Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Conjugation of cell-penetrating peptides with poly(lactic-co-glycolic acid)-polyethylene glycol nanoparticles improves ocular drug delivery
Authors Vasconcelos A, Vega E, Pérez Y, Gómara MJ ![]() , García ML, Haro I
, García ML, Haro I
Received 16 July 2014
Accepted for publication 24 September 2014
Published 27 January 2015 Volume 2015:10(1) Pages 609—631
DOI https://doi.org/10.2147/IJN.S71198
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Thomas Webster
Aimee Vasconcelos,1 Estefania Vega,2 Yolanda Pérez,3 María J Gómara,1 María Luisa García,2 Isabel Haro1
1Unit of Synthesis and Biomedical Applications of Peptides, Department of Biomedical Chemistry, Institute for Advanced Chemistry of Catalonia, Consejo Superior de Investigaciones Científicas (IQAC-CSIC), 2Department of Physical Chemistry, Institute of Nanoscience and Nanotechnology, Faculty of Pharmacy, University of Barcelona, 3Nuclear Magnetic Resonance Unit, IQAC-CSIC, Barcelona, Spain
Abstract: In this work, a peptide for ocular delivery (POD) and human immunodeficiency virus transactivator were conjugated with biodegradable poly(lactic-co-glycolic acid) (PGLA)–polyethylene glycol (PEG)-nanoparticles (NPs) in an attempt to improve ocular drug bioavailability. The NPs were prepared by the solvent displacement method following two different pathways. One involved preparation of PLGA NPs followed by PEG and peptide conjugation (PLGA-NPs-PEG-peptide); the other involved self-assembly of PLGA-PEG and the PLGA-PEG-peptide copolymer followed by NP formulation. The conjugation of the PEG and the peptide was confirmed by a colorimetric test and proton nuclear magnetic resonance spectroscopy. Flurbiprofen was used as an example of an anti-inflammatory drug. The physicochemical properties of the resulting NPs (morphology, in vitro release, cell viability, and ocular tolerance) were studied. In vivo anti-inflammatory efficacy was assessed in rabbit eyes after topical instillation of sodium arachidonate. Of the formulations developed, the PLGA-PEG-POD NPs were the smaller particles and exhibited greater entrapment efficiency and more sustained release. The positive charge on the surface of these NPs, due to the conjugation with the positively charged peptide, facilitated penetration into the corneal epithelium, resulting in more effective prevention of ocular inflammation. The in vitro toxicity of the NPs developed was very low; no ocular irritation in vitro (hen’s egg test–chorioallantoic membrane assay) or in vivo (Draize test) was detected. Taken together, these data demonstrate that PLGA-PEG-POD NPs are promising vehicles for ocular drug delivery.
Keywords: peptide for ocular delivery, flurbiprofen, controlled release, ocular tolerance, anti-inflammatory
Introduction
Cataract is one of the most common causes of partial loss of vision, with an estimated 16 million people affected worldwide. Surgical intervention is still the standard cataract treatment. Flurbiprofen (FB) is a nonsteroidal anti-inflammatory drug (NSAID), which has been introduced into ocular therapy recently, not only for the management of inflammatory diseases that affect ocular structures, but also for use during eye surgery.1 NSAIDs are widely used topically to inhibit intraoperative miosis, reduce postoperative inflammation, treat seasonal allergic conjunctivitis, and both prevent and treat cystoid macular edema.2 They have also recently been found to be useful in decreasing bacterial colonization of contact lenses and preventing bacterial adhesion to human corneal epithelial cells.3
The bioavailability of ophthalmic drugs in aqueous solutions is usually low due to their rapid elimination after mucosal instillation, a consequence of reflex blinking and tear drainage, as well as of the presence of the corneal barrier. In fact, only 5% of the applied dose reaches intraocular tissues after corneal penetration.4 Research into biomaterials has therefore included the use of biodegradable polymeric nanoparticles (NPs) in ocular drug delivery, one of the most promising applications of NPs, as they offer a controlled release profile of a drug which is entrapped in the polymeric matrix .5–7 These are advantages that suggest that the required therapeutic effects could easily be achieved.8 Over the years, the potential of a variety of synthetic biodegradable polymers such as polylactic acid, polyglycolic acid, and their copolymer, poly(lactic-co-glycolic acid) (PLGA), for the production of NPs, has been extensively explored due to their biocompatibility, biodegradability, and mechanical strength.9 It has been described that PLGA NPs are internalized in cells partly through fluid phase pinocytosis and also through clathrin-mediated endocytosis, and they rapidly escape the endolysosomes and enter the cytoplasm. The reticuloendothelial system eliminates the NPs from the blood stream. Subsequent PLGA hydrolysis leads to metabolite monomers (lactic and glycolic acids), which are easily metabolized via the Krebs cycle.10
Furthermore, they are easily formulated via several production methods such as solvent displacement,11 solvent diffusion, emulsification–evaporation, and phase-inversion procedures.12,13 These polymers, which are commercially available in a range of molecular weights and copolymer ratios,14,15 have been approved by the US Food and Drug Administration and the European Medicine Agency for use in drug delivery in humans.16
FB has previously been formulated in PLGA NPs by Vega et al17 who achieved good stability and appropriate physicochemical properties for ocular administration, without causing ocular irritation at any level. Recently, other authors have reported that by means of a suitable modification of the PLGA NPs, the stability of the encapsulated drug, its release profile, and delivery to a specific target can be optimized.18–20 PLGA has thus been combined with hydrophilic polymers such as polyethylene glycol (PEG) and polyethylene oxide. The modification of preformed NPs with PEG (ie, PEGylation) brings about substantial changes in their physicochemical properties.21 PEG is a hydrophilic, nonionic polymer that has been shown to be highly biocompatible, and it can be added to NPs via a great number of different routes including covalent binding, mixing during NP preparation, and surface adsorption. The presence of PEG on the surface of NPs may provide other advantages, such as increasing their systemic circulation half-life and mucoadhesion.22
Furthermore, cell-penetrating peptides (CPPs) can be conjugated with NPs to enhance the cellular and nuclear uptake of the cargo molecules. CPPs are short (<20 amino acids) cationic peptide sequences from a range of proteins that are capable of transporting molecules across the cell membrane. Despite the lack of a clear understanding of their mechanism of action, it has been demonstrated that CPPs can deliver different cargoes into cells, including peptides, proteins, viral particles, liposomes, and NPs.23,24 The most extensively studied CPPs are the human immunodeficiency virus transactivator protein (HIV-Tat),25 the herpes simplex virus type 1 protein VP2226 and the homeodomain transcription factor Antennapedia.27 Several authors have studied the potential use of HIV-Tat28 and herpes simplex virus-VP2229 in delivering recombinant proteins to human embryonic retinoblast in culture and to retinal tissues in vivo. It has been observed that while both peptides act efficiently in vitro, their performance in the retina in vivo is limited. Recently, a novel CPP for ocular delivery was reported (peptide for ocular delivery [POD]), that is capable of transporting both small and large molecules across the plasma membrane. Moreover, it enters the corneal epithelium, sclera, choroid, and the dura of the optic nerve via topical application. POD, is useful in a vehicle that targets ocular tissues and has bacteriostatic properties.30,31 Furthermore, the cornea possesses negative surface charges, and it is expected that cationic colloidal NPs may penetrate through the negatively charged ocular tissues more efficiently than anionic carriers.32 Read et al also demonstrated that PEGylated CPPs such as PEG-POD are promising candidates for gene delivery.33 The addition of a PEG moiety to the POD resulted in small, discrete, and spherical NPs, without any aggregation being observed in vitro. Moreover, they studied the gene transfer efficiency, with POD being more efficient for subretinal DNA delivery than two other CPPs: HIV-Tat 28 and CK30.34 Particularly, PEG-POD DNA NPs have been utilized to deliver a plasmid expressing various marker genes or glial cell line-derived neurotrophic factor to mice, enabling rescue of photoreceptor degeneration. Thus, PEG-POD NPs have been considered a therapeutically relevant nonviral DNA vehicle with potential application in therapy for retinal degeneration.35,36
The main aim of the current study is to improve the corneal epithelium penetration of NPs composed of PLGA-PEG by means of conjugating two different CPPs (POD and HIV-Tat), the final objective being to achieve a longer sustained release of FB, which has been used as an example of NSAID drug. Peptide-derivatized NPs were obtained by the solvent displacement method. Their physicochemical properties, in vitro release kinetics, ex vivo interaction with corneal permeation, cytotoxicity, and ocular tolerance (in vitro and in vivo) were studied. Anti-inflammatory efficacy assays were also performed to determine whether these novel formulations (PLGA-NPs-PEG-peptide and PLGA-PEG-peptide NPs) could form the basis of a suitable strategy for ocular delivery of NSAIDs.
Materials and methods
Materials
PLGA with a 50:50 monomer ratio (Resomer® RG 503H, 34 kDa, viscosity 0.32–0.44 dL/g) together with the PLGA 50:50 and PEG copolymer (Resomer® RGP type d5055 [PLGA-PEG®]) were purchased from Boehringer Ingelheim (Ingelheim, Germany). Maleimide-PEG-NH2 (2,000 Da) was obtained from Jenkem (Beijing, People’s Republic of China). Methoxy-PEG-NH2, N-hydroxysuccinimide (NHS), ethyl-3-(3-dimethylaminopropyl) carbodiimideydrochloride (EDC), N-diisopropylethylamine (DIEA), N-diisopropylcarbodiimide, 2-(1H-7-azabenzotriazol-1-yl)-1.1.3.3-tetramethyluroniumhexafluorophosphate (HATU), 1-hydroxybenzotriazole, and FB were purchased from Sigma (St Louis, MO, USA). Amino acids and NovaSyn TGR resin were purchased from Novabiochem (Hohenbrunn, Germany). N,N-Dimethylformamide (DMF) was obtained from Scharlau (Barcelona, Spain). Poloxamer 188 (Lutrol® F68) was from BASF (Barcelona, Spain) and 5(6)-carboxytetramethylrhodamine (Rho) was obtained from Merck (Hohenbrunn, Germany). Double-distilled water was used after filtration in a Millipore system. All other chemicals and reagents used in the study were of analytical grade.
Peptide synthesis
The POD (CGGG[ARKKAAKA]4) and HIV-Tat (CGGGGYGRKKRRQRRR) peptides were synthesized in the solid phase using fluorenylmethoxycarbonyl protected amino acids following procedures previously described.37,38 The polyglycine sequence is an adequate spacer while the N-terminal cysteine reacts with maleimide-activated PEG. Carboxamide peptides were obtained using NovaSyn TGR resin (0.25 eq/g). HATU was used as a coupling reagent in DMF activated with DIEA.
All the synthesized peptides were characterized by reversed-phase high-performance liquid chromatography (RP-HPLC). The RP-HPLC system consisted of an Agilent 1260 pump (Quat Pump VL) with a UV-Vis 1260 ALS detector set at 215 nm. A reversed-phase column (Kromasil C18 25×0.46 cm and 5 μM mean particle size) with a flow rate of 1 mL/min was used. Matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) mass spectrometry was used to determine the molecular weight. 2.5-Dihydroxybenzoic acid was used as the matrix, and the peptides were dissolved in acetonitrile (ACN)/water 30% v/v with 0.01% v/v trifluoroacetic acid at a concentration of 1 mg/mL. The sample was run in an AutoFlex III Smart Bean Bruker mass spectrometer, operating in reflector mode, and positive ions were monitored. A nitrogen laser beam at 200 nm was used, and 500 shots were averaged for each spectrum. The crude peptides were purified by semipreparative HPLC, yielding a purity of more than 95%.
