Back to Journals » Pediatric Health, Medicine and Therapeutics » Volume 7
Congenital central hypoventilation syndrome: diagnostic and management challenges
Authors Kasi A, Perez I, Kun S, Keens T
Received 18 March 2016
Accepted for publication 24 June 2016
Published 18 August 2016 Volume 2016:7 Pages 99—107
DOI https://doi.org/10.2147/PHMT.S95054
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Laurens Holmes, Jr
Ajay S Kasi,1 Iris A Perez,1,2 Sheila S Kun,1 Thomas G Keens1,2
1Division of Pediatric Pulmonology and Sleep Medicine, Children’s Hospital Los Angeles, 2Keck School of Medicine of the University of Southern California, Los Angeles, CA, USA
Abstract: Congenital central hypoventilation syndrome (CCHS) is a rare genetic disorder with failure of central control of breathing and of the autonomic nervous system function due to a mutation in the paired-like homeobox 2B (PHOX2B) gene. Affected patients have absent or negligible ventilatory sensitivity to hypercapnia and hypoxemia, and they do not exhibit signs of respiratory distress when challenged with hypercarbia or hypoxia. The diagnosis of CCHS must be confirmed with PHOX2B gene mutation. Generally, the PHOX2B mutation genotype can aid in anticipating the severity of the phenotype. They require ventilatory support for life. Home assisted ventilation options include positive pressure ventilation via tracheostomy, noninvasive positive pressure ventilation, and diaphragm pacing via phrenic nerve stimulation, but each strategy has its associated limitations and challenges. Since all the clinical manifestations of CCHS may not manifest at birth, periodic monitoring and early intervention are necessary to prevent complications and improve outcome. Life-threatening arrhythmias can manifest at different ages and a normal cardiac monitoring study does not exclude future occurrences leading to the dilemma of timing and frequency of cardiac rhythm monitoring and treatment. Given the rare incidence of CCHS, most health care professionals are not experienced with managing CCHS patients, particularly those with diaphragm pacers. With early diagnosis and advances in home mechanical ventilation and monitoring strategies, many CCHS children are surviving into adulthood presenting new challenges in their care.
Keywords: congenital central hypoventilation syndrome, PHOX2B, home mechanical ventilation, diaphragm pacing, CCHS
Introduction
Congenital central hypoventilation syndrome (CCHS) is a rare genetic disorder of the autonomic nervous system (ANS) due to a mutation in the paired-like homeobox 2B (PHOX2B) gene.1 CCHS patients usually present in the newborn period with apnea, hypoxemia, and hypercapnia without clinical signs of respiratory compromise that occur and are most severe during sleep1 particularly nonrapid eye movement sleep.2 CCHS was first reported in 1970 by Mellins et al3 in an infant with alveolar hypoventilation of central nervous origin in spite of a potent respiratory stimulus, like hypercapnia, after excluding primary diseases of the lungs, heart, thoracic cage, and neuromuscular system. Since that time, over 1,000 cases have been reported worldwide, although the incidence is expected to be higher. The estimated incidence is one in 200,000 live births in France and one in 148,000 in Japan.4,5 Older children and young adults have been identified where the presentation is severe respiratory compromise following general anesthesia or respiratory infections.6,7 With increased awareness of CCHS and early intervention, many CCHS children are now surviving into adulthood. In this review, we will discuss the clinical features, diagnostic and management challenges of patients with CCHS.
Genetics
In 2002, Sritippayawan et al reported a mother–daughter transmission of CCHS suggesting a dominant mode of inheritance.8 In 2003, PHOX2B was found to be the disease defining gene for CCHS. PHOX2B encodes a transcription factor that plays an important role in the early development of the ANS.1 PHOX2B normally contains a repeat sequence of 20 alanines in exon 3. The majority of CCHS patients have increased polyalanine repeat mutations (PARMs) in exon 3 of the PHOX2B gene in the range of 24–33.1,9 Approximately 10% have nonpolyalanine repeat mutations (NPARMs), that include missense, nonsense, or frameshift mutations. Different mutations in the PHOX2B gene lead to different levels and mechanisms of cellular dysfunction, which have implications for the severity of an individual CCHS patient’s phenotype. In general, milder disease courses are expected with fewer PARMs (20/25 or 20/26), and are not usually associated with Hirschsprung’s disease or neural crest tumors. Patients with genotypes 20/27–20/33 usually require continuous ventilatory support. The majority of NPARMs occur de novo and have been associated with severe phenotypes with Hirschsprung’s disease, need for continuous ventilatory support, and increased tumor risk in those over 1 year of age.1 However, the authors follow patients with 20/27 PARM through 20/33 and NPARM PHOX2B genotype who are ventilator-dependent only during sleep and without associated neural crest tumors.10 The 20/24 and 20/25 PARM genotypes and some of the NPARMs may be found in the germline of asymptomatic parents of CCHS patients. These findings indicate that these mutations are inherited in an autosomal dominant pattern, but with incomplete penetrance. Twins and siblings in addition to parent–child transmission can have variable presentations of the disease. Thus it is important to test both parents of an affected child for the PHOX2B mutations.
