Back to Journals » Drug Design, Development and Therapy » Volume 10
Computational and nonglycosylated systems: a simpler approach for development of nanosized PEGylated proteins
Authors Mirzaei H, Kazemi B, Bandehpour M, Shoari A, Asgary V ![]() , Shafiee Ardestani M
, Shafiee Ardestani M ![]() , Madadkar-Sobhani A, Cohan R
, Madadkar-Sobhani A, Cohan R ![]()
Received 14 October 2015
Accepted for publication 14 December 2015
Published 16 March 2016 Volume 2016:10 Pages 1193—1200
DOI https://doi.org/10.2147/DDDT.S98323
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Hadi Mirzaei,1 Bahram Kazemi,1 Mojgan Bandehpour,1 Alireza Shoari,2 Vahid Asgary,2 Mehdi Shafiee Ardestani,3 Armin Madadkar-Sobhani,4 Reza Ahangari Cohan2
1Department of Biotechnology, School of Medicine, Shahid Beheshti University of Medical Sciences, Tehran, Iran; 2Department of Pilot Nanobiotechnology, Pasteur Institute of Iran, Tehran, Iran; 3Department of Radiopharmacy, Pharmacy Faculty, Tehran University of Medical Sciences, Tehran, Iran; 4Faculty of Science, University of Ontario Institute of Technology, Oshawa, Canada
Abstract: Cysteine PEGylation includes several steps, and is difficult to manage in practice. In the current investigation, the cysteine PEGylation of erythropoietin analogs was examined using computational and nonglycosylated systems to define a simpler approach for specific PEGylation. Two model analogs (E31C and E89C) were selected for PEGylation based on lowest structural deviation from the native form, accessibility, and nucleophilicity of the free thiol group. The selected analogs were cloned and the expression was assessed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and Western blot using Coomassie blue staining and anti-His monoclonal antibody, respectively. PEGylation with 20 kDa mPEG-maleimide resulted in 79% and 82% conjugation yield for E31C and E89C nonglycosylated erythropoietin (ngEPO) analogs, respectively. The size distribution and charge analysis showed an increase in size and negative charge of the PEGylated forms compared with nonconjugated ones. Biological assay revealed that E31C and E89C mutations and subsequent PEGylation of ngEPO analogs have no deleterious effects on in vitro biological activity when compared to CHO-derived recombinant human erythropoietin. In addition, PEG-conjugated ngEPOs showed a significant increase in plasma half-lives after injection into rats when compared to nonconjugated ones. The development of the cysteine-PEGylated proteins using nonglycosylated expression system and in silico technique can be considered an efficient approach in terms of optimization of PEGylation parameters, time, and cost.
Keywords: site-specific PEGylation, nonglycosylated expression systems, computational methods, erythropoietin
Introduction
Recombinant human erythropoietin (rhEPO) is employed in the treatment of anemia, but like other therapeutic proteins, it suffer from short in vivo half-life. It must be injected 2–3 times/wk for the achievement of therapeutic effect. Erythropoietin (EPO) is structurally a glycoprotein, with 40% of its molecular weight made of glycan (N-glycosylation sites at N24, N38, N83 and O-glycosylation site at S126). Glycosylation plays an important role in maintaining EPO stability and increasing half-life as well as in vivo activity. Since human-like glycosylation only exists in eukaryotic cells, the first-generation of rhEPO was obtained from a CHO cell line transfected with the human erythropoietin gene. rhEPO produced in BHK cells has also been used clinically, in some European Union countries outside Europe and in North America.1,2
Human erythropoietin has been clinically used in the treatment of anemia caused by chronic kidney disease, chemotherapy, and acquired immune deficiency syndrome-related complications. Due to the pharmaceutical and therapeutic importance of EPO, many investigations have studied its in vivo half-life. PEGylation, one of the most promising tools, performed for in vivo half-life augmentation of EPO, is defined as the attachment of polyethylene glycol (PEG) to the molecule.3 For successful particular attachment of PEG polymer to a molecule, we need a reactive group on PEG polymer that reacts to a specific site of the molecule. PEGylation is also employed in chemistry for different purposes such as targeting and drug delivery systems, nanotechnology, sustained release of compounds, increasing hydrophilicity, and enhanced in vivo circulation time of therapeutic proteins in the body. PEGylation increases the size of the molecule, leads to lower renal clearance and shielding effect against degradable enzymes, and enhances in vivo half-life. In addition, PEGylation leads to better patient compliance, reduces usage frequency, has lesser side effects, and also decreases medical costs for injecting such drugs.4