Preparation of NPs
General procedure
NPs containing FB were prepared by the solvent displacement technique described by Fessi et al11 under the optimized conditions determined previously.17,39–41 Briefly, an organic solution of 90 mg of polymer PLGA 503H in 5 mL of acetone containing FB (10 mg) was poured, under moderate stirring, into 10 mL of an aqueous solution, adjusted to pH 3.5, containing 100 mg of Lutrol® F68. The resulting colloidal suspension was stirred for 5 minutes. The acetone was then evaporated, and the NP dispersion volume was concentrated under reduced pressure. Empty NPs were prepared using the same procedure but without FB being added to the organic phase.
The NPs were obtained by two different routes: one involving the preparation of PLGA NPs followed by PEG and peptide conjugation; and the other involving the self-assembly of PLGA-PEG and the PLGA-PEG-peptide copolymer followed by NP formulation (Figure 1).
 | Figure 1 Synthesis of (A) PLGA-NPs-PEG-peptide and (B) PLGA-PEG-peptide polymer followed NPs preparation. |
Synthesis of PEGylated-PLGA NPs and peptide conjugation: PLGA-NPs-PEG-peptide
PLGA NPs in suspension were incubated with 2.4 mg (21 μmol) of NHS, and then 4.1 mg (21 μmol) of EDC was added. The pH was adjusted to 3.5, and the mixture was incubated for 6 hours at room temperature with gentle stirring. The resulting PLGA-NHS-activated particles were covalently linked to 5.2 mg (2.6 μmol) NH2-PEG-maleimide. Next, PLGA-PEG NPs were covalently linked to 2.6 μmol of POD (9.4 mg) or HIV-Tat (5 mg) peptides. The reaction procedure is shown in Figure 1A. The presence of the peptides was confirmed by MALDI-TOF mass spectrometry under the same conditions as explained above. Finally, the unreacted PEG and peptide was separated from the NPs by centrifugation at 14,000 rpm and 4°C for 30 minutes.
The free PEG was quantified via a 2,4,6-trinitrobenzenesulphonic acid (TNBS) assay.42,43 In separate test tubes, 100 μL of aqueous solution containing free PEG or borate buffer was prepared (pH 8.0 for the blank) by mixing 900 μL of 0.1 M borate buffer (pH 9.3) and 40 μL of 0.03 M TNBS. The solution was incubated at room temperature for 30 minutes, and the absorption at 420 nm was determined spectrophotometrically. The amount of conjugated PEG was calculated by subtracting the amount of free PEG from the total amount added.
Unreacted POD and HIV-Tat were quantified using Ellman’s assay.44 A solution of 0.1−0.2 μmol of the peptides in sodium phosphate buffer was prepared and then 100 μL of DTNB (5.5 dithiobis[2-nitrobenzoic acid]) was added to the peptide solution, and the mixture was left to stand for 15 minutes. DTNB and the buffer were used as reference solutions. Absorbance was determined spectrophotometrically at 410 nm. The concentration of sulfhydryl groups, and thus of the corresponding free peptide, was calculated using the following equation:
SH = (A410 [sample] − A410 [reference])/13,650 | (1) |
Synthesis of PLGA-PEG and PLGA-PEG-peptide
As shown in Figure 1B, PLGA was preactivated before PEGylation with maleimide-PEG-amine. One gram (32.3 μmol) of PLGA, with an inherent viscosity of 0.32–0.44 dL/g was dissolved in 2 mL of chloroform with stirring in a tightly sealed vial. Then, 45 mg (234.7 μmol) of EDC and 27 mg (234.7 μmol) of NHS was added, and the mixture was stirred overnight in a tightly sealed vial. The preactivated PLGA-NHS was precipitated with 8 mL of diethyl ether and centrifuged at 4,000 rpmfor 10 minutes at room temperature. Following centrifugation, the supernatant was discarded and the polymer redissolved in 2 mL of chloroform. This precipitation/dissolution washing cycle was repeated three times before the activated PLGA-NHS ester was dried under vacuum and stored at −20°C. Then, 500 mg (16.2 μmol) of PLGA-NHS was dissolved in 2 mL of chloroform, and once it was fully dissolved, 35 mg (17.5 μmol) of maleimide-PEG-amine or metoxi-PEG-amine (nontargeted particles) and 20 μL (117.15 μmol) of DIEA was added, the vial was tightly sealed, and the mixture was stirred overnight. The product was precipitated with 8 mL of an ice-cold 80/20 mixture of diethyl ether/methanol and centrifuged at 4,000 rpmfor 10 minutes. After the supernatant was discarded, the product was redissolved in 2 mL of chloroform, and the washing cycle was repeated twice more. The PLGA-PEG copolymer was dried under vacuum and stored at 4°C. The percentage yield was calculated as follows:
|
|
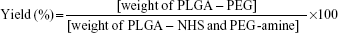
To conjugate the peptides with the PLGA-PEG-maleimide, 2.7 μmol of POD (9.8 mg) or HIV-Tat (5.2 mg) was dissolved in 250 μL of ACN/DMF and added to 100 mg (2.7 μmol) of the polymer dissolved in 1 mL of chloroform. The mixture was covered tightly and stirred overnight. The product was precipitated with 3 mL of an ice-cold 80/20 mixture of diethyl ether/methanol, centrifuged at 4,000 rpm for 10 minutes, the supernatant discarded, and the product redissolved in 1 mL of chloroform. This cycle was repeated twice more, and then the PLGA-PEG-peptide was dried under vacuum.
Proton nuclear magnetic resonance (1H-NMR) was used to assess the grafting of PEG to PLGA and the conjugation with the peptide. The PLGA-PEG was dissolved in deuterated chloroform and the PLGA-PEG-peptide in dimethyl sulfoxide (DMSO)-d6. The spectrum was recorded at 298 K on a Varian Inova 500 MHz spectrometer (Agilent Technologies, Santa Clara, CA, USA). Then, PLGA-PEG NPs and PLGA-PEG-peptide NPs containing FB were prepared following the general procedure described above.
Characterization of NPs
Particle size, polydispersity index, and zeta potential
The mean particle size, polydispersity index, and zeta potential (Zpot) were determined via dynamic light scattering using a Zetasizer nano ZS (Malvern Instruments, Malvern, UK) at 25°C. The measurements were performed on samples previously diluted in Milli-Q® water (Millipore Corporation, Billerica, MA, USA). Zpot is a measure of the electric charge at the surface of NPs, an indirect measure of their physical stability. Zpot values were calculated by determining the electrophoretic mobility using the Helmholtz–Smoluchowski equation, as previously reported.39 The values we report are the mean ± standard deviation (SD) of at least three different batches of each NP formulation.
Entrapment efficiency
The nonentrapped FB was separated by filtration/centrifugation using Amicon centrifugal filter devices with 100 kDa Ultracel membrane (Millipore Corporation) at 3,000 rpm for 10 minutes (Digicen 20-R centrifuge, Madrid, Spain). The samples were diluted (1:10) before injection with Milli-Q water to avoid deposition of free FB on the NPs. The concentration of FB in the supernatant was measured by RP-HPLC. The mobile phase was composed of ACN/water (65:35) acidified with phosphoric acid (pH 2.5). The column effluent was examined spectrophotometrically at 254 nm and the flow rate was set at 1 mL/min. The retention time of FB was 11.8 minutes. The calibration curve was linear in the range 0.0014–0.75 mg/mL with a correlation coefficient, r2, of 0.999. The percentage FB entrapment efficiency (EE) was calculated as indicated below:
|
|
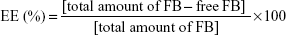
Morphological studies
The morphology of the NPs was studied by scanning electron microscopy (SEM). The NPs were centrifuged at 14,000 rpm at 4°C for 30 minutes. The supernatant was discarded and the pellet was dried under vacuum. Samples were attached to a metal stub, sputter coated with chrome, and viewed under Hitachi S43000 SEM apparatus.
In vitro drug release
In vitro release studies of FB from the NPs were carried out by applying the bulk equilibrium dialysis bag technique.45 The experiments were performed under ‘‘sink conditions’’ in order to avoid interference of FB solubility in the in vitro release. An NP formulation containing 1 mg/mL of FB, a control solution of FB (1 mg/mL), and 0.3 mg/mL commercial eye drops (Ocufen®) were placed into dialysis sacs (cellulose membrane with a molecular weight cut-off of 12,000 Da [Iberlabo, Madrid, Spain]) into 150 mL of phosphate buffered saline (PBS) (0.1 M, pH 7.4). The dialysis sacs were previously equilibrated in PBS. The temperature was maintained at 32°C (surface eye temperature) with moderate, continuous stirring. At specific time intervals, 1 mL was taken from the release medium and immediately replaced with 1 mL of fresh PBS. The concentration of the drug released was assessed as described above. Values are reported as the mean ± SD of three replicates.
Data obtained from in vitro release studies were fitted to the Korsmeyer–Peppas equation to determine the mechanism of drug release from the NPs.46 The Korsmeyer–Peppas model is,
Mt/M∞ = k · tn | (4) |
where Mt is the amount of drug released at time t, M∞ is the total amount released, Mt/M∞ is the fraction of drug released at time t, k is the release rate constant, and n is the diffusion exponent that indicates the mechanism of drug release. Several mechanisms may be involved in the release process from spherical matrices: drug diffusion from NPs (or the Fickian mechanism; n≤0.43), non-Fickian transport (or case-II transport, zero order; n≥0.85) or a combination of both processes (anomalous transport; 0.43<n<0.85). In order to fit only the Korsmeyer–Peppas empirical model, an initial 60% of drug released was used.
Fluorescence studies to visualize the NPs
Synthesis of PLGA-PEG-peptide-Rho
To evaluate the interaction of the PLGA-PEG-peptide NPs with the corneal epithelium, the peptides were prelabeled with Rho. A volume of 50 mg of peptidyl resin (containing POD or HIV-Tat) was added to 12.9 mg of Rho. The reaction was carried out using N-diisopropylcarbodiimide and 1-hydroxybenzotriazole in DMF medium for 24 hours in the dark. MALDI-TOF mass spectrometry was used to determine the molecular weight of the Rho-peptide. The degree of labeling was calculated separately by determining the peptide and Rho molar concentrations of the conjugate based on absorbance measurements; these concentrations were then expressed as a ratio (Rho:peptide molar ratio). Then, unlabeled peptides (2 mg [0.6 μmol]) POD or 1 mg [0.6 μmol] HIV-Tat) and the labeled Rho-peptides (0.9 mg [0.25 μmol]) Rho-POD or 0.4 mg [0.22 μmol] Rho-HIV-Tat) were conjugated with 30 mg (0.84 μmol) of polymer PLGA-PEG-maleimide, as detailed above. The new NPs not containing FB were prepared using the solvent displacement method as described above. The amount of Rho-peptides in the NPs was determined by fluorescence spectroscopy (excitation at 555 nm and emission at 580 nm). Standard curves were obtained by diluting Rho with blank PLGA-PEG NPs in the concentration range 0.1–1 μM. The PLGA-PEG-peptide-Rho NPs were analyzed by fluorescence microscopy. The particles were excited with a 572 nm laser and emissions registered between 570 and 590 nm. This channel was depicted as green to facilitate visualization on the computer screen.
