")
Back to Journals » Clinical Ophthalmology » Volume 16
Congenital Hypertrophy of the Retinal Pigment Epithelium (CHRPE) as a Screening Marker for Familial Adenomatous Polyposis (FAP): Systematic Literature Review and Screening Recommendations
Authors Bonnet LA , Conway RM, Lim LA
Received 18 December 2021
Accepted for publication 8 February 2022
Published 15 March 2022 Volume 2022:16 Pages 765—774
DOI https://doi.org/10.2147/OPTH.S354761
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Louis Antoine Bonnet, R Max Conway, Li-Anne Lim
University of Sydney, Sydney, New South Wales, Australia
Correspondence: Louis Antoine Bonnet, 83 Western Line, RD1, Whanganui, 4571, New Zealand, Tel +64 0273947946, Email [email protected]
Purpose: Familial adenomatous polyposis (FAP) has an almost 100% colorectal cancer risk warranting early detection in gene carriers. This study presents congenital hypertrophy of the retinal pigment epithelium (CHRPE) as a highly specific phenotypical marker for FAP that can be used in screening at-risk individuals. Screening recommendations including morphological subclassification were formulated with supporting literature.
Methods: A systematic literature review with a comprehensive search strategy was conducted using online databases. Manual searches of bibliographies and reference lists were also performed. Studies meeting inclusion criteria were graded with respect to their hierarchy of evidence and strength of recommendations according to the National Health and Medical Research Council (NHMRC) guidelines of Australia.
Results: Almost 4500 participants were analysed across 28 included studies. The mean specificity of CHRPE as a phenotypical screening marker of FAP was 89% (standard deviation (SD); 14) with a mean sensitivity of 79% (SD; 8). The mean prevalence of CHRPE amongst FAP participants; at-risk participants were found to be 76% (SD; 24) and 37% (SD; 21) respectively. Bilateralism and multiple lesion number ≥ 3 are features highly specific for FAP.
Conclusion: CHRPE was found to be a non-invasive, rapid, early phenotypical screening marker of FAP. Clinical recognition further allows increased gene analysis efficiency. The absence of CHRPE alone cannot exclude FAP. Our screening recommendations provide guidance to clinicians on evidence based CHRPE assessment. We would advocate inclusion of ocular examinations as part of a three-pronged approach, along with endoscopy and genetic testing, for efficient, timely FAP assessment in at-risk individuals.
Keywords: genetic disease, congenital anomalies, retinal examination, screening
Introduction
Familial adenomatous polyposis (FAP) is an inherited, highly penetrant genetic condition predisposing to an almost 100% colorectal cancer risk.1 Since FAP can be fatal if left untreated, early detection and diagnosis in gene carriers and at-risk individuals is vital. Currently there is no dependable haematological or biochemical serum marker identified that can reliably detect FAP in at-risk individuals.2 National screening guidelines for FAP currently consist of endoscopic colonic surveillance and gene linkage analysis.3 CHRPE is a congenital hamartoma of the retinal pigment epithelium (RPE), considered the first extracolonic manifestation of FAP. Colonic polyposis and CHRPE share a common mutation of the adenomatous polyposis colic (APC) gene, located on chromosome 5q21-22.4 This study presents congenital hypertrophy of the retinal pigment epithelium (CHRPE) as a specific, non-invasive, phenotypical screening marker for early recognition of FAP. Screening recommendations of FAP-associated CHRPE were produced with associated morphological subanalysis of highly specific lesion characteristics. These recommendations provide support for the inclusion of ocular examination in a multi-modal approach for efficient, timely FAP assessment in at-risk individuals.
Materials and Methods
Literature Search
The systematic literature review was completed using a comprehensive search strategy by a single reviewer. Multiple online databases were searched including; Cochrane library, Embase, PubMed and ProQuest. To ensure that all potentially relevant literature was not missed, both abbreviated and non-abbreviated terms and their related synonyms were used. These included the following terms; “CHRPE,” “congenital hypertrophy of the retinal pigment epithelium,” “typical,” “atypical,” “familial adenomatous polyposis,” “FAP,” “classic,” “CFAP,” “attenuated,” “AFAP,” “adenomatous polyposis coli,” “APC mutation,” “germline mutation,” “pigmented fundal lesion,” “pigmented ocular fundus lesions,” and “POFL.” No significant alternative spellings of terms were found during the search. Sentence quotations and word truncation were also used in an attempt to include as many studies within the broad search method as possible. Manual searches using bibliographies and reference lists of pertinent studies were also conducted.