Pathophysiology
To sustain adequate ventilation, ventilatory muscle power and central respiratory drive must be sufficient to overcome the respiratory load (Figure 1A). In CCHS, the central drive is decreased, more during sleep than wakefulness. Thus, the balance is tipped to the left. Central respiratory drive is inadequate to overcome the respiratory load, and inadequate ventilation or respiratory failure will result (Figure 1B). The physiologic ventilatory control abnormality in CCHS appears to be in the integration of chemoreceptor input to central ventilatory controllers, rather than abnormalities in the chemoreceptors themselves.11
 | Figure 1 The respiratory balance. Notes: (A) In order to sustain adequate ventilation, ventilatory muscle power and central respiratory drive need to be sufficient to overcome the respiratory load. Normally, this balance is tipped to the right, indicating adequate ventilation. (B) In CCHS, central respiratory drive is decreased, and thus it is inadequate to overcome the respiratory load. This balance is tipped to the left, and respiratory failure results. Abbreviation: CCHS, congenital central hypoventilation syndrome. |
The level of physical activity affects ventilation in CCHS.12,13 Shea et al showed that CCHS patients decrease ventilation when not moving, such as with reading, solving arithmetic problems, or playing video games.14 Silvestri et al showed severe gas exchange abnormalities in full-time ventilator-dependent CCHS patients during moderate exercise.15 However, Paton et al and Shea et al showed that exercise induced hyperpnea can occur in CCHS patients who only require ventilatory assistance during sleep.16,17 Paton et al found that these CCHS patients increased minute ventilation and respiratory rate with increasing exercise, but not as much as normal subjects. Gozal et al showed that passive leg movement also increased alveolar ventilation in awake and asleep CCHS children.12 These findings suggest that mechanoreceptors, sensing body movement, can stimulate breathing in CCHS. Thus, many CCHS patients have fewer problems with exercise than might have been predicted.
Clinical presentation
Most CCHS patients present in the neonatal period with apnea or hypercapnia requiring assisted ventilation. Some do not breathe at birth, which may be assumed to be due to perinatal asphyxia. However, these babies do not show global central nervous system depression. Many affected newborn do not have the classic sleep–wakefulness differences. Instead, they appear to have chronic intermittent duskiness, cyanosis, and hypercapnia.18 These symptoms are most apparent in nonrapid eye movement sleep, but are also seen in rapid eye movement sleep and sometimes during wakefulness. As their oxygen saturations fall and carbon dioxide levels increase, affected infants do not increase their respiratory rate or effort and usually do not arouse or appear distressed. Some older infants present with apnea, respiratory arrest upon falling asleep, apparent life-threatening events, or pulmonary hypertension due to prolonged periods of hypoxia or hypercapnia. Due to a lack of characteristic clinical response to hypoxia and hypercapnia, that stimulates the respiratory drive, these infants do not display signs of respiratory distress, such as tachypnea, chest retractions, or nasal flaring. Some CCHS patients with milder forms of the disease are not diagnosed until older childhood or adulthood, where a common presentation is an adverse reaction to general anesthesia or pneumonia such as prolonged apnea or hypoventilation.1
Alveolar hypoventilation is the hallmark of CCHS.1 CCHS is characterized by hypercapnia and/or hypoxemia, which is worse during sleep than during wakefulness. During both sleep and wakefulness, children with CCHS have absent or negligible ventilatory response to hypercapnia and hypoxemia.19 In addition, they lack arousal responses and sensations of dyspnea in response to hypercapnia and/or hypoxemia.20,21
In a normal person, clinical signs of respiratory compromise include tachypnea, nasal flaring, anxiety, and chest retractions. These signs are dependent on intact ventilatory control. Because CCHS patients have absent objective and subjective responses to hypoxemia and hypercapnia, these clinical signs are absent, and patients can have hypoxemia and hypercapnia without showing any signs of respiratory distress. CCHS patients appear normal even with profound hypercapnia and hypoxemia. The hypoxia and hypercapnia are often only detected at a later stage when severe cyanosis and central nervous system depression have ensued. This discrepancy between the gas exchange abnormalities and clinical presentation make the diagnosis and management of CCHS challenging.
CCHS is considered a generalized disorder of the ANS. Decreased heart rate variability, bradycardia, and transient abrupt asystole have been reported and some CCHS patients may require cardiac pacemaker implantation.1,22,23 Sudden death have been reported due to these life-threatening arrhythmias. Gronli et al showed that CCHS children with sinus pauses of ≥3 seconds are at high risk of life-threatening or fatal cardiac arrhythmias, and may require cardiac pacemaker implantation.23 In general, these are associated with 20/26 and longer PARM and NPARM. There are reports of adult CCHS patients with 20/25 PARM genotype with cardiac rhythm disturbances. This raises concern that individuals with 20/25 PARM genotype, that is considered a milder mutation, may be unaffected during childhood, but may manifest cardiac rhythm disturbances at a later age.1,23 This is an area of ongoing controversy. Currently, we do not know the best way to monitor cardiac status or when to implant cardiac pacemakers.