Ex vivo study by confocal laser scanning microscopy
In these studies, New Zealand white rabbits with no signs of abnormalities or ocular inflammation and weighing 1.8–2.2 kg were used. All experiments were performed according to the ARVO (Association for Research in Vision and Ophthalmology) resolution for the use of animals in research and the corresponding protocols were approved by the ethics committee for animal experimentation of the University of Barcelona. Volumes of 50 μL of each formulation were administered into the conjunctival sac of the rabbit. After 2 hours of instillation, the rabbits were killed. A corneal specimen, freshly extracted, was directly mounted on a glass slide, and both the membrane dye wheat germ agglutinin and the nucleus dye 2,5′-bi-1H-benzimidazole, 2′-(4-ethoxyphenyl)-5-(4-methyl-1-piperazinyl) (Hoechst) were added. Images were taken using confocal laser scanning microscopy (CLSM) with a Leika TCS SP2 AOTF laser, 40×/1.25 oil objective and background fluorescence were registered by laser excitation at 364, 488, and 561 nm.
Cytotoxicity assays
The cytotoxicity of the NPs was evaluated using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT) assay. HeLa and HepG2 cells were cultured in Dulbecco’s Modified Eagle’s Medium (D5796; Sigma) supplemented with 10% heat-inactivated fetal bovine serum (Invitrogen) and 1% penicillin-streptomycin (P11-010; PAA) under 5% CO2 at 37°C. The HeLa and HepG2 cells were cultured in 96-well plates by adding 100 μL of a suspension of 100,000 cells/mL (total density of 10,000 cells/well) and incubated for 24 hours to allow cell attachment. The medium was then replaced with 150 μL of medium containing FB-loaded PLGA-PEG-POD NPs and PLGA-PEG-HIV-Tat NPs at different concentrations (0.01–25 mg/mL) and incubated for 24 hours in triplicate. Then, the culture medium was removed from the wells to avoid interference and replaced with 100 μL of MTT (5 mg/mL in PBS was prepared and diluted 1:5 with fresh medium). The plates were again incubated for 3 hours under the same conditions. After this time, the medium was aspirated and replaced by 100 μL of DMSO to dissolve the crystals formed in the previous step and then incubated for 2 hours at room temperature in the dark. Finally, absorbance was measured spectrophotometrically at 570 nm using a SpectraMax M5 spectrophotometer (Molecular Devices, Sunnyvale, CA, USA). Untreated cells were used as a control with 100% viability. The results were processed using GraphPad Prism 5 software.
Ocular tolerance assays
In vitro HET-CAM
The risk of ocular irritation by the NPs was assessed using the hen’s egg test–chorioallantoic membrane (HET-CAM) assay.47 Hen’s eggs were incubated for 9 days and after this time defective eggs were discarded. The shell around the air cell was removed and the inner membranes extracted to reveal the CAM. NPs were added to the membrane and left in contact for 5 minutes. After proceeding with the positive control, one egg was treated with 0.1 M sodium hydroxide and six eggs were used for each sample tested. After exposing the CAM and rinsing it with PBS, 300 μL of the test solution was applied to the CAM. The intensity of the reactions was semiquantitatively assessed on a scale from 0 (no reaction) to 3 (strong reaction). The time of onset and intensity of reactions occurring within 5 minutes were recorded. The ocular irritation index (OII) was then calculated using the following expression:
OII = (301 - h) × 5/300 + (301 - l) × 7/300 + (301 - c) × 9/300 | (5) |
where his the time (in seconds) to the beginning of hemorrhage, l time to lysis, and c timeto coagulation. The following classification was used: OII ≤0.9, slightly irritating; 0.9< OII ≤4.9, moderately irritating; 4.9< OII ≤8.9, irritating; and 8.9< OII ≤21, severely irritating.
In vivo Draize test
An In vivo Draize test was performed to confirm the results obtained by the HET-CAM assay. The study was conducted using a modified Draize test.48 The irritation associated with the instillation of hypotonic and hypertonic suspensions may lead to reflex tearing or reflex blinking. The osmolality of the samples, measured with a Fiske osmometer, was adjusted with 5% (w/v) glucose to the tear osmolality (around 280 mOsm/kg).49 In order to avoid these effects in the ocular assays, the formulations tested were adjusted to tear fluid (pH 7.4) using 0.1 N sodium hydroxide. A single instillation of 50 μL of each NP was applied to one eye of the New Zealand white rabbits, and the other eye was used as a control. After 1 hour of exposure, the eyes were observed to evaluate possible injury to the cornea, conjunctiva, or iris, and subsequent readings were taken after 1, 2, 3, 4, and 7 days. The Draize score was determined by visual assessment of changes in the ocular structure involving cornea (turbidity or opacity), iris (inflammation degree), and conjunctiva (congestion, swelling, and discharge). Ocular lesions were quantified using the mean total score (MTS) equation:
MTS = ∑ X1(n) × 5 + ∑ X2(n) × 2 + ∑ X3(n) × 5 | (6) |
where X1(n), X2(n), and X3(n) are the cornea, conjunctiva, and iris scores, respectively, and n is the number of rabbits included in the ocular tolerance assay. Three animals were used for each formulation.
Anti-inflammatory efficacy assay
In order to study the prevention and treatment of ocular inflammation in cataract surgery by NPs, inflammation was induced by topical administration of sodium arachidonate (SA) (0.5% w/v) dissolved in PBS (pH 7.4). Following the experimental procedure described by Spampinato et al50 SA was inoculated in the right eye of five groups of six rabbits while the left eye was used as a control. The osmolarity and pH of each formulation were adjusted. The treatment protocol consisted of the induction of ocular inflammation by SA, and after 30 minutes the instillation of 50 μL of each formulation. In order to evaluate the prevention of inflammation (prophylaxis) by the NPs, 50 μL of each sample was instilled in the conjunctival sac of the right eye 30 minutes before induction of ocular inflammation by SA. Ocular inflammation was assessed from 60 minutes to 210 minutes, and the ocular inflammation score was calculated using the MTS as described above. Corneal transparency is not affected by SA, so this parameter was not considered. The total score of the conjunctiva and iris are expressed as mean ± SD.
Statistical analysis
All the data were presented as mean ± SD. Specific comparison between groups was carried out with an unpaired Student’s t-test (two tailed). For multiple-group comparison, one-way ANOVA was carried out to analyze the significant differences (P<0.05) between groups, followed by Tukey post hoc analysis after having confirmed normality and equal variance assumptions by the Kolmogorov–Smirnov statistical test. SPSS IBM21 for Windows was used throughout.
Results and discussion
Synthesis of POD and HIV-Tat
Two different CPPs (POD and HIV-Tat) containing cysteine at the N-terminus were synthesized using solid phase methods.51 The crude peptides were purified by semipreparative HPLC, yielding over 95% purity. The purified synthetic products were characterized by MALDI-TOF (Figure S1).
Characterization of PLGA-NPs-PEG-peptide
As previously described by Vega et al17,39 and Araújo et al40 PLGA NPs are promising systems for the delivery and controlled release of FB in ocular tissues. In this work, considering the results previously published by our group, optimized PLGA NPs incorporating FB were obtained using the solvent displacement technique. Lutrol® F68 was chosen as a surfactant to stabilize the NPs in aqueous suspension since, due to its nonionic character, it does not strongly irritate ocular tissues. Furthermore, it is accepted for ocular administration by regulatory authorities.52
POD and HIV-Tat were conjugated with PLGA NPs using PEG as a spacer by means of a maleimide-thiol coupling reaction. MALDI-TOF mass spectroscopy confirmed that the reactions went to completion. The mass spectra of the peptides (POD and HIV-Tat) as well as their conjugation with PLGA-PEG-maleimide NPs are shown in Figure S1. The molecular weights obtained correspond to a combination of PEG and POD (5,500–5,900 Da) or HIV-Tat (3,700–4,400 Da). The peptides and PEG located on the surface were determined after each reaction step. These concentrations were determined indirectly by quantifying the amount of peptides remaining in solution. The conjugation efficiency was 2.5%, and the percentages of POD and HIV-Tat peptides were 1.2% and 2.1%, respectively.
Characterization of PLGA-PEG and PLGA-PEG-peptide
PLGA-PEG (PEG, 2,000 Da) was synthesized with the primary aim of studying whether the form of presentation of PEG in PLGA-PEG® (PEG®, 5,000 Da) influences the NP properties in accordance with the different molecular weights. Furthermore, bifunctional PEG containing a maleimide group at one terminus and an amine group at the other was used to form PLGA-PEG-maleimide. With the aim of characterizing the synthetic PLGA-PEG, this copolymer was analyzed by 1H-NMR spectroscopy. The 1H-NMR analysis revealed four characteristic peaks at 1.5 ppm (CH3 lactide proton), 3.6 ppm (CH2-CH2 ethylene glycol protons), 4.8 ppm (CH2 glycolide proton), and 5.2 ppm (CH lactide proton) (Figure S2). A small peak was also observed at 6.7 ppm corresponding to the maleimide methine protons. These results confirm the presence of both PLGA and PEG domains in the PLGA-PEG synthetic copolymer. We also used 1H-NMR to determine the resulting molar ratio of PLGA to PEG and of PEG to maleimide by integration of the PLGA (CH3), PEG (CH2-CH2), and maleimide peaks, the conjugation efficiencies achieved being between 7%–15% and 2%–35% for PEG to PLGA and for maleimide to PEG, respectively. Then, POD and HIV-Tat were conjugated with the PLGA-PEG via cysteine/maleimide linkage. The 1H-NMR spectra of PLGA-PEG-POD and PLGA-PEG-HIV-Tat are shown in Figure 2, confirming the peptide coupling to PLGA-PEG-maleimide polymer. The amount of peptide (POD or HIV-Tat) in the polymer was expressed as a mass percentage, according to the following equation:53
|
|
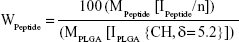
where MPeptide and MPLGA (130 g/mol) are, respectively, the molecular weight of the peptides and PLGA; IPEPTIDE is the peak integral of POD (amide/amine region, 7.0–8.2 ppm) and HIV-Tat (tyrosine [Tyr] signal at 6.64 ppm); IPLGA is the peak integral of the –CH groups in the polylactic acid unit of the PLGA corresponding to one proton; and n is number of protons (76 H for amide, amine, and guanidinium side chains in POD and 2 H for the isolated signal at 6.64 ppm corresponding to two aromatic protons of Tyr in HIV-Tat). Following this procedure, the calculated functionalization levels obtained were between 3.3%–3.9% and 2.3%–3.2% for PLGA-PEG-POD and PLGA-PEG-HIV-Tat, respectively.
 | Figure 2 The 1H-NMR spectra of PLGA-PEG-POD and PLGA-PEG-HIV-Tat confirm the peptide coupling to PLGA-PEG-maleimide polymer. |
Physicochemical characterization of FB-loaded NPs
For ocular administration drug delivery systems, particle size is an important parameter since it affects the risk of irritation and discomfort; certain sizes induce tearing and rapid drainage of the instilled dose thereby reducing the bioavailability and residence time of the drug in the conjunctival sac.54 In general, as shown in Tables 1 and 2, all the formulations we prepared had a suitable particle size for ocular administration (smaller than 250 nm diameter) and also the PI was lower than 0.1, indicating narrow particle size distributions.