One of the most important techniques of PEGylation is cysteine PEGylation. In this method, one of the amino acids in the protein, which is not crucial, neither in the activity nor the structure of the protein through genetic engineering, is replaced with cysteine. It is a critical step to define a proper site for the cysteine inside the protein. In the next step, the cysteine analog is expressed and subjected to PEGylation reaction with the PEG polymer that specifically reacts with the free thiol (–SH) group of replaced cysteine residue.5 As mentioned earlier, cysteine PEGylation includes several steps that are practically difficult to manage and are time-consuming and cost-consuming. Pitfalls include selection of PEG-attached sites (replacement of amino acids with cysteine) and the requirement of large amounts of protein to optimize the PEGylation parameters (eg, size of polymer, time, temperature, pH, and yield).6–8 The problem becomes more complicated when the protein is glycosylated. Despite advances in recombinant DNA technology, mammalian expression systems have not produced sufficient amount of protein. In addition, the complexity of attached glycans is not even precisely similar to human, leading to heterogeneity of the finished product.9,10 Therefore, to overcome the previously mentioned obstacles, a rational and effortless method for developing of nanosized PEGylated proteins is necessary. Hence, in this study, site-specific PEGylation (cysteine PEGylation) of EPO was examined using computational and nonglycosylated systems to define a simpler approach for site-specific PEGylation of proteins for both therapeutic and nontherapeutic goals.
Materials and methods
In silico analysis
Computational investigation was performed based on a previously described method.11 Briefly, three-dimensional (3D) structures of analogs were generated by homology modeling using MODELLER and confirmed using discrete optimized protein energy (DOPE) score profile and PROCHECK. The molecular dynamic (MD) simulation of modeled analogs was examined by GROMACS 3.3 package and Gromos96 force field at a constant temperature (300 K) and pressure (1 bar) for 5–15 ns. After calculating the average structures, cysteine analogs were subjected to screening based on the root-mean-square deviation (RMSD) from native EPO, surface area accessibility of engineered cysteine residues (cysteine-SAA), and pKa values of the thiol group using Qmol, PROPKA 2.0, and GETAREA, respectively. Finally, two cysteine analogs, E31C and E89C, which had better criteria were selected for experimental investigation.
Cloning of E31C and E89C ngEPO analogs
E31C and E89C analogs were selected for PEGylation on the basis of the results of the computational study. Cysteine analog 31 (E31C) cDNA was previously synthesized, using overlap polymerase chain reaction method,11 and cysteine analog 89 (E89C) cDNA was ordered to be synthesized by Bioneer Inc. (Daejeon, South Korea). Nde I and Hind III restriction sites and His-tag sequence were introduced using 5′-ACTCATATGGCCCCACCACGCCTCATCTGTGAC-3′and 5′-AAGCTTCAATGATGATGATGATGATGGTCCCCTGTCCTGCAG-3′ as forward and reverse primers. The constructs were digested and cloned into the pET-26b expression vector (Gene Bank, Pasteur Institute of Iran, Tehran, Iran). The accuracy of constructs were confirmed by restriction map analysis and sequencing.
Expression, identification, and purification of E31C and E89C ngEPO analogs
The constructs were transferred into the BL21 (DE3) Escherichia coli host cell (Gene Bank) using CaCl2 transformation method. The expression of analogs was induced using 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) (Sigma-Aldrich, Munich, Germany) and analyzed using 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) using Mini-PROTEIN® Tetra Cell system (Bio-Rad Laboratories, Hercules, CA, USA); the gel was developed using the Coomassie blue staining technique. The identification of expressed analogs was carried out by Western blot analysis, using a Mini Trans-Blot® electrophoretic transfer cell (Bio-Rad Laboratories), with anti-His mouse monoclonal antibody (1/1,000 dilution) and horseradish peroxidase (HRP)-conjugated rabbit anti-mouse IgG antibody (1/10,000 dilution) as primary and secondary antibodies (Qiagen Inc., Hilden, Germany) and 3,3′-diaminobenzidine as substrate (Qiagen Inc.). For purification, the cultured bacteria were centrifuged at 8,000× g for 15 minutes at 4°C, and the pellets were resuspended in 20 mM Tris–HCl (pH 8.0), containing 1 mM ethylenediaminetetraacetic acid (EDTA), and lysed using a Sonicator (Sartorius, Göttingen, Germany). The precipitates from the lysate samples were dissolved in 20 mM Tris–HCl (pH 8.0), containing 8 M urea, 1 mM EDTA, and 1% Triton X-100. Finally, the solutions were purified using Ni-NTA affinity chromatography as described by the manufacturer (Qiagen Inc.). A buffer containing 20 mM Tris–HCl, 80 mM imidazole, and 4 M urea (pH 8.0) and a buffer containing 20 mM Tris–HCl, 500 mM imidazole, and 2 M urea (pH 8.0) were used as washing and elution buffers, respectively.