Suboptimal school performance and/or neurodevelopmental impairment has been reported in CCHS patients as early as preschool and school age.1,24 It is unclear whether this is due to chronic unrecognized hypoxia and/or gene specific dysfunction.1,24 Other ANS dysfunction include glucose metabolism abnormalities, esophageal dysmotility, breath-holding spells, reduced basal body temperature, neuro-ophthalmological abnormalities, and altered perception to anxiety.1,25 Although Hirschsprung’s disease is more prevalent in patients with the NPARM mutations (87%–100% cases) than the PARM mutations (13%–20% cases), it can also occur in PARM patients with 20/27 and above. Tumors of neural crest origin occur more frequently among patients with NPARMs (50%) than among those with PARMs (1%).1
Diagnostic challenges
The diagnosis of CCHS is suspected when patients present with hypoventilation that is worse during sleep, in the absence of primary lung disease, ventilatory muscle weakness, or obvious neurological disorders. The diagnosis is confirmed by testing for mutations in the PHOX2B gene.1,18
When awaiting results of PHOX2B gene testing, other causes of hypoventilation should be ruled out. This evaluation may include: 1) chest radiograph; 2) diaphragm fluoroscopy; 3) electrocardiogram and echocardiogram; 4) magnetic resonance imaging and/or computed tomography scan of brain and brainstem for gross anatomic lesions; 5) tests for metabolic disorders; 6) comprehensive neurological evaluation; and 7) polysomnogram to establish the presence of hypoventilation and sleep-related breathing disorder. Noninvasive monitoring of oxygenation and ventilation by polysomnography is preferred, as intermittent blood gas sampling causes arousal, and therefore it is not an accurate assessment of gas exchange during sleep. Polysomnography is performed as per the American Academy of Sleep Medicine guidelines.26,27 The purpose of polysomnography is to assess the presence of sleep hypoventilation and sleep-related breathing disorders. Profound hypoxia and hypercapnia should be treated according to institutional policies and protocols. The diagnosis of CCHS is suspected when polysomnography shows hypoxia and hypercapnia worse during sleep than during wakefulness, and confirmed by a PHOX2B mutation testing, which is available in several commercial genetic laboratories. Once the diagnosis is made, polysomnography is generally used to assess the adequacy of ventilator settings and need not be repeated during spontaneous breathing. Barium enema and rectal biopsy can be considered in the event of abdominal distension or constipation due to the association with Hirschsprung’s disease.
CCHS can present beyond the newborn period, including later childhood, adolescence, and adulthood. The diagnosis of CCHS in these age groups should be considered in cases of alveolar hypoventilation, apnea, cyanosis, or seizures noted after administration of anesthetics or CNS depressants, or with relatively mild respiratory infections.1 Due to the rare incidence of this disease as well as presentation at a later age, this can be missed by many clinicians who are unaware of CCHS.
Management challenges
The goal of treatment in CCHS is to ensure adequate ventilation when patients are unable to achieve adequate gas exchange during spontaneous breathing, or simply put, to breathe for them. This requires assisted ventilation as no pharmacological respiratory stimulants have been shown to be effective, and they certainly do not prevent the need for ventilatory support.28 In our experience, there are some children with CCHS who require more than just overnight ventilatory support, but are not full-time ventilator-dependent. So the duration of ventilatory support in CCHS patients can be considered a spectrum anywhere between overnight ventilatory support to full-time ventilator dependence.
Children with hypercapnia and hypoxemia are at risk of developing progressive pulmonary hypertension. Supplemental oxygen alone is not sufficient for the treatment of hypoventilation, and it will not prevent pulmonary hypertension. Thus, CCHS patients require lifelong assisted ventilation. As children with CCHS do not have intrinsic severe lung disease, there are several options for assisted ventilatory support at home: 1) positive pressure ventilation (PPV) via tracheostomy; 2) noninvasive positive pressure ventilation (NIPPV); and 3) diaphragm pacing (DP).1 Although negative pressure ventilation can be effective, it is not portable and is now rarely used.
All CCHS patients require assisted ventilation for life at least during sleep. Thus, weaning these patients off ventilatory support should not be attempted. CCHS patients are not like other children on home mechanical ventilation. They must be managed with vigilance due to their absence of objective and subjective responses to hypoxemia and hypercapnia. With acute respiratory infections, children with CCHS may not show a fever, increase their respiratory rate, or dyspnea. The use of pulse oximetry or end-tidal CO2 (PETCO2) monitoring are invaluable aids in addition to the highly skilled and consistent caregivers at home.
