Back to Journals » Therapeutics and Clinical Risk Management » Volume 11
Comprehensive overview: efficacy, tolerability, and cost-effectiveness of clobazam in Lennox–Gastaut syndrome
Authors Faulkner M
Received 11 April 2015
Accepted for publication 16 May 2015
Published 8 June 2015 Volume 2015:11 Pages 905—914
DOI https://doi.org/10.2147/TCRM.S55930
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Garry Walsh
Michele A Faulkner1,2
1Department of Pharmacy Practice, Creighton University School of Pharmacy, 2Department of Neurology, Creighton University School of Medicine, Omaha, NE, USA
Abstract: Clobazam is the newest medication approved by the US Food and Drug Administration (FDA) for the treatment of Lennox–Gastaut syndrome (LGS) in patients at least 2 years of age, although the medication has been available in countries around the world to treat epilepsy and anxiety disorders for many years. Though classified as a benzodiazepine, the drug differs structurally from other drugs in the class as it possesses nitrogen atoms at the 1 and 5 positions within the heterocyclic ring rather than at the 1 and 4 positions. This difference and the classification of clobazam as a partial agonist are believed to be responsible for the decreased incidence of sedative effects compared to other benzodiazepines. Adverse events associated with clobazam use in clinical trials have generally been mild to moderate in nature. Data from an open-label extension trial have confirmed that clobazam is efficacious for the treatment of seizures associated with LGS, particularly atonic seizures (drop seizures), over the long term. Tolerance to the drug’s antiepileptic effects does not seem to be a common occurrence. The drug has proven to be a cost-effective option for therapy, particularly due to its ability to decrease the number of seizures that require medical treatment. Clobazam represents a welcome addition to the treatment options for LGS.
Keywords: clobazam, Lennox–Gastaut syndrome, drop seizures
Introduction
Lennox–Gastaut syndrome (LGS) is an epileptic encephalopathy associated with varying degrees of developmental delay.1 At the time of symptom onset, patients are usually younger than 8 years, and peak incidence is noted to be somewhere between ages 3 years and 5 years. The etiology varies from person to person. Brain damage or acquired destructive lesions may be the causative factor, as may a disorder of metabolism, a derangement of cortical development, or a genetic condition (eg, tuberous sclerosis).2 However, in up to one-third of patients no distinct etiology is identifiable.3 LGS itself is not associated with any specific biologic marker.4,5 Cognitive decline is common, and tends to increase with time, making participation in activities of daily living difficult.2,3
Patients with LGS experience a multitude of seizure types that are notoriously difficult to manage. It is estimated that between 1% and 10% of childhood epilepsies are a result of this syndrome.6 Previous data have suggested that only approximately 10% of LGS patients have seizures that are fully controlled with medication therapy.7 Seizure types vary from patient to patient, but the most commonly observed include tonic, atypical absence, and atonic (drop seizures [DS]), the latter of which is responsible for the elevated rate of injury noted in patients with this diagnosis.7 The mortality rate for patients with LGS is between 4% and 7% before the age of 11 years.8–10 Accidents stemming from neurologic causes, including DS, often contribute to patient death.11
Treatment of LGS-associated seizures
Most antiepileptic medications have been employed for the treatment of LGS-associated seizures at one time or another owing to the difficulty in gaining their control.5 Valproic acid, though not approved by the US Food and Drug Administration (FDA) for use in LGS, has traditionally been a favored medication as it possesses a broad spectrum of activity.12,13 Agents that have been granted FDA approval for the treatment of epilepsy in LGS include felbamate, lamotrigine, rufinamide, topiramate, and, most recently, clobazam.14 The side-effect profiles of many of these medications have somewhat limited their use. For instance, when felbamate was linked to elevated rates of aplastic anemia and liver toxicity, the number of patients receiving the drug declined by a factor of 10.15,16 Lamotrigine has been linked to elevated rates of life-threatening rash (eg, Stevens–Johnson syndrome and toxic epidermal necrolysis), with incidence in children noted to be higher than that in adults.17 Topiramate is often linked to psychomotor slowing as well as weight loss.18 Though no medication is without the potential for negative side effects, an efficacious medication with a high degree of acceptability is a welcome addition to the LGS drug arsenal. Clobazam appears to fit this profile.
A Medline search was conducted to identify recent clinical studies of clobazam (particularly those utilized by the FDA to determine eligibility for approval for use in LGS). This was supplemented with additional articles obtained from online sources and information provided by the FDA and manufacturers.
Clobazam
Benzodiazepines have been used for decades in the treatment of many ailments including anxiety, insomnia, panic disorders, alcohol withdrawal, epilepsy/status epilepticus, and spasticity, and also for sedation.19 Since its first approval in 1970 for the treatment of anxiety in Australia, clobazam has been used in countries around the world (known by the brand names Frisium® and Urbanol® outside the United States) both as a treatment for epilepsy and as an anxiolytic.20 The drug was originally granted orphan drug status in the United States in December 2008. Subsequently, in October 2011, it was approved for use as an adjunctive treatment of LGS in patients 2 years of age and older under the brand name Onfi® (Lundbeck, Deerfield, IL, USA).5 Clobazam is a member of the benzodiazepine class of medications, though it is structurally different from its marketed counterparts. While other benzodiazepines have nitrogen atoms in the 1 and 4 positions on the heterocyclic ring characteristic of the class, clobazam has nitrogen at the 1 and 5 positions.21 The medication was specifically developed in 1966 with the intent that it would have better efficacy than traditional benzodiazepines with fewer negative side effects related to differences in receptor affinity.22,23
Members of the benzodiazepine class bind to post-synaptic γ-aminobutyric acid type A (GABAA) receptors in a noncompetitive manner. Unlike 1,4 benzodiazepines, which tend to be nonselective full receptor agonists, clobazam is believed to be a partial agonist.19,24 The drug demonstrates lower affinity for the GABAA subunits that facilitate sedation.25 Stimulation of inhibitory GABAA receptors by clobazam results in more rapid opening of chloride channels and enhancement of the inward current, which ultimately results in membrane hyperpolarization. Clobazam is also believed to play a role in the upregulation of GABA transporters (GAT) 1 and 3.26
Efficacy
The efficacy of clobazam has been demonstrated and well documented. A review of 20 previously reported studies (completed before 2007) evaluating clobazam use in more than 300 patients with LGS demonstrated a cumulative reduction of >50% in the number of seizures experienced in 56.3% of those enrolled.27 The best efficacy was noted in decreasing the frequency of DS and myoclonic seizures. Prior to clobazam’s approval for the treatment of LGS in patients residing in the United States, additional studies were undertaken to satisfy FDA requirements.