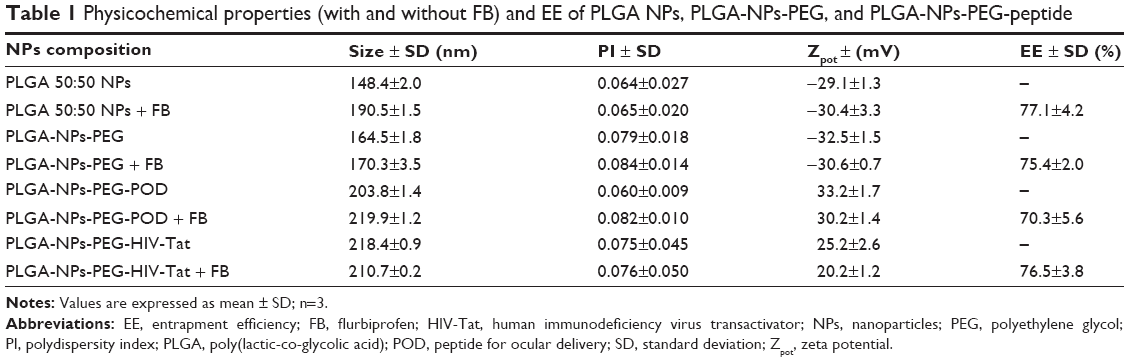 | Table 1 Physicochemical properties (with and without FB) and EE of PLGA NPs, PLGA-NPs-PEG, and PLGA-NPs-PEG-peptide |
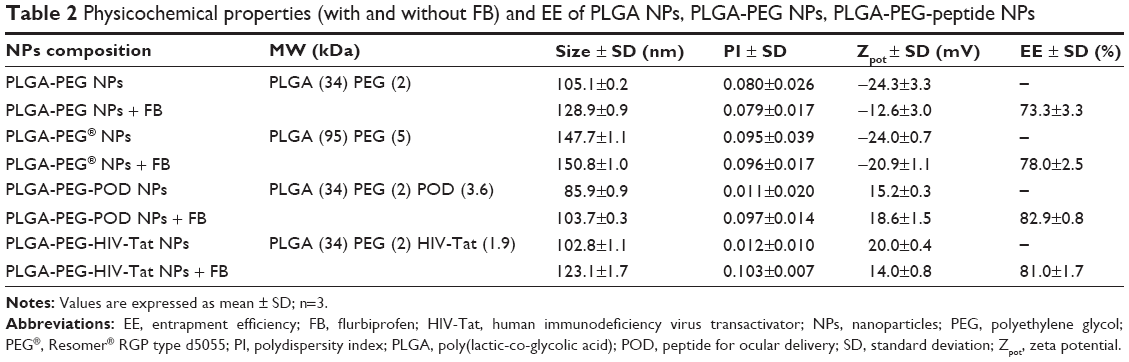 | Table 2 Physicochemical properties (with and without FB) and EE of PLGA NPs, PLGA-PEG NPs, PLGA-PEG-peptide NPs |
The results of the physicochemical characterization of PLGA-NPs-PEG-POD and PLGA-NPs-PEG-HIV-Tat are shown in Table 1. The results show that the final conjugations had slightly increased particle sizes, indicating that the attachment of PEG and peptides to the surface of PLGA NPs took place (Figure 1A). In contrast, the size of PLGA-NPs-PEG-POD and PLGA-NPs-PEG-HIV-Tat was similar, indicating that neither the choice of peptide nor the level of functionalization of the NPs affected their size. Moreover, high EE was obtained (between 70.3%±5.6% and 76.5%±3.8%), indicating that PEG and peptide surface conjugation did not lead to a dramatic loss of the drug. Nonsignificant differences in the EE of FB were observed for PLGA-NPs-PEG-HIV-Tat. The following P-values were obtained: 0.764 for PLGA-NPs-PEG-HIV-Tat versus PLGA NPs and 0.473 for PLGA-NPs-PEG-HIV-Tat versus PLGA-NPs-PEG. Thus, no release of initial loaded FB took place during these reaction steps. However, in the case of PLGA-NPs-PEG-POD, the EE of FB was up to 20% lower than that obtained for PLGA NPs (P=0.042). Table 2 shows the physicochemical properties of the PLGA-PEG-peptide NPs prepared from POD and HIV-Tat grafted onto the PLGA-PEG-maleimide copolymer, following the strategy illustrated in Figure 1B. The PLGA-PEG NPs obtained with the diblock copolymer that was synthesized in our laboratory were smaller than the NPs prepared from the commercial diblock copolymer (PLGA-PEG®). This may be due to the reduction of the molecular weight of the copolymer (PLGA segments).55,56 However, PLGA-PEG-peptide NPs (with greater molecular weight than PLGA-PEG NPs due to the peptide conjugation) were smaller in the case of PLGA-PEG-POD NPs than in the case of PLGA-PEG-HIV-Tat NPs. Finally, the PLGA-PEG-peptide NPs exhibited significantly higher EE values (81.0%±1.7%–82.9%±0.8%) than PLGA-PEG NPs (P=0.004 and P=0.007 for POD and HIV-Tat, respectively). These results indicate that a slight drug association exists when the peptides are within the polymer matrix.
Some different characteristics were observed depending on the preparation method. In general, PLGA-PEG-peptide NPs were smaller and with relatively higher EE values than PLGA-NPs-PEG-peptide.
The Zpot value is also an important NP physicochemical parameter, as it can influence the stability of the particles. In theory, larger magnitudes of Zpot values, whether positive or negative, tend to stabilize particle suspension. The electrostatic repulsion between NPs with the same electric charge prevents their aggregation.57 Our Zpot results show highly negative values for PLGA NPs and PLGA-PEG NPs due to the ionization of the carboxylic end groups of the polymer surface.58 Conjugation with positively charged POD or HIV-Tat changed the anionic Zpot of the resultant NPs to cationic; this effect was higher for PLGA-NPs-PEG-peptides, whereas PLGA-PEG-peptide NPs had lower Zpot values due to the presence of PEG and the peptides in the polymer matrix, which mask the PLGA charge. As previously reported, the presence of a positive charge is expected to promote ocular mucoadhesion due to electrostatic interactions with the negatively charged ocular mucosa.59
Morphology
The morphology of the NPs was determined by SEM (Figure S3). A spherical shape can be appreciated with a particle size of some 120 nm and a narrow size distribution. SEM analysis confirmed the dynamic light scattering data on NP size. Similar results were obtained for all the NPs involved in this study, indicating their suitability for ophthalmic applications.54
In vitro release profiles
Sustained release of FB over several hours might greatly improve its therapeutic efficacy. The in vitro release behavior of FB from NPs containing POD and HIV-Tat, a solution of FB (1 mg/mL) in 0.1 M PBS (pH 7.4), and the commercial eye-drops Ocufen® were therefore analyzed. We used the dialysis bag diffusion technique (Figure 3). As expected, the diffusion of FB from the PBS solution and Ocufen® was faster and was completed in 120 minutes. The NPs showed a biphasic pattern, ie, an immediate release (“burst effect”) followed by a slower release profile. The initial fast release was attributed to the fraction of the drug that is adsorbed or weakly bound to the NP surface. After this initial phase, the drug exhibited delayed release.
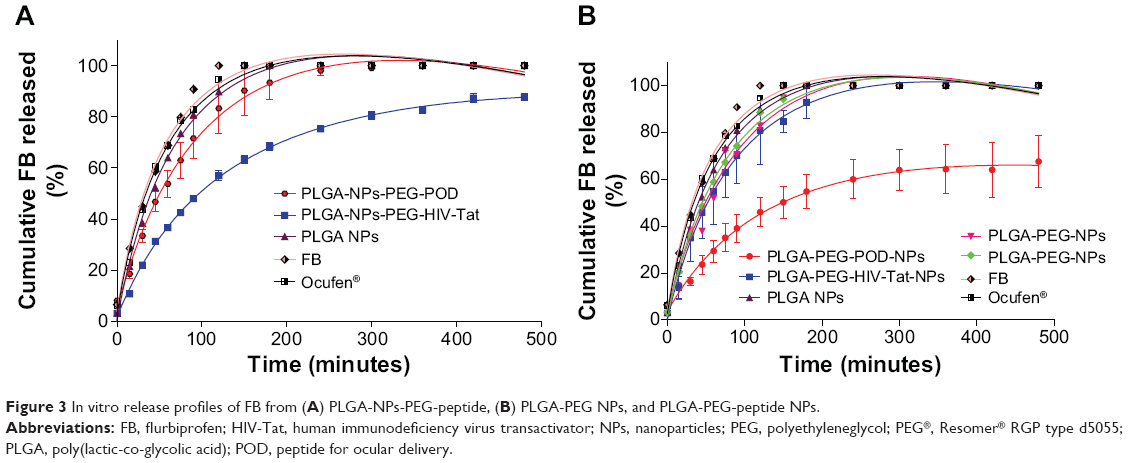 | Figure 3 In vitro release profiles of FB from (A) PLGA-NPs-PEG-peptide, (B) PLGA-PEG NPs, and PLGA-PEG-peptide NPs. |
As can be observed in Figure 3A, release of the drug from PLGA NPs was significantly faster than from NPs with conjugated peptides. Release of the drug was significantly slower for PLGA-NPs-PEG-HIV-Tat than from PLGA-NPs-PEG-POD (56.5% and 83.3%, respectively) after 120 minutes of follow-up. The more delayed release of FB from HIV-Tat (compared to POD) resulted in a maximum release profile of 90% over the whole study period (8 hours). This might be due to the greater amount of HIV-Tat peptide per unit of NP surface area, compared to POD, as described above. Figure 3B shows the release profiles of FB from PLGA-PEG NPs, PLGA-PEG® NPs, and PLGA-PEG-peptide NPs. No significant differences were observed between PLGA-PEG NPs and PLGA-PEG® NPs; and release of the drug was faster than observed from PLGA-PEG-peptide NPs, in agreement with previous results.41 In contrast to the results obtained for the PLGA-NPs-PEG-peptide formulations, the release of the drug was slower for PLGA-PEG-POD NPs than for PLGA-PEG-HIV-Tat NPs (46.1% and 80.9%, respectively). It seems that the presence of POD inside the polymeric matrix produced a more retarded release of the drug than for the other NP formulations, probably due to the greater FB-POD interaction, as observed in the EE results shown in Table 2. This interaction might introduce resistance to the diffusional release of the drug, thereby yielding a more sustained release of FB.