PPV via tracheostomy
There are two approaches to the long-term ventilation of infants: PPV via tracheostomy and noninvasive ventilation when possible. There is a lack of current literature at present demonstrating that one mode of ventilation is superior, thus this is an area of needed research. The authors of the ATS statement believe that PPV via tracheostomy in the first 6–8 years of life is associated with better oxygenation, and thus better neurologic development and function, than the use of NIPPV early in life.1
For children receiving PPV via tracheostomy, the authors prefer an uncuffed and relatively smaller tracheostomy tube, because it is less likely to cause tracheomalacia and it allows a large expiratory leak for phonation.29 Although small, uncuffed tracheostomy tubes are associated with leaks, which can be large and variable, the tracheostomy leak can be compensated for by using the ventilator in a pressure-control mode.29 Speaking valves may be used for phonation. It is preferable to initially ventilate infants for 24 h/day to ensure that oxygenation and ventilation remain adequate to optimize neurocognitive outcome. Ventilator settings are adjusted to achieve PETCO2 between 30–35 torr and SpO2 >95%. In our experience, children ventilated with these parameters have fewer complications, and they generally do better clinically. Allowing these children to be hypoxemic and/or hypercapnic defeats the purpose of chronic ventilatory support.
Ventilator settings should be assessed periodically to ensure adequate oxygenation and ventilation. This is best performed in a sleep laboratory. Periodic assessments of the airway and tracheostomy tube size should be conducted by an experienced pediatric otolaryngologist.
Between infancy and school age, some CCHS children gradually develop the ability to breathe adequately during wakefulness. These children, if stable, can be weaned from assisted ventilation while awake. This is best accomplished by sprint weaning. Sprint weaning is performed by removing the child from the ventilator for short periods of time. Between sprints, the child remains on full ventilatory support. Initially, these sprints may only last few minutes and supplemental oxygen may be required. The child must be carefully monitored noninvasively during sprints to prevent hypoxia or hypercapnia. These sprints are always started under medical observation, but may be gradually increased at home. However, CCHS children can never be weaned from ventilatory support during sleep.
Noninvasive positive pressure ventilation
Stable older children who require ventilatory support only during sleep can be ventilated with NIPPV delivered via nasal mask, nasal prongs, or face mask. Continuous positive airway pressure therapy should not be used in CCHS patients because it does not support ventilation. Spontaneous mode NIPPV cannot be used in CCHS because they cannot be trusted to increase their ventilatory rate in response to hypercapnia and hypoxemia on their own. NIPPV is provided via bi-level positive airway pressure or average volume assured pressure support30 in the spontaneous/timed mode or timed mode with appropriate rate. Children with CCHS characteristically have diminutive tidal volumes and monotonous respiratory rates awake and asleep. Only the timed mode guarantees breath delivery in children who cannot generate adequate spontaneous breaths to trigger the ventilator. The spontaneous/timed mode could lead to a variable breathing pattern. Hence the authors use timed mode only.1 Inspiratory positive airway pressure and expiratory positive airway pressure are adjusted independently to achieve an inspiratory positive airway pressure to expiratory positive airway pressure difference that provides optimal tidal volume. Although most CCHS patients do not require supplemental oxygen with their NIPPV, oxygen can be added during acute illness. The high flow of pressurized air with NIPPV can lead to the drying of nasal mucosa which may present as frequent nosebleeds or nasal congestion.31 NIPPV can be used with the attached humidifier to circumvent this problem.32
Mask ventilation has been associated with midface hypoplasia when instituted at an early age.31 NIPPV should not be used routinely outside of sleep time as the mask interferes with daily activities, social interaction, and increases the risk of skin breakdown. PPV via tracheostomy in children may avoid these complications.
During the first few years of life, CCHS patients can be very unstable. In general, NIPPV can be instituted in stable school-aged CCHS patients with a milder phenotype requiring ventilatory support only during sleep. In this group of patients, NIPPV allows for tracheostomy decannulation. When anticipating tracheostomy decannulation, we recommend an upper airway evaluation. Several CCHS centers prefer institution of NIPPV in the first few years of life. Some of the issues that may arise are finding an appropriate interface, prompt institution of NIPPV during daytime naps and development of midface hypoplasia. However, there are no comparative research studies on the effect of early NIPPV on brain development in CCHS.
Periodic assessment of NIPPV settings should be conducted in a sleep laboratory to ensure adequate oxygenation and ventilation.1 Polysomnography for adjustment of NIPPV settings is performed as per the American Academy of Sleep Medicine guidelines.26,33
Diaphragm pacing
DP is an attractive treatment option for some CCHS patients. It allows full-time ventilator-dependent patients to be free of PPV during the day, allowing mobility. In those who are stable and ventilator-dependent only during sleep, DP may permit tracheostomy decannulation.10
DP generates breathing using the child’s own diaphragm as the respiratory pump. A diaphragm pacer system has four components: 1) electrodes that are surgically implanted on the phrenic nerves; 2) receivers that are subcutaneously implanted bilaterally in the abdomen; 3) antennae that are taped on the skin over the subcutaneous receivers when pacing; and 4) a battery operated external transmitter (Figure 2). The external transmitter generates a train of pulses that are transmitted via the external antenna. The subcutaneous receivers convert the radiofrequency signal into an electrical current that is then conducted to the phrenic nerve electrodes. Electrical stimulation of the phrenic nerve electrodes causes diaphragmatic contraction, which generates the breath. The amount of electrical voltage is proportional to the diaphragm contraction or tidal volume. Since DP uses the child’s own diaphragm as a ventilator, an ideal candidate should have normal diaphragm function and intact phrenic nerves. The patient must also have little or no lung disease.
