Back to Archived Journals » Journal of Vascular Diagnostics and Interventions » Volume 4
Congenital vascular anomalies: current perspectives on diagnosis, classification, and management
Received 21 September 2015
Accepted for publication 7 December 2015
Published 14 July 2016 Volume 2016:4 Pages 23—37
DOI https://doi.org/10.2147/JVD.S63244
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Rahmi Oklu
Francine Blei,1 Mark E Bittman2
1Vascular Anomalies Program, Lenox Hill Hospital, Northwell Health, 2Department of Radiology, New York University Langone Medical Center, New York, NY, USA
Abstract: The term "congenital vascular anomalies" encompasses those vascular lesions present at birth. Many of these lesions may be detected in utero. This review serves to apprise the readership of newly identified diagnoses and updated classification schemes. Attention is focused on clinical features, patterns of presentation, clinical manifestations and behavior, diagnostic tools, and treatment modalities. It is an invigorating period for this field, with a surge in vascular anomalies-related basic and clinical research, genetics, pharmacology, clinical trials, and patient advocacy. A large number of professional conferences now include vascular anomalies in the agenda, and trainees in multiple specialties are gaining expertise in this discipline. We begin with a summary of classification schemes and introduce the updated classification adopted by the International Society for the Study of Vascular Anomalies. Disease entities are described, with liberal use of photographs, as many diagnoses can be established based on a thorough history and visual appearance and it is thus essential to develop a familiarity with diagnosis-specific physical features. Peripheral (non-central nervous system) vascular anomalies are the focus of this review. We focus on those entities in which diagnostic radiology is routinely used and accentuate when histologic confirmation is essential. We also underscore some differences in approach to the pediatric vs adolescent or adult patient. A list of Internet-based resources is included, with hyperlinks to informative sites. References are limited to seminal discoveries and review articles. We hope that our enthusiasm in writing this review will be shared by those who read this review.
Keywords: vascular anomalies, hemangiomas, vascular malformations, overgrowth syndromes
Introduction
The term “vascular anomalies” refers to a heterogeneous group of disorders with distinct clinical and radiologic features. Diagnoses can often be determined by incorporating the history, physical examination, imaging (for some diagnoses), and chronologic course. An updated classification provides structure and allows for clarity and consistency in terminology. Recent interest and advances in clinical and basic research have contributed to a more profound understanding of vascular anomalies and new treatment modalities.
The classification of anomalies has evolved based on anatomic and clinical features and is reviewed by Wassef and Blei.1 The most widely accepted classification is based on the original observations by Mulliken and Glowacki,2,3 whereby vascular anomalies are divided into those that are proliferative during all or part of the life cycle (vascular tumors – benign) and those that are predominantly structural anomalies (vascular malformations). Vascular malformations (VMs) are further characterized by the predominantly affected vessel (arterial, venous, lymphatic, capillary – individually or combined) and subclassified into truncular and extratruncular vessels. Staging systems based on escalating clinical symptoms or anatomic extent have been developed by Schöbinger4 (for arteriovenous malformations [AVMs]) and de Serres et al5 (lymphatic malformations [LMs]). The original Mulliken classification was recently expanded and updated to incorporate new entities, genetic information, and syndromes.6 Terminology in this review is aligned with the classification of the International Society for the Study of Vascular Anomalies (ISSVA) (Table 1), available at http://www.issva.org/content.aspx?page_id=22&club_id=298433&module_id=152904. Figure 1 provides a schematic representation of the development of the vasculature in vascular anomalies.
 | Table 1 ISSVA classification of vascular anomalies, 2014 |
 | Figure 1 Proposed mechanism of development of vascular malformations from fetal capillary plexus. |
Congenital vascular anomalies are those that are evident at birth and can be VMs or vascular tumors. If present at birth, it is likely that the lesion could be diagnosed prenatally, although this is not always appreciated. Vascular tumors that are present at birth include the congenital hemangiomas (rapidly involuting congenital hemangioma [RICH], noninvoluting congenital hemangioma [NICH]), Kaposiform hemangioendothelioma (KHE), and multifocal lymphangioendotheliomatosis. VMs present prenatally and at birth, however, may not be visible. Early detection of vascular anomalies is important, as this may affect childbirth decisions. Additionally, early awareness of vascular lesions and potentially associated syndromic features can inform evaluation and management strategies.7
Imaging for vascular anomalies has been well reviewed in several manuscripts. Nozaki et al,8,9 in a pictorial review titled “Syndromes associated with vascular tumors and malformations”, as well as an article titled “Imaging of vascular tumors with an emphasis on ISSVA classification”, impart a well-organized logical presentation based on the ISSVA classification (albeit the earlier version). Calvo-Garcia et al,10 in “Imaging of fetal vascular anomalies”, provide an update of prenatal diagnosis with ultrasonography and fetal magnetic resonance imaging (MRI). We also refer the readers to review published consensus statements regarding the diagnosis and treatment of VMs.7,11,12
Nozaki et al9 present a simple flowchart for radiologic evaluation of patients with vascular anomalies (Figure 2), and a more complex algorithm is presented by Tekes et al.13 Burrows14 and Rosen et al15 provide thorough reviews of the role of interventional radiology in VMs. Waner and O16 provide a review of the role of surgery in VMs.
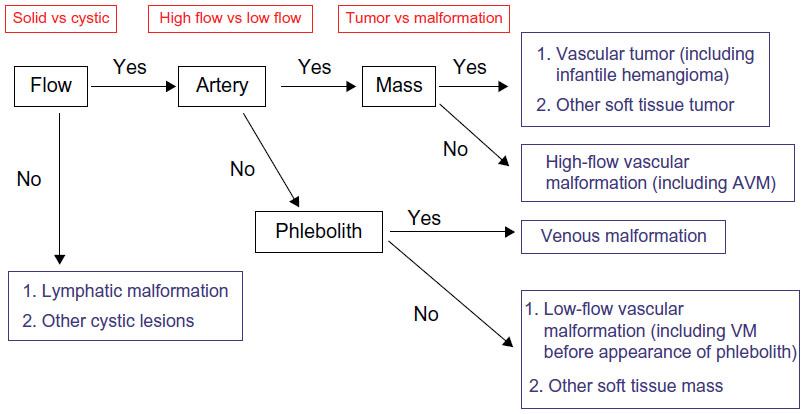 | Figure 2 Algorithm for radiologic evaluation of vascular malformations. |
Congenital hemangioma
Congenital hemangiomas include RICH, NICH, and partially involuting congenital hemangioma (PICH), although the latter two diagnoses are temporally determined, when the initial lesion involutes only partially (in the case of PICH) or not at all (as in NICH).17 In contrast to infantile hemangiomas, which are either undetectable at birth or minimally apparent with a precursor mark or discoloration, congenital hemangiomas are more easily discernable. Histologically, the congenital hemangiomas are negative for the glucose transporter type 1 (GLUT-1) marker, whereas hemangiomas of infancy stain strongly positive.18 Figure 3 demonstrates the stages of proliferation and involution of hemangiomas of infancy, as well as the associated features. RICH-type lesions can be associated with high flow, causing a fetal and postnatal high cardiac output state, and they may ulcerate and hemorrhage.19 There is one other presentation termed “rapidly involuting congenital hemangioma with fetal involution”, which has a distinctive appearance.20,21 Images of congenital hemangiomas are shown in Figure 4.