A randomized, double-blind, dose-ranging Phase II study (study OV-1002) of 68 LGS patients between the ages of 2 years and 26 years was undertaken to evaluate the change in the number of DS per week during the maintenance period (the primary endpoint) as compared to baseline (Table 1).7 For the purposes of this study, DS were defined as seizures that would likely have resulted in a fall without support. All patients experienced seizure onset prior to the age of 11 years, and were on up to three additional therapies for seizure control. The baseline rate of seizure activity was recorded during a 4-week period, which was followed by a 3-week dose titration. The final dose achieved was continued for additional 4 weeks (the maintenance period), after which the patients underwent a 3-week taper to drug discontinuation or enrollment in an open-label extension study. Participants were randomized into either a low-dose (0.25 mg/kg/day, maximum 5 mg twice daily) or high-dose (1 mg/kg/day, maximum 20 mg twice daily) cohort. For those individuals who were enrolled in the low-dose group who did not weigh enough to qualify for twice-daily administration, placebo was administered in place of the second dose. The efficacy analysis did not include seven of the enrollees, as there were no data available for the maintenance period.
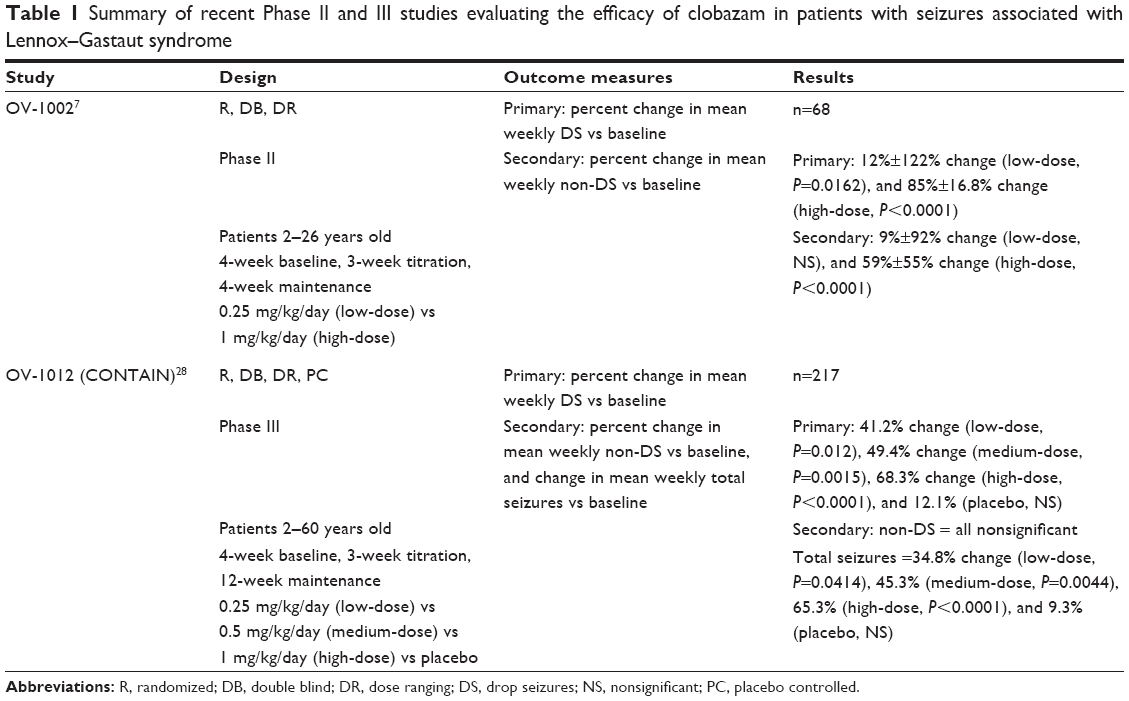 | Table 1 Summary of recent Phase II and III studies evaluating the efficacy of clobazam in patients with seizures associated with Lennox–Gastaut syndrome |
Individuals randomized into the low-dose group reported a percent change in DS frequency compared to baseline of 12%±122% (5–661 seizures per week vs 0–470 seizures per week, P=0.0162). Those enrolled in the high-dose group reported a decrease of 85%±16.8% (8–924 seizures per week vs 0–198 seizures per week, P<0.0001). There was a significant difference between the high- and low-dose groups (P=0.0001), with all patients in the former achieving a minimum 40% decrease in DS activity. Patients were further stratified into groups based on their individual response to clobazam therapy. Eighty-nine percent vs 56% of patients in the high- and low-dose groups experienced a minimum 25% decrease in DS (P=0.0025). A decrease in DS of 50% or greater was experienced by 83% of patients in the high-dose group and 38% in the low-dose group (P=0.0001), and a 75% or greater decrease was demonstrated by 67% and 25% of patients in the high- and low-dose groups, respectively (P=0.0006). A nonsignificant difference in the number of patients who achieved seizure freedom in the high-dose (22%) and low-dose (6%) groups was reported (P=0.0629). Changes in non-DS seizures were also evaluated. A decrease of 9%±92% in seizure activity was demonstrated by individuals in the low-dose cohort, compared to a decrease of 59%±55% in the high-dose cohort vs baseline (P=0.1466 and P<0.0001, respectively). The difference between the high- and low-dose groups was significant (P=0.0222).
