Back to Journals » Therapeutics and Clinical Risk Management » Volume 19
Congenital Athymia: Unmet Needs and Practical Guidance
Authors Howley E ![]() , Davies EG, Kreins AY
, Davies EG, Kreins AY ![]()
Received 14 December 2022
Accepted for publication 4 March 2023
Published 13 March 2023 Volume 2023:19 Pages 239—254
DOI https://doi.org/10.2147/TCRM.S379673
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Evey Howley,1 E Graham Davies,1 Alexandra Y Kreins1,2
1Department of Immunology and Gene Therapy, Great Ormond Street Hospital for Children NHS Foundation Trust, London, UK; 2Infection, Immunity and Inflammation Research & Teaching Department, University College London, London, UK
Correspondence: Alexandra Y Kreins, Email [email protected]
Abstract: Inborn errors of thymic stromal cell development and function which are associated with congenital athymia result in life-threatening immunodeficiency with susceptibility to infections and autoimmunity. Athymic patients can be treated by thymus transplantation using cultured donor thymus tissue. Outcomes in patients treated at Duke University Medical Center and Great Ormond Street Hospital (GOSH) over the past three decades have shown that sufficient T-cell immunity can be recovered to clear and prevent infections, but post-treatment autoimmune manifestations are relatively common. Whilst thymus transplantation offers the chance of long-term survival, significant challenges remain to optimise the outcomes for the patients. In this review, we will discuss unmet needs and offer practical guidance based on the experience of the European Thymus Transplantation programme at GOSH. Newborn screening (NBS) for severe combined immunodeficiency (SCID) and routine use of next-generation sequencing (NGS) platforms have improved early recognition of congenital athymia and increasing numbers of patients are being referred for thymus transplantation. Nevertheless, there remain delays in diagnosis, in particular when the cause is genetically undefined, and treatment accessibility needs to be improved. The majority of athymic patients have syndromic features with acute and chronic complex health issues, requiring life-long multidisciplinary and multicentre collaboration to optimise their medical and social care. Comprehensive follow up after thymus transplantation including monitoring of immunological results, management of co-morbidities and patient and family quality-of-life experience, is vital to understanding long-term outcomes for this rare cohort of patients. Alongside translational research into improving strategies for thymus replacement therapy, patient-focused clinical research will facilitate the design of strategies to improve the overall care for athymic patients.
Keywords: athymia, thymus transplantation, immunodeficiency, DiGeorge syndrome, SCID, rare diseases, quality of life
Introduction
The thymus is a lymphoid organ in which bone marrow-derived T-cell progenitors complete their maturation into functional, self-tolerant T-cells. Congenital athymia, due to complete DiGeorge syndrome (cDGS) or rare monogenic disorders affecting thymus development, is associated with profound T-cell lymphopaenia and susceptibility to opportunistic infections.1 Left untreated, congenital athymia is incompatible with long-term survival. It can be treated successfully by thymus transplantation, using cultured postnatal thymic tissue obtained from infant donors in whom a partial or total thymectomy is necessary during cardiac surgery. This technique was pioneered at Duke University Hospital and more than 100 patients have been treated in the United States of America (USA).2 Outside the USA, the only centre offering this treatment, since 2009, is Great Ormond Street Hospital (GOSH) in London, United Kingdom (UK).3 The procedure involves lymphodepletion of donor thymus tissue over a culture period of 2–3 weeks, after which the cultured thymus tissue is implanted bilaterally into the quadriceps muscles of athymic recipients.4 Immune reconstitution starts around 6 to 9 months after thymus transplantation after recipient-derived precursor T-cells repopulate the donor thymus tissue and undergo thymopoiesis. This T-cell maturation process is mainly dependent on lympho-stromal crosstalk mediated by interactions between T-cell receptors (TCR) on developing T-cell progenitors and human leukocyte antigens (HLA) on antigen-presenting cells (APC).1 The key APCs in normal thymus tissue are thymic epithelial cells (TEC), therefore it remains unclear how, in the absence of tissue type matching between donors and recipients, thymopoiesis is supported in thymic allografts (this has been discussed elsewhere)5,6 Nevertheless, successful recovery of T-cell immunity has been reported after thymus transplantation, testifying to the ability of these HLA-mismatched thymic allografts to support T-cell development with the generation of self-tolerant naïve T-cells showing a diverse TCR repertoire.2,3
More than 150 patients with congenital athymia have been transplanted to date across both centres, including 105 patients in the USA between 1993 and 2020 and 12 patients in the UK between 2009 and 20142,3 (and unreported from GOSH Thymus Transplantation programme). For both cohorts, a similar overall survival of around 75% has been reported. Mortality is closely related to pre-existing infections and is usually seen in the first year after transplantation whilst awaiting immune reconstitution. In the remaining patients, T-cell counts, including counts of naïve T-cells, have been reported to steadily increase over a duration of 2 years after thymus transplantation.2,3 Longer term immune reconstitution results have only been reported in a small number of patients, with T-cell counts seeming to stabilise after peaking during this initial 2-year follow up.3,7 Though the absolute T-cell counts achieved usually remain below those seen in age-matched healthy individuals, the levels are sufficient to clear both pre-existing infections and newly acquired infections.2,3 In addition to discontinuation of the previously required antimicrobial prophylaxis and isolation precautions, the majority of patients no longer require immunoglobulin replacement therapy. They maintain normal immunoglobulin levels and, upon immunisation, have been shown to produce satisfactory antibody titres to tetanus toxoid and, to a lesser extent, to pneumococcal vaccine serotypes.2,3 Autoimmunity has been reported in a significant number of patients after thymus transplantation, including autoimmune cytopaenias which are mostly transient, and autoimmune thyroiditis, as well as other less frequently occurring autoimmune manifestations.2,3 Overall, even though immune reconstitution after thymus transplantation is suboptimal, the procedure is life-saving and has become the standard of care for congenital athymia, resulting in an improved quality of life (QOL) not restricted by infection.2,3
Congenital athymia is a rare condition and patients often have acute and chronic complex care needs, requiring extraordinary specialist care with close collaboration across several teams and centres.8 Understanding the natural history of the condition and comprehensively recording outcomes after thymus transplantation, including the QOL reported by the patients and their families, are crucial to improving the long-term health and social care services available to this cohort of patients. Analysis of the clinical outcomes after thymus transplantation demonstrates the ability of patients to live a near normal life from an immunological perspective, yet significant challenges remain in terms of optimising overall outcomes for many patients with congenital athymia (see Table 1). These include the timely recognition of congenital athymia, treatment accessibility, systematic long-term follow up, patient and family engagement and translation of research into the clinic. In this review, we will discuss the unmet needs of patients with congenital athymia, address the challenges encountered and offer practical guidance based on the experience of the European Thymus Transplantation programme at GOSH.