Inclusion and Exclusion Criteria
Study selection was based on publication date, quantitative analysis and direct CHRPE correlation with FAP using specified fundoscopy. Studies published between 1970–2021 were included, encapsulating the last 50 years of research within the field. Descriptive studies lacking quantitative analysis were excluded. Studies which did not involve patient fundus examination were reviewed but excluded from the quantitative analysis used for formulating screening recommendations. Case-series lacking a comparison group were similarly reviewed but excluded from the quantitative analysis. There were no exclusion criteria placed on geographical location of populations sampled nor participant ethnicity. Studies where full English-translations of manuscripts were not obtainable were excluded. Animal and in-vitro studies were both excluded.
Studies meeting the predetermined inclusion criteria were graded with respect to their hierarchy of study design and level of evidence. Using the National Health and Medical Research Council (NHMRC) of Australia framework,5 each study was assigned a grade between I and IV. Individual study recommendations were further assessed by applying the NHMRC grading of evidence matrix (grade A–D).5 The consistency, degree of bias, clinical impact, generalisability and applicability of the evidence presented were all considered when grading individual studies.
Quantitative Analysis and Formulation of Screening Recommendations
Each included study was tabled with respect to its sample size and NHMRC grading. Results were weighted against the relative quality of evidence presented (Table 1). Descriptive analysis of CHRPE morphology, size, location and laterality were recorded in addition to study prevalence, sensitivity, specificity and predictive value. Screening recommendations, including highly specific FAP-associated CHRPE characteristics, were presented along with lesion subclassification based on the weighted results. The screening recommendations (Figure 1) were designed in an algorithmic style with reference to the supporting literature reviewed. Multiple iterations were constructed after case-scenario testing of multiple different potential patient presentations and outcomes. It was intended to be constructed in a manner whereby the main target audience would be ophthalmologists and ophthalmology trainees, however it would still be interpretable to general medical practitioners.
 |
Table 1 Included Literature Listed by Year of Publication with Associated Study Size, NHMRC Level of Evidence and Grading |
 |
Figure 1 FAP-associated CHRPE screening recommendations. |
Results
Literature Search Results (Figure 2)
There were 104 positive search results using the database search methods listed. Of the positive results, 44 full texts were digitally or physically accessible and reviewed. 34 of these studies met the inclusion criteria and were appraised according to the NHMRC grading system.5 Low strength studies, either by design or applicability of recommendations, were excluded. Of the excluded studies, 5 were either duplicates, untranslatable non-English texts or abstracts without complete full-text studies. A further 12 were observational-only, describing CHRPE without direct fundoscopic analysis and 6 case-series without any comparison group. 28 studies were included in the quantitative analysis and formation of screening recommendations as demonstrated in Figure 2.
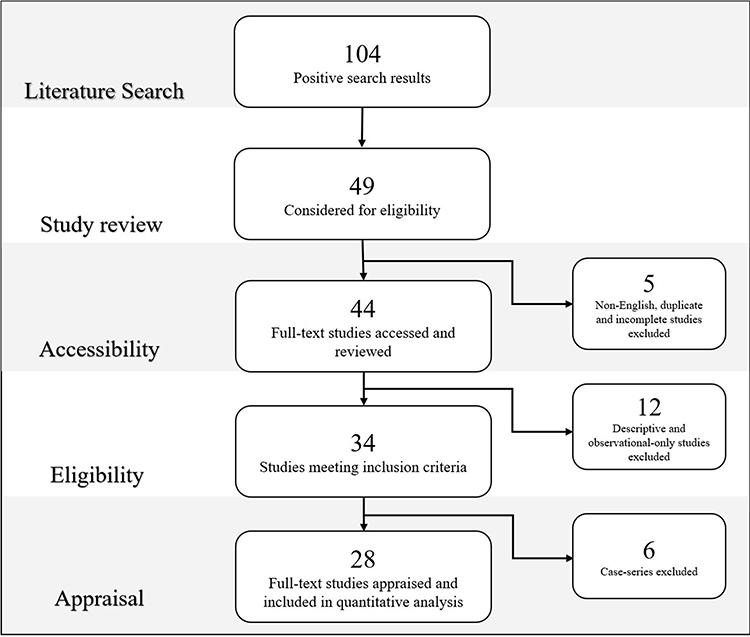 |
Figure 2 Literature review search, selection and appraisal of studies. |
Study Hierarchy and Grading of Evidence (Table 1)
All 28 studies presented evidence equivalent to NHMRC level III. The majority of studies were level III-C (n = 18), with 9 level III-B and 1 level III-1. There were no studies with level I or II evidence. All study recommendations were either grade B (n = 14) or C (n = 14). There were no grade A recommendations, which corresponds to the lack of level I evidence studies. All grade D recommendations and level IV evidence from case-series were excluded (n=6).