Study OV-1012 (also known as the CONTAIN trial) is a double-blind, placebo-controlled Phase III efficacy and safety study designed to evaluate the percent change in average weekly DS with clobazam compared to baseline (primary outcome) as well as the percent change in non-DS and total seizures (secondary outcomes).28 Two-hundred and thirty-eight patients between the ages of 2 years and 60 years (mean 12.5 years) who experienced the onset of LGS prior to the age of 11 years were randomized to receive either placebo, or clobazam stratified into low-dose (0.25 mg/kg/day, maximum 10 mg/day), medium-dose (0.5 mg/kg/day, maximum 20 mg/day), or high-dose (1 mg/kg/day, maximum 40 mg/day) groups. Total daily doses were administered in divided doses in the morning and evening. The study design included a 4-week baseline period, 3 weeks of titration, and a 12-week maintenance period. When the trial was initiated, patients were allowed to discontinue treatment and transition into an open-label extension trial before completion of OV-1012. This led to multiple premature withdrawals in the initial weeks of the study (23 individuals transitioned to the open-label trial). A protocol revision was implemented after the first 81 patients had already been enrolled, disallowing additional premature discontinuations for this purpose.
Compared to baseline, DS occurrence decreased by 12.1%, 41.2%, 49.4%, and 68.3% in the placebo, low-, medium-, and high-dose groups, respectively. All of the active treatment groups achieved significant changes in DS frequency (P=0.0120 [low-dose], P=0.0015 [medium-dose], and P<0.0001 [high-dose]), clearly demonstrating a linear trend toward better efficacy with increasing doses. This trend was further demonstrated by taking the 50% responder rate into account, as DS frequency decreased by 43.4%, 58.6%, and 77.6% in the low-, medium-, and high-dose groups. However, only medium (P=0.0159) and high (P<0.0001) doses resulted in significant changes compared to baseline. Two patients receiving placebo became DS-free compared to four, seven, and 12 patients in the low-, medium-, and high-dose groups, the latter representing nearly a quarter of all patients receiving high-dose clobazam therapy. When considering non-DS seizures experienced per week, only the high-dose group demonstrated a mean percent decrease (40%, nonsignificant). Regarding total seizures (DS and non-DS), all three doses of clobazam resulted in significant decreases in frequency compared to baseline. The demonstrated mean decreases in total seizure frequency were 34.8% (low dose, P=0.0414), 45.3% (medium dose, P=0.0044), and 65.3% (high dose, P<0.0001). Those receiving placebo did not demonstrate a decrease in total seizures that was significant vs baseline (9.3%). It should be noted that this study was not designed to evaluate the efficacy of clobazam for the control of non-DS, and, because no baseline rate of non-DS seizures was specified for inclusion, these cohorts were smaller.
The results of the extension trial (OV-1004) that enrolled patients from OV-1002 and OV-1012 have now been published.29,30 This extension study provides much-needed long-term clobazam data in patients with LGS. Interim data for patients enrolled from December 2005 through June 2010 was published in 2012, and the final results were published in 2014.29,30 The study itself concluded in March 2012. Patients who enrolled in the extension study (n=267) had to do so within 14 days of receiving their final clobazam doses in the other studies. For those individuals who enrolled at centers outside the United States (n=60), participation was limited to 24 months, while those enrolled in the United States were allowed to continue in the trial up to the point when clobazam became commercially available (some for as long as 6 years). In comparison to the earlier trials, patients enrolled in OV-1004 had their clobazam doses adjusted according to clinical response and tolerability. Doses of 2 mg/kg (divided into two daily doses) were allowed, though no daily dose could exceed 80 mg. The majority of those enrolled received doses between 0.375 and 1.25 mg/kg/day. Concomitant treatments (including vagus nerve stimulation [VNS] and a ketogenic diet) were allowed and adjustable as a patient’s circumstance dictated. The most commonly utilized medications were valproic acid (52%), lamotrigine (36%), levetiracetam (36%), and topiramate (30%). The primary outcome remained consistent with the previous studies (percent decrease in average weekly DS vs baseline).
The median decrease in DS activity reported in the interim dataset, irrespective of the total duration of clobazam therapy, was 71.1% at the 3-month data collection point and 91.6% at the 24-month point.29 The number of total seizures recorded up to those time points demonstrated a decrease of 64.8% and 81.5% vs baseline. These decreases remained consistent for the entire study duration with a mean percent decrease in weekly DS and total seizures reported to be between 85%–91% and 85%, respectively, at study conclusion.30 The interim results demonstrated that 61.5% and 79.5% of enrollees achieved the 50% responder rate threshold for DS at 3 months and 24 months, respectively. For total seizures, the responder rates were 61.5% and 70.3% at the same time points.30 In total, over a 5-year time period, between 62% and 69% of patients achieved at least a 75% reduction in weekly DS, while 50%–65% achieved reductions in total seizure activity of the same magnitude. Seizure freedom was achieved in 32% (DS only) and 18% (total seizures) of patients.30 The extension study proved that patients who were initially responders tended to remain responders with long-term clobazam exposure.
Due to the low prevalence of LGS and the challenges associated with conducting studies in children, there is a paucity of data with regard to head-to-head comparisons of medications used for the control of seizures in this population. A 2013 study utilizing a method for indirect comparison of antiepileptic agents used in the treatment of LGS was published in an attempt to fill this gap.31 Medications included in the analysis were clobazam, felbamate, lamotrigine, rufinamide, and topiramate. Utilizing Cohen’s-d to provide a common metric by which to compare effects of the various treatment modalities, the authors evaluated data from 716 patients. Considering all cohorts, the highest overall effect size (0.8) was seen in studies utilizing high doses of clobazam. This was followed (in descending order) by lower doses of clobazam, rufinamide, topiramate, lamotrigine, and felbamate. Each of the medications produced an effect size >0.2, indicating that the benefits of each were clinically relevant and detectable. When considering individual seizure types, clobazam’s effect size remained highest for DS, tonic/atonic seizures, and total seizures. Rufinamide demonstrated similar efficacy for decreasing DS rates when compared to medium and high doses of clobazam. The odds of achieving the 50% responder rate threshold for DS for clobazam was double that of lamotrigine, topiramate, and rufinamide, though only the difference between clobazam and lamotrigine reached significance. There are understandably several limitations with regard to this study, the most obvious being differences in study designs and measurement of primary outcomes. It should also be noted that, with the exception of clobazam, all of the other drugs were studied using dose ranges as opposed to specific, defined doses.31
One other study was undertaken to determine whether a specific concomitant treatment altered outcomes with clobazam. A post hoc analysis of OV-1012 and OV-1004 aimed to determine whether exposure to VNS had an effect on clobazam efficacy.32 Consideration was given to the groups with the highest number of DS (median 86–1,007 [OV-1012], median 111–1,147 [OV-1004]) and total seizures (median 125–1,465 [OV-1012], median 159–1,413 [OV-1004]). Regardless of the presence of VNS therapy, weekly seizures decreased significantly compared to placebo (P<0.01). Though the number of patients evaluated was small (54 and 38 in the placebo and active treatment groups, respectively), the authors concluded that clobazam efficacy did not appear to be altered in the presence of VNS.