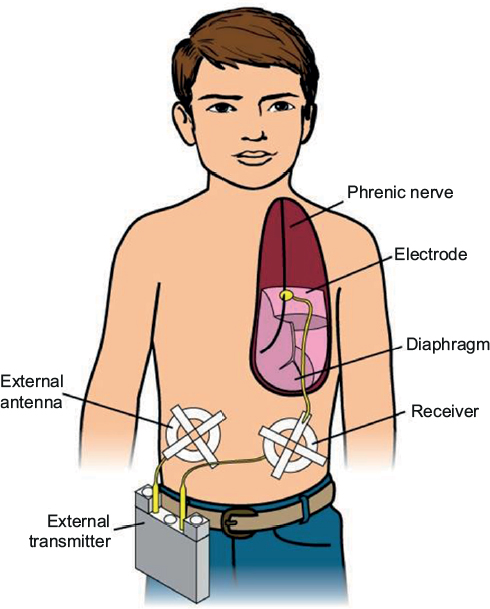 | Figure 2 Diaphragm pacing. Notes: A phrenic nerve electrode is surgically implanted. It is connected by a lead wire to a receiver implanted subcutaneously in the abdomen. A battery operated transmitter transmits electrical energy, via an antenna taped on the skin over the receiver, to the receiver. The receiver converts this to a standard electrical current, which causes contraction of the diaphragm. (Figure by Bill Franz.) |
Bilateral phrenic nerve electrodes are surgically implanted on the phrenic nerves thoracoscopically.34 Once implanted, the pacers are tested intraoperatively, but DP is not used right away. DP is initiated ~4–6 weeks later to allow for healing and stabilization of tissue reaction around the electrodes.35
When DP is used for the first time, we begin with the child awake in a friendly, supportive, and nonthreatening setting. The child is not on the ventilator when on DP. We begin with the lowest setting on each side, and increase the tidal volume settings until a reasonable diaphragm contraction is observed. Continuous pulse oximetry and PETCO2 monitoring are used to objectively assess SpO2 and PETCO2. The authors start pacing at 60–90 min/day because DP can result in fatigue (decreasing diaphragmatic contraction for the same electrical stimulus). The duration of DP is gradually increased by 30 min/day each week. “Aerobic training” of these muscle fibers is required to achieve full pacing. During this period, DP is used for a part of the day/night and baseline ventilator support for the rest of the day/night. With gradual diaphragm training, it takes ~2–3 months to establish full overnight pacing in those who are ventilator-dependent only during sleep.10,35 In general, DP is limited to 12–14 h/day. Diaphragm pacer equipment can probably last indefinitely, but can be damaged and may need to be replaced.36
The greatest benefit of DP is portability and freedom from being tethered to a ventilator circuit in children who are full-time ventilator-dependent. Tracheostomy decannulation can be considered in children who are ventilator-dependent only during sleep after successful overnight DP for at least 3 months. Assessment of upper airway anatomy by a pediatric otolaryngologist and polysomnography with a capped tracheostomy tube should be performed prior to considering decannulation.10 Upper airway obstruction can occur with DP due to the absence of synchronous upper airway skeletal muscle contraction with diaphragm contraction. This can be surpassed by changes in diaphragm pacer settings and other medical management facilitating tracheostomy decannulation.10 Obesity is an obstacle to effective DP as the adipose tissue increases distance between the antenna and the receiver which may result in decreased diaphragm contractions.10 Therefore, we do not recommend DP in obese patients.
Once diaphragm pacers are placed, the patient cannot have magnetic resonance imaging due to the magnetic field. DP causes artifacts in transthoracic impedance-based cardiorespiratory monitors, and should not be used at home. DP may not provide enough ventilatory support if CCHS patients become acutely ill or have major surgical procedures and these children may need other ventilatory support on a temporary basis in these situations. DP in children with CCHS has been established as an effective method of mechanically assisted ventilation, and allows for maximum mobility. In our center, we have CCHS patients who have been using DP for 40 years without phrenic nerve or diaphragm damage.
Periodic noninvasive assessment and titration of diaphragm pacer settings should be conducted in a sleep lab to ensure adequate oxygenation and ventilation. This can be a challenge, since many sleep laboratories do not monitor PETCO2 and do not have experience adjusting diaphragm pacers. CCHS centers with expertise in DP are available for management or consultation of these patients.