Study Cohort Demographics
Across the 28 studies, 4451 subjects were included. 1137 with FAP, 794 at increased risk of FAP and 2262 controls. At-risk individuals included first-degree relatives of family members with known APC gene mutation. The mean age of FAP subjects across the 28 studies was 31.2 years with a range of 8–79 years. At-risk individuals ranged from 7–83 years with a mean age of 21.5 years. Of the studies which included controls as their comparative group; the mean age was 38.5 years with a range of 24–88 years. Few studies distinguished variation in gender between comparison, test and control groups. Of the studies which did, there were approximately equal numbers of males and females with no statistically significant difference.
Reliability and Accuracy
Across the 28 studies the mean specificity (Sp) of CHRPE as a phenotypical marker of FAP was 89% with a standard deviation (SD) of 14. The mean sensitivity (Sn) was 79% (SD; 8). The mean prevalence of CHRPE amongst FAP participants was found to be 76% (SD; 24) and 37% (SD; 21) in at-risk participants. Giardiello et al had the highest reported CHRPE specificity (100%),27 while Mirinezhad et al reported the lowest CHRPE specificity (61.5%).6 The sensitivity of CHRPE was consistently lower than its relative specificity amongst most authors. The highest reported sensitivity was by Morton et al (84%).25 Tiret et al reported the lowest CHRPE sensitivity (65%).13 Few authors included predictive values of CHRPE for FAP. Of the studies which did, Giardiello et al recorded the highest positive predictive value (PPV) (100%) but the lowest negative predictive value (NPV) (40%).27 The highest NPV was described by Mirinezhad et al (76.2%).6 Gelisken et al and Lam et al had the highest prevalence of CHRPE amongst FAP individuals (100%).11,12 Rushwurm et al identified CHRPE prevalence of 54% in all examined patients and 75% of those diagnosed with FAP.10 Hunt et al recorded the lowest CHRPE prevalence amongst FAP subjects (33.3%).18 With respect to at-risk individuals, Heyen et al described the highest CHRPE prevalence (80.4%).19,30 Rossato et al recorded one of the lowest CHRPE prevalences amongst study controls (5.5%, n = 160) while Chen et al demonstrated no CHRPE-positive lesions in their control group (n = 37).8,15
Total Lesion Number
The mean number of CHRPE lesions across all included literature was 10.9 (FAP group) and 4.8 (at-risk group). Multiple lesions were found in 59.6% of FAP individuals. Tourino et al recorded the greatest mean lesion number in FAP individuals (11.9 lesions, SD; 9.2) followed by Olea et al (11.06, SD; 8.7) and Rushwurm et al (10.9).9,10,16 Tiret et al found no significant difference in the number of lesions between either eye (27).13 In at-risk individuals, there was a significantly lower total CHRPE lesion count. The lowest mean lesion numbers were recorded by Olea et al (1.4, SD; 0.7) and Tourino et al (2.8, SD; 1.5).9,16
Laterality
Bilateral lesions were seen on average in 69.4% of FAP individuals across all included literature. Polkinghorne et al, had the highest prevalence of CHRPE bilateralism in FAP individuals (97%).28 Tourino et al, found bilateralism to be the most sensitive and specific feature of FAP-associated CHRPE (Sn 83%, Sp 100%).9
Size
Across the 28 studies; 78.5% of FAP individuals had CHRPE lesions measuring <0.5 DD compared to 82% in at-risk individuals. Heyen et al demonstrated the strongest size relationship with 83% of CHRPE lesions in FAP individuals being <0.5 DD in comparison to 11% (0.5–1.0 DD) and 6% (>1.0 DD) respectively.30 Conversely, Rushwurm et al and Olea et al described large CHRPE size association with the FAP disease genotype.10,16 Rushwurm et al reported 80% of CHRPE >0.5 DD in FAP individuals compared to 66% in the at-risk group.10 Olea et al, reported 96% of CHRPE >0.125 DD associated with intestinal disease in 96% of FAP individuals with a sensitivity of 82%, specificity 97%, PPV 96% and NPV 88%.16