 | Figure 3 Schematic growth and regression curve of hemangioma of infancy. |
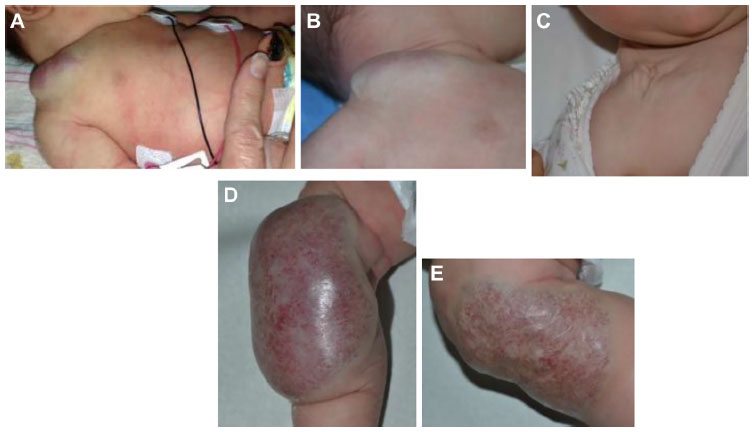 | Figure 4 Rapidly involuting congenital hemangioma (RICH). |
Imaging of RICH and NICH
Case reports and studies predominantly demonstrate ultrasound and MRI findings in congenital hemangiomas. RICH can occur in many locations, often sparing the face. When RICH occurs in the liver, it is important to characterize the lesion and differentiate it from other liver lesions. RICH should be suspected in a neonate with a solitary liver lesion and normal-for-age serum alpha-fetoprotein levels. On ultrasonography, RICH has a variable appearance, although typically characterized as a solid mass with a heterogeneous echotexture. A minority of cases contain small calcifications. On color Doppler imaging, prominent vessels are often detected in the periphery of the lesion with a relatively hypovascular center.22 RICH are markedly hyperintense on T2-weighted MRI, and larger lesions are often heterogeneous due to central thrombosis, necrosis, and/or fibrosis. Small lesions may be homogeneous on T2-weighted imaging. On T1-weighted imaging, they are hypointense relative to normal liver parenchyma and may contain foci of bright signal due to internal hemorrhage. On arterial phase postgadolinium images, the enhancement pattern is peripheral and nodular, which follows the signal of the blood pool. Portal venous and delayed phase imaging shows progressive centripetal enhancement.23 Computed tomography (CT) is not the preferred modality due to the potential harmful effects of ionizing radiation to the newborn. Findings with noncontrast CT include a well-defined mass that is hypodense to the adjacent liver. The postcontrast enhancement pattern is similar to that observed with MRI, ie, peripheral nodular enhancement and centripetal enhancement on delayed phase images. Serial ultrasonography of the liver is commonly performed to ensure progressive shrinkage of the lesion. If there is interval growth, then a biopsy is recommended.
Congenital hemangiomas, in contrast to the more commonly seen hemangiomas of infancy, do not typically appear in full form prenatally; at birth, there may be small red spots or a pale white or pink precursor area. Postnatally, hemangiomas of infancy proliferate for several months and then naturally involute. Small retrospective imaging studies have attempted to distinguish infantile hemangioma from congenital hemangiomas. On sonography, RICH and NICH are more often heterogeneous in echotexture compared with infantile hemangioma, which is usually homogeneously echogenic. Congenital hemangiomas more commonly have detectible vessels compared with the infantile form. Calcifications have not reported in infantile hemangioma but have been identified in a subset of congenital hemangiomas.24
Location, size, morphology, and any functional impairment caused by the hemangioma influence decisions regarding imaging and/or treatment. Despite the now-historical temptation to leave hemangiomas alone because they will “go away on their own”, in fact, early treatment allows for an optimal esthetic and functional outcome. Segmental hemangiomas of the cervicofacial and upper arm regions prompt evaluation for possible PHACES syndrome (Posterior fossa/other central nervous system [CNS] structural malformations, segmental Hemangiomas, Arterial anomalies, Cardiovascular anomalies, Eye abnormalities, and Sternal cleft/supraumbilical raphe or other midline abnormality).25 Similarly, segmental hemangiomas on the lower back/lumbosacral area, buttocks, and/or perianal area dictate an evaluation for possible LUMBAR association (Lower body segmental hemangioma, Urogenital anomalies and ulceration, Myelopathy, Bony abnormalities, Anorectal and/or Arterial anomalies, and Renal anomalies).26
Representative images of patients with PHACE and LUMBAR symptoms are shown in Figures 5 and 6. Imaging for PHACE includes MRI with and without contrast to evaluate CNS structure, as well as magnetic resonance angiography of the brain, neck, and upper chest for evaluation of arteriopathies, which usually occur on the side ipsilateral to the facial hemangioma. Arteriopathies may include presistent embryonic arteries, atretic dysplastic tortuous vessels, and/or those with abnormal branching, as well as aortic arch anomalies. There is a high incidence of a funnel-shaped enlarged internal auditory canal, many of which have a hemangioma within their enlarged structure.11,27–29 Dandy–Walker malformation (which is characterized by vermian hypoplasia, cystic dilatation of the fourth ventricle, and an enlarged posterior fossa with torcular–lambdoid inversion) is the most common posterior fossa malformation associated with PHACES syndrome. Large cervicofacial hemangiomas are the hallmarks of PHACES, classically with a segmental distribution. Although rare, stroke is a reported complication of the arterial cerebrovascular anomalies associated with PHACES syndrome. Aortic coarctation is the most common cardiovascular abnormality identified in this patient population. Other cardiac and aortic anomalies described in PHACES include patent ductus arteriosis, septal defects, pulmonary stenosis, aberrant subclavian artery, tetralogy of Fallot, and aortic aneurysms.
 | Figure 5 Segmental hemangioma/PHACE syndrome. |
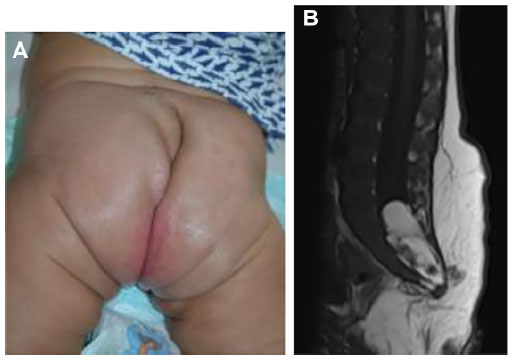 | Figure 6 LUMBAR associations. |
Prenatal PHACE may be suspected with ultrasonographic or MRI detection of unilateral cerebellar hypoplasia, other posterior fossa anomalies, sternal clefting, aortic arch, or other vascular dysplasias.30–35 Adults have been diagnosed retrospectively with PHACE (ie, questioning if the patients had facial segmental hemangiomas in infancy), after incidental findings of arteriopathies, cerebellar hypoplasia, and other abnormalities are found on CNS MRIs (obtained for other reasons).36–38
Imaging of LUMBAR association
Ultrasonography of the lower spine, abdomen, and pelvis is indicated for midline segmental vascular lesions of the lower back, perineum, and buttock areas. The optimal age to perform a spinal ultrasound investigation in a neonate is less than 3–4 months due to lack of ossification of the posterior elements of the spine. Sonography of the spine beyond 6 months of age is often limited.39 MRI with contrast confirms suspected abnormalities after an abnormal ultrasound result and is the first-line study in elderly patients for whom ultrasonography is technically limited.