Potential for tolerance
A fair amount of attention has been given to the possibility that extended clobazam use results in tolerance to the drug’s effects on seizure activity. Gastaut and Low claimed that in one-third of patients receiving clobazam, its positive effects on seizure activity are exhausted within a matter of weeks.33 Studies have reported tolerance rates as high as 87%.12 There are several challenges associated with determining whether a return of seizure activity is in fact due to tolerance. First, no universal definition of tolerance to antiepileptic medication has been put forth and accepted. In addition, the possibility of worsening disease needs to be taken into account. The possibility that coadministered medications may affect a drug’s efficacy also exists.12,21 Data generated by both OV-1012, and in particular, OV-1004 do not seem to support the idea that tolerance to clobazam merits much concern.20 Both studies boast high rates of participant retention.28,30 In OV-1012, tolerance was defined as a return to the previously established baseline seizure rate after initially reaching a response rate of at least 50%.28 The number of patients meeting this parameter was low, and there was no difference compared to placebo (5.3% vs 5.6%, respectively). For the study OV-1004, the mean dosages through the 5th year of the study increased by a mere 8%.30 The stability of the mean dose and the fact that the response rate did not change are evidence that tolerance to clobazam efficacy was not a major factor. In addition, only 6% of patients stopped the drug citing a lack of efficacy as the reason, and 15% of patients were able to discontinue concomitant antiepileptic therapies and maintain seizure control on clobazam monotherapy.30
Pharmacokinetics and drug interactions
Clobazam displays linear, dosage-independent pharmacokinetics.34 The drug’s bioavailability is high at approximately 87%, and peak concentrations are achieved at median time of 1 hour (range: 1–4 hours).34,35 Metabolism occurs mainly via the cytochrome P450 (CYP450) enzymes. Demethylation by CYP3A4 is the primary metabolic pathway for clobazam, with CYP2C19 playing a role as well.35 Metabolism of clobazam results in the formation of N-desmethylclobazam (N-CLB), an active metabolite with antiepileptic potency approximately equivalent to that of diazepam but less than its parent compound.36 This metabolite is formed, slowly reaching Tmax in approximately 60 hours.35 The half-life (t1/2) of clobazam appears to be shortened in patients experiencing seizures. In healthy adults, the t1/2 is approximately 24 hours compared to 12 hours in adults with epilepsy.37 Children with epilepsy display a t1/2 of approximately 16 hours. The N-CLB t1/2 is also affected by a diagnosis of epilepsy (49 hours in epileptic adults vs 57 hours in healthy volunteers).37 There are no data demonstrating significant alterations based on patient race, sex, age, weight, or renal function. There is some evidence that CYP2C19 polymorphisms may increase both the concentration of clobazam and the concentration of N-CLB. However, in a group of Japanese patients identified as either poor or extensive metabolizers, no differences in the rate of adverse events were noted in comparison to individuals displaying normal rates of clobazam and N-CLB metabolism.38
Neither clobazam nor N-CLB appear to induce or inhibit CYP450 enzymes to any significant extent.34 However, the manufacturer recommends the use of nonhormonal contraceptive additions to existing hormonal contraceptives owing to a weak induction of CYP3A4.39 The manufacturer also suggests that caution be exercised when clobazam in given with drugs metabolized by CYP2D6, stating that the drug is an inhibitor. However, no specific changes are suggested, and no drugs are mentioned by name.39 Drug interactions between clobazam and other members of the antiepileptic drug class appear to be insignificant, including those with other substrates of CYP2C19 (carbamazepine, phenytoin, and phenobarbital).35,40 An analysis of seven clinical trials also demonstrated that coadministration of clobazam and valproic acid resulted in no significant changes to the pharmacokinetic profile of either medication.35 Similarly, lamotrigine pharmacokinetics have not been found to have an effect on, or be affected by, clobazam.28
A prospective trial that enrolled 36 patients was completed with the goal of determining whether interactions with clobazam were likely when patients were given concurrent ketoconazole (an inhibitor of CYP3A4 and CYP2C19) or omeprazole (a CYP2C19 inhibitor).35 Blood samples were drawn for analysis at nine time points over a 22-day time period. The administration of ketoconazole resulted in a delay to maximum clobazam concentration of 1–2 hours. However, the mean t1/2 remained stable. The time to N-CLB maximum concentration was lengthened by approximately 24 hours, but no changes were noted with regard to the maximum concentration achieved. For omeprazole, a slight increase in the t1/2 of clobazam (32 hours vs 37 hours) was observed, as well as a 15% increase in the maximum N-CLB concentration. None of the pharmacokinetic changes with either ketoconazole or omeprazole was determined to be of substantial clinical significance.35 Even so, the drug labeling for clobazam suggests that consideration be given to clobazam dose adjustments when administered with these medications, particularly because coadministration may result in elevated concentrations of N-CLB.39
Another 18 individuals, all healthy males without LGS, were administered a drug cocktail containing 4 mg of the benzodiazepine midazolam, 200 mg of caffeine, 500 mg of tolbutamide, and 30 mg of dextromethorphan to study the effects of clobazam on CYP3A4, CYP1A2, CYP2C9, and CYP2D6, respectively.35 As with the previous study, blood samples were drawn at various time intervals at baseline and post-administration of the medications. Midazolam concentrations were found to be lowered by clobazam coadministration (27%), while its metabolite, 1-hydroxymidazolam, demonstrated a fourfold increase in concentration. For dextromethorphan, the area under the concentration–time curve (extrapolated to infinity, AUC0–∞) increased by 95%, and the maximum serum concentration was 59% higher than at baseline with concomitant clobazam. No clinically meaningful adverse events were observed with these changes, though the authors concede that dosage adjustment may be required when clobazam and CYP2D6 substrates are given simultaneously, which is consistent with product labeling.39 No significant changes were noted with caffeine or with tolbutamide.