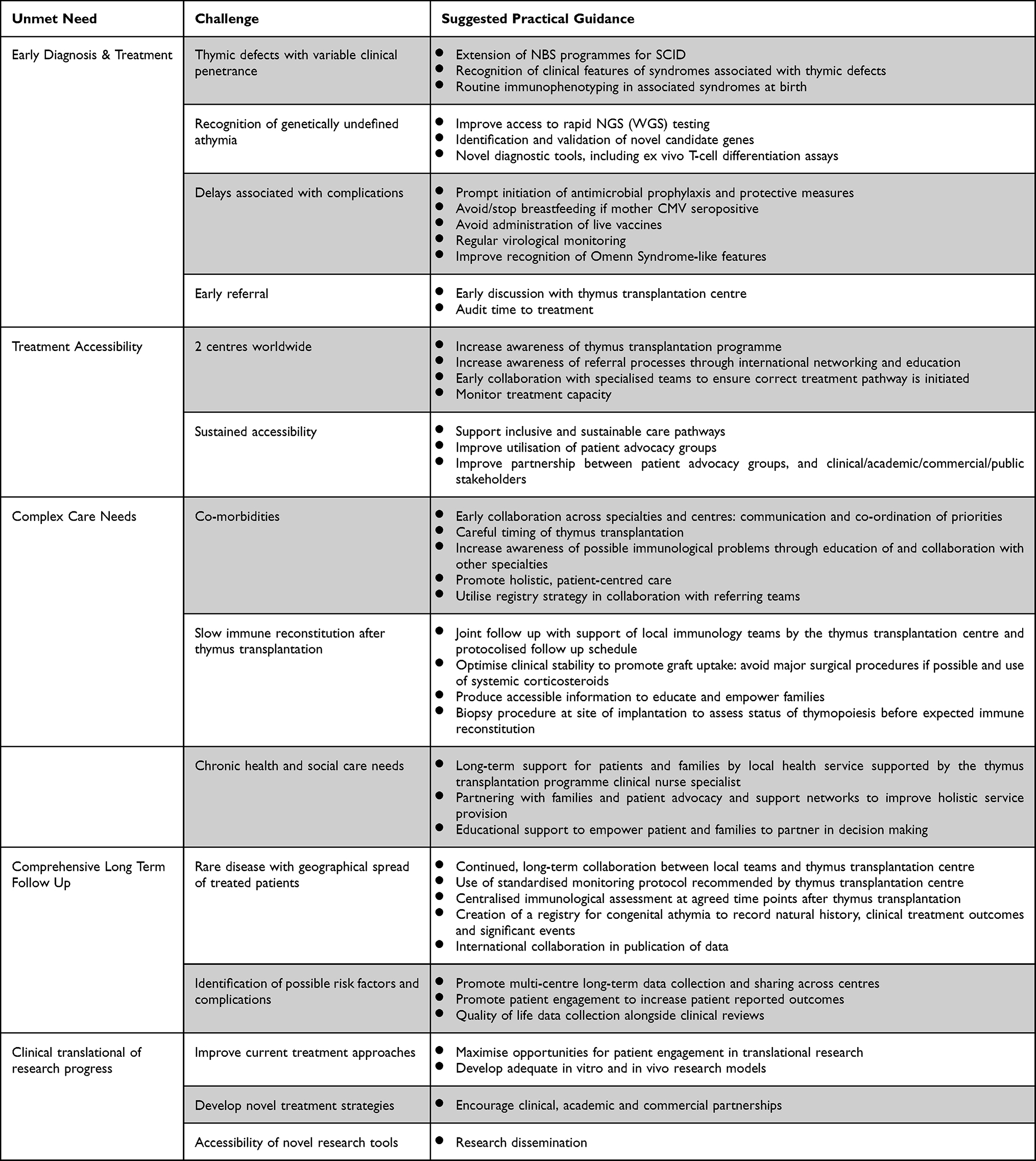 |
Table 1 Practical Guidance to Overcome Challenges Resulting in Unmet Needs for Patients with Congenital Athymia |
Timely Recognition of Congenital Athymia and Referral for Thymus Transplantation
Inborn errors of thymic stromal cell development are associated with aberrant 3rd pharyngeal pouch (PP) patterning during early embryogenesis with impaired or absent development of thymic stroma, respectively, resulting in thymic hypoplasia or aplasia.1 The development of other structures which originate from the 3rd PP, such as the parathyroid glands and the cardiac outflow tract, can also be affected. The triad of congenital thymic hypoplasia/aplasia, hypoparathyroidism and conotruncal heart defect constitutes the hallmark features of DiGeorge Syndrome (DGS).9 The most common cause of DGS is 22q11.2 deletion syndrome (22q11.2DS).10,11 This usually occurs de novo following chromosomal misalignment with non-allelic homologous recombination during meiosis, though in 5–10% this is inherited from a parent.12 There is no correlation between the severity of the DGS phenotype and the size of the microdeletions, but the more distal deletions which do not include the gene TBX1 have been associated with better T-cell numbers.13 DGS has also been reported in patients with monogenic TBX1 haploinsufficiency.1 TBX1 encodes a key transcription factor regulating the expression of thousands of genes and interacting with several other proteins, impacting PP development during embryogenesis through a complex network of factors.14 For example, TBX1 interacts with CHD7, which equally has been implicated in 3rd PP patterning and thymus organogenesis.1 CHD7 haploinsufficiency underlies CHARGE syndrome, which has overlapping DGS features with 22q11.2DS.15 Other less frequent genetic aetiologies1 associated with DGS include TBX2 deficiency,16 partial monosomy 10p17 and FOXI3 haploinsufficiency.18,19 Non-genetic aetiologies, specifically foetal exposure to retinoic acid, alcohol and maternal diabetes, have also been linked to DGS.1 Regardless of the underlying cause, DGS is characterised by incomplete and variable penetrance across its clinical features, and no correlation has been established between the severity of the overall clinical phenotype and the degree of immunodeficiency.1,11,12 Only a minority of DGS patients, approximately 1%, have cDGS with life-threatening immunodeficiency due to thymic aplasia.1,12 Increasingly, the implementation of newborn screening (NBS) programmes for severe combined immunodeficiency (SCID), based on the identification of profoundly T-cell lymphopaenic infants with low levels of TCR excision circles (TREC),20 facilitates the early diagnosis of cDGS. In the absence of NBS, it remains crucial to rule out severe immunodeficiency through immunophenotyping of all patients with DGS features.
Congenital athymia has also been recognised in non-DGS patients with a SCID-like immunophenotype.21 It underlies the immunodeficiency in patients with otofaciocervical syndrome type 2 (OTFCS2)1,22 and Nude SCID,1,23 respectively, caused by bi-allelic mutations in PAX1 and FOXN1, both genes that encode transcription factors regulating thymus organogenesis and TEC development and function.1 In the absence of NBS for SCID, these PAX1- and FOXN1-deficient patients also present syndromic features at birth which can alert to the need for investigating a possible life-threatening immunodeficiency.24–27 Additionally, a significant number of SCID patients remain without a genetic diagnosis despite the expanding access to comprehensive gene panels and next-generation-sequencing (NGS),28 and some of these genetically undefined patients may suffer from congenital athymia. Overall, it is not uncommon that thymic stromal cell defects are only suspected after failed immune reconstitution following haematopoietic stem cell transplantation (HSCT). HSCT outcomes in athymic patients are extremely poor, with a high risk of graft-versus-host-disease (GVHD) and immune reconstitution in surviving patients solely depending on the transfer of mature donor T-cells.29 Whilst a “rescue” thymus transplantation can be attempted,2 a second procedure is not always feasible if the patient develops severe complications after HSCT.30 Timely recognition of congenital athymia, even in genetically undefined T-B+NK+ SCID, is thus necessary to ensure patients receive the correct definitive treatment.30,31 Research assays that allow the ex vivo study of T-cell differentiation have been proposed as novel tools to assist in distinguishing genetically undefined thymic stromal cell defects from defects intrinsic to the haematopoietic stem cell.32,33 These assays are based on the co-culture of CD34+ haematopoietic stem and progenitor cells (HSPC) with murine stromal cell lines expressing the Notch Delta-like ligand (DLL)-4 in a three-dimensional artificial thymic organoid (ATO).34 If HSPCs from a genetically undefined T-B+NK+ SCID patient show in vitro potential to differentiate into T-cells, a thymic stromal cell defect should be considered, possibly requiring referral for treatment with thymus transplantation. For genetically undefined patients referred to GOSH for thymus transplantation, we have used a two-dimensional assay similarly based on the co-culture of HSPCs and murine stromal cells expressing DLL-1.35 These research assays may however not be readily accessible and have some limitations, including variable cell yield and seeding efficiency.36,37 Additionally, in absence of a molecular diagnosis, the interpretation of ex-vivo T cell differentiation results should be made with caution if there are no clinical features suggesting 3rd PP patterning defects, as successful in vitro T-cell differentiation has been reported for patients with haematopoietic cell-intrinsic defects affecting late T-cell development.32,33 Even if a (novel) candidate gene is identified, plausibly underlying a thymic stromal cell defect, uncertainty on the best therapeutic approach can remain due to the limited data available on disease progression or on treatment outcome if thymus transplantation is completed. On occasion, an initial “watch and wait” approach is indicated to assess the severity of the disease over time, before determining the need and type of intervention. This is well illustrated by the recently reported NBS-based identification of infants with heterozygous FOXN1 mutations in whom, despite absent TRECs and profound lymphopaenia at birth, conservative management seems to be the most appropriate approach with patients rarely suffering significant infections and T-cell counts increasing and even normalising over time.38,39
Patients with congenital athymia present profound T-cell lymphopaenia with absolute CD3+ T-cell counts below 50x106/L.30 Over time, a significant number of athymic patients develop atypical features due to the oligoclonal expansion of dysfunctional T-cells which cause an Omenn Syndrome (OS)-like phenotype, with desquamating erythema, enteropathy, lymphadenopathy and hepatosplenomegaly.40 In these patients, T-cell counts will be higher or even normal, but absent thymic activity is reflected in the negligible TREC levels and the very low proportions of naïve CD45RA+CD27+ (or CD45RA+CD62L+) T-cells and CD45RA+CD31+ recent thymic emigrants, typically below 5% of total CD4+ T-cells.2,30 Early identification through NBS programmes together with NGS facilitates earlier referral for and treatment with thymus transplantation. Despite protective measures for the prevention of opportunistic infections, patients remain susceptible to infections and are at risk of developing OS while awaiting corrective treatment. These two types of complications make treatment by thymus transplantation more challenging. Infections are associated with increased mortality before thymus transplantation can be arranged, as well as after thymus transplantation given that immune reconstitution is slow. As is systematically done in infants with a SCID diagnosis, appropriate recommendations need to be made regarding breastfeeding depending on maternal CMV status and close monitoring through regular viral PCRs is required.3,41 In athymic patients, CMV and other systemic viral infections can be extremely difficult to control before and after thymus transplantation whilst awaiting immune reconstitution.2,3 Therefore, if a matched sibling donor (MSD) is available, treatment with HCT may lead to a more favourable outcome than proceeding with thymus transplantation in patients with severe viral infections.42,43 Additionally, infections can drive inflammatory complications at the time of early immune reconstitution, as seen in patients with infections including BCG, (vaccine strain) rotavirus and Norovirus.3,44 These immune reconstitution inflammatory response-type complications can be life-threatening, requiring the use of high-dose systemic steroids which significantly compromise developing thymopoiesis. Patients developing atypical OS-like features before thymus transplantation will require immunosuppression, including Cyclosporin A (CSA) and, in the days just before the transplantation procedure, lympho-ablative treatment with anti-thymocyte globulin.30 Overall outcomes after thymus transplantation are similar for patients with and without atypical features,2,3 but the use of immunosuppression often requires a longer admission in the transplantation centre and the treatment with CSA needs to be continued after thymus transplantation until thymopoiesis is well established in the allograft. In summary, every possible effort should be made to proceed with thymus transplantation as soon as feasible, reducing the overall risk of infections and OS. Early referral and discussion with one of the centres offering this treatment are important in order to achieve this. It is to be expected, that similar to reported HSCT outcomes in SCID, early diagnosis and treatment are synonymous with improved clinical outcomes.20
Treatment Accessibility
Thymus transplantation is currently only offered at Duke University Medical Centre in the USA and GOSH in the UK. Though tissue preparation and implantation techniques are largely similar in both programmes, there are important differences when accessing the care pathways. In the USA, thymus transplantation has recently been approved as a medicinal product by the Food and Drugs Administration (FDA) and is now known as Rethymic® (allogeneic processed thymus tissue-agdc).45 In the UK, the treatment is considered a tissue transplantation, and not a medicinal product. It remains known as thymus transplantation and is offered through a nationally commissioned transplantation service, regulated by the UK’s Human Tissue Authority (HTA). Through this process the HTA controls the safe and ethical use of human tissue and organs and ensures proper consent for use.46 With only two centres, there are challenges to worldwide accessibility to this treatment, with patients and their families having to travel long distances and across borders. Nevertheless, the thymus transplantation programme at GOSH has provided treatment and ongoing advice for patients from more than 40 centres in the UK and Europe, Oceania, the Middle East, India, North and South America over the last 10+ years (personal communication by the GOSH Thymus Transplantation programme). Despite the UK’s departure from the European Union (EU) in 2019, thymus transplantation has remained available to EU patients through continued reciprocal health-care agreements, and treatment has never been declined for funding reasons. For referring centres outside of the UK and the EU, GOSH offers treatment as international and private care, whereby patients are funded by their health-care insurance or by country’s health authority or government given that thymus transplantation is widely recognised as the most appropriate treatment for congenital athymia. Even so, economic and geographical reasons play a role in limiting awareness of and access to treatment, especially for patients from low- and middle-income countries who also face barriers in access to specialised immunology centres, early diagnostic testing and essential therapies.47,48 The programme at GOSH adopts an inclusive policy and the current care pathway remains relatively affordable, in particular, in comparison with the price for the commercial treatment now on offer in the USA, where the medicinal product is estimated to cost approximately ten times more than the complete GOSH care package. Nevertheless, even in high-income countries, economic hurdles in accessing life-saving novel therapies are a growing concern for patients with rare diseases, especially for those treatments that may not necessarily be workable economically for commercial manufacturers unless pricing is escalated.49,50 Initiatives, bringing together clinicians, researchers, patient advocates and commercial partners, to promote sustained accessibility to gene therapies for rare disorders have been endorsed by our centre50,51 and may also hold valuable lessons for ensuring treatment accessibility for athymic patients.