Subcutaneous hemangiomas often warrant evaluation to confirm reassuring radiologic findings. If any concerns arise clinically or radiologically, histologic confirmation should be considered. The differential diagnosis includes pilomatrixoma, pericytoma, sarcoma, lymphoma, rhabdomyosarcoma, neuroblastoma, and other diagnoses.
KHE, which may be seen in the newborn period, can present as a boggy, red/purple mass with ill-defined borders (Figure 7). Kasabach–Merritt phenomenon (with profound thrombocytopenia and hypofibrinogenemia) may accompany this diagnosis, although several cases without a hematologic abnormality have been described.40 Patients with Kasabach–Merritt syndrome can be quite ill, with hemorrhage into the lesion or elsewhere. This is generally treated medically with vincristine, sirolimus, steroids, or other agents.41–45 A study comparing the efficacy of sirolimus vs vincristine is under way (NCT02110069).
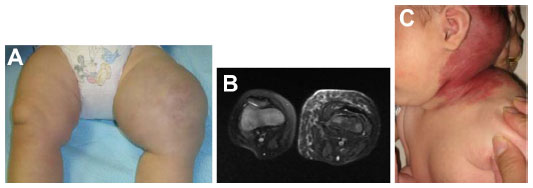 | Figure 7 Kaposiform hemangioendothelioma. |
Imaging of KHE
KHE involves a spectrum of lesions ranging from small superficial lesions to large infiltrative lesions with potential life-threatening complications. KHE is usually unifocal and is most commonly located in the extremity. The torso (thoracic cavity and retroperitoneum) and cervicofacial locations are the next most commonly affected locations. MRI is the most useful modality to assess KHE. On MRI, KHE is characterized as an infiltrative lesion with poorly defined borders. It often crosses tissue planes and involves the skin, subcutaneous soft tissues, with deep extension into the muscles, and possibly bone. Due to this appearance, it can mimic an infiltrative neoplasm such as sarcoma. On T1-weighted imaging, it is hypointense or isointense relative to skeletal muscle. On T2-weighted imaging, KHE is hyperintense in signal, with reticular edema-like signal within the subcutaneous fat, often referred to as “stranding”.8,43,46 In contrast, infantile hemangioma does not typically demonstrate an infiltrative growth pattern. After contrast administration, KHE demonstrates marked enhancement. Flow voids are often depicted along the periphery of the lesion, although these are less prominent compared with common hemangiomas.47
CT shows an infiltrative soft tissue mass with ill-defined margins, skin thickening, and fat stranding within the subcutaneous fat. There is often homogeneous postcontrast enhancement. Ultrasonography often shows a solid mass with a heterogeneous echotexture with poorly defined borders. Color Doppler imaging demonstrates hypervascularity with high-resistance arterial waveforms. Sonography may be limited in determining the full extent of the lesion; therefore, MRI is the preferred modality.
Multifocal lymphangioendotheliomatosis ± thrombocytopenia (MLT) is a rare but important vascular anomaly.48,49 There are cutaneous (macular and popular red/purple) lesions of variable size, which may crust and bleed, as well as gastrointestinal, lung, or other parenchymal vascular lesions. Patients with MLT are at risk of severe gastrointestinal hemorrhage.50
Vascular malformations
Capillary malformation
Imaging is not usually necessary to make a diagnosis of capillary malformation (CM), which is diagnosed clinically. When imaging is performed, subtle signal abnormality is often seen superficially within the subcutaneous fat. CM may be a clue to an underlying structural malformation, with imaging playing an important role in its detection and characterization. Patients at risk for Sturge–Weber syndrome (SWS) represent a particular case, wherein patients with CMs should undergo neuroimaging.
Two recent studies have challenged the previously accepted notion that CMs over the distribution of the trigeminal nerve places patients at highest risk of SWS.51,52 In these papers, upper facial port-wine stains (forehead and temple areas) are correlated with neurologic involvement. A somatic mutation in the GNAQ gene in port-wine stains of patients with and without SWS has been identified.53 It is believed that the earlier the mutation occurs, the more the cell types will be affected (eg, SWS), suggesting that SWS occurs due to an early somatic mosaicism.
Arteriovenous malformation
On sonography, low-resistance arterial flow is depicted within the arteries of an AVM. Sampling of the veins shows arterial waveforms due to shunting. The characteristic findings of an AVM on MRI include tortuous and enlarged vascular channels with a dilated draining vein. AVMs may have perilesional edema, but are not associated with a soft tissue mass. The high-flow vessels seen with AVM are hypointense curvilinear flow voids on spin-echo imaging. MRA is helpful in showing the high-flow nature of the malformation and can identify the supplying vessel.
Venous malformation
Although radiographs often have a limited role in the workup for suspected VM, radiographs may reveal pathognomonic phleboliths caused by thrombosis and calcification. When VMs reach a significant size, bony distortion from mass effect may occur. The most commonly observed bony finding is cortical thickening involving the bone adjacent to the VM due to venous stasis and venous hypertension, which leads to organized periosteal reaction.54 VMs involving the bone result in hypoplasia, cortical thinning, demineralization, or osteolysis.
On grayscale ultrasound imaging, VMs appear heterogeneous and are typically hypoechoic relative to adjacent tissue. Tubular hypoechoic channels are seen in <50% of cases.55 Although rarely identified on ultrasonography, phleboliths appear as well-defined echogenic foci with posterior acoustic shadowing. When superficial, VMs are compressible when pressure is applied to the transducer. Color Doppler imaging typically demonstrates venous monophasic flow within the malformation. In a minority of cases, flow is not demonstrated, which may be related to thrombosis or flow below a detectible level.55
CT plays a limited role in the workup for VMs mainly due to poor contrast resolution and due to the effects of ionizing radiation. When a VM is encountered on CT, it is typically hypodense on noncontrast imaging with internal calcified phleboliths. After contrast administration, there is tubular enhancement of the malformation because of the opacification of the venous channels.
VMs are best demonstrated and characterized on MRI (Figure 8). VMs may be small and localized or diffuse and extensive involving an entire limb or body part. Most VMs are hyperintense on T2-weighted imaging and hypointense on T1-weighted imaging and typically have a lobulated margin. Phleboliths are rounded hypointense signal voids, best demonstrated on T2-weighted imaging. Fluid–fluid levels are uncommon.56 Enhancement is typically present within the low-flow vascular channels. The presence of enhancement is an important differentiating feature from nonenhancing cystic lesions such as LM.