Other challenges
Since CCHS patients lack the normal physiological responses to hypoxemia and hypercapnia, noninvasive monitoring of oxygenation and ventilation using pulse oximetry and PETCO2 monitoring are ideal in addition to skilled nursing for ventilator management.1 However, our center has had difficulty obtaining PETCO2 monitors for all patients due to the expense and insurance authorization process. Apnea/bradycardia monitor alone is insufficient, as many patients hypoventilate but are not apneic. In children who use DP, the pacer artifacts will interfere with heart rate and respiratory rate monitors.1
Pulmonary hypertension and cor pulmonale can occur in children with CCHS as a result of recurrent hypoxemia due to inadequate ventilator settings, unrecognized hypoventilation during spontaneous breathing when awake, or suboptimal compliance with assisted ventilation. Therefore, annual polysomnography should be performed to rule out hypoventilation. Echocardiogram should be performed periodically to evaluate for pulmonary hypertension and cor pulmonale.1
Children with CCHS exhibit an increased frequency of arrhythmias, particularly sinus bradycardia and transient asystole which can result in sudden death.23 These cardiac rhythm disturbances may manifest at different ages and even in adulthood. Extended cardiac monitoring performed periodically may detect cardiac rhythm disturbances.1 Cardiac pacemaker and diaphragm pacer have been successfully used together.37 CCHS patients can have alterations in blood pressure due to autonomic dysfunction. CCHS patients can have ophthalmologic abnormalities.25 Therefore, eye examination should be performed when indicated to allow for intervention and avoid interference with learning. Serial chest and abdominal imaging is recommended for children with NPARM and with PARM 20/29–20/33 genotype for neural crest tumor surveillance. However, there is no consensus on when or how often to perform these studies.
Children with CCHS, with or without a tracheostomy may swim only under close supervision. Competitive underwater swimming contests is discouraged as these children may not perceive the asphyxia or dyspnea and are at increased risk of drowning.
Sedative medications and central nervous system depressants worsen hypoventilation. Therefore, CCHS patients should be on full-time ventilatory support during surgery and postoperatively.
Upper respiratory infections in younger patients, which are otherwise mild, can cause ventilatory control to deteriorate temporarily during the infection. Thus, increased ventilator settings and/or increased time of assisted ventilation is frequently required. In children with CCHS, the most common problem is inadequate ventilation, which occurs both during sleep and wakefulness. If a child with CCHS has any explained problem such as seizure or lethargy, they should be stabilized with hyperventilation with 100% oxygen until the source of the problem can be identified. The etiology of any such problem is likely to be hypoventilation until proven otherwise. A brief period of hyperventilation will not be harmful, but may be lifesaving if the child has hypoventilation.
Many CCHS patients do not respond to ventilator alarms, and therefore may not be able to live alone. It may be necessary for another person knowledgeable about CCHS, to be available to assist the patient in the event of ventilator malfunction or disconnection.
Challenges in the management of adolescents and young adults
Due to improved outcomes and survival of CCHS patients into adolescence and adulthood, they are faced with the normal adolescent temptations of alcohol and drug abuse. Alcohol and many illicit substances are known respiratory depressants. Drinking alcohol at parties has been associated with coma and death in CCHS patients.38 At our center, we start counseling CCHS children to abstain from alcohol use starting at 12 years of age. CCHS patients cannot count on their friends at parties to help them. They may not know that a person has CCHS; and even if they do, they will likely not know the implications and the need for ventilatory support. Further, if a CCHS patient has been drinking to the point of passing out, his friends are likely also impaired, and would not have adequate judgment to act. Hence, adolescent CCHS patients should be counseled regarding alcohol and drug abstinence at every clinic visit.
Due to improved survival of CCHS patients into adulthood, there is a need for adult health care professionals with expertise in the management of CCHS and DP. Due to the rare incidence of CCHS, many health care professionals are not experienced with managing CCHS patients, particularly with diaphragm pacers. It has been a challenge to educate the majority of health care professionals about the proper management of CCHS patients.
Pregnancy may decrease ventilation due to the increased respiratory load from an enlarging uterus. Therefore, frequent monitoring to assess for adequate oxygenation and ventilation during both sleep and wakefulness is recommended in pregnancy. This is especially important in patients utilizing DP. We have successfully used DP in three-term pregnancies of two CCHS mothers without compromised ventilation.8 If a cesarean section delivery is planned in a pregnant woman with CCHS using DP, arrangements should be made to use NIPPV in the postpartum period, as DP is painful and will not be tolerated after an abdominal incision.
As CCHS is inherited in an autosomal dominant manner, the delivery should be planned in a tertiary care center prepared for intubation and mechanical ventilation of the newborn. We recommend prenatal testing for CCHS in the fetus to plan for the delivery accordingly.
There is a need to create a centralized national and international database of CCHS patients to gain a deeper insight into the genotypes, phenotypes, and long-term outcomes. This would also aid in creating a forum to address the issues of young adults with CCHS, for peer-to-peer interaction and networking opportunities.