The development of advanced drug delivery systems relies on the judicious and careful selection of components, configurations, and geometries, which can be facilitated through mathematical modeling of controlled release systems. Therefore, data obtained from the in vitro release studies were modeled by the Korsmeyer–Peppas kinetics equation to establish the mechanism of FB release from PLGA-NPs-PEG-peptide and PLGA-PEG-peptide NPs. The diffusion exponent (n) and the release rate constant (k) obtained are indicated in Table S1. All the NPs had n values in the range 0.47–0.74; with a coefficient, r2, of between 0.948 and 0.989. These results indicated that the release of FB from these systems follows a non-Fickian diffusion mechanism, which is probably governed by a combination of drug diffusion and polymer chain relaxation during polymer swelling.60
As already demonstrated, the PLGA-PEG-peptide NPs derived from the modified polymer showed the best results; specifically, PLGA-PEG-POD NPs were the smallest NPs, and they exhibited both the highest EE and the most sustained release. So, these NPs should be considered as candidates for drug delivery into intraocular tissues, and they were selected for further biological studies.
Ex vivo CLSM study of the interaction of PLGA-PEG-peptide NPs with the corneal epithelium
In order to evaluate the NPs as delivery carrier to uptake the corneal epithelium, the peptides were labeled with Rho and were used to prepare fluorescent NPs. The MALDI-TOF spectra revealed the presence of the labeled peptides and, furthermore, revealed the absence of unlabeled peptide molecules (Figure S4). The degree of peptide labeling was determined by measuring the absorbance of free and labeled peptides, yielding 0.003 mol Rho/mol POD and 0.04 mol Rho/HIV-Tat. Then, PLGA-PEG-Rho-peptide NPs were produced, and the results showed that the PLGA-PEG-Rho-POD and PLGA-PEG-Rho-HIV-Tat NPs contained similar concentrations of labeled peptide (approximately 0.25 μM). The physicochemical parameters (Table S2) were similar to those obtained for the unlabeled NPs. An optimal size for ocular delivery (≤100 nm) and the positive charge was maintained. Fluorescence microscopy was used to visualize the labeled NPs (Figure S5). The fluorescence images were also similar for both NPs and illustrate that a homogeneous particle size distribution was obtained.
Cornea is the major route of anterior drug absorption. There are three corneal layers, namely epithelium, stroma, and endothelium, and all of them have a distinct role in transcorneal drug permeability. The corneal epithelium is the major limiting barrier in transcorneal drug absorption. CLSM images showed the corneal epithelium of the rabbit once it had been exposed to PLGA-PEG-Rho-POD NPs and PLGA-PEG-Rho-HIV-Tat NPs (Figure 4). Although both peptides increase the interaction with the mucin (data not shown), the ex vivostudy by CLSM show that PLGA-PEG-POD NPs have a better interaction than PLGA-PEG-HIV-Tat NPs with epithelial cells. More fluorescent spots can be seen in the images that correspond to the NPs containing POD than those containing HIV-Tat. This suggests that the PLGA-PEG-Rho-POD NPs interact more with the corneal epithelium. As previously reported in the literature, the penetration rate was dependent on the coating composition.61 Kompella et al62 demonstrated that corneal epithelial uptake of 20 nm NPs can be elevated from approximately 2%–16% by surface coating or functionalizing NPs.
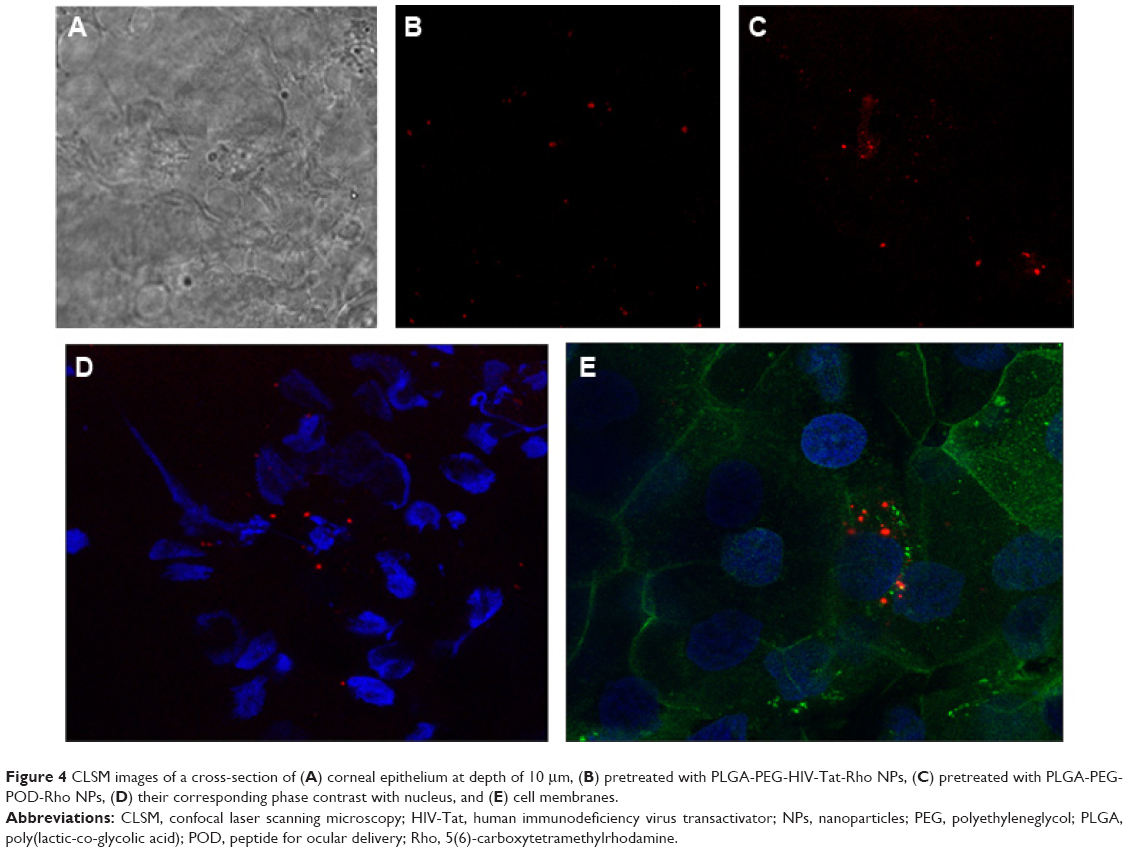 | Figure 4 CLSM images of a cross-section of (A) corneal epithelium at depth of 10 μm, (B) pretreated with PLGA-PEG-HIV-Tat-Rho NPs, (C) pretreated with PLGA-PEG-POD-Rho NPs, (D) their corresponding phase contrast with nucleus, and (E) cell membranes. |
Hoechst dyes and wheat germ agglutinin were used to distinguish the boundaries of the nucleus and the cell membrane, respectively. We observed that the fluorescent spots were mainly distributed around the nucleus of the cells, indicating that the NPs penetrated the corneal epithelium through a transcellular pathway. These results are in agreement with those reported by other authors for PLGA-PEG,41 poly-ε-caprolactone,63 and poly(alkyl cyanoacrylate)64 NPs.
In vitro cytotoxicity of PLGA-PEG-peptide NPs
To assess the cellular cytotoxicity of the NPs, HeLa and HepG2 cell lines were used. Cytotoxic activity was evaluated at concentrations ranging from 0.1 to 25 mg/mL. Following 24 hours of exposition to the NPs, cell viability was assessed using the MTT assay. The results were plotted onto a fitted curve to obtain median lethal dose (LD50) values (NP concentrations at which cell viability is 50%). PLGA-PEG NPs without FB were used as a control. While the FB solution showed higher cytotoxicity, the experimental LD50 values for PLGA-PEG-HIV-Tat NPs were 10.8 mg/mL in HepG2 cells and 8.4 mg/mL in HeLa cells, whereas the PLGA-PEG-POD NPs rendered values of 4.4 mg/mL and 3.9 mg/mL, respectively (Figure 5). These results correspond to approximately 200–300 mg/kg of body weight in an adult human, suggesting low in vitro cytotoxicity, as this would be a much higher intravenous dose than required for in vivo drug delivery.65 Representative photographs of HeLa and HepG2 cells are shown, with no NPs added (left column), incubated with NPs at LD50 values (center column), and incubated with NPs at lethal dose values (right column) (Figure 5C).
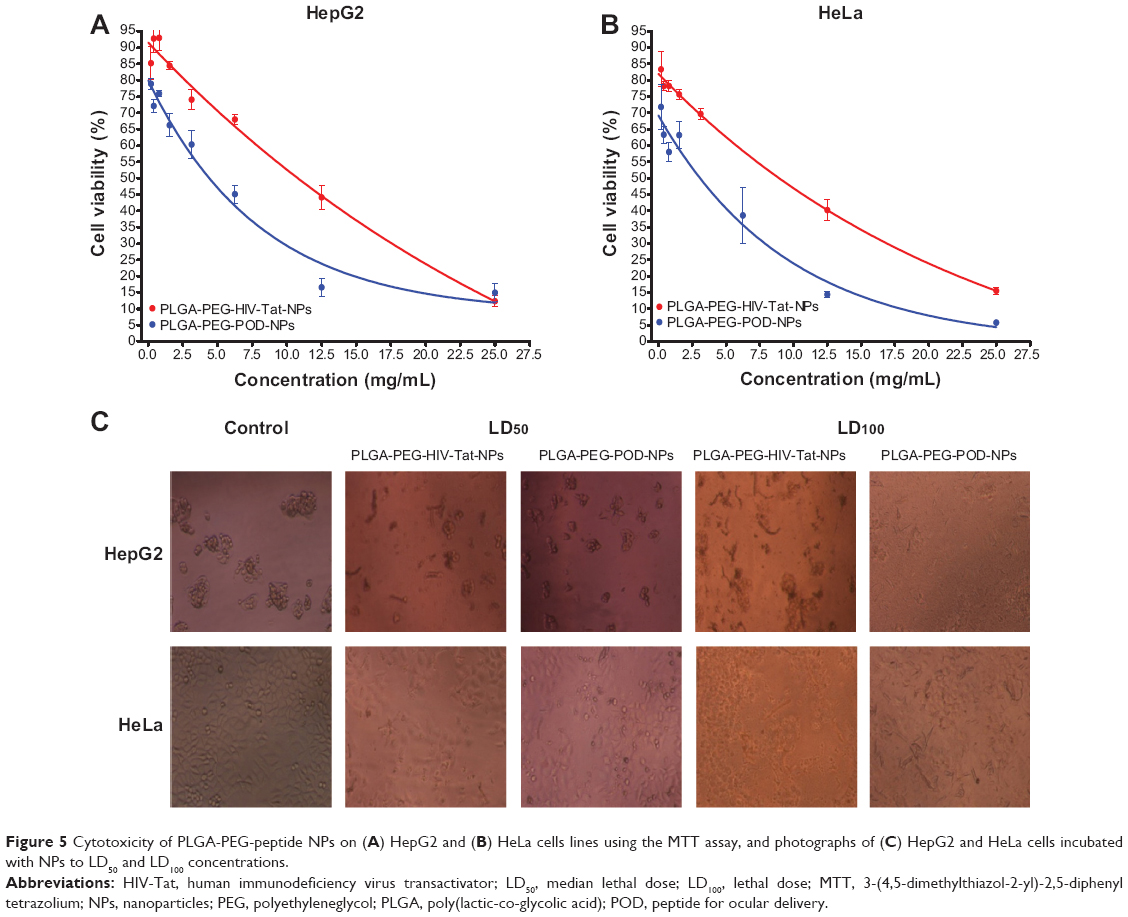 | Figure 5 Cytotoxicity of PLGA-PEG-peptide NPs on (A) HepG2 and (B) HeLa cells lines using the MTT assay, and photographs of (C) HepG2 and HeLa cells incubated with NPs to LD50 and LD100 concentrations. |
Ocular tolerance assays
For an ocular instillation drug delivery system, it is extremely important to test ocular tolerability. In accordance with the concept of the 3Rs (reduction, refinement and, replacement), several techniques are available to reduce and replace animal testing. In vitro screening serves as a preliminary method to assess possible risk in animal studies. Since a single in vitro test cannot mimic complete in vivo behavior, eg, the entire systemic mechanism including inflammation, penetration/permeability, and tolerance, the assays were also carried out on male albino rabbits.