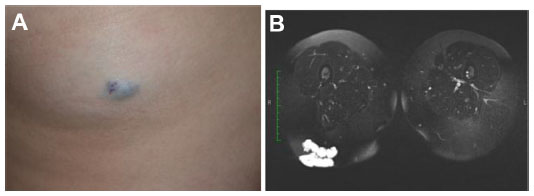 | Figure 8 Venous malformation. |
Lymphatic malformation
Clinically, LMs (Figure 9) may present as soft compressible masses with normal overlying skin. A diagnostic feature is the transillumination of these lesions (Figure 9B). LMs may be classified as macrocystic or microcystic depending on their imaging appearance. On sonography, macrocystic LMs appear as hypoechoic cystic spaces with thin internal septations. Although ultrasonography may be adequate to define subcutaneous cystic nonvascular prominent masses, MRI with contrast is the preferred method to define the anatomic extent of the process and, importantly, its juxtaposition with other structures. This will also help in treatment planning. Microcystic LMs appear as echogenic areas on soft tissue thickening.56 The cysts in a microcystic LM are usually too small to identify as discrete structures on MRI and usually appear as diffuse areas of low T1 signal and increased signal on T2-weighted imaging, with absence of or mild postcontrast enhancement. On MRI, macrocystic LMs appear as well-defined cysts characterized by hypointense signal on T1-weighted imaging and marked hyperintense signal on T2-weighted imaging. Fluid–fluid levels are common, due to internal hemorrhage or proteinaceous material, although a feature that is not pathognomonic for LM as they can be seen in a minority of VMs. After gadolinium administration, there is septal and peripheral enhancement, but the cystic spaces are not internally enhanced, which is a helpful differentiating feature from VM. Edema and enhancement of the adjacent soft tissue may be present in the setting of an inflamed LM.
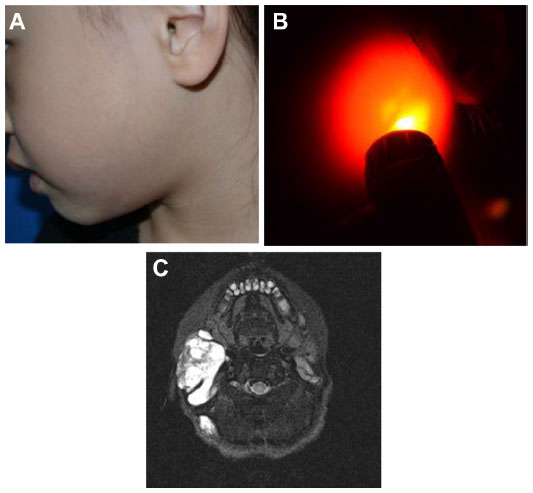 | Figure 9 Lymphatic malformation. |
Syndromic VMs
Syndromic hemangiomas and VMs are summarized in Tables 2 and 3, respectively. Many patients with VMs have skeletal, soft tissue, adipose, and/or parenchymal overgrowth. They are phenotypically distinct, and causative genetic mutations have been identified for many. In most cases, the identified mutation presents mosaic expression due to a somatic (postzygotic) mutational event; thus, these syndromes are not familial. These patients in particular require ongoing surveillance, collaboration among multiple specialists, and – in some cases – early screening for potential malignancies. Syndromes which include slow- and fast-flow lesions are: blue rubber bleb nevus syndrome (Figure 10), Proteus syndrome, Klippel–Trenaunay syndrome (CM, VM, and/or LM, soft tissue hypertrophy and/or skeletal anomalies), RASA-1 AVM-CM (AVMs with multiple cutaneous CMs), and hereditary hemorrhagic telangiectasia (HHT), the latter which is inherited as an autosomal dominant trait (Figure 11), Parkes Weber syndrome (CM, VM with arteriovenous fistula), CLOVES (congenital lipomatous overgrowth, VMs, epidermal nevus, and spinal/skeletal scoliosis – Figure 12), and SWS. Many new treatments are derived from basic and genetic studies, which have identified signaling pathways that are operative in the evolution of vascular anomaly syndromes (Figure 13).
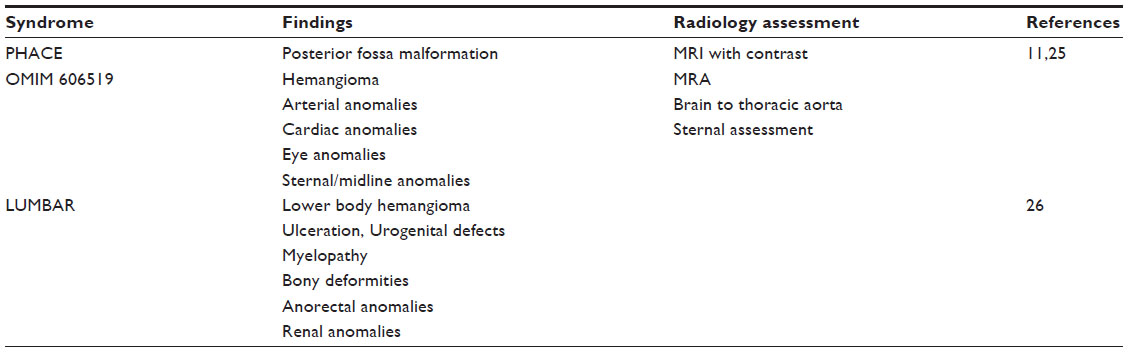 | Table 2 Syndromic vascular anomalies: hemangioma syndromes |
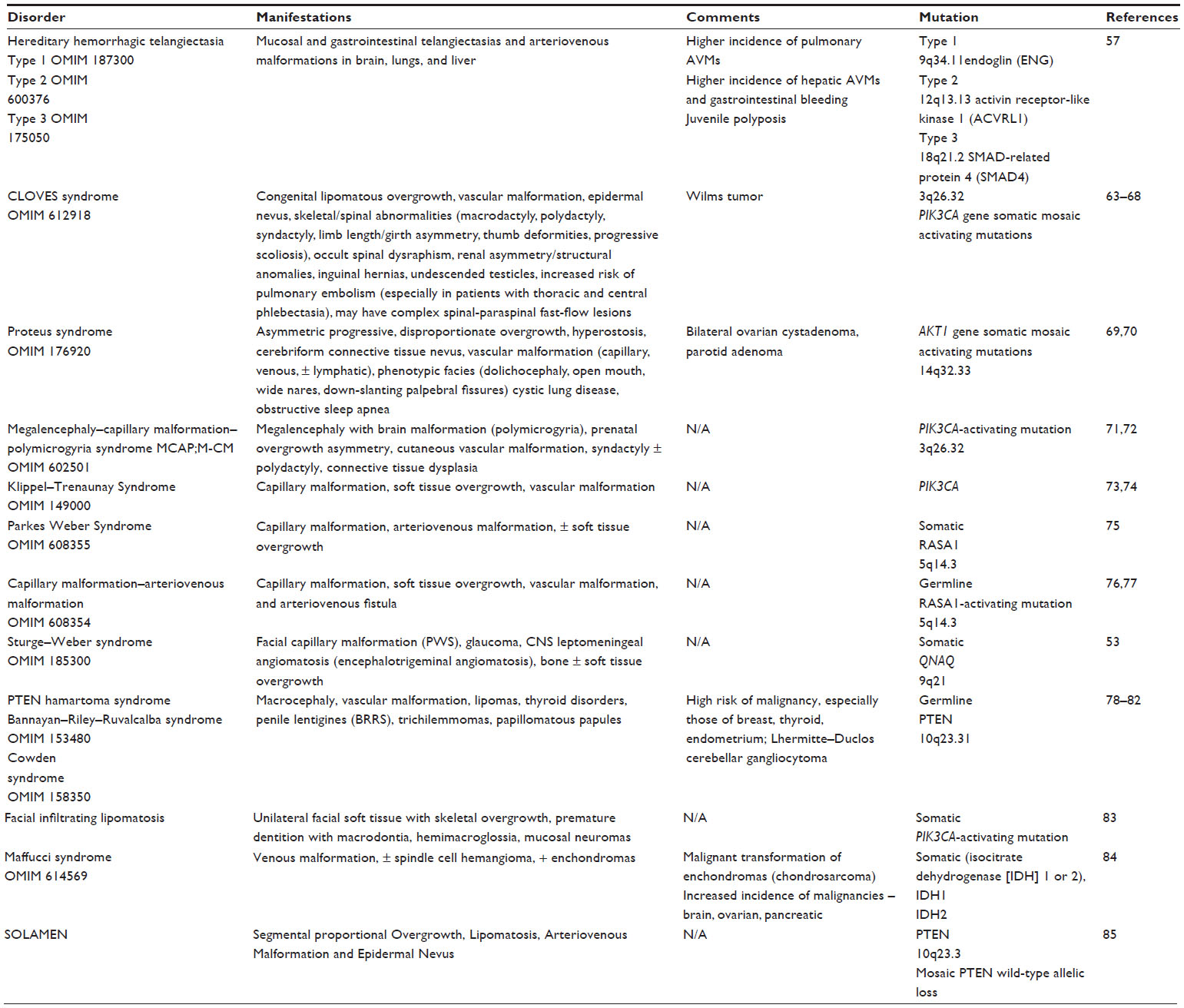 | Table 3 Syndromic vascular anomalies: vascular malformation syndromes |
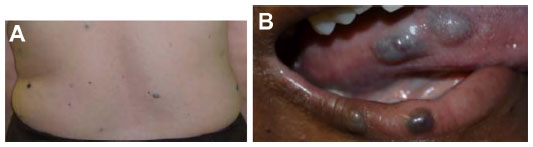 | Figure 10 Blue rubber bleb nevus syndrome. |
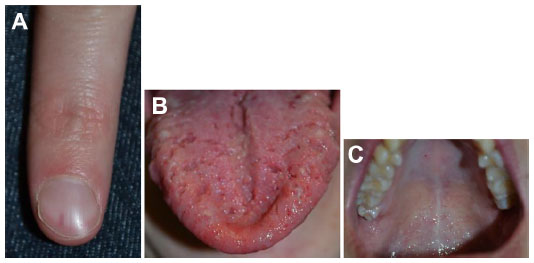 | Figure 11 Hereditary hemorrhagic telangiectasia. |
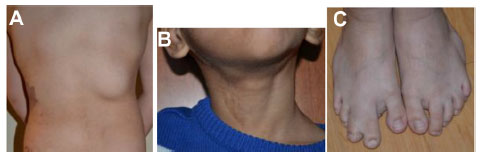 | Figure 12 CLOVES syndrome in two patients. |
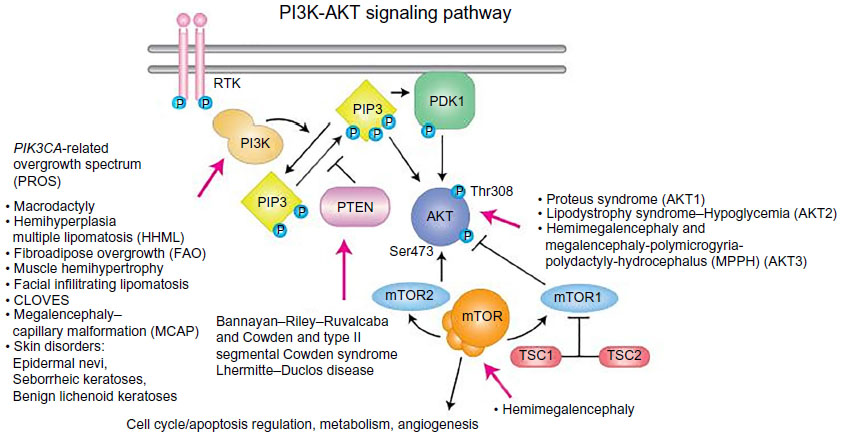 | Figure 13 PIK3CA-related overgrowth spectrum (PROS): diagnostic and testing eligibility criteria, differential diagnosis, and evaluation. |
Hereditary hemorrhagic telangiectasia