PEG-conjugation and purification of PEGylated E31C and E89C ngEPO analogs
Thirtyfold molar excess of 20 kDa methoxypoly(ethylene glycol (mPEG)-maleimide (SunBio Inc.) was added to each analog solution (0.5 mg/mL) in 50 mM Tris buffer pH 8.0, incubated for 3 hours at room temperature. The reaction mixture was then diluted with 20 mM phosphate buffer (pH 7.0) and loaded onto a 6 mL pre-equilibrated cation exchange Resource S column (GE Healthcare, Piscataway, NJ, USA). After washing with 20 mM phosphate buffer (pH 7.0), bonded proteins were eluted using a gradient 20 mM phosphate buffer containing 500 mM NaCl adjusted to pH 7.0. Finally, PEGylated forms were analyzed by SDS-PAGE, Coomassie blue staining, using Mini-PROTEIN® Tetra Cell system (Bio-Rad Laboratories).
Characterization of PEGylated E31C and E89C ngEPOs
The purity and yield of the conjugates were also determined using size-exclusion high-performance liquid chromatography (using a Shodex column PROTEIN KW-802.5, Showa Denko, Kanagawa, Japan). A solution of 50 mM sodium phosphate buffer (pH 7.0) containing 150 mM NaCl was used as an equilibration and elution buffer. The biological activity was assessed based on the proliferation of UT-7 cells. UT-7 cells (ACC 137; DMSZ, Braunschweig, Germany) were grown in α-minimum essential medium supplemented with 20% fetal bovine serum, 40 μg/mL gentamycin, 2 mM glutamine, and 5 ng/mL granulocyte-macrophage colony-stimulating factor (Sigma-Aldrich). UT-7 cells (5×103) were seeded in each well of a 96-well plate and incubated at 37°C in a humidified 5% CO2 incubator. Twofold serial dilutions of the protein samples and standard were prepared in assay media. Approximately 100 μL of the diluted protein samples was added in triplicate in each well of a 96-well microplate. Phosphate-buffered saline and 200 ng/mL rhEPO (R&D Systems, Minneapolis, MN, USA) were added to the cultured cells as a negative and a positive control, respectively. The proliferation of cells was measured by MTT assay at 570 nm, using Epoch Microplate Spectrophotometer (BioTek, Winooski, VT, USA). In addition, size distribution and the charge of macromolecules were assessed using dynamic light scattering technique (Zetasizer Nano ZS, Malvern Instruments, Malvern, UK).
Pharmacokinetic investigation
Animal experiments were carried out once ethical clearance was obtained from the Animal Ethics Committee of the Pasteur Institute of Iran. All animals were housed at room temperature (20–23°C) on a 12 h light/dark cycle, with unlimited access to food and water and adapted for one week before the test. Animal experiments were performed based on the ethical standards in the Declaration of Helsinki. The five Sprague Dawley rats (300±30 g of weight) in each group subcutaneously received a single dose of 100 μg protein/kg. The blood samples were collected from the lateral tail vein, and the plasma was analyzed by enzyme-linked immunosorbent assay method (Roche Diagnostics GmbH, Penzberg, Germany). The terminal half-life and the area under the curve were determined using linear regression of the last three plasma concentrations and a linear trapezoidal method with extrapolation to infinity, respectively.
Molecular drawing and statistical analysis
Statistical analysis was made using Student’s t-test and one-way multivariate analysis of variance to compute the level of significance (P-value), and P<0.05 was considered statistically significant. The schematic representation of cysteine analogs was identified using VMD software version 1.8.7, and Microsoft Excel 2007 was used to create the RMSD and DOPE graphs.