Location
Overall, 84% of CHRPE lesions were located in the retinal periphery compared to 16% within the posterior pole. Tourino et al described the most common location for CHRPE amongst FAP individuals was in the retinal equator.9 92% of FAP individuals had CHRPE present proximal to the retinal vessels compared to 7% in controls (p < 0.001).9
Pigmentation
CHRPE lesions were found to be predominantly pigmented (61.5%) compared to depigmented (8.0%) and mixed pigmentation (30.5%). Baba et al, described large pigmentation, 0.5–2.0 times the papillary diameter, having a sensitivity and specificity of 65.2% and 99.9% respectively.1 Small pigmentation, less than 0.5 times the papillary diameter, had a sensitivity and specificity of 78.3% and 99.7% respectively.1
Discussion
Our screening recommendations were produced to assist clinicians in the assessment of CHRPE as a phenotypical screening marker for FAP. In particular we wanted to address 4 main questions which were extensions of our initial research objectives; (1) What are the specific characteristics of CHRPE associated with FAP? (2) Who should receive an ocular examination and at what age should it commence? (3) What is the reliability and accuracy of CHRPE as a FAP screening marker? (4) What are the relative limitations and clinical implications of CHRPE as a screening tool?
CHRPE Nomenclature
Chen et al, suggest that the term “CHRPE” amongst individuals of the general public without FAP is incorrect.8 FAP individuals have CHRPE lesions which are histologically distinct, consisting of focal malformations of the melanin granules within RPE cells which microscopically resemble neoplasms (but are clinically benign).8 Lesions amongst individuals of the general population are characterised by macromelanin granule accumulation within a single, enlarged layer of RPE cells.8 Hennessy et al, proposed changing the term to “multiple retinal pigment epithelial hamartomata (MRPEH)” when referring to CHRPE in FAP individuals, while Traboulsi et al, referred to these lesions as “pigmented ocular fundus lesions (POFL).”14,33 Shields et al proposed the term “RPE hamartomas associated with FAP (RPEH-FAP).”34 Alternative terms such as “multi-focal CHRPE,” referring to large sectoral lesions with surrounding smaller satellite lesions or “bear tracks” have also be reported to distinguish from “solitary CHRPE” referring to a single, pigmented well-demarcated lesion commonly with a depigmented halo.35 In our screening recommendations we recognised the importance of using nomenclature which accurately correlates the lesion’s genetics, histopathology and clinical presentation. In our screening recommendations, we use the term “FAP-associated CHRPE” when referring to fundus lesions associated with FAP, and POFL when referencing lesions within the general population without FAP.
CHRPE Definition
There was no consensus on a single CHRPE definition and subclassification method amongst authors. Berk et al presented one of the earliest case-control studies on CHRPE which was used to varying degrees by subsequent authors.9,15,19,22,25,29,32 One limitation of the classification system presented by Berk et al is that it only subclassified CHRPE relative to its morphology.32 Laterality, total lesion number and retinal location were not considered. Classification was largely descriptive with little quantitative consideration. Subsequent subclassification systems by Rossato et al and Valanzano et al utilised morphology and lesion size while most other authors included total lesion number in their CHRPE-positive criteria.15,17 Multiple authors considered bilaterality to be a highly specific characteristic for FAP-associated CHRPE.9,15,19,22,25,29,32 The CHRPE definition and subclassification system used in our screening recommendations were adapted from Berk et al with the additional consideration of total lesion number, laterality, size, location and degree of pigmentation.