Finally, the same authors reported the results of a population pharmacokinetics analysis of seven studies, two of which were conducted in an LGS population.35 The total number of individuals included in the analysis was 171. Clobazam pharmacokinetics were not significantly affected by any antiepileptic medication, including coadministration with CYP3A4 inducers (phenobarbital, phenytoin, and carbamazepine), CYP2C9 inducers (valproic acid, phenobarbital, phenytoin, and carbamazepine), or CYP2C9 inhibitors (felbamate, oxcarbazepine). When patients with LGS were considered separately, pharmacokinetic changes remained insignificant.
Dosing
Dosing of clobazam is based on body weight, with those individuals weighing ≤30 kg starting with a dose of 5 mg once daily, with dose escalation occurring at weekly intervals to a maximum of 20 mg/day in divided doses. Individuals who weigh >30 kg may start with 10 mg/day in divided doses, which may be titrated to a maximum of 40 mg/day. More rapid titration is not advised due to the long t1/2 of the drug and the corresponding length of time that must pass in order for steady state to be reached.
Safety and tolerability
Adverse events believed to be medication-related, which occurred in at least 5% of patients enrolled in OV-1002, included somnolence, lethargy, sedation, salivary hypersecretion, constipation, aggressive behavior, hypomania, and insomnia.7 Treatment-emergent adverse events were experienced with equal frequency in both the low- and high-dose groups (84% vs 86%). Additionally, there was no difference in the stratification of adverse event severity between the two groups. Patients in the low-dose group discontinued therapy due to convulsions, aggression, and sedation that led to a decrease in oral intake and drooling. In the high-dose group, two patients stopped therapy due to somnolence. Chorea, defiant behavior, encephalopathy, and sedation were responsible for the withdrawal of an additional one patient in each group. Interestingly, four of the patients who withdrew from OV-1002 due to adverse events opted into the extension study. Four patients were reported to have experienced serious adverse events (severe aspiration with questionable association with clobazam use, sleep apnea, constipation + pyrexia, and severe respiratory distress possibly related to tonsillar hypertrophy). Each of these events resolved. In addition, nine new seizure types were identified in patients taking clobazam, but a relationship to the medication could not be definitively established.
In OV-1012, 67.8%, 72.4%, 88.7%, and 76.3% of individuals in the placebo, low-, medium-, and high-dose groups experienced one or more adverse events during the trial.28 The majority of these events were reported to be of mild or moderate severity. Specific adverse events experienced by at least 10% of those enrolled included somnolence, pyrexia, lethargy, drooling, and constipation. Of those, only somnolence and drooling appeared to be dose-related. Twenty-nine individuals had their clobazam dose decreased due to adverse events, and ten (one in the low-dose group, four in the medium-dose group, and five in the high-dose group) were among those who discontinued their participation in the study. Discontinuations due to adverse events totaled 27 (two in the placebo group, and four, eight and 13 in the low-, medium-, and high-dose groups, respectively). Specific adverse events that led to at least two individuals withdrawing from the study included lethargy, somnolence, aggressive behavior, ataxia, insomnia, and fatigue. There were two serious adverse events related to seizure activity. The first occurred in a patient randomized to the medium-dose group (myoclonic seizures), and the second was a tonic–clonic seizure (defined as “grand mal”) in an individual receiving high-dose clobazam. Sixteen additional serious adverse events were reported in nine patients. In each of these cases (two, three, six, and five instances in the placebo, low-, medium-, and high-dose groups, respectively), the recorded event was pneumonia. Thrombocytopenia and an increased eosinophil count, possibly related to drug administration, were identified during the study, though the number of patients in whom this occurred was not specified. There were no deaths, and no patients displayed an exacerbation of seizure activity during the end-of-study drug taper.
Results from the extension trial showed that the primary reason for study discontinuation was request from the patient or caregiver.30 This is not unexpected due to the fact that patients who experienced adverse events significant enough to lead to discontinuation would likely have experienced them during their initial study involvement, and would have been unlikely to sign on for additional medication exposure. Even so, 60% of participants reported experiencing one or more adverse events while enrolled. The most commonly reported events occurring in at least 10% of patients included upper respiratory tract infections, pneumonia, somnolence, falls, otitis media, urinary tract infections, constipation, convulsions, sinusitis, insomnia, nasopharyngitis, viral infections, and lethargy. As with the Phase III study, the majority of events were mild or moderate in severity. The severe events that were reported included pneumonia (non-aspiration, 6%), seizure-related events (status epilepticus and convulsions, each experienced by 3% of participants), dehydration (2%), and aspiration pneumonia (2%). Eighteen enrollees discontinued participation due to adverse events. Over the course of the study, ten deaths were recorded, nine of which were classified as not likely to be related to clobazam therapy. The etiology of those deaths included three cases of pneumonia, one case of pneumonia with acute respiratory distress syndrome, one case of pneumonia with cardiac arrest, one case of increasing seizure activity in conjunction with atelectasis and respiratory failure, and one case of acute respiratory distress syndrome in a patient with a hematoma in the leg. The one case that was deemed possibly related to clobazam administration was documented as a convulsion. The causes of two of the deaths were not reported.