In part thanks to the ongoing expansion of universal and pilot NBS programmes for SCID across Europe and elsewhere,52–55 increasing numbers of athymic patients are being referred to GOSH in consideration for thymus transplantation.52,56 For example, in the first 2.5 years of universal NBS for SCID in Germany, 7 patients with congenital athymia have been referred for thymus transplantation,52 whereas in the 10 preceding years we have only treated a total of 3 patients from Germany (unpublished, GOSH Thymus Transplantation programme). One core rationale for NBS programmes is to ensure early treatment and given patients with athymic disorders are being equally identified, it is important that beyond early recognition and initiation of prophylaxis and isolation, we also aim for early corrective treatment. Most NBS programmes recommend treating SCID patients within 4 months.57 This time frame is not always achievable for athymic patients, as they often have co-morbidities requiring complex care (see below) in order to achieve the clinical stability necessary prior to proceeding with thymus transplantation.3,44 To date, the GOSH thymus transplantation programme continues to offer timely treatment, in particular to patients from the UK and Europe, thanks to a strengthened care pathway.56 Waiting times at GOSH are primarily dependent upon the clinical status of the patients, with the most stable children often being transplanted well within the SCID recommendation of 4 months.52,56
In the future, it may be desirable to establish thymus transplantation programmes at additional centres. Whether and where to do so, however, will need careful consideration based on monitoring of treatment capacity at existing centres, given that tissue preparation is a very specialised process and patient care is highly complex, both requiring significant levels of expertise. While there are several renowned Paediatric Immunology centres across Europe, there may be only one or two athymic children identified every year even in larger European countries,58,59 demonstrating the rareness of the condition. A thymus transplantation programme requires specialist medical and laboratory staff, and provision of laboratories and equipment needs to be such that donor tissue is consistently processed according to a strict set of quality standards. Staff training and maintenance of specialised equipment carry an economic burden, and must be considered, when analysing whether it is plausible to deliver safe and economically viable care at several sites for such an ultra-rare cohort of patients. In the USA, the FDA license for Rethymic® is currently restricted to treatment at Duke Medical Centre only.45 At GOSH, our programme’s capacity and capability are enhanced by the shared use of services, facilities and personnel with other programmes delivering cell and gene therapies at our site.60–65 The delivery and success of the thymus transplantation programme at GOSH additionally relies on a much wider workforce across sub-specialities with vital knowledge, skills and experience in caring for patients with such complex conditions. Also essential to our programme delivery is the presence of a large cardiac surgery department, necessary to ensure continuous access to donated thymus tissue.
Inborn errors of immunity (IEI) are still going undiagnosed and undertreated across the world.47,66 International patient organisations such as the International Patient Organisation for Primary Immunodeficiencies (IPOPI), national primary immunodeficiency charities and patient advocacy groups have a vital role in raising awareness and promoting a culture of shared learning by providing accurate up to date information, supporting the education of patients, families and multi-disciplinary clinicians alike.47,48,67,68 Such positive partnerships have shown value from the involvement and engagement of patients and families in both clinical and strategic protocol research and design, with improved patient opportunity in study design acceptability and recruitment, and also in the dissemination and evaluation of results.67–70 However, it remains important to implement supportive guidance to safeguard ethical relationships and to manage bias and incentive for experience.68,70 Surprisingly, even in countries from which patients are being referred for thymus transplantation, a lack of awareness and access to thymus transplantation has been reported.48 While there is no registered advocacy group specifically for patients with congenital athymia, patient advocates have independently been using social media to raise the profile and success of thymus transplantation, often joining up families across geographical boundaries. The use of social media to raise awareness is not without risk,71,72 however in an ever-changing digital world, not utilising such resources could be considered a missed opportunity. These platforms provide a space to share pioneering breakthroughs and innovation in practice, while educating and networking important stakeholders, including health professionals, policy makers, funders and patient and families.71,72 Used correctly, respecting institutional restrictions and patient confidentiality, they can promote equitable access to information, disseminated from reliable and trusted sources. Families with athymic children treated at GOSH have highlighted the lack of patient-focused resources in Europe providing guidance when considering treatment options for their child and education around acute and long-term care. Comprehensible leaflets and booklets are known to be an effective method of communicating with and teaching families73 while empowering them to be partners in decision-making,67,74 which is especially important when families are consenting to novel treatments. A current project involving the families who received thymus transplantation at GOSH has been undertaken to co-create a digital resource outlining the journey for children receiving thymus transplantation for congenital athymia at our centre.75 Families have engaged with the clinical nurse specialist of the thymus transplantation programme to offer their opinion on matters relating to both design and content, and to answer questions relating to readability and suitability. Engaging patients and families in their care pathways is known to improve the patient experience with better service design and therapeutic options leading to elevated health and wellbeing and a health system which is more resourcefully efficient.67
Complex Care
Athymic infants often have syndromic features, some of which are associated with complex medical and surgical needs, requiring immediate attention from a variety of specialities. The most common ones in the context of DGS for example are congenital heart defects (CHD), which may have been diagnosed prenatally and/or become apparent shortly after birth, breathing and/or feeding difficulties due to anatomical anomalies, and neonatal seizures due to hypocalcaemia.44 As such, the diagnosis of cDGS or congenital athymia is regularly made as a diagnosis within a diagnosis, typically only recognised after the acute manifestation of these other clinical symptoms. The management of these co-morbidities plays a fundamental role in the timing of thymus transplantation once congenital athymia has been diagnosed, as clinical stability is required for safe transfer to the transplantation centre but also for the months after the procedure to facilitate establishment of thymopoiesis in the allograft. It is therefore essential to attain a stable cardiopulmonary status prior to proceeding with implantation of the thymic graft to avoid graft failure. In this scenario, it is not uncommon that thymus transplantation needs to be delayed in athymic patients until after correction of haemodynamically relevant CHD or upper airway stabilisation. Optimal planning and sequencing of these procedures is best agreed in multi-speciality and multicentre discussions including the referring clinical team, the thymus transplantation team and appropriate specialists from both centres to balance the risks related to timing of these procedures against those related to delaying thymus transplantation and recovery of T-cell immunity. Another pre-requisite for thymus transplantation is sufficient weight gain, as the thymus tissue is implanted into quadriceps muscle tissue which should not be wasted.2 Due to their complex co-morbidities, athymic patients are frequently at risk of failure to thrive and adequate nutritional and clinical support measures need to be in place to support weight gain.
In the months following thymus transplantation, all the protective measures remain in place unchanged until protective levels of immunity slowly develop.2,3 Patients carry on with anti-microbial prophylaxis and social isolation. This unavoidably limits their access to supportive services, including additional health-care resources, and can impact on the management of their other complex needs. For example, it is crucial to avoid surgical procedures requiring prolonged anaesthetics in the months following thymus transplantation and to avoid certain medications until thymopoiesis is well established in the thymic graft.8,30,76 Systemic steroids in particular will have a negative effect on the developing thymus tissue. These are standard treatments in the management of respiratory exacerbations in patients with complex airways problems and for the treatment of autoimmune cytopaenias, in particular autoimmune haemolytic anaemia (AIHA), which can occur after thymus transplantation.3,8,77 Whenever possible, steroid-sparing strategies should be explored until proof of satisfactory thymic output has been obtained. For patients treated at GOSH, a biopsy procedure at the site of thymus tissue implantation is typically scheduled at approximately 3 months after transplantation in order to evaluate the degree of thymopoiesis in the graft3,78 as this can help in therapeutic decision-making should complications arise before thymic output can be detected in the peripheral blood.