Screening Recommendations
Our screening recommendations were designed to be used supplementary to the current endoscopic and genetic testing guidelines already established by the Cancer Council of Australia (Figure 1).3
Who Should Undergo Ocular Screening?
There was a consensus within the literature that ocular screening for FAP should be offered to at-risk individuals rather than the general population. This corresponds to the current Cancer Council of Australia’s recommendations with respect to endoscopy and genetic testing alike.3 There is limited evidence to suggest that FAP-associated CHRPE occurs in non-polyposis colorectal cancers which lack APC gene mutation. It is further considered an inappropriate marker for attenuated forms of FAP (AFAP). Ocular examination will add little diagnostic or prognostic value in FAP individuals who have already undergone definitive genetic screening. For FAP individuals who know their disease status, endoscopic colonic surveillance is recommended over ocular fundus monitoring. Our screening recommendations are therefore targeted towards FAP at-risk individuals defined as first-degree relatives of an index case with a known APC gene mutation.
At What Age Should Ocular Screening Commence?
The mean age of diagnosis of FAP is between the second and fourth decade of life.36 FAP-associated CHRPE is considered the earliest extracolonic manifestation of FAP, usually present prior to the onset of detectable colonic polyps.36 There have been pigmented ocular fundus lesions observed in neonates as early as 3 months of age.1,14 The youngest study participant recorded with FAP was 8 years of age. As FAP-associated CHRPE is a phenotypical marker rather than a definitive test, ocular examination age selection should be considered in association with the age-appropriate commencement of follow-up diagnostic testing. The accepted age to start colonoscopy currently is 10–11 years of age.3 Both patient and familial anxiety from a CHRPE-positive result would be significantly reduced if it is accompanied promptly by a definitive test. Furthermore, selecting a screening age prior to the onset of clinically detectable polyposis would allow the greatest prognostic benefit to the individual. In our screening recommendations we suggest ocular examination to screen for FAP-associated CHRPE from 10 years of age, or prior to endoscopic evaluation.
Genotype and Phenotype Correlation
The long arm of chromosome 5 (5q21-22), codons 463–1393, has been regarded as the most frequent location of the APC gene mutation related to CHRPE.4 Mutations of exons 10–15 have been associated with clinically significant numbers of FAP-associated CHRPE lesions (≥3).14 Mutations outside the 463–1387 codon sequence, such as 1597, have been linked with other extraintestinal manifestations, such as desmoid tumours, without the presence of CHRPE.7 An individual’s FAP-associated CHRPE status is valuable with respect to genomic sequencing as it can help locate the mutated sequence in gene linkage analysis allowing for greater testing efficiency. Furthermore, clinically relevant information can be obtained through knowledge of the exact location of the mutation. In particular; age of onset and polyposis extent are highly linked with specific gene loci.2,8
Reliability and Accuracy of FAP-Associated CHRPE
Although the sensitivity and specificity vary among authors, the majority consensus is that CHRPE is a reliable indicator of FAP carrier status. Baba et al postulated that the differing reliability values between authors relates to the method of ophthalmologic examination as well as sampling errors in comparatively small groups of patients studied.1 We found a mean specificity of 89% and sensitivity of 79% for FAP-associated CHRPE across all literature reviewed. This indicates that as a marker, it is less likely to produce false-positive results but is more prone to false-negatives. FAP-associated CHRPE is not considered a diagnostic test, but rather a phenotypical marker of a single genetic process. Its relatively low sensitivity would result in a higher number of false negatives, and thereby considered inappropriate to exclude FAP based on the absence of CHRPE. This sentiment is further supported by its positive and negative predictive value. Hickey-Dwyer et al suggested that CHRPE is an unreliable maker of APC gene mutation due to its less than desirable sensitivity.22 Rushwurm et al reported intrafamilial variation of CHRPE expression indicating that negative results amongst at-risk individuals does not exclude FAP.10 In kindreds which do not carry the trait, eye examination may not help in predicting the presence of disease.30 To the contrary, there is literature with strong supporting evidence to suggest that FAP-associated CHRPE is an appropriate disease marker for FAP.1,9,16,28,32,34,37 With the current body of literature quantitatively assessed and considered as a whole, we would promote FAP-associated CHRPE as an effective phenotypical marker for FAP. Lesion subclassification with particular attention to “high risk” features, such as; bilateralism and multiple lesion number, has shown to increase the relative specificity of CHRPE as a marker. We would suggest ocular examination complimentary to endoscopic examination and genetic testing rather than in isolation. It is apparent that the absence of FAP-associated CHRPE cannot exclude polyposis and should not preclude further definitive testing.