In vitro HET-CAM
The CAM is a noninnervated complete tissue containing arteries, veins, and capillaries, and it is technically easy to study. It responds to injury via an inflammatory process similar to that observed in the conjunctival tissue of a rabbit eye. The well-developed CAM vascularization provides an ideal model for ocular irritation studies.
The HET-CAM, a suitable alternative to animal testing, is based on direct application onto the CAM and the subsequent reactions, such as hemorrhage, intravasal coagulation, or lysis of blood vessels, which are microscopically assessed along a time-course.47 These irritancy effects may occur within 5 minutes of mucosal administration of the sample onto the HET-CAM, according to the INVITTOX protocol.66
Ocular tolerance assays for free FB in PBS showed slight irritancy. In contrast, when the different NP formulations developed in this work were added, no signs of ocular irritancy were detected within 5 minutes; the irritancy index was zero (Figure S6). With respect to the effect of particle size on ocular irritancy, as described by Schoenwald and Stewart,54 only particles of 20 μm mean diameter induced irritation. Thus, the FB-loaded NPs seem to be suitable systems for ocular administration, and the observed long-term changes in their average size do not reach the critical range.
In vivo Draize test
Topical application of the NPs to rabbit eyes showed no signs of irritation in the cornea, conjunctiva, or iris (MTS =0) (Figure S7). These results are in accordance with those obtained in the HET-CAM test.
Anti-inflammatory efficacy assay
Two studies were performed to determine the anti-inflammatory efficacy of the NPs in order to probe their usefulness for preventing and treating inflammation during cataract surgery.
Conjunctival inflammation with significant hyperemia was induced by SA, peaking at 30 minutes. In general, NPs containing FB (Table 2) and Ocufen® significantly decreased the ocular inflammation, showing no significant differences among them (Figure 6). Only PLGA-PEG-POD NPs exhibited better anti-inflammatory response than the other formulations at 120 minutes in the prevention of inflammation (Figure 6B), and the SA-induced inflammation was inhibited 210 minutes after SA instillation. These results are in accordance with those obtained in the ex vivostudy by CLSM, where it was demonstrated that PLGA-PEG-POD NPs had a better interaction than PLGA-PEG-HIV-Tat NPs with epithelial cells. Thus, the in vivo results might be explained on the basis of the prolonged retention in the precorneal area and also in the uptake/internalization into the corneal epithelium.
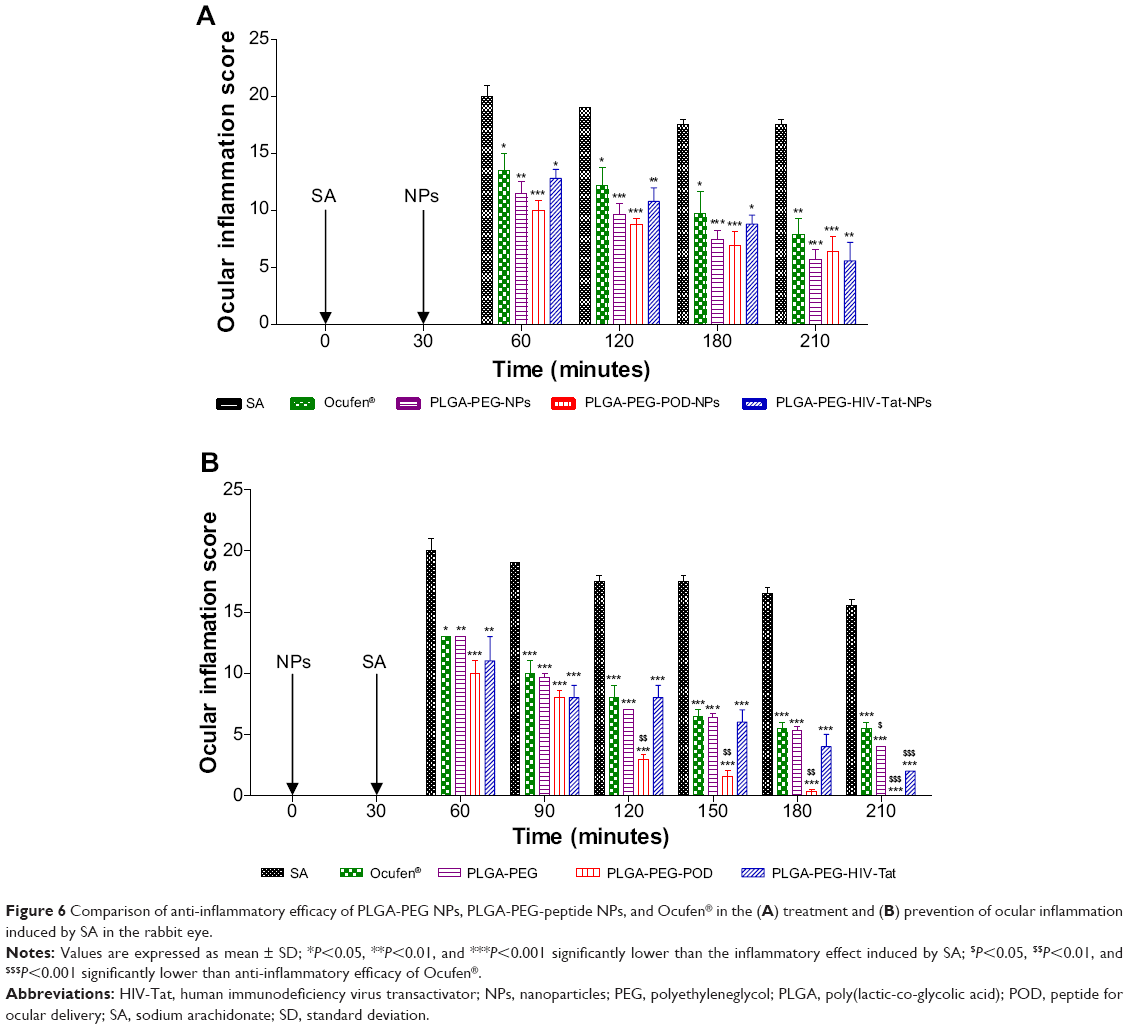 | Figure 6 Comparison of anti-inflammatory efficacy of PLGA-PEG NPs, PLGA-PEG-peptide NPs, and Ocufen® in the (A) treatment and (B) prevention of ocular inflammation induced by SA in the rabbit eye. |
As can be observed in Figure 6A, in the case of treatment, no improvement was achieved using PLGA-PEG-POD-NPs, while a better anti-inflammatory efficacy was observed in the prevention of inflammation (Figure 6B and Figure S8). These results could be related to the different absorption of NPs in healthy and inflamed tissues. The instillation of SA prior to administration of NPs led to enhanced lacrimation and thus increasing precorneal loss and clearance of NPs. Our results showed that PLGA-PEG-POD NPs enhance the ocular tissue residence time and the anti-inflammatory efficacy of FB.
Conclusion
In this study, we proposed PLGA-PEG NPs modified with POD or HIV-Tat as an effective ocular drug delivery system to reduce inflammation after cataract surgery. Of the formulations devised, PLGA-PEG-POD NPs showed the best results: uniformly spherical shape with a particle size of 103.7±0.3 nm. Their morphology was confirmed by SEM and fluorescence microscopy. Moreover, their positive Zpot (18.6±1.5 mV) promotes ocular mucoadhesion. Furthermore, these NPs present higher EE and more sustained release of FB, the anti-inflammatory drug studied. PLGA-PEG-POD NPs enhanced the delivery of drugs to the corneal epithelium and represent an effective system for increasing the bioavailability of FB. The NPs were tolerated well by human cell line models (HeLa and HepG2) and did not induce any sign of irritation during ocular tolerance studies (either in vitro or in vivo). Their anti-inflammatory efficacy suggests that PLGA-PEG-POD NPs may be employed to prevent ocular inflammatory reactions in which the SA cascade is activated. These results indicated that PLGA-PEG-POD NPs could offer an effective ocular drug delivery system that would facilitate permeation of FB via topical application.
Acknowledgments
This work was supported by the Cooperation Research Program CSIC-CITMA and a project (MAT2011-26994) funded by the Spanish Ministry of Science and Technology. AV is a recipient of a PhD grant from the CSIC. The authors thank Nacho Pérez of the IQAC-CSIC for performing the cytotoxicity assays.
Disclosure
The author reports no conflicts of interest in this work.