Outcome
CCHS is a lifelong disorder. With early detection and modern techniques for home ventilation and monitoring, most children with CCHS have prolonged survival into adolescence and adulthood with a good quality of life.39 As more children with CCHS are surviving into adulthood, there is a necessity for establishing care with adult health care professionals experienced in managing CCHS.
Disclosure
The authors report no conflicts of interest in this work.
References
Weese-Mayer DE, Berry-Kravis EM, Ceccherini I, Keens TG, Loghmanee DA, Trang H; ATS Congenital Central Hypoventilation Syndrome Subcommittee. An official ATS clinical policy statement: congenital central hypoventilation syndrome – genetic basis, diagnosis, and management. Am J Respir Crit Care Med. 2010;181(6):626–644. | ||
Fleming PJ, Cade D, Bryan MH, Bryan AC. Congenital central hypoventilation and sleep state. Pediatrics. 1980;66(3):425–428. | ||
Mellins RB, Balfour HH Jr, Turino GM, Winters RW. Failure of automatic control of ventilation (Ondine’s curse). Report of an infant born with this syndrome and review of the literature. Medicine (Baltimore). 1970;49:487–504. | ||
Trang H, Dehan M, Beaufils F, Zaccaria I, Amiel J, Gaultier C; French CCHS Working Group. The French Congenital Central Hypoventilation Syndrome Registry: general data, phenotype, and genotype. Chest. 2005;127(1):72–79. | ||
Shimokaze T, Sasaki A, Meguro T, et al. Genotype–phenotype relationship in Japanese patients with congenital central hypoventilation syndrome. J Hum Genet. 2015;60(9):473–477. | ||
Mahmoud M, Bryan Y, Gunter J, Kreeger RN, Sadhasivam S. Anesthetic implications of undiagnosed late onset central hypoventilation syndrome in a child: from elective tonsillectomy to tracheostomy. Paediatr Anaesth. 2007;17(10):1001–1005. | ||
Mahfouz AKM, Rashid M, Khan MS, Reddy P. Late onset congenital central hypoventilation syndrome after exposure to general anesthesia. Can J Anaesth. 2011;58(12):1105–1109. | ||
Sritippayawan S, Hamutcu R, Kun SS, Ner Z, Ponce M, Keens TG. Mother–daughter transmission of congenital central hypoventilation syndrome. Am J Respir Crit Care Med. 2002;166(3):367–369. | ||
Perez IA, Keens TG. Peripheral chemoreceptors in congenital central hypoventilation syndrome. Respir Physiol Neurobiol. 2013;185(1):186–193. | ||
Diep B, Wang A, Kun S, et al. Diaphragm pacing without tracheostomy in congenital central hypoventilation syndrome patients. Respiration. 2015;89(6):534–538. | ||
Spengler CM, Gozal D, Shea SA. Chemoreceptive mechanisms elucidated by studies of congenital central hypoventilation syndrome. Respir Physiol. 2001;129(1–2):247–255. | ||
Gozal D, Marcus CL, Ward SL, Keens TG. Ventilatory responses to passive leg motion in children with congenital central hypoventilation syndrome. Am J Respir Crit Care Med. 1996;153(2):761–768. | ||
Gozal D, Simakajornboon N. Passive motion of the extremities modifies alveolar ventilation during sleep in patients with congenital central hypoventilation syndrome. Am J Respir Crit Care Med. 2000;162(5):1747–1751. | ||
Shea SA, Andres LP, Paydarfar D, Banzett RB, Shannon DC. Effect of mental activity on breathing in congenital central hypoventilation syndrome. Respir Physiol. 1993;94(3):251–263. | ||
Silvestri JM, Weese-Mayer DE, Flanagan EA. Congenital central hypoventilation syndrome: cardiorespiratory responses to moderate exercise, simulating daily activity. Pediatr Pulmonol. 1995;20(2):89–93. | ||
Paton JY, Swaminathan S, Sargent CW, Hawksworth A, Keens TG. Ventilatory response to exercise in children with congenital central hypoventilation syndrome. Am Rev Respir Dis. 1993;147(5):1185–1191. | ||
Shea SA, Andres LP, Shannon DC, Banzett RB. Ventilatory responses to exercise in humans lacking ventilatory chemosensitivity. J Physiol. 1993;468:623–640. | ||
Chen ML, Keens TG. Congenital central hypoventilation syndrome: not just another rare disorder. Paediatr Respir Rev. 2004;5(3):182–189. | ||
Paton JY, Swaminathan S, Sargent CW, Keens TG. Hypoxic and hypercapnic ventilatory responses in awake children with congenital central hypoventilation syndrome. Am Rev Respir Dis. 1989;140(2):368–372. | ||
Shea SA, Andres LP, Shannon DC, Guz A, Banzett RB. Respiratory sensations in subjects who lack a ventilatory response to CO2. Respir Physiol. 1993;93(2):203–219. | ||