A summary report of adverse events related to drug discontinuation in clobazam trials conducted in patients with LGS was published in 2014.41 In Phase I trials, study discontinuation was reported as being associated with headache, insomnia, tremor, and anxiety in 14%, 12.6%, 10.1%, and 8.7% of patients, respectively. All of these events were classified as being mild to moderate in severity, and each resolved completely. In these trials, clobazam was typically discontinued abruptly rather than being tapered over an extended time period. As such, it has been hypothesized that the symptoms may have been related to too rapid withdrawal of medication, since 57% were reported within 7 days of drug discontinuation. In contrast, no withdrawal-related adverse events, including seizures, were reported in Phase II or III trials that included a drug taper as part of the protocol. These results are consistent with the known adverse events associated with rapid withdrawal of 1,4 benzodiazepines.42–44
Several small studies have been undertaken specifically to compare clobazam-related adverse events to those associated with other benzodiazepines, given the belief that clobazam demonstrates selectivity toward the GABAA subunits that are responsible for anxiolytic and anticonvulsant effects rather than those that are associated with the sedative effects of the drug class.45 The first of these was a study of five healthy males to compare clobazam 20 mg with the 1,4 benzodiazepines chlordiazepoxide (20 mg) or diazepam (10 mg), or placebo.46 The study was a randomized, double-blind, four-period crossover study. Each medication was administered for 2 days followed by a 4-week washout before the next medication exposure. Adaptive tracking and reaction times were measured at several time points post-administration beginning at 30 minutes and ending at 9.5 hours. Diazepam administration resulted in deficits in adaptive tracking at 30 minutes and 2.5 hours, while clobazam and chlordiazepoxide administration did not produce a negative effect. Reaction time was impaired by both diazepam and chlordiazepoxide, whereas clobazam did not differ from placebo at any time point. A second small study with 24 healthy enrollees measured reaction time to use the brake while driving.47 Administration of placebo, clobazam 20 mg/day, or diazepam 10 mg/day was completed for a total of 3 days. Individuals receiving diazepam had an extended time to react in traffic situations compared to clobazam and placebo (P<0.01). In addition, the volunteers reported increased lethargy only after receiving diazepam. The final benzodiazepine comparison measured various cognitive and psychomotor attributes in ten healthy individuals receiving clobazam 20 mg or 30 mg, and lorazepam 2 mg or 3 mg.48 Individuals receiving lorazepam experienced significant differences in attention, concentration, motor speed, reaction accuracy, mood, and psychomotor activity (which was increased) in comparison to clobazam. Anterograde amnesia was also worsened by lorazepam but not by clobazam.
Cost-effectiveness
The cost-effectiveness of clobazam in LGS was studied using a trial-based economic model constructed to compare the medication with rufinamide, topiramate, and lamotrigine.49 The duration used for each therapy was determined by the data available in existing studies. The model itself was based on patient characteristics from OV-1012. Maximum daily doses for each drug were set at 45 mg/kg for rufinamide, 6 mg/kg for topiramate, and 5 mg/kg/day or 15 mg/kg/day for lamotrigine (the lower dose used for patients taking valproic acid due to the risk of Stevens–Johnson syndrome development). Utilizing a retrospective analysis of the available claims data from January 2009 to September 2010, the mean health care cost for seizures experienced by LGS patients requiring medical intervention was estimated to be $5501 (2013 US dollars). The cost of medication therapy was estimated using the average doses that had been effective in clinical trials and calculating the corresponding wholesale acquisition cost utilizing 2013 dollars. Quality-adjusted life years (QALYs) gained with each medication was based on the reduction in DS categorized into groups (<50%, 50%–75%, 76%–99%, or seizure freedom). The 3-month model demonstrated a higher number of QALYs gained with clobazam vs the other medications. The rate of seizure freedom with clobazam was 21.5% vs <5% for the other three drugs. The mean decrease in DS was 64% with clobazam, 51% with rufinamide, 46% with topiramate, and 44% with lamotrigine. While the drug cost alone was significantly higher for clobazam ($2017) compared to topiramate and lamotrigine ($11 each), the overall cost of care with clobazam ($30,147) was less than that associated with the care of patients receiving the other medications ($35,378 for rufinamide, $34,223 for topiramate, and $34,970 for lamotrigine). The estimated cost per QALY for clobazam was $73,300. In the 2-year model, clobazam and rufinamide were evaluated. DS reduction with clobazam was 53% associated with a total per-patient cost of $177,068. For rufinamide, a lower rate of seizure reduction was reported (33%) at a cost of $265,814 per patient. DS freedom was again higher with clobazam (26.7% vs 5.7%). Overall, due to inferior efficacy in controlling DS compared to clobazam, the other medications were all associated with higher overall medical costs. This remained true when the estimated number of DS requiring medical attention was set at 2.3% using data from a survey of physicians in the United Kingdom in conjunction with a claims database from the United States.50 At a lower threshold of 0.76%, topiramate would become the most cost-effective medication in the group.
A second study undertaken to evaluate the 2-year budgetary impact of adding clobazam therapy to other medications utilized in the treatment of LGS has been also completed.51 As with the previous study, the target population was modeled after the participants in OV-1012. Also, consistent with the previous study, the comparators were rufinamide, topiramate, and lamotrigine at the described target doses. Based on the midpoint of the estimated United States prevalence of LGS, the number of persons in the health care plan expected to be diagnosed was set at 0.4%.52,53 Utilizing data from OV-1004, the average number of DS experienced per month by each patient was assumed to be 132.9 All other parameters (estimated percent of DS medically attended, mean total cost, cost of medication) were consistent with the previous study.49 The addition of clobazam to the formulary saved the private payer $98,059 in the first year and $131,690 in the second year. The per-member, per-month cost savings (considering all enrollees) was calculated to be $0.08 in year 1 and $0.11 in year 2. These values were superior to the other therapies. Multiple scenario adjustments (assumption of maintained DS reduction with clobazam discontinuation, increases in DS reduction with other medications, dropping the number of medically treated DS and sequelae to 0.5%, and decreasing the number of eligible LGS patients in the health plan to 0.01%) did not change the superiority of cost savings associated with clobazam vs the other medications.