Several genetic mutations (most commonly affecting one of two genes in the transforming growth factor-beta/bone morphogenetic protein [BMP] signaling family: endoglin or activin receptor-like kinase-1 [ACVRL1], or less commonly SMAD4 [Mothers Against Decapentaplegic, Drosophila Homolog of 4] or growth/differentiation factor [GDF2]; BMP9]) have been identified in this disorder, which has been recently reviewed by McDonald et al.57 Patients experience epistaxis due to telangiectatic vessels, as well as gastrointestinal and parenchymal AVMs in the gastrointestinal tract, lungs, brain, and/or liver. There may be subungual, facial, lip, tongue, and/or intraoral telangiectasias (Figure 11).
On radiography, pulmonary AVMs demonstrate a well-defined lobulated pulmonary nodule with a draining vein. CT with contrast demonstrates an enhancing well-circumscribed mass with an enhancing feeding artery and draining vein.
Blue rubber bleb nevus syndrome
Patients with blue rubber bleb nevus syndrome have multiple small soft blue VMs of variable size in the skin, gastrointestinal tract, and other organs (Figure 10). On imaging, gastrointestinal tract VMs are characteristic, with the small bowel most commonly affected. Fluoroscopic examinations demonstrate multiple polypoid filling defects, potentially mimicking a polyposis syndrome. Radiographs and CT images demonstrate multiple calcifications, reflecting phleboliths. MRI is useful in detecting the VMs in the musculoskeletal system and solid organ involvement. As detailed in the section on VMs, VMs are typically hypointense on T1-weighted imaging, hyperintense on T2-weighted sequences, and enhanced after gadolinium administration. Phleboliths are common, manifesting as round T2 hypointense filling defects.
Maffucci syndrome
Maffucci syndrome is a nonhereditary enchondromatosis characterized by multiple enchondromas and soft tissue spindle cell hemangioendotheliomas.58 On the basis of the Mulliken and Glowacki classification scheme,3 the “hemangioendotheliomas” may represent a form of VM as they often contain phleboliths. Patients with enchondromatoses have an increased risk of chondrosarcomatous transformation, seen in up to 15%–30% of cases.59,60 Radiographs are often pathognomonic, showing enchondromas of the small bones of the hands and feet and soft tissue calcifications representing phleboliths. On radiographs, the enchondromas are defined, expansile lucent bony lesions with endosteal scalloping and cortical thinning. There is often a mineralized matrix with an ring-and-arc pattern. On MRI, enchondromas have similar signal intensity as cartilage on all pulse sequences; they are hypointense or isointense on T1-weighted imaging, hyperintense on both T2-weighted and cartilage-sensitive sequences, and may demonstrate peripheral enhancement.
Prenatal diagnosis of vascular anomalies
Antenatal detection of vascular anomalies can be important for parental anticipation, fetal monitoring, pre-and postnatal management, and obstetrical decision-making. Marler et al61 reported 29 patients in a 12-month period prenatally diagnosed with a vascular anomaly, 42% of whom were incorrectly diagnosed. In this series, patients correctly diagnosed had optimal decision-making with regard to in utero therapy and mode of delivery (vaginal, cesarean section, ex utero intrapartum treatment procedure).61
Radiologic evaluation provides noninvasive detection of prenatally diagnosed vascular anomalies, including dysmorphic or asymmetrical organs/limbs or abnormal growth patterns, which may be detected with prenatal ultrasonography. Fetal MRI can confirm and refine diagnoses suspected on ultrasonography and assist in arranging the most appropriate type of delivery.10,62
For disorders with identified genomic mutations in a proband, molecular genetic testing may be utilized. Preimplantation genetic diagnosis, amniocentesis, or chorionic villus sampling may be feasible for future offspring.