Limitations and Implications
We found limited published literature and quantitative data addressing this research area. Our sample size and the statistical power of our results are a reflection of FAP-associated CHRPE research encapsulating the last 50 years. The quality of evidence included in our systematic literature review, based on the NHMRC grading criteria, was moderate to low.5 There were no grade A or level I evidence present, which may not reflect poorly on the individual studies, but rather the nature of CHRPE as a disease marker. The highest quality study design for the research question presented were case-control and cohort studies which have a greater risk of bias. Nusliha et al, described the presence of recall bias in their case-control study where response rates amongst FAP-affected individuals (68%) was more than double that of unaffected individuals at-risk (32%).7 A further limitation with respect to study design was the inclusion of non-representative controls. Tiret et al had a control group consisting of 32 subjects which had alternative colorectal diseases including HNPCC (n = 30), hyperplastic polyposis (n = 1) and Peutz-Jeghers syndrome (n = 1).13 Olea et al, had an age difference of greater than 20-years between his control and FAP groups.16 Further general limitations across studies included sampling errors, systematic bias with single examiners and relatively small sample sizes less than 100 participants. The low sample sizes may have been influenced by the rare nature of FAP. Variation in reported data and methods of assessment by different authors made the collated data more difficult to standardise. We conducted our systematic literature review with pre-determined research objectives, which were addressed in the majority of studies, but not all. Some studies only reported FAP-associated CHRPE prevalence and incidence without providing sensitivity or specificity data. All quantitative analysis of data was assessed by a single examiner which was further checked and verified by two other independent researchers.
Implementation of our screening recommendations in clinical practice would be a reasonable progression of the research presented. Applying these guidelines through a FAP registry, or equivalent, with quantitative feedback would aid in its appraisal and refinement. There is further potential that FAP-associated CHRPE assessment could be an area augmented by artificial intelligence (AI). There has been significant utility of AI currently incorporated in the screening of other retinal disease, most notably diabetic retinopathy. The ability to accurately localise, quantify and correlate pathological features to a pre-set algorithm would be a powerful tool. Incorporating the FAP-associated CHRPE subclassification system and high-risk features presented into such an algorithm may be of great clinical benefit. It would allow more efficient FAP-associated CHRPE assessment, particularly in non-specialist primary care areas where ophthalmic equipment is deficient or unavailable.
Conclusion
CHPRE is a rapid, non-invasive and specific phenotypical marker for FAP. Multiple lesion number and bilateralism are CHRPE features with the greatest specificity for FAP. Combining ophthalmic assessment with endoscopy and genetic testing has the greatest accuracy when assessing at-risk individuals. Our screening recommendations have been established to aid clinicians in FAP-associated CHRPE assessment. In kindreds carrying the trait, a positive eye examination has predictive value in familial polyposis diagnosis and could further facilitate patient management and genetic counselling.
Acknowledgments
We acknowledge the contributions of Prof. R. Max Conway and Dr. Li-Anne Lim, consultant ophthalmologists from the Sydney Eye Hospital and Save Sight Institute, affiliated with the University of Sydney, New South Wales, Australia. Prof. Conway and Dr. Lim have provided academic guidance and supervision throughout the completion of this body of work. They are recognised as contributory authors of this systematic literature review and screening recommendation.
Funding
No external funding received.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Baba S, Tsuchiya M, Watanabe I, Machida H. Importance of retinal pigmentation as a subclinical marker in familial adenomatous polyposis. Dis Colon Rectum. 1990;33(8):660–664.
2. Rehan S, Kyaw A. In patients with positive family history of familial adenomatous polyposis can the condition be diagnosed from the presence of congenital hypertrophy of the retinal pigment epithelium detected via an eye examination: a systematic review. Clin Exp Ophthalmol. 2020;48(1):98–116.