References
Ahuja M, Dhake AS, Sharma SK, Majumdar DK. Topical ocular delivery of NSAIDs. AAPS J. 2008;10(2):229–241. | ||
Brown RM, Roberts CW. Preoperative and postoperative use of nonsteroidal antiinflammatory drugs in cataract surgery. Insight. 1996;21(1):13–16. | ||
Bandara BM, Sankaridurg PR, Willcox MD. Non-steroidal anti inflammatory agents decrease bacterial colonisation of contact lenses and prevent adhesion to human corneal epithelial cells. Curr Eye Res. 2004;29(4–5):245–251. | ||
Zhang W, Prausnitz MR, Edwards A. Model of transient drug diffusion across cornea. J Control Release. 2004;99(2):241–258. | ||
Pignatello R, Bucolo C, Spedalieri G, Maltese A, Puglisi G. Flurbiprofen-loaded acrylate polymer nanosuspensions for ophthalmic application. Biomaterials. 2002;23(15):3247–3255. | ||
Pignatello R, Bucolo C, Ferrara P, Maltese A, Puleo A, Puglisi G. Eudragit RS100 nanosuspensions for the ophthalmic controlled delivery of ibuprofen. Eur J Pharm Sci. 2002;16(1–2):53–61. | ||
Bucolo C, Maltese A, Puglisi G, Pignatello R. Enhanced ocular anti-inflammatory activity of ibuprofen carried by an Eudragit RS100 nanoparticle suspension. Ophthalmic Res. 2002;34(5):319–323. | ||
Dillen K, Weyenberg W, Vandervoort J, Ludwig A. The influence of the use of viscosifying agents as dispersion media on the drug release properties from PLGA nanoparticles. Eur J Pharm Biopharm. 2004;58(3):539–549. | ||
Deshpande AA, Heller J, Gurny R. Bioerodible polymers for ocular drug delivery. Crit Rev Ther Drug Carrier Syst. 1998;15(4):381–420. | ||
Shive MS, Anderson JM. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv Drug Delivery Rev. 1997;28(1):5–24. | ||
Fessi H, Puisieux F, Devissaguet JP, Ammoury N, Benita S. Nanocapsule formation by interfacial polymer deposition following solvent displacement. Int J Pharm. 1989;55(1):R1–R4. | ||
Soppimath KS, Aminabhavi TM, Kulkarni AR, Rudzinski WE. Biodegradable polymeric nanoparticles as drug delivery devices. J Control Release. 2001;70(1–2):1–20. | ||
Pinto Reis C, Neufeld RJ, Ribeiro AJ, Veiga F. Nanoencapsulation I. Methods for preparation of drug-loaded polymeric nanoparticles. Nanomedicine. 2006;2(1):8–21. | ||
Prokop A, Davidson JM. Nanovehicular intracellular delivery systems. J Pharm Sci. 2008;97(9):3518–3590. | ||
Vert M, Mauduit J, Li S. Biodegradation of PLA/GA polymers: increasing complexity. Biomaterials. 1994;15(15):1209–1213. | ||
Jain RA. The manufacturing techniques of various drug loaded biodegradable poly(lactide-co-glycolide) (PLGA) devices. Biomaterials. 2000;21(23):2475–2490. | ||
Vega E, Egea MA, Valls O, Espina M, García ML. Flurbiprofen loaded biodegradable nanoparticles for ophtalmic administration. J Pharm Sci. 2006;95(11):2393–2405. | ||
Cui Y, Xu Q, Chow PK, Wang D, Wang CH. Transferrin-conjugated magnetic silica PLGA nanoparticles loaded with doxorubicin and paclitaxel for brain glioma treatment. Biomaterials. 2013;34(33):8511–8520. | ||
Zeng X, Tao W, Mei L, Huang L, Tan C, Feng SS. Cholic acid-functionalized nanoparticles of star-shaped PLGA-vitamin E TPGS copolymer for docetaxel delivery to cervical cancer. Biomaterials. 2013;34(25):6058–6067. | ||
Zhu H, Chen H, Zeng X, et al. Co-delivery of chemotherapeutic drugs with vitamin E TPGS by porous PLGA nanoparticles for enhanced chemotherapy against multi-drug resistance. Biomaterials. 2014;35(7):2391–2400. | ||
Essa S, Rabanel JM, Hildgen P. Effect of polyethylene glycol (PEG) chain organization on the physicochemical properties of poly(D, L-lactide) (PLA) based nanoparticles. Eur J Pharm Biopharm. 2010;75(2):96–106. | ||
Owens DE, Peppas NA. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm. 2006;307(1):93–102. | ||
Drin G, Cottin S, Blanc E, Rees AR, Temsamani J. Studies on the internalization mechanism of cationic cell-penetrating peptides. J Biol Chem. 2003;278(33):31192–31201. | ||
Brooks NA, Pouniotis DS, Tang CK, Apostolopoulos V, Pietersz GA. Cell-penetrating peptides: application in vaccine delivery. Biochim Biophys Acta. 2010;1805(1):25–34. | ||
Frankel AD, Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 1988;55(6):1189–1193. | ||
Elliott G, O’Hare P. Intercellular trafficking and protein delivery by a herpesvirus structural protein. Cell. 1997;88(2):223–233. | ||
Derossi D, Joliot AH, Chassaing G, Prochiantz A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J Biol Chem. 1994;269(14):10444–10450. | ||
Cashman SM, Morris DJ, Kumar-Singh R. Evidence of protein transduction but not intercellular transport by proteins fused to HIV tat in retinal cell culture and in vivo. Mol Ther. 2003;8(1):130–142. | ||
Cashman SM, Sadowski SL, Morris DJ, Frederick J, Kumar-Singh R. Intercellular trafficking of adenovirus-delivered HSV VP22 from the retinal pigment epithelium to the photoreceptors – implications for gene therapy. Mol Ther. 2002;6(6):813–823. | ||
Johnson LN, Cashman SM, Kumar-Singh R. Cell-penetrating peptide for enhanced delivery of nucleic acids and drugs to ocular tissues including retina and cornea. Mol Ther. 2008;16(1):107–114. | ||
Johnson LN, Cashman SM, Read SP, Kumar-Singh R. Cell penetrating peptide POD mediates delivery of recombinant proteins to retina, cornea and skin. Vision Res. 2010;50(7):686–697. | ||
Nagarwal RC, Kant S, Singh PN, Maiti P, Pandit JK. Polymeric nanoparticulate system: a potential approach for ocular drug delivery. J Control Release. 2009;136(1):2–13. | ||
Read SP, Cashman SM, Kumar-Singh R. A poly(ethylene) glycolylated peptide for ocular delivery compacts DNA into nanoparticles for gene delivery to post-mitotic tissues in vivo. J Gene Med. 2010;12(1):86–96. | ||
Shen WC, Ryser HJ. Conjugation of poly-L-lysine to albumin and horseradish peroxidase: a novel method of enhancing the cellular uptake of proteins. Proc Natl Acad Sci U S A. 1978;75(4):1872–1876. | ||
Read SP, Cashman SM, Kumar-Singh R. POD nanoparticles expressing GDNF provide structural and functional rescue of light-induced retinal degeneration in an adult mouse. Mol Ther. 2010;18:1917–1926. | ||
Binder C, Cashman SM, Kumar-Singh R. Extended duration of transgene expression from pegylated POD nanoparticles enables attenuation of photoreceptor degeneration. PLoS One. 2013;8(11):e82295. | ||
Herrera E, Tenckhoff S, Gómara MJ, et al. Effect of synthetic peptides belonging to E2 envelope protein of GB virus C on human immunodeficiency virus type 1 infection. J Med Chem. 2010;53(16):6054–6063. | ||
Malakoutikhah M, Gómara MJ, Gómez-Puerta JA, Sanmartí R, Haro I. The use of chimeric vimentin citrullinated peptides for the diagnosis of rheumatoid arthritis. J Med Chem. 2011;54(21):7486–7492. | ||