Results
Selection of cysteine analogs
The 3D structures of 115 selected analogs based on literature review (Supplementary materials 1 and 2) were generated using homology modeling, and the quality of models was verified by Ramachandran plot and DOPE score assessments (Supplementary materials 3–5). RMSD during MD was plotted versus time for stability and average structure calculations (Supplementary material 5). Frothy stable cysteine analogs during MD simulations were then subjected to screening based on the SAA of replaced cysteine residue (Supplementary material 6), pKa values of the thiol group, and whole RMSD versus native EPO (Supplementary material 7). Finally, based on the minimum whole RMSD and maximum cysteine-SSA and pKa, the E31C and E89C analogs were chosen for experimental analysis (Table 1, Figure 1).
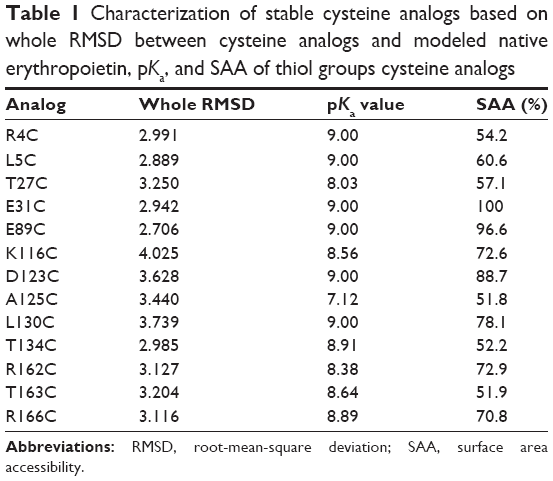 | Table 1 Characterization of stable cysteine analogs based on whole RMSD between cysteine analogs and modeled native erythropoietin, pKa, and SAA of thiol groups cysteine analogs |
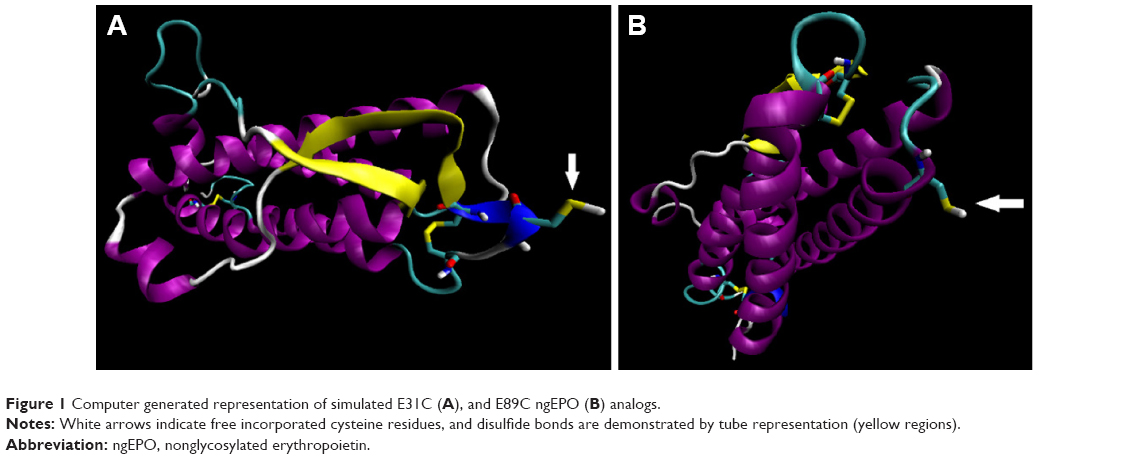 | Figure 1 Computer generated representation of simulated E31C (A), and E89C ngEPO (B) analogs. |
Expression, identification, and purification of E31C and E89C ngEPO analogs
The expression was found to be optimum at 4 hours after induction with 1 mM IPTG. The expression was confirmed by observing a band approximately 18.4 kDa on 12% SDS-PAGE, using Coomassie blue staining (Figure 2A). The bands were identified by Western blot analysis, using anti-His monoclonal antibody (Figure 2B).