3. Leggett B, Poplawski B, Pachter N, Rosty C, Norton I, Rosty C. Cancer Council Australia Colorectal Cancer Guidelines. Colorectal Cancer/Familial Adenomatous Polyposis: Clinical Practice Guidelines for the Prevention, Early Detection and Management of Colorectal Cancer. [Internet]. Sydney: Cancer Council Australia; 2017. Available from: https://wiki.cancer.org.au/australia/Guidelines:Colorectal_cancer/Familial_adenomatous_polyposis#cite_note-Citation:Cancer_Institute_NSW_2016-1.
4. Mirchils G, Tejpar S, Thoelen R, et al. Large deletions of the APC gene in 15 percent of mutation-negative patients with classical polyposis (FAP): a Belgian study. Hum Mutat. 2005;25(2):125.
5. National Health and Medical Research Council. NHMRC Levels of Evidence and Grades for Recommendation for Developers of Guidelines. Canberra: Australian Government, National Health and Medical Research Council; 2009.
6. Mirinezshad S, Mousavi F, Baghri M, Sepehri B, Ghavidel A, Ghojazadeh M. Congenital hypertrophy of the retinal pigment epithelium for diagnosis of familial adenomatous polyposis – the first FAP registry in Iran. Asian Pac J Cancer Prev. 2018;19(1):167–169.
7. Nusliha A, Dalpatadu U, Amarasinghe B, Chandrasinghe P, Deen K. Congenital hypertrophy of the retinal pigment epithelium (CHRPE) in patients with familial adenomatous polyposis (FAP): a polyposis registry experience. BMC Res Notes. 2014;7:734.
8. Chen C, Phillips K, Grist S. Congenital hypertrophy of the retinal pigment epithelium (CHRPE) in familial colorectal cancer. Fam Cancer. 2006;5(4):397–404.
9. Tourino R, Conde-Freire R, Cabezas-Agricola J, et al. Value of the congenital hypertrophy of the retinal pigment epithelium in the diagnosis of familial adenomatous polyposis. Int Ophthalmol Clin. 2004;25(2):101–112.
10. Rushwurm I, Zehetmayer M, Dejaco C, Wolf B, Karner-Hanusch J. Ophthalmic and genetic screening in pedigrees with familial adenomatous polyposis. Am J Ophthalmol. 1998;125(5):680–686.
11. Gelisken O, Yucel A, Guler K, Zorluoglu A. Ocular findings in familial adenomatous polyposis. Int Ophthalmol. 1998;21(4):205–208.
12. Lam D, Kwok S, Kwok A, Liew C, Lau J, Pang C. Incidence and predictive value of congenital hypertrophy of retinal pigment epithelium in Chinese familial adenomatous polyposis patients. Chin Med J (Engl). 1998;111(3):278–281.
13. Tiret A, Taiel-Sartral M, Tiret E, Laroche L. Diagnostic value of fundus examination in familial adenomatous polyposis. Br J Ophthalmol. 1997;81(9):755–758.
14. Traboulsi E, Apostolides J, Giardiello F, et al. Pigmented ocular fundus lesions and APC mutations in familial adenomatous polyposis. Ophthalmic Genet. 1996;17(4):167–174.
15. Rossato M, Rigotti M, Grazia M, Turco AE, Bonomi L. Congenital hypertrophy of the retinal pigment epithelium (CHRPE) and familial adenomatous polyposis (FAP). Acta Ophthalmol Scand. 1996;74(4):338–342.
16. Olea J, Mateos J, Llompart A, Obrador A. Frequency of congenital hypertrophy of the retinal pigment epithelium in familial adenomatous polyposis. Acta Ophthalmol Scand. 1996;74(1):48–50.
17. Valanzano R, Cama A, Volpe R, et al. Congenital hypertrophy of the retinal pigment epithelium in familial adenomatous polyposis; novel criteria of assessment and correlations with constitutional adenomatous polyposis coli gene mutations. Cancer. 1996;78(1):2400–2410.
18. Hunt L, Robinson M, Hugkulstone C, et al. Congenital hypertrophy of the retinal pigment epithelium and mandibular osteomata as markers in familial colorectal cancer. Br J Cancer. 1994;70(1):173–176.
19. Hodgson S, Bishop D, Jay B. Genetic heterogeneity of congenital hypertrophy of the retinal pigment epithelium (CHRPE) in families with familial adenomatous polyposis. J Med Genet. 1994;31(1):44–48.