The management of co-morbidities and complications require significant healthcare resource utilization. The US experience has been well documented with comprehensive guidance for clinicians providing care for children with syndromic congenital athymia.8 The associated heavy economic burden has also been estimated within the American health system for children receiving supportive care only in the first 3 years of life.79 A comparable burden has been reported with many rare diseases in which the nature of their complexity and intense need for supportive health-care measures create both direct and indirect economic burden.80 Whilst timely curative treatment can reduce the burden of prolonged supportive care directly related to congenital athymia alone,8,81 many transplanted patients will continue to have complex health-care needs, requiring specialised and co-ordinated health-care service provision even when satisfactory T-cell immunity has been attained. Patients with anatomical abnormalities of the respiratory tract and/or chronic lung damage resulting from pre-transplantation complications often remain at risk of respiratory infections, but the overall infectious risks will be alleviated after successful thymus transplantation and patients will be able to access support services more readily. Nevertheless, their medical complexity can be considered to amount to a primary determinant of unmet needs in society, contributing to disadvantage and burden.82,83 As DGS patients mature, there is also the additional risk of mental health disorders,84–86 which is not always disclosed to families until later in their child’s development.84,87,88
As discussed, under the umbrella diagnosis of DGS or other rare syndromes associated with congenital athymia, patients have multiple diagnoses, and while there are clinical and social support networks focused on the management of individual specialist areas, for example CHD, hypoparathyroidism, neurodevelopmental delay or autism, they may not be designed to support a patient with several co-morbidities,89 increasing family burden to seek additional support elsewhere and in some cases, families report feeling the need to fight for services to help their child.88 Advocacy groups specifically for 22q11.DS or CHARGE syndrome exist but remain sparse and, regardless of geographic location and economic status, athymic patients and their families do not have access to a singular joined up network of support, therefore creating a gap in the co-ordinated management and treatment potential for these patients.89 To help mitigate this, clinicians and health-care providers can offer additional support by partnering with patient advocacy and support networks to increase the engagement of patients and families in service design, creating the opportunity to learn from real life patient experience.67–69 The European thymus transplantation programme has a dedicated clinical nurse specialist, who acts as a single point of contact from diagnosis, through admission and ongoing long-term follow up for families. This role includes patient advocacy and signposting for support services, as well as specialist immunological education and liaison between teams at GOSH and abroad.
Long-Term Outcomes After Thymus Transplantation and Impact on Health-Related Quality of Life
While thymus transplantation is recognised as the only known effective, life-saving option for patients with congenital athymia, limited data are available on long-term immunological outcomes. Mortality most frequently occurs in the first year after thymus transplantation and is associated with pre-existing infections or infections occurring before immune reconstitution.2,3 Increase in T-cell counts seems to peak in the second year after thymus transplantation2,3 and in a few patients thymic output has been shown to be sustained over several years.3,7 While the US experience confirms patient survival into adulthood, no detailed clinical and immunological data are available for this cohort beyond 2 years after thymus transplantation.2 The European cohort of patients treated at GOSH is younger and patients are yet to transition into adulthood. Early autoimmune complications after thymus transplantation mainly consist of transient cytopaenias, including AIHA, thrombocytopaenia and neutropaenia. While there is variability in the evaluation of treatment-related adverse effects, later and long-term autoimmune manifestations have been reported in a significant number of patients in both cohorts (2, 3; these publications provide the complete lists of reported adverse effects so far). This later autoimmunity largely affects the thyroid, which can relatively easily be treated. Other than autoimmune manifestations, significant late adverse events, such as severe infections or malignancies, have not been reported to date.2,3
Learning from long-term outcomes after HSCT for IEI, it is evident that careful data collection over time is important in order to recognise late adverse effects and their risk factors.90 Patients treated at GOSH are geographically dispersed and after thymus transplantation their long-term follow up is managed by their local immunologists. The programme employs a standardised monitoring protocol advising on which analyses to perform and at what intervals. Some of these investigations are not routinely performed in most clinical immunology laboratories, such as thymus donor T-cell engraftment studies and TREC levels in sorted peripheral T-cells. In an attempt to achieve comprehensive long-term follow up for this unique and small cohort of patients, the GOSH thymus transplantation team and most local centres have continued to closely collaborate. Nevertheless, consistent measuring of immunological and clinical parameters, including adverse events and quality of life, can prove challenging and can falter over time, particularly if post-transplantation patients are considered to be well and thriving.
More accurate long-term follow up data could be achieved by creating a registry for congenital athymia. Registries are well recognised as an innovative and patient-centred method to improve pattern recognition, diagnostics, medical treatments, and service design.47,48,69,91,92 National and international registries exist for IEI and have proven to be extremely valuable for sharing information, learning from multicentre experience and improving overall clinical practice in order to optimise outcomes for this group of rare disorders.93–96 Within IEI registries, congenital athymia does not feature as its own entity in any current version, making it difficult to establish prevalence and missing the opportunity to profile details specific to this cohort. Publications presenting patients with congenital athymia mainly relate to treatment outcomes; however, in our experience, there is an unreported cohort of infants who are referred but do not make it to transplantation for a variety of reasons, including uncontrollable infection or a decision to treat with palliation only due to severe co-morbidities. A dedicated registry for athymic patients, who are underrepresented and geographically spread, would provide a convenient, cost-effective, easily accessible documentation tool, with which to build a central foundation to further understanding this disease, its progression and comprehensively record outcomes after thymus transplantation.
Development of a registry is not without challenges, including restricted funding, multi-professional training, harmonisation of protocols and purpose, scientific methods and data structures, sustainability, transparency, and quality assurance.48,92,97 However, we believe with the growing recognition of congenital athymia and thymus transplantation treatment, interest and support for a central reference hub will be well supported by the international network of teams caring for these patients and by their families. Extensive collection and analysis of uniform data sets before and after thymus transplantation, including long-term clinical results, will undoubtedly contribute to optimising treatment modalities, monitoring and overall outcomes for athymic patients. Examples of excellent use of registries have been reported in other disciplines.98,99 Health services are poorly designed to cope with increasing populations of complex patients with multiple chronic health and social care needs, such as 22q11DS,87 comprehensive analysis of registry data would have the capacity to pre-empt care needs and costs. A registry may also promote unified working within the wider multi-specialist teams to view the child as a whole and not as a patient with multiple differing system disorders, benefiting a smoother transition of patients with congenital athymia and syndromic health needs to adult health services. Establishing long term, consistent follow up care for athymic patients into adulthood across multiple specialities is key to maximising health potential and reducing familial and economic burdens.8
Understanding the impact of rare diseases on a patient’s physical, emotional, and psychosocial development, with subjective perspective, cannot be fully captured through studying mortality and morbidity data alone. Incorporating QOL measurements is becoming increasingly recognised as an essential component to holistic investigation, leading to meaningful discussions between patients, families and their health-care providers when evaluating and planning treatments and interventions.100,101 Children living with an IEI, who have not received curative treatment, report consistently lower health-related QOL (HRQOL) than healthy children,102,103 and children with comparable chronic conditions.104–107 Life for children with DGS, often examined in the context of the family unit due to developmental difficulties, is on a parallel85,89,108 with increased challenges associated with the severity of symptoms, interventions required and paucity of support systems available.86 Collectively, this suggests that syndromic patients with congenital athymia are expected to experience poorer HRQOL than their healthy peers. One initial study indeed reports that athymic children, receiving supportive care only, face significant burden across all HRQOL domains, resulting in lower HRQOL scores.81 HRQOL has not been explored for patients with congenital athymia who received thymus transplantation and the patients’ and their families’ lived experience following thymus transplantation is not reflected in published data. To understand how this burden is affected after corrective treatment from a patient-reported perspective, we suggest that HRQOL data is collected alongside physical health reviews, subsequent to utilising patient advocacy groups in instrument design.69 By providing a unique insight into patient and family lived experience, this knowledge would have the potential to enhance and strengthen patient care by improving shared decision-making and offering opportunities for appropriate protective and preventative interventions, as reported by the patient and their families.89,100
Translational Research
Thymus transplantation using cultured, postnatal donor thymus tissue has been undertaken without significant changes over the past decades. It is evident that alongside comprehensive analysis of outcome data for patients having received thymus transplantation, translational research is crucial for improving the current treatment modalities and for developing new treatment strategies.5 Better understanding of the mechanisms by which allogeneic thymus transplantation supports thymopoiesis in the absence of tissue type matching between recipient and donor remains is essential. In normal thymus tissue, developing T-cell progenitors or thymocytes that have successfully rearranged their TCR are positively selected and migrate from the thymic cortex into the medulla, where their TCR specificities are tested against tissue-restricted self-antigens (TRSA) which are promiscuously expressed by medullary TEC.1 Evidence from murine studies suggests that negative selection may also be mediated by expression of TRSA on cells of haematopoietic origin, particularly dendritic cells.109,110 This interaction results in the negative selection or elimination of thymocytes carrying TCR that strongly recognise self-antigens, while those with TCR with intermediate affinity for self-antigens differentiate into regulatory T-cells. Upon completion of this developmental programme, thymocytes with low affinity for self-antigens mature into functional, self-tolerant T-cells. The thymic allograft can successfully educate recipient T-cells towards tolerance to self and donor tissues111,112 and the role of recipient-derived cells of haematopoietic origin in negative selection may be important here. Nevertheless, it is to be expected that the preparatory pre-implantation tissue culture together with the lack of tissue type matching may impair lympho-stromal crosstalk in the graft, perhaps explaining the suboptimal recovery of T-cell immunity and relatively common autoimmune manifestations after thymus transplantation.2,3 The increasing application of genomic and transcriptomic approaches to biological samples may be of use here to decipher cellular heterogeneity and cell–cell interactions,113,114 and patients treated at GOSH are given the opportunity to participate in research. In theory, partial tissue type matching may improve outcomes after thymus transplantation, as SCID patients treated by haploidentical HSCT develop normal T-cell numbers and do not suffer significant autoimmune complications.115 Achieving partial tissue type matching would necessitate the creation of a biobank for thymus tissue upon confirmation that cryopreserved tissue is equally capable of supporting thymopoiesis. In a mouse model of thymus transplantation, it was demonstrated that previously cryopreserved human thymus tissue can support mouse T-cell development.116 Histological assessment of cultured, previously frozen thymus slices shows that viable thymic epithelium is retained upon thawing.117 Compared to the current use of fresh cultured thymus tissue for implantation, which requires rapid transfer of the recipient to the transplantation centre upon tissue collection, the use of cryopreserved tissue would make it possible to proceed following a more predictable treatment schedule, simplifying the logistics at the referring centre and at the transplantation site, as well as for the patients’ families.