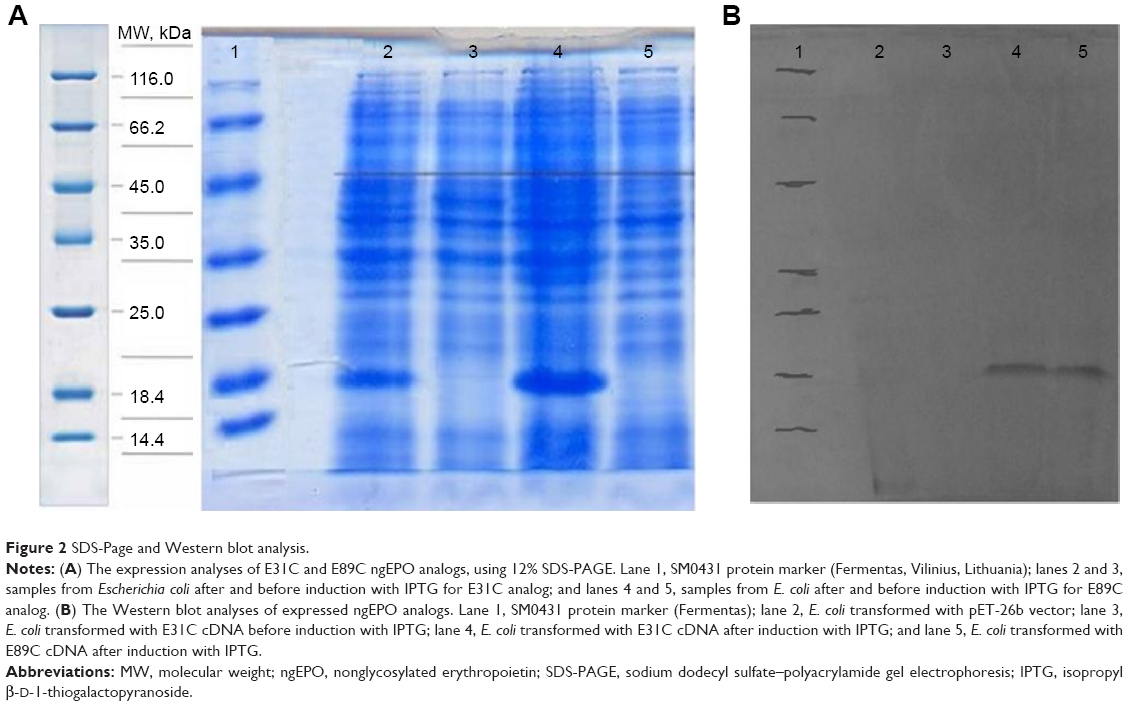 | Figure 2 SDS-Page and Western blot analysis. |
In the purification step, cellular proteins were passed through the column and only the E31C and E89C nonglycosylated erythropoietin (ngEPO) analogs containing His-tag attached to the column. The bonded proteins were eluted using elution buffer containing 500 mM imidazole (Figure 3).
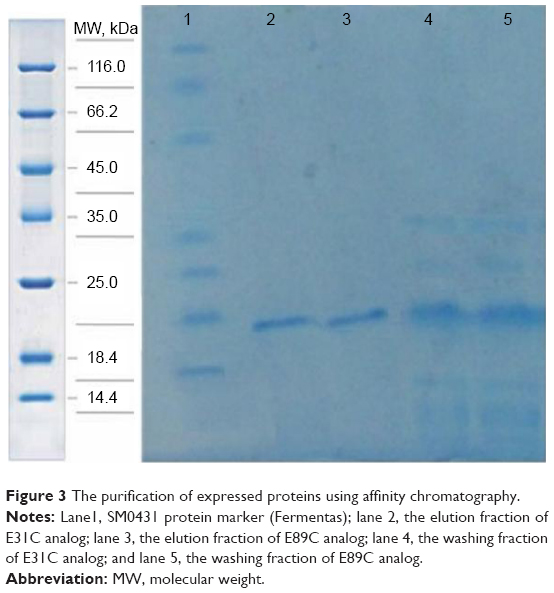 | Figure 3 The purification of expressed proteins using affinity chromatography. |
Characterization of PEGylated E31C and E89C ngEPOs
Conjugated E31C and E89C ngEPO analogs were analyzed using 12% SDS-PAGE, and the bands were visualized by Coomassie blue staining. As shown in Figure 4, bands approximately 40 kDa were attributed to PEGylated ngEPO analogs.
