of the akt-smad3-shp1 axis in lung adenocarcinoma. Cancer Res. 2023. doi:10.1158/0008-5472.CAN-22-3009
47. Boutilier AJ, Elsawa SF. Macrophage polarization states in the tumor microenvironment. Int J Mol Sci. 2021;22(13). doi:10.3390/ijms22136995
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
A Novel lncRNA Panel for Risk Stratification and Immune Landscape in Breast Cancer Patients

Li C, Wang X, Chen T, Li W, Yang Q
International Journal of General Medicine 2022, 15:5253-5272
Published Date: 27 May 2022
RuleFit-Based Nomogram Using Inflammatory Indicators for Predicting Survival in Nasopharyngeal Carcinoma, a Bi-Center Study
Luo C, Li S, Zhao Q, Ou Q, Huang W, Ruan G, Liang S, Liu L, Zhang Y, Li H
Journal of Inflammation Research 2022, 15:4803-4815
Published Date: 24 August 2022
Significance of Pyroptosis in Immunoregulation and Prognosis of Patients with Acute Respiratory Distress Syndrome: Evidence from RNA-Seq of Alveolar Macrophages
Liu B, Li Y, Xiang J, Li Y, Zhou M, Ren Y, Fu Z, Ding F
Journal of Inflammation Research 2023, 16:3547-3562
Published Date: 21 August 2023
Assessment of Immune Status in Patients with Mismatch Repair Deficiency Endometrial Cancer
Ma J, Lin J, Lin X, Ren Y, Liu D, Tang S, Huang L, Xu S, Mao X, Sun P
Journal of Inflammation Research 2024, 17:2039-2050
Published Date: 2 April 2024
Utility of Alpha-Fetoprotein and Ultrasound in the Diagnosis and Prognosis of Patients with Hepatocellular Liver Cancer
Huang D, Zhang J, Xu J, Niu Q, Zhou D
Journal of Multidisciplinary Healthcare 2024, 17:1819-1826
Published Date: 23 April 2024