Whilst the use of cultured postnatal thymus tissue is likely to remain the standard treatment for athymic patients for years to come, increasing levels of research are being dedicated to developing alternative approaches for thymus replacement therapy.5 Re-aggregate thymus organ cultures (RTOCs) using murine stromal cells have been developed and have been shown to support T-cell differentiation ex vivo, as well as in vivo upon transplantation into athymic mice.34 These artificial thymic organoids using murine stroma are relatively small and are not suitable for clinical translation for the treatment of congenital athymia. In normal thymus tissue, TECs are arranged within a complex three-dimensional (3D) environment supported by the extracellular matrix (ECM), which is important for their function.118 The ECM proteins, such as collagen, are produced by mesenchymal cells. Recent studies in a Tbx1-deficient murine model suggest that thymic hypoplasia and aplasia in 22q11.2DS (and possibly in other thymic stromal cell defects) are due to mesenchymal cell defects and can be corrected by mesenchymal cell replacement.119 Novel tools have been developed for delivery of thymic stromal cells in a more appropriate 3D structure than the one achieved in RTOCs by using artificial collagen scaffolds120 and decellularized thymuses from rodents,121,122 yet both approaches are still not applicable in a translational setting.5 A significant breakthrough towards successful human thymus tissue engineering has been reached recently by the optimisation of a whole-organ perfusion system facilitating the retrieval of decellularized ECM from human thymuses, which has been shown to enable reorganisation of expanded human TECs into recognizable thymic stroma and to support thymopoiesis ex vivo and in vitro.123 In the future, such an artificial scaffold may also make it possible to deliver gene-corrected TECs and other stromal cells differentiated from patient-derived induced pluripotent stem cells (iPSCs). While iPSC-based differentiation protocols124,125 need further optimisation for the generation of functional, mature iTECs able to support thymopoiesis, this is an exciting future avenue for autologous thymus tissue replacement which could reduce the incidence of autoimmune manifestations after thymus transplantation.5 Currently, these novel tools such as RTOCs and iTECs already play an important role in disease modelling and characterisation of novel thymic stromal cell defects,19,22,32,33 with direct benefits in the current diagnostic and therapeutic management of athymic patients.
Conclusion
Congenital athymia is a rare, life-threatening disease. Thymus transplantation using postnatal, cultured donor thymus tissue is a life-saving procedure which is recognised as the best treatment for congenital athymia. In this review, we have discussed hurdles leading to unmet needs for athymic patients and have proposed practical guidance to overcoming these (summarised in Figure 1). We have done so based on our experience at GOSH, which has established a successful thymus transplantation programme providing this essential care in Europe. Improving early diagnosis of congenital athymia is the first step toward improving outcomes for athymic patients. NBS for SCID and T-cell lymphopaenia increasingly makes early referral for thymus transplantation possible, but further efforts are required to accelerate the identification of novel genetic aetiologies. While this specialised treatment is only available in two centres worldwide, limited awareness of the European Thymus Transplantation programme and its inclusive referral process further restrict timely treatment for this rare disease, highlighting the need to strengthen avenues of communication across specialities and international centres. Detailed recording of outcomes after thymus transplantation, integrating the patients’ immunological results, as well as data relating to their co-morbidities and HRQOL, is crucial for the adequate identification of risks and challenges that need to be tackled through high-quality patient-centred care delivered in partnership with patients, families and patient organisations, in parallel to clinical and translational research aiming at improving outcomes for patients with congenital athymia.
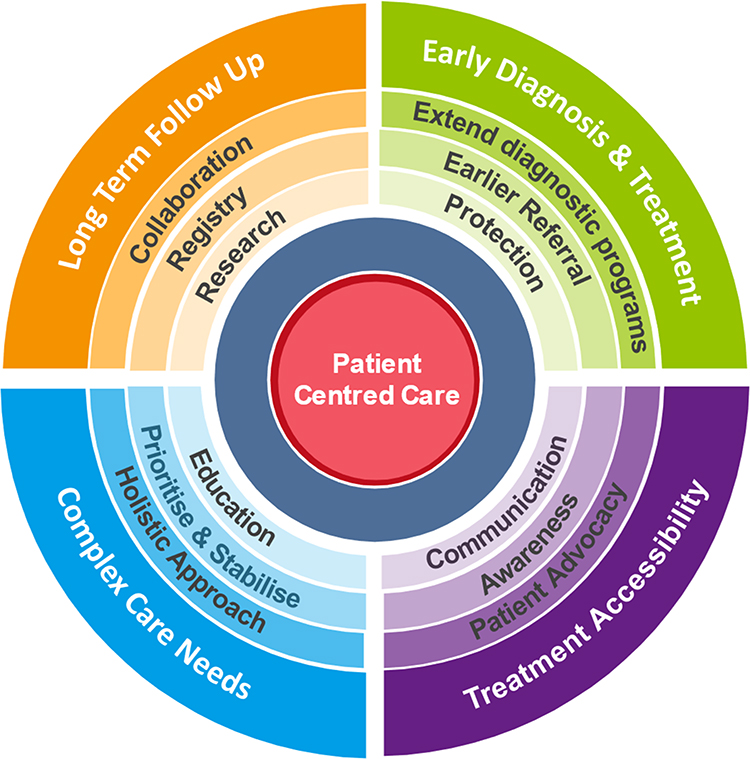 |
Figure 1 Optimising outcomes for patients with congenital athymia: Diagram reflecting the layering of patient-centred practical guidance to systematically and comprehensively address the challenges encountered and unmet needs experienced by athymic patients. |
Acknowledgments
EH, EGD and the UCL GOSH Thymus Transplantation programme are supported by LetterOne in conjunction with GOSH Children’s Charity. AYK is supported by the Wellcome Trust (222096/Z/20/Z). This work is supported by the National Institute of Health Research and the GOSH Biomedical Research Centre (NIHR GOSH BRC). The views expressed are those of the authors and not necessarily those of GOSH, the NHS, the NIHR or the Department of Health. We thank Dr Zainab Golwala for careful reading and feedback.