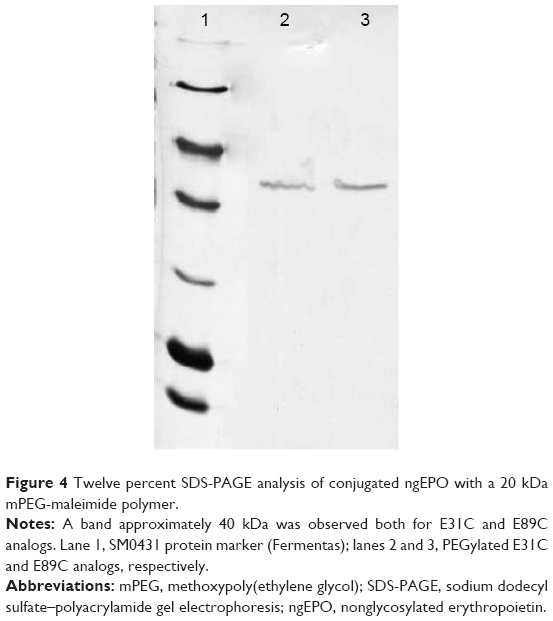 | Figure 4 Twelve percent SDS-PAGE analysis of conjugated ngEPO with a 20 kDa mPEG-maleimide polymer. |
The PEGylated and nonPEGylated EPO cysteine analogs’ biological activity were analyzed using MTT assay method. UT-7 cells were cultured in the presence of different concentrations of PEGylated and nonPEGylated analogs. The glycosylated rhEPO (R&D Systems) was used as positive control in the assay (Table 2). EC50 (ng/mL) was defined as the protein concentration that stimulates 50% of maximal stimulation. The conjugation yield was measured to be 79% and 82% for E31C and E89C analogs, respectively. In addition, PEG-conjugated ngEPOs showed approximately tenfold increase in plasma half-lives after injection into rats when compared to nonconjugated ones (Table 2).
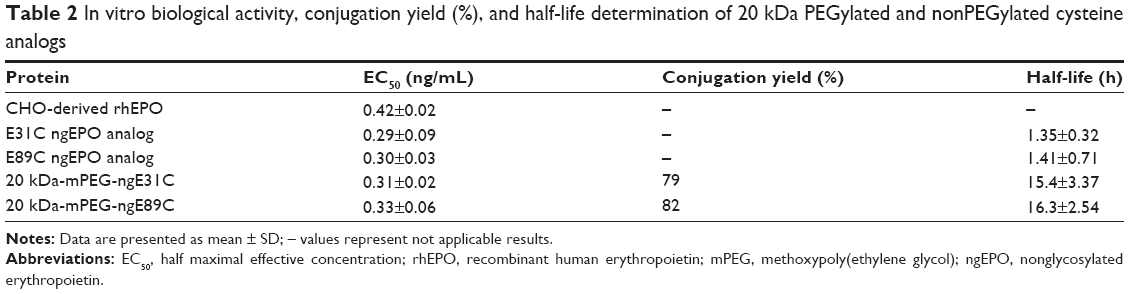 | Table 2 In vitro biological activity, conjugation yield (%), and half-life determination of 20 kDa PEGylated and nonPEGylated cysteine analogs |
The size distribution and the charge of conjugated and nonconjugated ngEPO analogs were also investigated. As seen in Figure 5, the zeta potential of the E89C ngEPO analog was switched to the negative range due to attachment of 20 kDa PEG polymer. In addition, the size of E89C ngEPO analog was increased when a 20 kDa mPEG polymer attached to the analog due to an increase in hydrodynamic radius of conjugated form (Figure 5). The results were the same for conjugated and nonconjugated form of E31C ngEPO analog (data not shown).