Resources
Currently, there are several vascular anomalies-related therapeutic trials, registries, and observational and genetic studies. A database of clinical trials and other studies in the USA and internationally, including those open to recruitment, is available at https://clinicaltrials.gov. At this time, pharmacologic studies for vascular anomalies include A Study to Compare Vincristine to Sirolimus for Treatment of High Risk Vascular Tumors (NCT02110069), Sirolimus for VMs with or without hypertrophy syndromes (NCT00975819, NCT02509468, NCT01811667, NCT02428296, and NCT02443818), Laser and oral or topical sirolimus vs laser alone for port-wine stain–type CMs (NCT00830466, NCT02214706), Bevacizumab or Timolol Nasal Spray for HHT-related epistaxis (NCT02106520, NCT02157987, NCT02484716, and NCT01752049), Thalidomide or Pazopanib for HHT (NCT01485224, NCT02204371), as well as several registries and imaging and sclerotherapy studies.
Instructive Web sites are listed in Table 4 and include the following: Online Mendelian Inheritance in Man (OMIM®; http://www.omim.org), which provides OMIM numbers for each disorder as well as academic summaries and links to relevant publications. Genetics Home Reference (http://ghr.nlm.nih.gov) provides, in layman’s terms, medical and genetic information. The Genetic Testing Registry® is particularly robust, synthesizing information from various Web sites to include available clinical and research tests, diagnoses/phenotypes, genetic mutations, and approved state and Clinical Laboratory Improvement Amendments (CLIA)-approved laboratories worldwide. GeneTests (https://www.genetests.org) links to OMIM and other sites, as well as providing a comprehensive database of international laboratories performing genetic testing, including availability for research and/or clinical test method and availability of prenatal and carrier testing. GeneReviews® provides peer-reviewed concise summaries of clinical and genetic information with links to PubMed; however, this may not be as updated (last viewed August 9, 2015) as other sites. The Vascular Anomaly and Lymphedema Mutation Database, maintained by the Laboratory of Human Molecular Genetics, de Duve Institute, Brussels (Belgium) is an excellent resource for recognized mutations for several vascular anomalies-related disorders, with links to PubMed references and an analysis of types of variants reported.
 | Table 4 Online resources for genetic and further information |
Patient- /family-initiated foundations and support groups provide patient/family education, advocacy, guidelines, direct research funding, links to outside funding opportunities, and financial assistance (Table 5). Many funding agencies now encourage patient advocates as panel members and grant reviewers. Patient advocacy groups have become extremely important in moving the field forward, with patient/family education, support, activism, guidelines, fundraising, direct research funding, links to outside funding opportunities, and liaisons with professional organizations and government agencies.
 | Table 5 Disease-specific advocacy groups |
Summary and conclusion
It should be apparent that the term “vascular anomalies” does indeed represent a spectrum of vascular disorders, and that evaluation, diagnosis, and management is a multidisciplinary process. Experts in medical and surgical subspecialties (otolaryngology, plastic/reconstructive surgery, dermatology, orthopedics, ophthalmology, neurosurgery, urology, and general medicine), as well as geneticists, physiatrists, and others, work as a team.
It is important to recognize the clinical presentation and to establish an appropriate diagnosis to most appropriately evaluate and manage the patients. Some congenital vascular anomalies require no or minimal intervention, while others require a cohesive multidisciplinary approach. Understanding these nuances and complexities is essential for the most comprehensive care and optimal clinical outcomes.
Acknowledgment
The authors acknowledge the patients, families, and staff involved in the care of patients with vascular anomalies.
Disclosure
The authors report no conflicts of interest in this work.
References
Wassef M, Blei F. Classification of vascular anomalies. In: Persky M, Waner M, Blei F, Berenstein A, editors. Vascular Lesions of the Head and Neck: Diagnosis and Management. Vol 1. New York, NY: Thieme; 2015;1–9. | |
Mulliken JB, Glowacki J. Hemangiomas and vascular malformations in infants and children: a classification based on endothelial characteristics. Plast Reconstr Surg. 1982;69(3):412–422. | |
Mulliken JB, Glowacki J. Classification of pediatric vascular lesions. Plast Reconstr Surg. 1982;70(1):120–121. | |
Schöbinger R. Schöbinger Classification of Arteriovenous Malformations. Rome: International Society for the Study of Vascular Anomalies Congress; 1996. | |
de Serres LM, Sie KC, Richardson MA. Lymphatic malformations of the head and neck. A proposal for staging. Arch Otolaryngol Head Neck Surg. 1995;121(5):577–582. | |
Wassef M, Blei F, Adams D, et al. Vascular anomalies classification: recommendations from the international society for the study of vascular anomalies. Pediatrics. 2015;136(1):e203–e214. | |
Lee BB, Baumgartner I, Berlien P, et al. Diagnosis and treatment of venous malformations. Consensus document of the international union of phlebology (IUP): updated 2013. Int Angiol. 2015;34(2):97–149. | |
Nozaki T, Matsusako M, Mimura H, et al. Imaging of vascular tumors with an emphasis on ISSVA classification. Jpn J Radiol. 2013; 31(12):775–785. | |
Nozaki T, Nosaka S, Miyazaki O, et al. Syndromes associated with vascular tumors and malformations: a pictorial review. Radiographics. 2013;33(1):175–195. | |
Calvo-Garcia MA, Kline-Fath BM, Adams DM, et al. Imaging evaluation of fetal vascular anomalies. Pediatr Radiol. 2015;45(8):1218–1229. | |
Metry D, Heyer G, Hess C, et al. Consensus statement on diagnostic criteria for PHACE syndrome. Pediatrics. 2009;124(5):1447–1456. | |
Biesbroeck L, Brandling-Bennett HA. Propranolol for infantile haemangiomas: review of report of a consensus conference. Arch Dis Child Educ Pract Ed. 2014;99(3):95–97. | |
Tekes A, Koshy J, Kalayci TO, et al. SE Mitchell vascular anomalies flow chart (SEMVAFC): a visual pathway combining clinical and imaging findings for classification of soft-tissue vascular anomalies. Clin Radiol. 2014;69(5):443–457. | |
Burrows PE. Endovascular treatment of slow-flow vascular malformations. Tech Vasc Interv Radiol. 2013;16(1):12–21. | |