Disclosure
Dr E Graham Davies reports personal fees from Videregen Limited, Leeds, UK, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Kreins AY, Maio S, Dhalla F. Inborn errors of thymic stromal cell development and function. Semin Immunopathol. 2021;43(1):85–100. doi:10.1007/s00281-020-00826-9
2. Markert ML, Gupton SE, McCarthy EA. Experience with cultured thymus tissue in 105 children. J Allergy Clin Immunol. 2022;149(2):747–757. doi:10.1016/j.jaci.2021.06.028
3. Davies EG, Cheung M, Gilmour K, et al. Thymus transplantation for complete DiGeorge syndrome: European experience. J Allergy Clin Immunol. 2017;140(6):1660–70 e16. doi:10.1016/j.jaci.2017.03.020
4. Markert ML, Kostyu DD, Ward FE, et al. Successful formation of a chimeric human thymus allograft following transplantation of cultured postnatal human thymus. J Immunol. 1997;158(2):998–1005. doi:10.4049/jimmunol.158.2.998
5. Kreins AY, Bonfanti P, Davies EG. Current and future therapeutic approaches for thymic stromal cell defects. Front Immunol. 2021;12:655354. doi:10.3389/fimmu.2021.655354
6. Markert ML, Sarzotti M, Ozaki DA, et al. Thymus transplantation in complete DiGeorge syndrome: immunologic and safety evaluations in 12 patients. Blood. 2003;102(3):1121–1130. doi:10.1182/blood-2002-08-2545
7. Albuquerque AS, Marques JG, Silva SL, et al. Human FOXN1-deficiency is associated with alphabeta double-negative and FoxP3+ T-cell expansions that are distinctly modulated upon thymic transplantation. PLoS One. 2012;7(5):e37042. doi:10.1371/journal.pone.0037042
8. Gupton SE, McCarthy EA, Markert ML. Care of children with DiGeorge before and after cultured thymus tissue implantation. J Clin Immunol. 2021;41(5):896–905. doi:10.1007/s10875-021-01044-0
9. Di George AM, Lischner HW, Dacou C, Arey JB. Absence of the thymus. Lancet. 1967;1(7504):1387. doi:10.1016/S0140-6736(67)91808-9
10. Driscoll DA. Genetic basis of DiGeorge and velocardiofacial syndromes. Curr Opin Pediatr. 1994;6(6):702–706. doi:10.1097/00008480-199412000-00016
11. Giardino G, Radwan N, Koletsi P, et al. Clinical and immunological features in a cohort of patients with partial DiGeorge syndrome followed at a single center. Blood. 2019;133(24):2586–2596. doi:10.1182/blood.2018885244
12. Ryan AK, Goodship JA, Wilson DI, et al. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: a European collaborative study. J Med Genet. 1997;34(10):798–804. doi:10.1136/jmg.34.10.798
13. Crowley B, Ruffner M, McDonald McGinn DM, Sullivan KE. Variable immune deficiency related to deletion size in chromosome 22q11.2 deletion syndrome. Am J Med Genet A. 2018;176(10):2082–2086. doi:10.1002/ajmg.a.38597
14. Du Q, De la morena MT, van Oers NSC. The Genetics and Epigenetics of 22q11.2. Deletion Syndrome Front Genet. 2019;10:1365. doi:10.3389/fgene.2019.01365
15. Pagon RA, Graham JM, Zonana J, Yong SL. Coloboma, congenital heart disease, and choanal atresia with multiple anomalies: CHARGE association. J Pediatr. 1981;99(2):223–227. doi:10.1016/S0022-3476(81)80454-4
16. Liu N, Schoch K, Luo X, et al. Functional variants in TBX2 are associated with a syndromic cardiovascular and skeletal developmental disorder. Hum Mol Genet. 2018;27(14):2454–2465. doi:10.1093/hmg/ddy146
17. Daw SC, Taylor C, Kraman M, et al. A common region of 10p deleted in DiGeorge and velocardiofacial syndromes. Nat Genet. 1996;13(4):458–460. doi:10.1038/ng0896-458
18. Bernstock JD, Totten AH, Elkahloun AG, et al. Recurrent microdeletions at chromosome 2p11.2 are associated with thymic hypoplasia and features resembling DiGeorge syndrome. J Allergy Clin Immunol. 2020;145(1):358–67e2. doi:10.1016/j.jaci.2019.09.020
19. Ghosh R, Bosticardo M, Singh S, et al. FOXI3 haploinsufficiency contributes to low T-cell receptor excision circles and T-cell lymphopenia. J Allergy Clin Immunol. 2022;150:1556–1562. doi:10.1016/j.jaci.2022.08.005
20. van der Burg M, Mahlaoui N, Gaspar HB, Pai SY. Universal newborn screening for severe combined immunodeficiency (SCID). Front Pediatr. 2019;7:373. doi:10.3389/fped.2019.00373
21. Bousfiha A, Moundir A, Tangye SG, et al. The 2022 update of IUIS phenotypical classification for human inborn errors of immunity. J Clin Immunol. 2022;42(7):1508–1520. doi:10.1007/s10875-022-01352-z
22. Yamazaki Y, Urrutia R, Franco LM, et al. PAX1 is essential for development and function of the human thymus. Sci Immunol. 2020;5(44). doi:10.1126/sciimmunol.aax1036
23. Markert ML, Marques JG, Neven B, et al. First use of thymus transplantation therapy for FOXN1 deficiency (nude/SCID): a report of 2 cases. Blood. 2011;117(2):688–696. doi:10.1182/blood-2010-06-292490
24. Gallo V, Cirillo E, Giardino G, Pignata C. FOXN1 deficiency: from the discovery to novel therapeutic approaches. J Clin Immunol. 2017;37(8):751–758. doi:10.1007/s10875-017-0445-z
25. Patil SJ, Das Bhowmik A, Bhat V, Satidevi Vineeth V, Vasudevamurthy R, Dalal A. Autosomal recessive otofaciocervical syndrome type 2 with novel homozygous small insertion in PAX1 gene. Am J Med Genet A. 2018;176(5):1200–1206. doi:10.1002/ajmg.a.38659
26. Paganini I, Sestini R, Capone GL, et al. A novel PAX1 null homozygous mutation in autosomal recessive otofaciocervical syndrome associated with severe combined immunodeficiency. Clin Genet. 2017;92(6):664–668. doi:10.1111/cge.13085
27. Frank J, Pignata C, Panteleyev AA, et al. Exposing the human nude phenotype. Nature. 1999;398(6727):473–474. doi:10.1038/18997
28. Dvorak CC, Haddad E, Buckley RH, et al. The genetic landscape of severe combined immunodeficiency in the United States and Canada in the current era (2010–2018). J Allergy Clin Immunol. 2019;143(1):405–407. doi:10.1016/j.jaci.2018.08.027
29. Janda A, Sedlacek P, Honig M, et al. Multicenter survey on the outcome of transplantation of hematopoietic cells in patients with the complete form of DiGeorge anomaly. Blood. 2010;116(13):2229–2236. doi:10.1182/blood-2010-03-275966
30. Kreins AY, Graham Davies E. Replacing defective thymus function. Curr Opin Allergy Clin Immunol. 2020;20(6):541–548. doi:10.1097/ACI.0000000000000695
31. Blom M, Zetterstrom RH, Stray-Pedersen A, et al. Recommendations for uniform definitions used in newborn screening for severe combined immunodeficiency. J Allergy Clin Immunol. 2022;149(4):1428–1436. doi:10.1016/j.jaci.2021.08.026
32. Bifsha P, Leiding JW, Pai SY, et al. Diagnostic assay to assist clinical decisions for unclassified severe combined immune deficiency. Blood Adv. 2020;4(12):2606–2610. doi:10.1182/bloodadvances.2020001736
33. Bosticardo M, Pala F, Calzoni E, et al. Artificial thymic organoids represent a reliable tool to study T-cell differentiation in patients with severe T-cell lymphopenia. Blood Adv. 2020;4(12):2611–2616. doi:10.1182/bloodadvances.2020001730
34. Montel-Hagen A, Tsai S, Seet CS, Crooks GM. Generation of artificial thymic organoids from human and murine hematopoietic stem and progenitor cells. Curr Protoc. 2022;2(4):e403. doi:10.1002/cpz1.403
35. Six EM, Benjelloun F, Garrigue A, et al. Cytokines and culture medium have a major impact on human in vitro T-cell differentiation. Blood Cells Mol Dis. 2011;47(1):72–78. doi:10.1016/j.bcmd.2011.04.001
36. de Pooter R, Zuniga-Pflucker JC. T-cell potential and development in vitro: the OP9-DL1 approach. Curr Opin Immunol. 2007;19(2):163–168. doi:10.1016/j.coi.2007.02.011
37. Schmitt TM, Zuniga-Pflucker JC. Induction of T cell development from hematopoietic progenitor cells by delta-like-1 in vitro. Immunity. 2002;17(6):749–756. doi:10.1016/S1074-7613(02)00474-0
38. Bosticardo M, Yamazaki Y, Cowan J, et al. Heterozygous FOXN1 variants cause low TRECs and severe T cell lymphopenia, revealing a crucial role of FOXN1 in supporting early thymopoiesis. Am J Hum Genet. 2019;105(3):549–561. doi:10.1016/j.ajhg.2019.07.014
39. Giardino G, Sharapova SO, Ciznar P, et al. Expanding the nude SCID/CID phenotype associated with FOXN1 homozygous, compound heterozygous, or heterozygous mutations. J Clin Immunol. 2021;41(4):756–768. doi:10.1007/s10875-021-00967-y
40. Markert ML, Devlin BH, Chinn IK, McCarthy EA. Thymus transplantation in complete DiGeorge anomaly. Immunol Res. 2009;44(1–3):61–70. doi:10.1007/s12026-008-8082-5
41. Markert ML, Devlin BH, McCarthy EA. Thymus transplantation. Clin Immunol. 2010;135(2):236–246. doi:10.1016/j.clim.2010.02.007
42. Ip W, Zhan H, Gilmour KC, Davies EG, Qasim W. 22q11.2 deletion syndrome with life-threatening adenovirus infection. J Pediatr. 2013;163(3):908–910. doi:10.1016/j.jpeds.2013.03.070
43. Chitty-Lopez M, Duff C, Vaughn G, et al. Case report: unmanipulated matched sibling donor hematopoietic cell transplantation in TBX1 congenital athymia: a lifesaving therapeutic approach when facing a systemic viral infection. Front Immunol. 2021;12:721917. doi:10.3389/fimmu.2021.721917
44. Collins C, Sharpe E, Silber A, Kulke S, Hsieh EWY. Congenital athymia: genetic etiologies, clinical manifestations, diagnosis, and treatment. J Clin Immunol. 2021;41(5):881–895. doi:10.1007/s10875-021-01059-7
45. FDA. Rethymic; 2021. Available from: https://www.fda.gov/vaccines-blood-biologics/rethymic.
46. HTA. Human tissue act 2004; 2004. Available from: https://www.hta.gov.uk/guidance-professionals/hta-legislation/human-tissue-act-2004.
47. Condino-Neto A, Espinosa-Rosales FJ. Changing the lives of people with primary immunodeficiencies (PI) with early testing and diagnosis. Front Immunol. 2018;9:1439. doi:10.3389/fimmu.2018.01439
48. Nordin J, Solis L, Prevot J, et al. The PID principles of care: where are we now? A global status report based on the PID life index. Front Immunol. 2021;12:780140. doi:10.3389/fimmu.2021.780140
49. Aiuti A, Pasinelli F, Naldini L. Ensuring a future for gene therapy for rare diseases. Nat Med. 2022;28(10):1985–1988. doi:10.1038/s41591-022-01934-9
50. Fox T, Bueren J, Candotti F, et al. Access to gene therapy for rare diseases when commercialization is not fit for purpose. Nat Med. 2023. doi:10.1038/s41591-023-02208-8
51. GOSH. Europe-wide group launched to boost access to life-saving gene therapies; 2022. Available from: https://www.gosh.nhs.uk/news/euro-group-to-boost-access-to-life-saving-gene-therapies/.