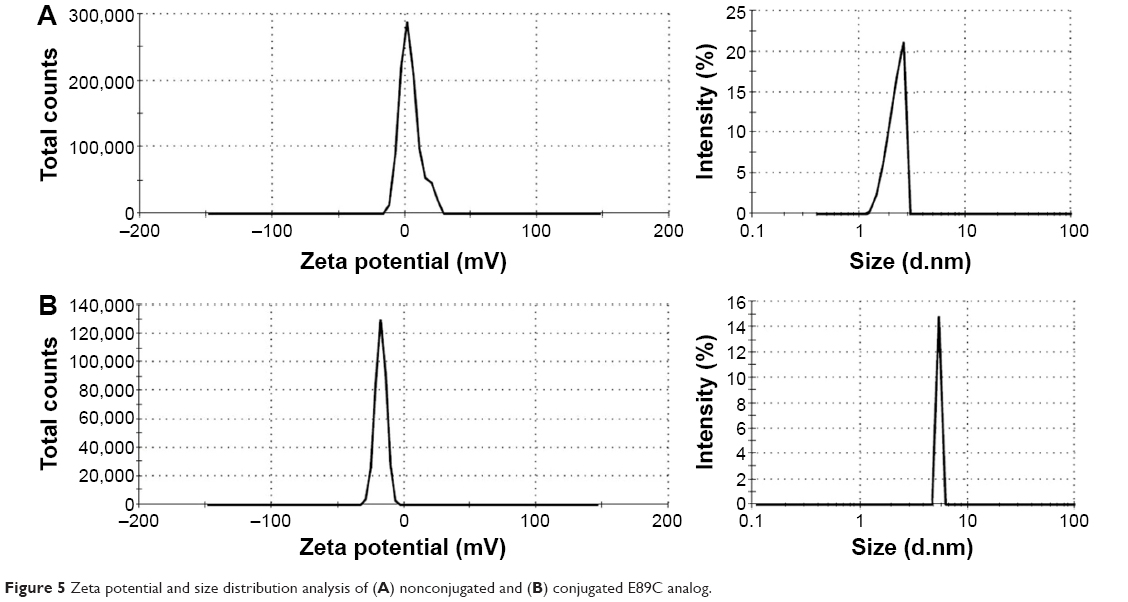 | Figure 5 Zeta potential and size distribution analysis of (A) nonconjugated and (B) conjugated E89C analog. |
Discussion
Recombinant therapeutic proteins have more limitations for application as a drug than chemical ones, such as sensitivity to temperature, pH, organic solvents, and degradable enzymes that lead to lower activity and stability either in vitro or in vivo. Therefore, protein engineering plays undoubtedly a remarkable role in biopharmaceutical development to improve pharmacodynamic and pharmacokinetic properties of biotechnological products.12,13 The effects of manipulations applied by protein engineering are usually unclear on the structure and function. Moreover, despite the progress in recombinant DNA technology, the production of sufficient quantity of protein(s) for chemical engineering acts as a bottleneck in the engineering process. This limitation becomes clearer when a complex biopharmaceutical drug with posttranslational modifications (PTMs) is desired.14 For example, in our previous study, the site-specific PEGylation of EPO was investigated. It should be noted that the possible sites for the conjugation (even on consideration of experimental information in the literature) are very numerous.11,15 The condition is more complicated with variation in PEG sizes and shapes that could employed for PEGylation.16,17 Therefore, the examination of all possible sites is not practically achievable in a lab as it requires considerable time and cost. On one hand, the analysis of engineered protein is a problem. On the other hand, the amount of engineered protein must be sufficient for all in-process quality control tests.
In this study, two analogs, previously purposed by computational analysis for cysteine PEGylation, were subjected to conjugation with 20 kDa mPEG polymer. Two new conjugated ngEPO cysteine analogs had the same bioactivity, better physicochemical properties compared to nonconjugated ones, as well as high yield of PEGylation. Computational systems are the more feasible and less laborious way for high-throughput screening of desired changes in the protein. Our more recent study on glyco-engineering of interferon β reveals that computational systems are reliable, if the condition parameters for engineering are fully known. Computational calculation can decrease the possible sites for modification of proteins, especially for PEGylation and glyco-engineering as it is shown.11,18
In addition, the expression of the protein in eukaryotic cells, like CHO, is difficult, and PTMs made by such systems create many variations in final product. Therefore, the use of simpler expression system, like E. coli, for producing recombinant proteins needed in developmental steps of protein engineering is a promising approach. In this way, the homogenous protein is prepared faster and more efficiently as a starting material for engineering. Although, proteins derived from E. coli lack PTMs, like glycan chains, but screening at this level is easier than most onerous systems. Moreover, the obtained results are extensible for such systems, as we showed for E31C analog, which investigated in both prokaryotic and eukaryotic systems. In case of PEGylation, PTM deficiency can be compensated by a covalent binding of PEG polymer. As shown, by attachment of 40 kDa PEG polymer to ngEPOs, the half-life of ngEPO increases even more than CHO-expressed EPO.19 Also, the increase in size due to attachment of a 20 kDa mPEG polymer enhances the hydrodynamic radius of the macromolecule, potentially incurring repulsion against Bowman’s capsule, leading to lower kidney excretion, and longer in vivo half-life.20
Conclusion
The strategy of creating new forms of protein through computational and nonglycosylated approach can be considered an easy and economical solution to develop new biopharmaceutical products in a more time- and cost-effective manner. In addition, usage of nonglycosylated form of protein is a simpler method for the PEGylation reaction parameter optimization, such as temperature, pH, time, and PEG/protein ratio at industrial scale.
Acknowledgment
The authors wish to express their deep gratitude to all who provided support during the course of this research.