20. Campbell W, Spence R, Parks T. The role of congenital hypertrophy of the retinal pigment epithelium in screening for familial adenomatous polyposis. Int J Colorect Dis. 1994;9:191–196.
21. Chagas C, Fidalgo P, Marins A, et al. Vale of congenital hypertrophy of the retinal pigment epithelium as diagnostic marker in familial adenomatous polyposis. Acta Med Port. 1993;6(7):303–306.
22. Hickey-Dwyer M, Willoughby C. Assessment of the value of congenital hypertrophy of the retinal pigment epithelium as an ocular marker for familial adenomatous polyposis coli. Eye (Lond). 1993;7:562–564.
23. Bertario L, Russo A, Sala P, et al. Multiple approach to the exploration of genotype-phenotype correlations in familial adenomatous polyposis. J Clin Oncol. 2003;21(9):1698–16707.
24. Romania A, Zakov Z, Church J, Jagelman D. Retinal pigment epithelium lesions as a biomarker of disease in patients with familial adenomatous polyposis. A follow-up report. Ophthalmology. 1992;99(6):911–913.
25. Morton D, Gibson J, Macdonald F, et al. Role of congenital hypertrophy of the retinal pigment epithelium in the predictive diagnosis of familial adenomatous polyposis. Br J Surg. 1992;79(7):689–693.
26. Moore A, Maher E, Koch D, Charles S. Incidence and significance of congenital hypertrophy of the retinal pigment epithelium (CHRPE) in familial adenomatous polyposis coli (FAPC). Ophthalmic Paediatr Genet. 1992;13(6):67–71.
27. Giardiello F, Offerhaus G, Traboulsi E, et al. Value of combined phenotypic markers in identifying inheritance of familial adenomatous polyposis. Gut. 1991;32(10):1170–1174.
28. Polkinghorne P, Ritchie S, Neale K, Schoeppner G, Thomson J, Jay B. Pigmented lesions of the retinal pigment epithelium and familial adenomatous polyposis. Eye (Lond). 1990;4:216–221.
29. Iwama T, Mishima Y, Okamoto N, Inoue J. Association of congenital hypertrophy of the retinal pigment epithelium with familial adenomatous polyposis. Br J Surg. 1990;77(3):273–276.
30. Heyen F, Jagelman D, Romania A, et al. Predictive value of congenital hypertrophy of the retinal pigment epithelium as a clinical marker for familial adenomatous polyposis. Colon Rectum. 1990;33:1003–1008.
31. Chapman P, Church W, Burn J, Gunn A. Congenital hypertrophy of retinal pigment epithelium: a sign of familial adenomatous polyposis. BMJ. 1989;298(6670):353–354.
32. Berk T, Cohen Z, Mcleod RS, Parker JA. Congenital hypertrophy of the retinal pigment epithelium as a marker for familial adenomatous polyposis. Dis Colon Rectum. 1988;31(4):253–257.
33. Hennessy M, Collins F, Coroneo M. The distinction between multiple retinal pigment epithelial hamartomata (MRPEH) in familial adenomatous polyposis (FAP) and congenital hypertrophy of the retinal pigment epithelium (CHRPE). Aust N Z J Ophthalmol. 1993;21(4):275–276.
34. Shields J, Shields C. Tumors and Related Lesions of the Pigment Epithelium.
35. Kiernan D, Ortiz-Morales G, Stuart K, Bhagat N, Lim J. Congenital Hypertrophy of the Retinal Pigment Epithelium. American Academy of Ophthalmology. Eyewiki; 2021. Available from: https://eyewiki.aao.org/Congenital_Hypertrophy_of_the_Retinal_Pigment_Epithelium.
36. Chung DC, Rodgers LH Clinical manifestations and diagnosis of familial adenomatous polyposis; 2020. Available from: https://www.uptodate.com/contents/clinical-manifestations-and-diagnosis-of-familial-adenomatous-polyposis?Search=familial%20adenomatous%20polyposis&topicref=2485&source=see_link.
37. Nielsen M, Infante E, Brand R MUTYH Polyposis. GeneReviews; 2021. Available from: https://www.ncbi.nlm.nih.gov/books/NBK107219/.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.