Marcus CL, Bautista DB, Amihyia A, Ward SL, Keens TG. Hypercapneic arousal responses in children with congenital central hypoventilation syndrome. Pediatrics. 1991;88(5):993–998. | ||
Woo MS, Woo MA, Gozal D, Jansen MT, Keens TG, Harper RM. Heart rate variability in congenital central hypoventilation syndrome. Pediatr Res. 1992;31(3):291–296. | ||
Gronli JO, Santucci BA, Leurgans SE, Berry-Kravis EM, Weese-Mayer DE. Congenital central hypoventilation syndrome: PHOX2B genotype determines risk for sudden death. Pediatr Pulmonol. 2008;43(1):77–86. | ||
Charnay AJ, Antisdel-Lomaglio JE, Zelko FA, et al. Congenital central hypoventilation syndrome: neurocognition already reduced in preschool-aged children. Chest. 2016;149(3):809–815. | ||
Patwari PP, Stewart TM, Rand CM, et al. Pupillometry in congenital central hypoventilation syndrome (CCHS): quantitative evidence of autonomic nervous system dysregulation. Pediatr Res. 2012;71(3):280–285. | ||
Iber C, Ancoli-Israel S, Chesson A, Quan SF. The AASM Manual for the Scoring of Sleep and Associated Events: Rules, Terminology and Technical Specifications. 1st ed. Illinois: American Academy of Sleep Medicine; 2007. | ||
Berry RB, Budhiraja R, Gottlieb DJ, et al. Rules for scoring respiratory events in sleep: update of the 2007 AASM Manual for the Scoring of Sleep and Associated Events. Deliberations of the Sleep Apnea Definitions Task Force of the American Academy of Sleep Medicine. J Clin Sleep Med. 2012;8(5):597–619. | ||
Straus C, Similowski T. Congenital central hypoventilation syndrome and desogestrel: a call for caution: addendum to “C. Straus, H. Trang, M.H. Becquemin, P. Touraine, T. Similowski, Chemosensitivity recovery in Ondine’s curse syndrome under treatment with desogestrel” [Respir. Physiol. Neurobiol. 171 (2010) 171–174]. Respir Physiol Neurobiol. 2011;178(2):357–358. | ||
Keens TG, Kun SS, Ward SLD. Chronic respiratory failure. In: Shaffner DH, Nichols DG, editors. Roger’s Textbook of Pediatric Intensive Care. 5th ed. Alphen aan den Rijn, the Netherlands: Wolters Kluwer; 2015:794–807. | ||
Vagiakis E, Koutsourelakis I, Perraki E, Roussos C, Mastora Z, Zakynthinos S, Kotanidou A. Average volume-assured pressure support in a 16-year-old girl with congenital central hypoventilation syndrome. J Clin Sleep Med. 2010;6(6):609–612. | ||
Amin R, Al-Saleh S, Narang I. Domiciliary noninvasive positive airway pressure therapy in children. Pediatr Pulmonol. 2016;51(4):335–348. | ||
Nilius G, Franke KJ, Domanski U, Schroeder M, Ruhle KH. Effect of APAP and heated humidification with a heated breathing tube on adherence, quality of life, and nasopharyngeal complaints. Sleep Breath. 2016;20(1):43–49. | ||
Berry RB, Chediak A, Brown LK, et al; NPPV Titration Task Force of the American Academy of Sleep Medicine. Best clinical practices for the sleep center adjustment of noninvasive positive pressure ventilation (NPPV) in stable chronic alveolar hypoventilation syndromes. J Clin Sleep Med. 2010;6(5):491–509. | ||
Shaul DB, Danielson PD, McComb JG, Keens TG. Thoracoscopic placement of phrenic nerve electrodes for diaphragmatic pacing in children. J Pediatr Surg. 2002;37(7):974–978. | ||
Chen ML, Tablizo MA, Kun S, Keens TG. Diaphragm pacers as a treatment for congenital central hypoventilation syndrome. Expert Rev Med Devices. 2005;2(5):577–585. | ||
Nicholson KJ, Nosanov LB, Bowen KA, Kun SS, Perez IA, Keens TG, Shin CE. Thoracoscopic placement of phrenic nerve pacers for diaphragm pacing in congenital central hypoventilation syndrome. J Pediatr Surg. 2015;50(1):78–81. | ||
Kolb C, Eicken A, Zrenner B, Schmitt C. Cardiac pacing in a patient with diaphragm pacing for congenital central hypoventilation syndrome (Ondine’s curse). J Cardiovasc Electrophysiol. 2006;17(7):789–791. | ||
Chen ML, Turkel SB, Jacobson JR, Keens TG. Alcohol use in congenital central hypoventilation syndrome. Pediatr Pulmonol. 2006;41(3):283–285. | ||
Marcus CL, Jansen MT, Poulsen MK, Keens SE, Nield TA, Lipsker LE, Keens TG . Medical and psychosocial outcome of children with congenital central hypoventilation syndrome. J Pediatr. 1991;119(6):888–895. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.