Summary
Clobazam, though not a new medication, is the latest drug to be approved by the FDA for the treatment of seizures associated with LGS. No other medications approved by the FDA have demonstrated the degree of efficacy that has been noted with clobazam use (frequently >50% reduction in DS). The decrease in DS with clobazam compared to other seizure types is particularly robust, and is primarily responsible for the cost-effectiveness of the drug since use is expected to result in fewer seizure-related injuries. Clobazam is less likely to be associated with the cognitive and psychomotor adverse events associated with 1,4 benzodiazepines due to differences in binding and receptor affinity. It is also less likely to be associated with life-threatening adverse reactions than some of its counterparts. Tolerance, though once a frequently discussed phenomenon with clobazam, appears to be of minimal concern given the long-term data that were made available from OV-1004. Because the LGS population is small, it is unlikely that a head-to-head medication trial will be conducted in this population. However, available data suggest that clobazam is at least as efficacious as other medications used in the treatment of LGS-related seizure activity, and in many cases, may be superior.
Disclosure
The author reports no conflicts of interest in this work.
References
Arzimanoglou A, Resnick T. All children who experience epileptic falls do not necessarily have Lennox-Gastaut syndrome…but many do. Epileptic Disord. 2011;13(suppl 1):S3–S13. | ||
Arzimanoglou A, French J, Blume WT, et al. Lennox-Gastaut syndrome: a consensus approach on diagnosis, assessment, management and trial methodology. Lancet Neurol. 2009;8:82–93. | ||
Ioannidis JP, Haidich AB, Pappa M, et al. Comparison of evidence of treatment effects in randomized and non-randomized studies. JAMA. 2001;286:821–830. | ||
Bourgeois BF, Douglass LM, Sankar R. Lennox-Gastaut syndrome: a consensus approach to differential diagnosis. Epilepsia. 2014;55(suppl 4):4–9. | ||
Giarratano M, Standley K, Benbadis SR. Clobazam for treatment of epilepsy. Expert Opin Pharmacother. 2012;13(2):227–233. | ||
Neih SE, Sherr EH. Epileptic encephalopathies: new genes and pathways. Neurotherapeutics. 2014;11:796–806. | ||
Conry JA, Ng YT, Paolicchi JM, et al. Clobazam in the treatment of Lennox-Gastaut syndrome. Epilepsia. 2009;50(5):1158–1166. | ||
Beaumanoir A, Blume W. The Lennox-Gastaut syndrome. In: Roger J, Bureau M, Dravet CH, Genton P, Tossinari CA, Wolf P, editors. Epileptic Syndromes in Infancy, Childhood and Adolescence. 3rd ed. London: John Libbey & Co Ltd.; 2002:113–135. | ||
Crumrine PK. Lennox-Gastaut syndrome. J Child Neurol. 2002;17(suppl 1):S70–S75. | ||
Markand ON. Lennox-Gastaut syndrome (childhood epileptic encephalopathy). J Clin Neurophysiol. 2003;20:426–441. | ||
Autry AR, Trevathan E, Van Naarden Braun K, Yeargin-Allsopp M. Increased risk of death among children with Lennox-Gastaut syndrome and infantile spasm. J Child Neurol. 2010;25:441–447. | ||
Leahy JT, Chu-Shore CJ, Fisher JL. Clobazam as an adjunctive therapy in treating seizures associated with Lennox-Gastaut syndrome. Neurophsychiatr Dis Treat. 2011;7:673–681. | ||
Wheless JW, Clarke DF, Arzimanoglou A, Carpenter D. Treatment of pediatric epilepsy: European expert opinion, 2007. Epileptic Disord. 2007;9:353–412. | ||
Schmidt D, Bourgeois B. A risk-benefit assessment of therapies for Lennox-Gastaut syndrome. Drug Saf. 2000;22:467–477. | ||
French J, Smith M, Faught E, et al. The use of felbamate in the treatment of patients with intractable epilepsy-report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Epilepsia. 1999;40:803–808. | ||
World Health Organization regulatory matters. WHO Drug Info. 1994;8:152–160. | ||
Guberman AH, Besag FM, Brodie MJ, et al. Lamotrigine-associated rash: risk/benefit considerations in adults and children. Epilepsia. 1999;40:985–991. | ||
Glauser TA. Behavioral and psychiatric adverse events associated with antiepileptic drugs commonly used in pediatric patients. J Child Neurol. 2004;19(suppl 1):S25–S38. | ||
Sankar R. GABAA receptor physiology and its relationship to the mechanism of action of the 1,5 benzodiazepine clobazam. CNS Drugs. 2012;26(3):229–244. | ||
Wheless JW, Phelps SJ. Clobazam: a newly approved but well-established drug for the treatment of intractable epilepsy syndromes. J Child Neurol. 2013;28(2):219–229. | ||
Robertson MM. Current status of the 1,4- and 1,5-benzodiazepines in the treatment of epilepsy: the place of clobazam. Epilepsia. 1986;27(suppl 1):S27–S41. | ||
Wieck HH, Blaha L, Heerklotz B. A clinical view of clobazam. Br J Clin Pharmacol. 1979;7(suppl 1):1098–1128. | ||
Chapman AG, Horton RW, Meldrum BS. Anticonvulsant action of a 1,5-benzodiazepine, clobazam, in reflex epilepsy. Epilepsia. 1979;19(3):293–299. | ||
Jensen AA, Hammer H, Ebert B, et al. Clobazam and its active metabolite, N-desmethylclobazam, are partial benzodiazepine receptor agonists at cloned GABAA receptors expressed in Xenopus laevis oocytes. Neurology. 2010;78(Meeting Abstracts 1):P05.085. | ||