52. Speckmann C, Nennstiel U, Honig M, et al. Prospective newborn screening for SCID in Germany: a first analysis by the Pediatric Immunology Working Group (API). J Clin Immunol. 2023. doi:10.1007/s10875-023-01450-6
53. Argudo-Ramirez A, Martin-Nalda A, Gonzalez de Aledo-Castillo JM, et al. Newborn screening for SCID. Experience in Spain (Catalonia). Int J Neonatal Screen. 2021;7(3):46. doi:10.3390/ijns7030046
54. Heather N, de Hora M, Brothers S, Grainger P, Knoll D, Webster D. Introducing newborn screening for severe combined immunodeficiency-the New Zealand experience. Int J Neonatal Screen. 2022;8(2):33. doi:10.3390/ijns8020033
55. Lev A, Sharir I, Simon AJ, et al. Lessons learned from five years of newborn screening for severe combined immunodeficiency in Israel. J Allergy Clin Immunol Pract. 2022;10(10):2722–31 e9. doi:10.1016/j.jaip.2022.04.013
56. Howley E, Buckland M, Giuliani S, et al. Impact of the COVID-19 pandemic on the delivery of the European thymus transplantation programme. Arch Dis Child. 2021;106:A455.2–A6.
57. Pai SY, Logan BR, Griffith LM, et al. Transplantation outcomes for severe combined immunodeficiency, 2000–2009. N Engl J Med. 2014;371(5):434–446. doi:10.1056/NEJMoa1401177
58. Mahlaoui N, Picard C, Bach P, et al. Genetic diagnosis of primary immunodeficiencies: a survey of the French national registry. J Allergy Clin Immunol. 2019;143(4):1646–9 e10. doi:10.1016/j.jaci.2018.12.994
59. Shai S, Perez-Becker R, Andres O, et al. Incidence of SCID in Germany from 2014 to 2015 an ESPED* Survey on Behalf of the API*** Erhebungseinheit fur Seltene Padiatrische Erkrankungen in Deutschland (German Paediatric Surveillance Unit) ** Arbeitsgemeinschaft Padiatrische Immunologie. J Clin Immunol. 2020;40(5):708–717. doi:10.1007/s10875-020-00782-x
60. Elfeky R, Shah RM, Unni MNM, et al. New graft manipulation strategies improve the outcome of mismatched stem cell transplantation in children with primary immunodeficiencies. J Allergy Clin Immunol. 2019;144(1):280–293. doi:10.1016/j.jaci.2019.01.030
61. Kreins AY, Velasco HF, Cheong KN, et al. Long-term immune recovery after hematopoietic stem cell transplantation for ADA deficiency: a single-center experience. J Clin Immunol. 2022;42(1):94–107. doi:10.1007/s10875-021-01145-w
62. Kohn DB, Booth C, Shaw KL, et al. Autologous ex vivo lentiviral gene therapy for adenosine deaminase deficiency. N Engl J Med. 2021;384(21):2002–2013. doi:10.1056/NEJMoa2027675
63. Magnani A, Semeraro M, Adam F, et al. Long-term safety and efficacy of lentiviral hematopoietic stem/progenitor cell gene therapy for Wiskott-Aldrich syndrome. Nat Med. 2022;28(1):71–80. doi:10.1038/s41591-021-01641-x
64. Ottaviano G, Georgiadis C, Gkazi SA, et al. Phase 1 clinical trial of CRISPR-engineered CAR19 universal T cells for treatment of children with refractory B cell leukemia. Sci Transl Med. 2022;14(668):eabq3010. doi:10.1126/scitranslmed.abq3010
65. Cordoba S, Onuoha S, Thomas S, et al. CAR T cells with dual targeting of CD19 and CD22 in pediatric and young adult patients with relapsed or refractory B cell acute lymphoblastic leukemia: a phase 1 trial. Nat Med. 2021;27(10):1797–1805. doi:10.1038/s41591-021-01497-1
66. Meyts I, Bousfiha A, Duff C, et al. Primary immunodeficiencies: a decade of progress and a promising future. Front Immunol. 2020;11:625753. doi:10.3389/fimmu.2020.625753
67. Ambrosini A, Quinlivan R, Sansone VA, et al. ”Be an ambassador for change that you would like to see”: a call to action to all stakeholders for co-creation in healthcare and medical research to improve quality of life of people with a neuromuscular disease. Orphanet J Rare Dis. 2019;14(1):126. doi:10.1186/s13023-019-1103-8
68. Nguyen CQ, Alba-Concepcion K, Palmer EE, Scully JL, Millis N, Farrar MA. The involvement of rare disease patient organisations in therapeutic innovation across rare paediatric neurological conditions: a narrative review. Orphanet J Rare Dis. 2022;17(1):167. doi:10.1186/s13023-022-02317-6
69. Cowan MJ. The Primary Immune Deficiency Treatment Consortium: how can it improve definitive therapy for PID? Expert Rev Clin Immunol. 2016;12(10):1007–1009. doi:10.1080/1744666X.2016.1216317
70. Stein S, Bogard E, Boice N, et al. Principles for interactions with biopharmaceutical companies: the development of guidelines for patient advocacy organizations in the field of rare diseases. Orphanet J Rare Dis. 2018;13(1):18. doi:10.1186/s13023-018-0761-2
71. Bernhardt JM, Alber J, Gold RS. A social media primer for professionals: digital dos and don’ts. Health Promot Pract. 2014;15(2):168–172. doi:10.1177/1524839913517235
72. Ventola CL. Social media and health care professionals: benefits, risks, and best practices. P T. 2014;39(7):491–520.
73. PIF. Co-production matters; 2021. Available from: https://pifonline.org.uk/download/file/562/.
74. Ashcraft LE, Asato M, Houtrow AJ, Kavalieratos D, Miller E, Ray KN. Parent empowerment in pediatric healthcare settings: a systematic review of observational studies. Patient. 2019;12(2):199–212. doi:10.1007/s40271-018-0336-2
75. Howley E, Kreins, A, Worth A, Davies EG. Educating families through purposefully designed patient information for the thymus transplant pathway. Arch Dis Child. 2019;104:A17.2–A.
76. Markert ML, Devlin BH, Chinn IK, McCarthy EA, Li YJ. Factors affecting success of thymus transplantation for complete DiGeorge anomaly. Am J Transplant. 2008;8(8):1729–1736. doi:10.1111/j.1600-6143.2008.02301.x
77. Lee JH, Markert ML, Hornik CP, et al. Clinical course and outcome predictors of critically ill infants with complete DiGeorge anomaly following thymus transplantation. Pediatr Crit Care Med. 2014;15(7):e321–6. doi:10.1097/PCC.0000000000000219
78. Markert ML, Li J, Devlin BH, et al. Use of allograft biopsies to assess thymopoiesis after thymus transplantation. J Immunol. 2008;180(9):6354–6364. doi:10.4049/jimmunol.180.9.6354
79. Collins C, Kim-Chang JJ, Hsieh E, et al. Economic burden of congenital athymia in the United States for patients receiving supportive care during the first 3 years of life. J Med Econ. 2021;24(1):962–971. doi:10.1080/13696998.2021.1962129
80. Angelis A, Tordrup D, Kanavos P. Socio-economic burden of rare diseases: a systematic review of cost of illness evidence. Health Policy (New York). 2015;119(7):964–979. doi:10.1016/j.healthpol.2014.12.016
81. Hsieh EWY, Kim-Chang JJ, Kulke S, Silber A, O’Hara M, Collins C. Defining the clinical, emotional, social, and financial burden of congenital athymia. Adv Ther. 2021;38(8):4271–4288. doi:10.1007/s12325-021-01820-9
82. Kuo DZ, Goudie A, Cohen E, et al. Inequities in health care needs for children with medical complexity. Health Aff. 2014;33(12):2190–2198. doi:10.1377/hlthaff.2014.0273
83. Toomey SL, Chien AT, Elliott MN, Ratner J, Schuster MA. Disparities in unmet need for care coordination: the national survey of children’s health. Pediatrics. 2013;131(2):217–224. doi:10.1542/peds.2012-1535
84. Martin N, Mikhaelian M, Cytrynbaum C, et al. 22q11.2 deletion syndrome: attitudes towards disclosing the risk of psychiatric illness. J Genet Couns. 2012;21(6):825–834. doi:10.1007/s10897-012-9517-7
85. Allen TM, Hersh J, Schoch K, Curtiss K, Hooper SR, Shashi V. Association of the family environment with behavioural and cognitive outcomes in children with chromosome 22q11.2 deletion syndrome. J Intellect Disabil Res. 2014;58(1):31–47. doi:10.1111/jir.12054
86. Cortes-Martin J, Penuela NL, Sanchez-Garcia JC, Montiel-Troya M, Diaz-Rodriguez L, Rodriguez-Blanque R. Deletion syndrome 22q11.2: a systematic review. Children. 2022;9(8):1168. doi:10.3390/children9081168
87. Fung WL, Butcher NJ, Costain G, et al. Practical guidelines for managing adults with 22q11.2 deletion syndrome. Genet Med. 2015;17(8):599–609. doi:10.1038/gim.2014.175
88. Vo OK, McNeill A, Vogt KS. The psychosocial impact of 22q11 deletion syndrome on patients and families: a systematic review. Am J Med Genet A. 2018;176(10):2215–2225. doi:10.1002/ajmg.a.38673
89. Joyce P, O’Rourke C, McDermott B, Heussler H. Health-related quality of life in 22q11.2 deletion syndrome: the child’s perspective. J Paediatr Child Health. 2018;54(3):311–315. doi:10.1111/jpc.13746
90. Day JW, Elfeky R, Nicholson B, et al. Retrospective, landmark analysis of long-term adult morbidity following allogeneic HSCT for inborn errors of immunity in infancy and childhood. J Clin Immunol. 2022;42(6):1230–1243. doi:10.1007/s10875-022-01278-6
91. McGettigan P, Alonso Olmo C, Plueschke K, et al. Patient registries: an underused resource for medicines evaluation: operational proposals for increasing the use of patient registries in regulatory assessments. Drug Saf. 2019;42(11):1343–1351. doi:10.1007/s40264-019-00848-9
92. Boulanger V, Schlemmer M, Rossov S, Seebald A, Gavin P. Establishing patient registries for rare diseases: rationale and challenges. Pharmaceut Med. 2020;34(3):185–190. doi:10.1007/s40290-020-00332-1
93. Shillitoe B, Bangs C, Guzman D, et al. The United Kingdom Primary Immune Deficiency (UKPID) registry 2012 to 2017. Clin Exp Immunol. 2018;192(3):284–291. doi:10.1111/cei.13125
94. Scheible R, Rusch S, Guzman D, Mahlaoui N, Ehl S, Kindle G. The NEW ESID online database network. Bioinformatics. 2019;35(24):5367–5369. doi:10.1093/bioinformatics/btz525
95. El-Helou SM, Biegner AK, Bode S, et al. The German National Registry of Primary Immunodeficiencies (2012–2017). Front Immunol. 2019;10:1272. doi:10.3389/fimmu.2019.01272
96. Ryan P, Redenbaugh V, McGucken J, et al. Inborn errors of immunity on the Island of Ireland - a cross-jurisdictional UKPID/ESID registry report. J Clin Immunol. 2022;42(6):1293–1299. doi:10.1007/s10875-022-01274-w
97. EMA. Patient registries; 2015. Available from: https://www.ema.europa.eu/en/human-regulatory/post-authorisation/patient-registries.