Rosen RJ, Nassiri N, Drury JE. Interventional management of high-flow vascular malformations. Tech Vasc Interv Radiol. 2013; 16(1):22–38. | |
Waner M, O TM. The role of surgery in the management of congenital vascular anomalies. Tech Vasc Interv Radiol. 2013;16(1):45–50. | |
Nasseri E, Piram M, McCuaig CC, et al. Partially involuting congenital hemangiomas: a report of 8 cases and review of the literature. J Am Acad Dermatol. 2014;70(1):75–79. | |
North PE, Waner M, James CA, et al. Congenital nonprogressive hemangioma: a distinct clinicopathologic entity unlike infantile hemangioma. Arch Dermatol. 2001;137(12):1607–1620. | |
Vildy S, Macher J, Abasq-Thomas C, et al. Life-threatening hemorrhaging in neonatal ulcerated congenital hemangioma: two case reports. JAMA Dermatol. 2015;151(4):422–425. | |
Ozcan UA. Rapidly involuting congenital hemangioma: a case of complete prenatal involution. J Clin Ultrasound. 2010;38(2):85–88. | |
Maguiness S, Uihlein LC, Liang MG, et al. Rapidly involuting congenital hemangioma with fetal involution. Pediatr Dermatol. 2015; 32(3):321–326. | |
Roebuck D, Sebire N, Lehmann E, et al. Rapidly involuting congenital haemangioma (RICH) of the liver. Pediatr Radiol. 2012; 42(3):308–314. | |
Chung EM, Cube R, Lewis RB, et al. From the archives of the AFIP: pediatric liver masses: radiologic-pathologic correlation part 1. Radiographics. 2010;30(3):801–826. | |
Gorincour G, Kokta V, Rypens F, et al. Imaging characteristics of two subtypes of congenital hemangiomas: rapidly involuting congenital hemangiomas and non-involuting congenital hemangiomas. Pediatr Radiol. 2005;35(12):1178–1185. | |
Frieden IJ, Reese V, Cohen D. PHACE syndrome. The association of posterior fossa brain malformations, hemangiomas, arterial anomalies, coarctation of the aorta and cardiac defects, and eye abnormalities. Arch Dermatol. 1996;132(3):307–311. | |
Iacobas I, Burrows PE, Frieden IJ, et al. LUMBAR: association between cutaneous infantile hemangiomas of the lower body and regional congenital anomalies. J Pediatr. 2010;157(5):.e1–.e7. | |
Heyer GL, Dowling MM, Licht DJ, et al. The cerebral vasculopathy of PHACES syndrome. Stroke. 2008;39(2):308–316. | |
Oza VS, Wang E, Berenstein A, et al. PHACES association: a neuroradiologic review of 17 patients. AJNR Am J Neuroradiol. 2008; 29(4):807–813. | |
Meltzer DE, Robson CD, Blei F, et al. Enlargement of the internal auditory canal and associated posterior fossa anomalies in PHACES association. AJNR Am J Neuroradiol. 2015;36(11):2159–2162. | |
Carles D, Pelluard F, Alberti EM, et al. Fetal presentation of PHACES syndrome. Am J Med Genet A. 2005;132A(1):110. | |
Thebault N, Le Guern H, Le Fiblec B, et al. Prenatal diagnosis of a complete sternal cleft in a child with PHACES syndrome-a case report. Prenat Diagn. 2009;29(2):179–181. | |
Recio-Rodriguez M, Martin Fernandez-Mayoralas D, Jimenez-de-la-Pena M, et al. Síndrome PHACES (Pascual Castroviejo tipo II): resonancia magnética pre y posnatal. [PHACES syndrome (Pascual Castroviejo type II): prenatal and postnatal magnetic resonance imaging]. Radiologia. 2013;55(6):537–540. Spanish. | |
Massoud M, Cagneaux M, Garel C, et al. Prenatal unilateral cerebellar hypoplasia in a series of 26 cases: significance and implications for prenatal diagnosis. Ultrasound Obstet Gynecol. 2014;44(4):447–454. | |
Fernandez-Mayoralas DM, Recio-Rodriguez M, Fernandez-Perrone AL, et al. In utero diagnosis of PHACE syndrome by fetal magnetic resonance imaging (MRI). J Child Neurol. 2014;29(1):118–121. | |
Chiappa E, Greco A, Fainardi V, et al. Aortic arch interruption and persistent fifth aortic arch in phace syndrome: prenatal diagnosis and postnatal course. Echocardiography. 2015;32(9):1441–1443. | |
Arora SS, Plato BM, Sattenberg RJ, et al. Adult presentation of PHACES syndrome. Interv Neuroradiol. 2011;17(2):137–146. | |
Burch EA, Garzon MC, Parikh A, et al. A 65-year-old woman diagnosed with PHACE syndrome. Pediatr Dermatol. 2013;30(6):e153–e156. | |
Conley L, Malhotra A, Blei F, Shatzkes D. The Adult Face of PHACES. In: 20th Workshop of the International Society for the Study of Vascular Anomalies; 2014; Melbourne, VIC. | |
Dick EA, Patel K, Owens CM, et al. Spinal ultrasound in infants. Br J Radiol. 2002;75(892):384–392. | |
Gruman A, Liang MG, Mulliken JB, et al. Kaposiform hemangioendothelioma without Kasabach-Merritt phenomenon. J Am Acad Dermatol. 2005;52(4):616–622. | |
Haisley-Royster C, Enjolras O, Frieden IJ, et al. Kasabach-merritt phenomenon: a retrospective study of treatment with vincristine. J Pediatr Hematol Oncol. 2002;24(6):459–462. | |
Hammill AM, Wentzel M, Gupta A, et al. Sirolimus for the treatment of complicated vascular anomalies in children. Pediatr Blood Cancer. 2011;57(6):1018–1024. | |
Croteau SE, Liang MG, Kozakewich HP, et al. Kaposiform hemangioendothelioma: atypical features and risks of Kasabach-Merritt phenomenon in 107 referrals. J Pediatr. 2013;162(1):142–147. | |
Wang Z, Li K, Yao W, et al. Steroid-resistant kaposiform hemangioendothelioma: a retrospective study of 37 patients treated with vincristine and long-term follow-up. Pediatr Blood Cancer. 2015; 62(4):577–580. | |
Kai L, Wang Z, Yao W, et al. Sirolimus, a promising treatment for refractory Kaposiform hemangioendothelioma. J Cancer Res Clin Oncol. 2014;140(3):471–476. | |
Drolet BA, Trenor CC 3rd, Brandao LR, et al. Consensus-derived practice standards plan for complicated Kaposiform hemangioendothelioma. J Pediatr. 2013;163(1):285–291. | |
Konez O, Burrows PE. Magnetic resonance of vascular anomalies. Magn Reson Imaging Clin N Am. 2002;10(2):363–388, vii. | |
North PE, Kahn T, Cordisco MR, et al. Multifocal lymphangioendotheliomatosis with thrombocytopenia: a newly recognized clinicopathological entity. Arch Dermatol. 2004;140(5):599–606. | |
Droitcourt C, Boccara O, Fraitag S, et al. Multifocal lymphangioendotheliomatosis with thrombocytopenia: clinical features and response to sirolimus. Pediatrics. 2015;136(2):e517–e522. | |
Campbell CM, Beckum KM, Hammers YA, et al. Multiple congenital red-brown macules, thrombocytopenia, and gastrointestinal bleeding. Diagnosis: multifocal lymphangioendotheliomatosis with thrombocytopenia (MLT). Pediatr Dermatol. 2010;27(4):395–396. | |
Waelchli R, Aylett SE, Robinson K, et al. New vascular classification of port wine stains: improving prediction of Sturge-Weber risk. Br J Dermatol. 2014;171(4):861–867. | |