Vega E, Gamisans F, García ML, Chauvet A, Lacoulonche F, Egea MA. PLGA nanospheres for the ocular delivery of flurbiprofen: drug release and interactions. J Pharm Sci. 2008;97(12):5306–5317. | ||
Araújo J, Vega E, Lopes C, Egea MA, Garcia ML, Souto EB. Effect of polymer viscosity on physicochemical properties and ocular tolerance of FB-loaded PLGA nanospheres. Colloids Surf B Biointerfaces. 2009;72(1):48–56. | ||
Vega E, Egea MA, Calpena AC, Espina M, García ML. Role of hydroxypropyl-β-cyclodextrin on freeze-dried and gamma-irradiated PLGA and PLGA-PEG diblock copolymer nanospheres for ophthalmic flurbiprofen delivery. Int J Nanomedicine. 2012;7:1357–1371. | ||
Habeeb AF. Determination of free amino groups in proteins by trinitrobenzenesulfonic acid. Anal Biochem. 1966;14(3):328–336. | ||
Zhang N, Chittasupho C, Duangrat C, Siahaan TJ, Berkland C. PLGA nanoparticle – peptide conjugate effectively targets intercellular cell-adhesion molecule-1. Bioconjug Chem. 2008;19(1):145–152. | ||
Riddles PW, Blakeley RL, Zerner B. Ellman’s reagent: 5,5′-dithiobis(2-nitrobenzoic acid) – a reexamination. Anal Biochem. 1979;94(1):75–81. | ||
Magenheim B, Levy MY, Benita S. A new in vitro technique for the evaluation of drug release profile from colloidal carriers-ultrafiltration technique at low pressure. Int J Pharm. 1993;94:115–123. | ||
Korsmeyer RW, Gurny R, Doelker E, Buri P, Peppas NA. Mechanisms of solute release from porous hydrophilic polymers. Int J Pharm. 1983;15:25–35. | ||
Tavaszi J, Budai P. The use of HET-CAM test in detecting the ocular irritation. Commun Agric Appl Biol Sci. 2007;72(2):137–141. | ||
Kay JH, Calandra JC. Interpretation of eye irritation tests. J Soc Cosmet Chem. 1962;13:281–289. | ||
López Bernal D, Ubels JL. Quantitative evaluation of the corneal epithelial barrier: effect of artificial tears and preservatives. Curr Eye Res. 1991;10(7):645–656. | ||
Spampinato S, Marino A, Bucolo C, Canossa M, Bachetti T, Mangiafico S. Effects of sodium naproxen eye drops on rabbit ocular inflammation induced by sodium arachidonate. J Ocul Pharmacol. 1991;7(2):125–133. | ||
Atherton E, Sheppard R. Solid Phase Peptide Synthesis: A Practical Approach. Oxford: Oxford University Press. 1989;216. | ||
Singh-Joy SD, McLain VC. Safety assessment of poloxamers 101, 105, 108, 122, 123, 124, 181, 182, 183, 184, 185, 188, 212, 215, 217, 231, 234, 235, 237, 238, 282, 284, 288, 331, 333, 334, 335, 338, 401, 402, 403, and 407, poloxamer 105 benzoate, and poloxamer 182 dibenzoate as used in cosmetics. Int J Toxicol. 2008;27(Suppl 2):93–128. | ||
Martín-Banderas L, Alvarez-Fuentes J, Durán-Lobato M, et al. Cannabinoid derivate-loaded PLGA nanocarriers for oral administration: formulation, characterization, and cytotoxicity studies. Int J Nanomedicine. 2012;7:5793–5806. | ||
Schoenwald RD, Stewart P. Effect of particle size on ophthalmic bioavailability of dexamethasone suspensions in rabbits. J Pharm Sci. 1980;69(4):391–394. | ||
Dalwadi G, Sunderland B. An ion pairing approach to increase the loading of hydrophilic and lipophilic drugs into PEGylated PLGA nanoparticles. Eur J Pharm Biopharm. 2009;71(2):231–242. | ||
Song CX, Labhasetwar V, Murphy H, et al. Formulation and characterization of biodegradable nanoparticles for intravascular local drug delivery. J Control Release. 1997;43(1–2):197–212. | ||
Feng S, Huang G. Effects of emulsifiers on the controlled release of paclitaxel (Taxol) from nanospheres of biodegradable polymers. J Control Release. 2001;71(1):53–69. | ||
Govender T, Stolnik S, Garnett MC, Illum L, Davis SS. PLGA nanoparticles prepared by nanoprecipitation: drug loading and release studies of a water soluble drug. J Control Release. 1999;57(2):171–185. | ||
Vandervoort J, Ludwig A. Ocular drug delivery: nanomedicine applications. Nanomedicine (Lond). 2007;2(1):11–21. | ||
Peppas NA. Analysis of Fickian and non-Fickian drug release from polymers. Pharm Acta Helv. 1985;60(4):110–111. | ||
De Campos AM, Sánchez A, Gref R, Calvo P, Alonso MJ. The effect of a PEG versus a chitosan coating on the interaction of drug colloidal carriers with the ocular mucosa. Eur J Pharm Sci. 2003;20(1):73–81. | ||
Kompella UB, Sundaram S, Raghava S, Escobar ER. Luteinizing hormone-releasing hormone agonist and transferrin functionalizations enhance nanoparticle delivery in a novel bovine ex vivo eye model. Mol Vis. 2006;12:1185–1198. | ||
Calvo P, Thomas C, Alonso MJ, Vila Jato JL, Robinson JR. Study of the mechanism of interaction of poly(ε-caprolactone) nanocapsules with the cornea by confocal laser scanning microscopy. Int J Pharm. 1994;103(3):283–291. | ||
Fresta M, Fontana G, Bucolo C, Cavallaro G, Giammona G, Puglisi G. Ocular tolerability and in vivo bioavailability of poly(ethylene glycol) (PEG)-coated polyethyl-2-cyanoacrylate nanosphere-encapsulated acyclovir. J Pharm Sci. 2001;90(3):288–297. | ||
Chan JM, Zhang L, Yuet KP, et al. PLGA-lecithin-PEG core-shell nanoparticles for controlled drug delivery. Biomaterials. 2009;30(8):1627–1634. | ||
Marquardt C, Matuschek E, Bölke E, et al. Evaluation of the tissue toxicity of antiseptics by the hen’s egg test on the chorioallantoic membrane (HETCAM). Eur J Med Res. 2010;15(5):204–209. |
Supplementary materials
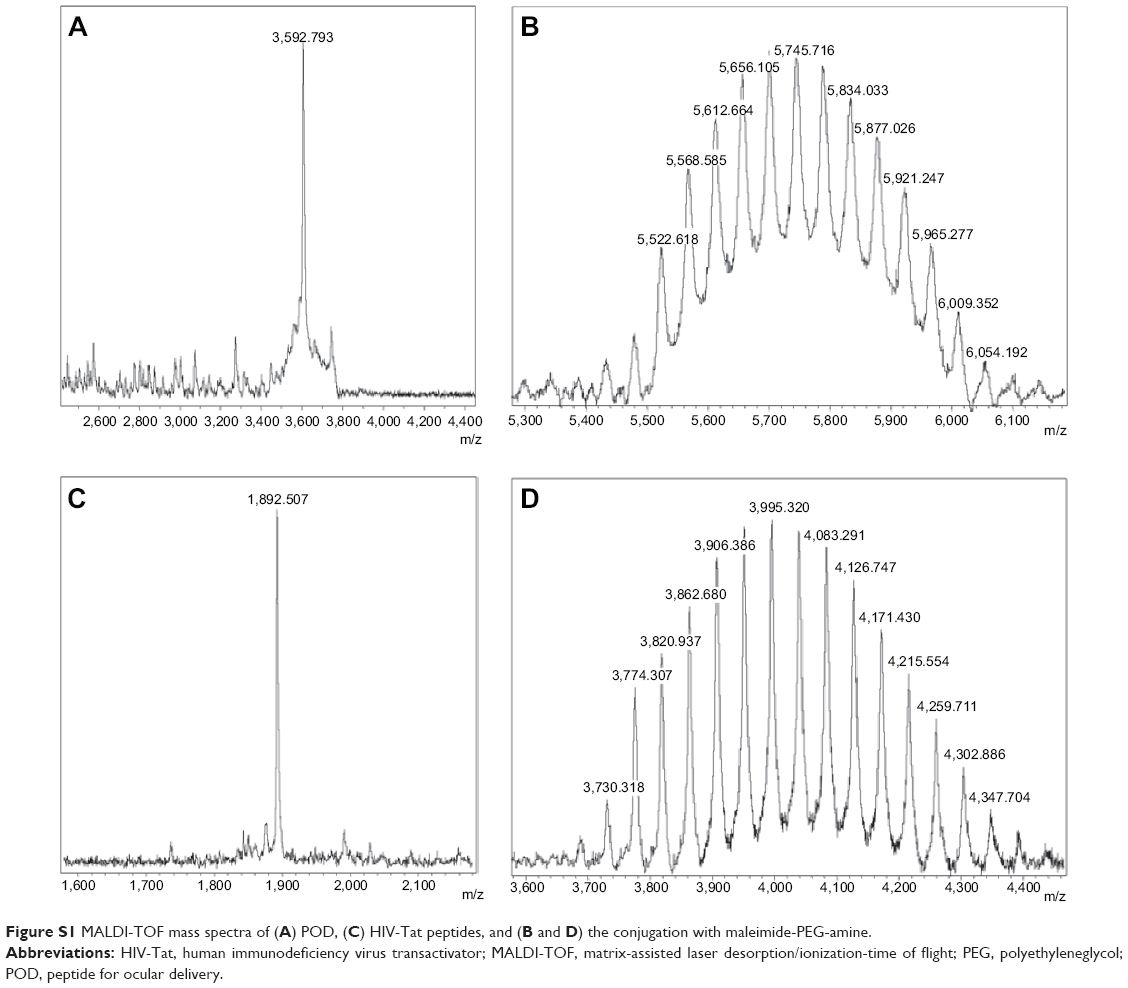 | Figure S1 MALDI-TOF mass spectra of (A) POD, (C) HIV-Tat peptides, and (B and D) the conjugation with maleimide-PEG-amine. |
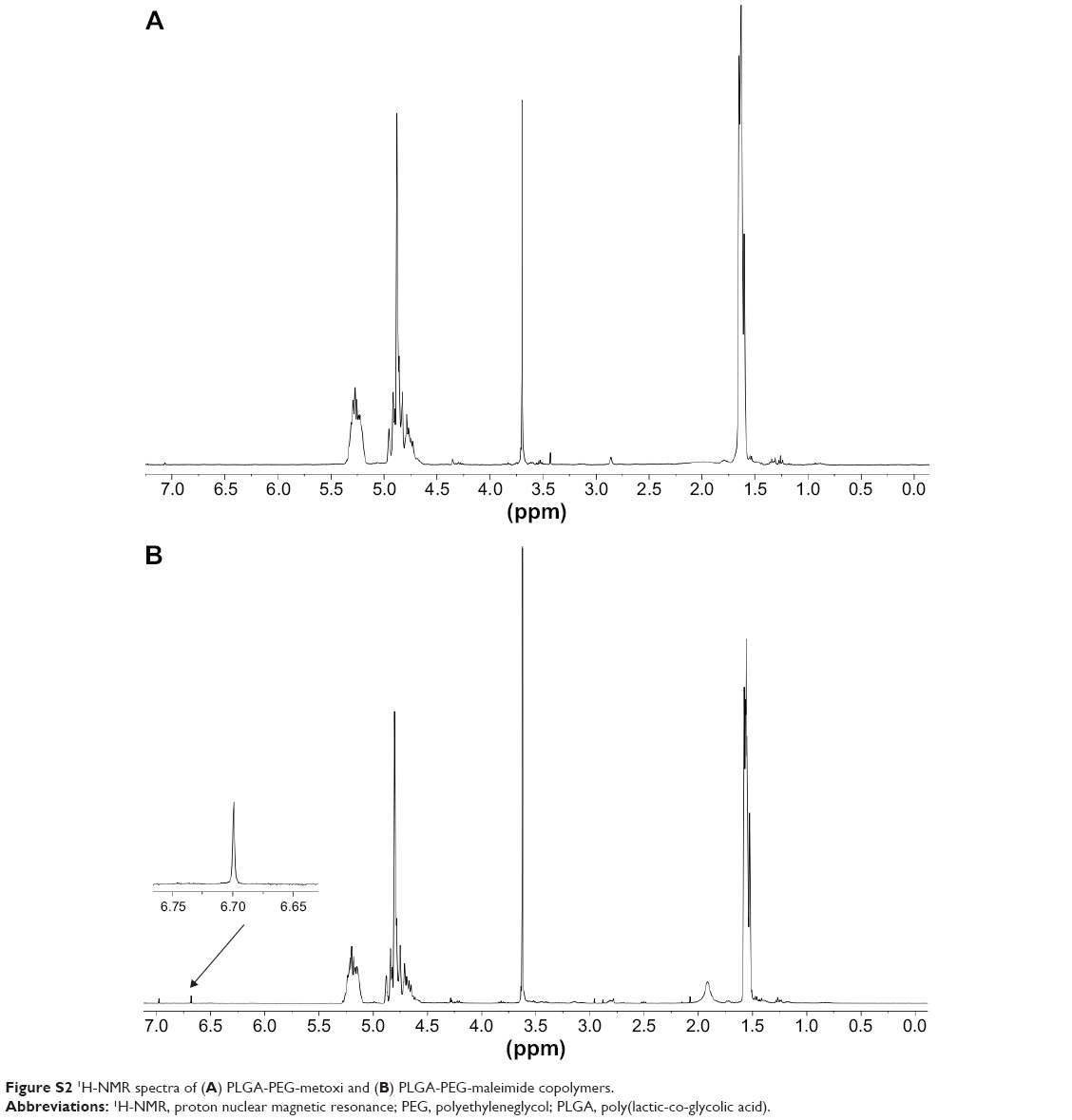 | Figure S2 1H-NMR spectra of (A) PLGA-PEG-metoxi and (B) PLGA-PEG-maleimide copolymers. |
 | Figure S3 Scanning electron microscopy analysis of PLGA-PEG-POD NPs. |
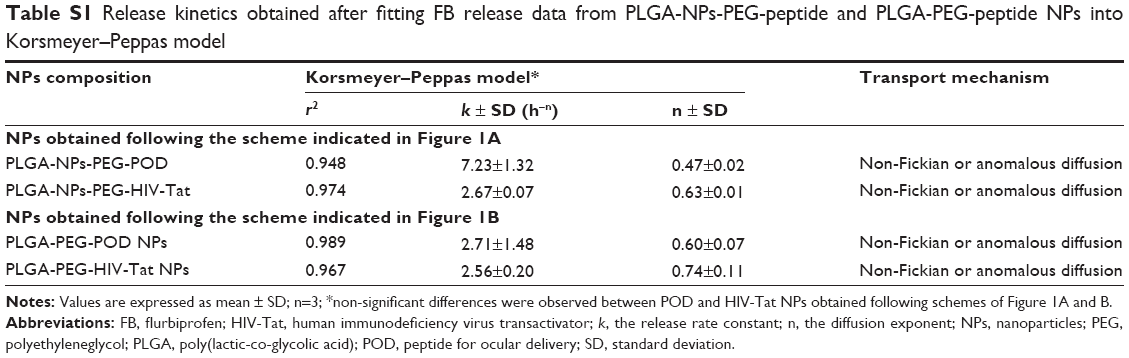 | Table S1 Release kinetics obtained after fitting FB release data from PLGA-NPs-PEG-peptide and PLGA-PEG-peptide NPs into Korsmeyer–Peppas model |
 | Figure S4 MALDI-TOF spectra of (A) Rho-POD and (B) Rho-HIV-Tat peptides. |
 | Table S2 Physicochemical properties of PLGA-PEG-peptide-Rho NPs |
 | Figure S5 Fluorescence microscopy images of (A) PLGA-PEG-peptide-Rho NPs and (B) green emission of all scanned NPs. |
 | Figure S6 Photographs of CAM 5 minutes after the addition of NPs containing FB: (A) PLGA NPs, (B) PLGA-PEG NPs, (C) PLGA-PEG-POD NPs, (D) PLGA-PEG-HIV-Tat NPs, and (E) positive control (0.1 M sodium hydroxide). |
 | Figure S7 Draize test after instillation of PLGA-PEG-POD NPs (A) and inflammation induced by SA (B). |
 | Figure S8 Anti-inflammatory effect of PLGA-PEG-POD NPs in the prevention of ocular inflammation induced by SA. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.