Disclosure
The authors report no conflicts of interest in this work.
References
Debeljak N, Sytkowski AJ. Erythropoietin and erythropoiesis stimulating agents. Drug Test Anal. 2012;4(11):805–812. | ||
Lee JS, Ha TK, Lee SJ, Lee GM. Current state and perspectives on erythropoietin production. Appl Microbiol Biotechnol. 2012;95(6):1405–1416. | ||
Esposito C, Abelli M, Sileno G, et al. Effects of continuous erythropoietin receptor activator (CERA) in kidney transplant recipients. Transplant Proc. 2012;44(7):1916–1917. | ||
Gonzalez MT, Ramos R, Vera M, et al. Monthly CERA treatment maintains stable hemoglobin levels in routine clinical practice of peritoneal dialysis patients. Ren Fail. 2013;35(3):314–319. | ||
Roberts MJ, Bentley MD, Harris JM. Chemistry for peptide and protein PEGylation. Adv Drug Deliv Rev. 2002;54(4):459–476. | ||
Gaberc-Porekar V, Zore I, Podobnik B, Menart V. Obstacles and pitfalls in the PEGylation of therapeutic proteins. Curr Opin Drug Discov Devel. 2008;11(2):242–250. | ||
Wang Y-S, Youngster S, Grace M, Bausch J, Bordens R, Wyss DF. Structural and biological characterization of pegylated recombinant interferon α-2b and its therapeutic implications. Adv Drug Deliv Rev. 2002;54(4):547–570. | ||
Dou H, Zhang M, Zhang Y, Yin C. Synthesis and purification of mono-PEGylated insulin. Chem Biol Drug Des. 2007;69(2):132–138. | ||
Porowinska D, Wujak M, Roszek K, Komoszynski M. Prokaryotic expression systems. Postepy Hig Med Dosw (Online). 2013;67:119–129. Polish. | ||
Sodoyer R. Expression systems for the production of recombinant pharmaceuticals. BioDrugs. 2004;18(1):51–62. | ||
Cohan RA, Madadkar-Sobhani A, Khanahmad H, et al. Design, modeling, expression, and chemoselective PEGylation of a new nanosize cysteine analog of erythropoietin. Int J Nanomedicine. 2011;6:1217–1227. | ||
Bayram Akcapinar G, Venturini A, Martelli PL, Casadio R, Sezerman UO. Modulating the thermostability of Endoglucanase I from Trichoderma reesei using computational approaches. Protein Eng Des Sel. 2015;28(5):127–135. | ||
Vertesy S, Michalak M, Miller MC, et al. Structural significance of galectin design: impairment of homodimer stability by linker insertion and partial reversion by ligand presence. Protein Eng Des Sel. 2015;28(7):199–210. | ||
Jenkins N, Murphy L, Tyther R. Post-translational modifications of recombinant proteins: significance for biopharmaceuticals. Mol Biotechnol. 2008;39(2):113–118. | ||
Maleki A, Madadkar-Sobhani A, Roohvand F, et al. Design, modeling, and expression of erythropoietin cysteine analogs in Pichia pastoris: improvement of mean residence times and in vivo activities through cysteine-specific PEGylation. Eur J Pharm Biopharm. 2012;80(3):499–507. | ||
Bonora GM, Drioli S. Reactive PEGs for protein conjugation. In: Veronese FM, editor. PEGylated Protein Drugs: Basic Science and Clinical Applications Milestones in Drug Therapy. 1st ed. Basel, Switzerland: Birkhäuser Basel Publications; 2009:33–45. | ||
Pasut G, Veronese FM. State of the art in PEGylation: the great versatility achieved after forty years of research. J Control Release. 2012;161(2):461–472. | ||
Samoudi M, Tabandeh F, Minuchehr Z, et al. Rational design of hyper-glycosylated interferon beta analogs: a computational strategy for glycoengineering. J Mol Graph Model. 2015;56:31–42. | ||
Wang YJ, Hao SJ, Liu YD, et al. PEGylation markedly enhances the in vivo potency of recombinant human non-glycosylated erythropoietin: a comparison with glycosylated erythropoietin. J Control Release. 2010;145(3):306–313. | ||
Jevševar S, Kunstelj M. Half-life extension through PEGylation. In: Kontermann R, editor. Therapeutic Proteins: Strategies to Modulate Their Plasma Half-Lives. 1st ed. Weinheim, Germany: John Wiley & Sons Publications; 2012:41–56. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.