McKernan RM, Rosahl TW, Reynolds DS, et al. Sedative but not anxiolytic properties of benzodiazepines are mediated by the GABA(A) receptor alpha1 subtype. Nat Neurosci. 2000;3:587–592. | ||
Doi T, Ueda Y, Tokumaru J, Willmore LJ. Molecular regulation of glutamate and GABA transporter proteins by clobazam during eliptogenesis in FE(+++)-induced epileptic rats. Brain Res Mol Brain Res. 2005;142:91–96. | ||
Ng YT, Collins SD. Clobazam. Neurotherapeutics. 2007;4:138–144. | ||
Ng YT, Conry JA, Drummond R, Stolle J, Weinberg MA; OV-1012 Study Investigators. Randomized, phase III study results of clobazam in Lennox-Gastaut syndrome. Neurology. 2011;77:1473–1481. | ||
Ng YT, Conry J, Paolicchi J, et al; OV-1004 Study Investigators. Long-term safety and efficacy of clobazam for Lennox-Gastaut syndrome: interim results of an open-label extension study. Epilepsy Behav. 2012;25:687–694. | ||
Conry JA, Ng YT, Kernitsky L, et al; OV-1004 Study Investigators. Stable dosages of clobazam for Lennox-Gastaut syndrome are associated with sustained drop-seizure and total-seizure improvements over 3 years. Epilepsia. 2014;55(4):558–567. | ||
Cramer JA, Sapin C, François C. Indirect comparison of clobazam and other therapies for Lennox-Gastaut syndrome. Acta Neurol Scand. 2013;128:91–99. | ||
Wheless JW, Isojarvi J, Lee D, Drummond R, Benbadis SR. Clobazam is efficacious for patients across the spectrum of disease severity of Lennox-Gastaut syndrome: post hoc analyses of clinical trial results by baseline seizure-frequency quartiles and VNS experience. Epilepsy Bahav.2014;41:47–52. | ||
Gastaut H, Low MD. Antiepileptic properties of clobazam, a 1-5 benzodiazepine, in man. Epilepsia. 1979;20:437–446. | ||
Rupp W, Badian M, Christ O, et al. Pharmacokinetics of single and multiple doses of clobazam in humans. Br J Clin Pharmacol. 1979;7(suppl 1):51S–57S. | ||
Walzer M, Bekersky I, Blum RA, Tolbert D. Pharmacokinetic drug interactions between clobazam and drugs metabolized by cytochrome P450 isoenzymes. Pharmacotherapy. 2012;32(4):340–353. | ||
Fisher JL. Interactions between modulators of the BABAA receptor: stiripentol and benzodiazepines. Eur J Pharmacol. 2011;654:160–165. | ||
Bun H, Monjanel-Mouterde S, Noel F, Durand A, Cano JP. Effects of age and antiepileptic drugs on plasma levels and kinetics of clobazam and N-desmethylclobazam. Pharmacol Toxicol. 1990;67(2):136–140. | ||
Seo T, Nagata R, Ishitsu T, et al. Impact of CYP2C19 polymorphisms on the efficacy of clobazam therapy. Pharmacogenomics. 2008;9:527–537. | ||
ONFI® (clobazam) [package insert]. Deerfield, IL: Lundbeck; 2014. | ||
Bardy AH, Seppälä T, Salokorpi T, Granström ML, Santavuori P. Monitoring of concentrations of clobazam and norclobazam in serum and saliva of children with epilepsy. Brain Dev. 1991;13:174–179. | ||
Tolbert D, Harris SI, Bekersky I, Lee D, Isojarvi J. Withdrawal-related adverse events from clinical trials of clobazam in Lennox-Gastaut syndrome. Epilepsy Behav. 2014;37:11–15. | ||
Denis C, Fatséas M, Lavie E, Auriacombe M. Pharmacological interventions for benzodiazepine mono-dependence management in outpatient settings. Cochrane Database Syst Rev. 2006;3:CD005194. | ||
Petursson H, Lader MH. Withdrawal from long-term benzodiazepine treatment. Br Med J (Clin Res Ed). 1981;283:643–645. | ||
Petursson HJ, Lader MH. Withdrawal reaction from clobazam. Br Med J (Clin Res Ed). 1981;282:1931–1932. | ||
Rudolph U, Crestani F, Benke D, et al. Benzodiazepine actions mediated by specific gamma-aminobutyric acid (A) receptor subtypes. Nature. 1999;401:796–800. | ||
Borland RG, Nicholson AN. Immediate effects on human performance of a 1,5-benzodiazepine (clobazam) compared with the 1,4-benzodiazepines, chlordiazepoxide bydrochloride and diazepam. Br J Clin Pharmacol. 1975;2(3):215–221. | ||
Biehl B. Studies of clobazam and car-driving. Br J Clin Pharmacol. 1979;7(suppl 1):85S–90S. | ||
Saletu B, Grunberger J, Berner P, et al. On differences between 1,5 and 1,4-benzodiazepines: pharmaco-EEG and psychometric studies with clobazam and lorazepam. In: Hindmarker I, Stonier PD, Trimble MR, editors. Clobazam: Human Psychopharmacology and Clinical Applications. International Congress and Symposium Series 74. London: Royal Society of Medicine; 1985:23–46. | ||
Clements KM, Skornicki M, O’ullivan AK. Cost-effectiveness analysis of antiepileptic drugs in the treatment of Lennox-Gastaut syndrome. Epilepsy Behav. 2013;29:184–189. | ||
Benedict A, Verdian I, Maclaine G. The cost effectiveness of rufinamide in the treatment of Lennox-Gastaut syndrome in the UK. Pharmacoeconomics. 2010;28:185–199. | ||
Skornicki M, Clements KM, O’Sullivan AK. Budget impact analysis of antiepileptic drugs for Lennox-Gastaut syndrome. J Manag Care Spec Pharm. 2014;20(4):400–406. | ||
Camfield PR. Definition and natural history of Lennox-Gastaut syndrome. Epilepsia. 2011;52(suppl 5):S3–S9. | ||
Kobau R, Zahran H, Thurman DJ, et al; Centers for Disease Control and Prevention (CDC). Epilepsy surveillance among adults – 9 states. Behavioral Risk Factor Surveillance System, 2005. MMWR Surveill Summ. 2008;57(6):1–20. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.