Dutkiewicz AS, Ezzedine K, Mazereeuw-Hautier J, et al. A prospective study of risk for Sturge-Weber syndrome in children with upper facial port-wine stain. J Am Acad Dermatol. 2015;72(3): 473–480. | |
Shirley MD, Tang H, Gallione CJ, et al. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med. 2013;368(21):1971–1979. | |
Legiehn GM, Heran MK. Classification, diagnosis, and interventional radiologic management of vascular malformations. Orthop Clin North Am. 2006;37(3):435–474, vii–viii. | |
Trop I, Dubois J, Guibaud L, et al. Soft-tissue venous malformations in pediatric and young adult patients: diagnosis with Doppler US. Radiology. 1999;212(3):841–845. | |
Burrows PE, Laor T, Paltiel H, et al. Diagnostic imaging in the evaluation of vascular birthmarks. Dermatol Clin. 1998;16(3):455–488. | |
McDonald J, Wooderchak-Donahue W, VanSant Webb C, et al. Hereditary hemorrhagic telangiectasia: genetics and molecular diagnostics in a new era. Front Genet. 2015;6:1–8. | |
Fanburg JC, Meis-Kindblom JM, Rosenberg AE. Multiple enchondromas associated with spindle-cell hemangioendotheliomas. An overlooked variant of Maffucci’s syndrome. Am J Surg Pathol. 1995; 19(9):1029–1038. | |
Lewis RJ, Ketcham AS. Maffucci’s syndrome: functional and neoplastic significance. Case report and review of the literature. J Bone Joint Surg Am. 1973;55(7):1465–1479. | |
Kaplan RP, Wang JT, Amron DM, et al. Maffucci’s syndrome: two case reports with a literature review. J Am Acad Dermatol. 1993;29(5 pt 2):894–899. | |
Marler JJ, Fishman SJ, Upton J, et al. Prenatal diagnosis of vascular anomalies. J Pediatr Surg. 2002;37(3):318–326. | |
Saleem SN. Fetal MRI: an approach to practice: a review. J Adv Res. 2014;5(5):507–523. | |
Sapp JC, Turner JT, van de Kamp JM, et al. Newly delineated syndrome of congenital lipomatous overgrowth, vascular malformations, and epidermal nevi (CLOVE syndrome) in seven patients. Am J Med Genet A. 2007;143A(24):2944–2958. | |
Alomari AI. Characterization of a distinct syndrome that associates complex truncal overgrowth, vascular, and acral anomalies: a descriptive study of 18 cases of CLOVES syndrome. Clin Dysmorphol. 2009; 18(1):1–7. | |
Alomari AI, Burrows PE, Lee EY, et al. CLOVES syndrome with thoracic and central phlebectasia: increased risk of pulmonary embolism. J Thorac Cardiovasc Surg. 2010;140(2):459–463. | |
Kurek KC, Luks VL, Ayturk UM, et al. Somatic mosaic activating mutations in PIK3CA cause CLOVES syndrome. Am J Hum Genet. 2012;90(6):1108–1115. | |
Group CSW. Clinical Practice Guidelines for CLOVES Syndrome. 2014. Available from: http://www.clovessyndrome.org/sites/default/files/CLOVES_Syndrome_Management_Guidelines_For_Families_6-21-2014-2.pdf. Accessed July 09, 2015. | |
Alomari AI, Chaudry G, Rodesch G, et al. Complex spinal-paraspinal fast-flow lesions in CLOVES syndrome: analysis of clinical and imaging findings in 6 patients. AJNR Am J Neuroradiol. 2011;32(10):1812–1817. | |
Lindhurst MJ, Sapp JC, Teer JK, et al. A mosaic activating mutation in AKT1 associated with the proteus syndrome. N Engl J Med. 2011;365(7):611–619. | |
Wee JS, Mortimer PS, Lindhurst MJ, et al. A limited form of proteus syndrome with bilateral plantar cerebriform collagenomas and varicose veins secondary to a mosaic AKT1 mutation. JAMA Dermatol. 2014;150(9):990–993. | |
Mirzaa GM, Conway RL, Gripp KW, et al. Megalencephaly-capillary malformation (MCAP) and megalencephaly-polydactyly-polymicrogyria- hydrocephalus (MPPH) syndromes: two closely related disorders of brain overgrowth and abnormal brain and body morphogenesis. Am J Med Genet A. 2012;158A(2):269–291. | |
Mirzaa GM, Riviere JB, Dobyns WB. Megalencephaly syndromes and activating mutations in the PI3K-AKT pathway: MPPH and MCAP. Am J Med Genet C Semin Med Genet. 2013;163C(2):122–130. | |
Jacob AG, Driscoll DJ, Shaughnessy WJ, et al. Klippel-Trenaunay syndrome: spectrum and management. Mayo Clin Proc. 1998; 73(1):28–36. | |
Luks VL, Kamitaki N, Vivero MP, et al. Lymphatic and other vascular malformative/overgrowth disorders are caused by somatic mutations in PIK3CA. J Pediatr. 2015;166(4):.e1–.e5. | |
Revencu N, Boon LM, Mulliken JB, et al. Parkes Weber syndrome, vein of Galen aneurysmal malformation, and other fast-flow vascular anomalies are caused by RASA1 mutations. Hum Mutat. 2008;29(7):959–965. | |
Eerola I, Boon LM, Mulliken JB, et al. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet. 2003;73(6):1240–1249. | |
Boon LM, Mulliken JB, Vikkula M. RASA1: variable phenotype with capillary and arteriovenous malformations. Curr Opin Genet Dev. 2005;15(3):265–269. | |
Eng C. PTEN Hamartoma Tumor Syndrome (PHTS). In: Pagon RA, Adam MP, Ardinger HH, et al, editors. GeneReviews®. Seattle, WA; 2001 [Updated January 23, 2014]. | |
Orloff MS, Eng C. Genetic and phenotypic heterogeneity in the PTEN hamartoma tumour syndrome. Oncogene. 2008;27(41):5387–5397. | |
Pilarski R, Burt R, Kohlman W, et al. Cowden syndrome and the PTEN hamartoma tumor syndrome: systematic review and revised diagnostic criteria. J Natl Cancer Inst. 2013;105(21):1607–1616. | |
Nieuwenhuis MH, Kets CM, Murphy-Ryan M, et al. Cancer risk and genotype-phenotype correlations in PTEN hamartoma tumor syndrome. Fam Cancer. 2014;13(1):57–63. | |
Tan WH, Baris HN, Burrows PE, et al. The spectrum of vascular anomalies in patients with PTEN mutations: implications for diagnosis and management. J Med Genet. 2007;44(9):594–602. | |
Maclellan RA, Luks VL, Vivero MP, et al. PIK3CA activating mutations in facial infiltrating lipomatosis. Plast Reconstr Surg. 2014;133(1):12e–19e. | |
Amary MF, Damato S, Halai D, et al. Ollier disease and Maffucci syndrome are caused by somatic mosaic mutations of IDH1 and IDH2. Nat Genet. 2011;43(12):1262–1265. | |
Caux F, Plauchu H, Chibon F, et al. Segmental overgrowth, lipomatosis, arteriovenous malformation and epidermal nevus (SOLAMEN) syndrome is related to mosaic PTEN nullizygosity. Eur J Hum Genet. 2007;15(7):767–773. | |
Storch CH, Hoeger PH. Propranolol for infantile haemangiomas: insights into the molecular mechanisms of action. Br J Dermatol. 2010;163(2):269–274. | |
Keppler-Noreuil KM, Rios JJ, Parker VE, et al. PIK3CA-related overgrowth spectrum (PROS): diagnostic and testing eligibility criteria, differential diagnosis, and evaluation. Am J Med Genet A. Feb 2015;167A(2):287–295. | |
ISSVA International Society for the Study of Vascular Anomalies [webpage on the Internet]. Available from: http://www.issva.org/content.aspx?page_id=22&club_id=298433&module_id=152904. Accessed January 29, 2016. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.