98. Rondeau E, Cataland SR, Al-Dakkak I, Miller B, Webb NJA, Landau D. Eculizumab safety: five-year experience from the global atypical hemolytic uremic syndrome registry. Kidney Int Rep. 2019;4(11):1568–1576. doi:10.1016/j.ekir.2019.07.016
99. Volkova N, Moy K, Evans J, et al. Disease progression in patients with cystic fibrosis treated with ivacaftor: data from national US and UK registries. J Cyst Fibros. 2020;19(1):68–79. doi:10.1016/j.jcf.2019.05.015
100. Haverman L, Limperg PF, Young NL, Grootenhuis MA, Klaassen RJ. Paediatric health-related quality of life: what is it and why should we measure it? Arch Dis Child. 2017;102(5):393–400. doi:10.1136/archdischild-2015-310068
101. Germain N, Aballea S, Toumi M. Measuring the health-related quality of life in young children: how far have we come? J Mark Access Health Policy. 2019;7(1):1618661. doi:10.1080/20016689.2019.1618661
102. Abd Hamid IJ, Slatter MA, McKendrick F, Pearce MS, Gennery AR. Long-term health outcome and quality of life post-HSCT for IL7Ralpha-, Artemis-, RAG1- and RAG2-deficient severe combined immunodeficiency: a single center report. J Clin Immunol. 2018;38(6):727–732. doi:10.1007/s10875-018-0540-9
103. Shah AJ, Sokolic R, Logan B, et al. Quality of life of patients with wiskott Aldrich syndrome and X-linked thrombocytopenia: a study of the Primary Immune Deficiency Consortium (PIDTC), Immune Deficiency Foundation, and the Wiskott-Aldrich Foundation. J Clin Immunol. 2019;39(8):786–794. doi:10.1007/s10875-019-00689-2
104. Cole T, McKendrick F, Titman P, et al. Health related quality of life and emotional health in children with chronic granulomatous disease: a comparison of those managed conservatively with those that have undergone haematopoietic stem cell transplant. J Clin Immunol. 2013;33(1):8–13. doi:10.1007/s10875-012-9758-0
105. Titman P, Allwood Z, Gilmour C, et al. Quality of life in children with primary antibody deficiency. J Clin Immunol. 2014;34(7):844–852. doi:10.1007/s10875-014-0072-x
106. Jiang F, Torgerson TR, Ayars AG. Health-related quality of life in patients with primary immunodeficiency disease. Allergy Asthma Clin Immunol. 2015;11:27. doi:10.1186/s13223-015-0092-y
107. Peshko D, Kulbachinskaya E, Korsunskiy I, et al. Health-related quality of life in children and adults with primary immunodeficiencies: a systematic review and meta-analysis. J Allergy Clin Immunol Pract. 2019;7(6):1929–57 e5. doi:10.1016/j.jaip.2019.02.013
108. Goodwin J, McCormack L, Campbell LE. “You don’t know until you get there”: the positive and negative “lived” experience of parenting an adult child with 22q11.2 deletion syndrome. Health Psychol. 2017;36(1):45–54. doi:10.1037/hea0000415
109. Zinkernagel RM, Althage A. On the role of thymic epithelium vs. bone marrow-derived cells in repertoire selection of T cells. Proc Natl Acad Sci U S A. 1999;96(14):8092–8097. doi:10.1073/pnas.96.14.8092
110. Liston A, Lesage S, Wilson J, Peltonen L, Goodnow CC. Aire regulates negative selection of organ-specific T cells. Nat Immunol. 2003;4(4):350–354. doi:10.1038/ni906
111. Kreins AY, Junghanns F, Mifsud W, et al. Correction of both immunodeficiency and hypoparathyroidism by thymus transplantation in complete DiGeorge syndrome. Am J Transplant. 2020;20(5):1447–1450. doi:10.1111/ajt.15668
112. Kwun J, Li J, Rouse C, et al. Cultured thymus tissue implantation promotes donor-specific tolerance to allogeneic heart transplants. JCI Insight. 2020;5(11):65.
113. Haque A, Engel J, Teichmann SA, Lonnberg T. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med. 2017;9(1):75. doi:10.1186/s13073-017-0467-4
114. Rood JE, Maartens A, Hupalowska A, Teichmann SA, Regev A. Impact of the Human Cell Atlas on medicine. Nat Med. 2022;28:2486–2496. doi:10.1038/s41591-022-02104-7
115. Haddad E, Hoenig M. Hematopoietic stem cell transplantation for severe combined immunodeficiency (SCID). Front Pediatr. 2019;7:481. doi:10.3389/fped.2019.00481
116. Furmanski AL, O’Shaughnessy RF, Saldana JI, et al. T-cell reconstitution after thymus xenotransplantation induces hair depigmentation and loss. J Invest Dermatol. 2013;133(5):1221–1230. doi:10.1038/jid.2012.492
117. Kelly P, Marriott C, Howley E, et al. Cryopreservation and recovery of thymus tissue prior to transplantation into paediatric patients with DiGeorge syndrome. Arch Dis Child. 2019;104:A13.1–A.
118. Bhalla P, Su DM, van Oers NSC. Thymus functionality needs more than a few TECs. Front Immunol. 2022;13:864777. doi:10.3389/fimmu.2022.864777
119. Bhalla P, Du Q, Kumar A, et al. Mesenchymal cell replacement corrects thymic hypoplasia in murine models of 22q11.2 deletion syndrome. J Clin Invest. 2022;132(22). doi:10.1172/JCI160101
120. Bortolomai I, Sandri M, Draghici E, et al. Gene modification and three-dimensional scaffolds as novel tools to allow the use of postnatal thymic epithelial cells for thymus regeneration approaches. Stem Cells Transl Med. 2019;8(10):1107–1122. doi:10.1002/sctm.18-0218
121. Fan Y, Tajima A, Goh SK, et al. Bioengineering thymus organoids to restore thymic function and induce donor-specific immune tolerance to allografts. Mol Ther. 2015;23(7):1262–1277. doi:10.1038/mt.2015.77
122. Hun M, Barsanti M, Wong K, Ramshaw J, Werkmeister J, Chidgey AP. Native thymic extracellular matrix improves in vivo thymic organoid T cell output, and drives in vitro thymic epithelial cell differentiation. Biomaterials. 2017;118:1–15. doi:10.1016/j.biomaterials.2016.11.054
123. Campinoti S, Gjinovci A, Ragazzini R, et al. Reconstitution of a functional human thymus by postnatal stromal progenitor cells and natural whole-organ scaffolds. Nat Commun. 2020;11(1):6372. doi:10.1038/s41467-020-20082-7
124. Chhatta AR, Cordes M, Hanegraaf MAJ, et al. De novo generation of a functional human thymus from induced pluripotent stem cells. J Allergy Clin Immunol. 2019;144(5):1416–9 e7. doi:10.1016/j.jaci.2019.05.042
125. Zeleniak A, Wiegand C, Liu W, et al. De novo construction of T cell compartment in humanized mice engrafted with iPSC-derived thymus organoids. Nat Methods. 2022;19(10):1306–1319. doi:10.1038/s41592-022-01583-3
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.