Back to Journals » OncoTargets and Therapy » Volume 10
Comprehensive analysis of differential co-expression patterns reveal transcriptional dysregulation mechanism and identify novel prognostic lncRNAs in esophageal squamous cell carcinoma
Authors Li Z, Yao Q, Zhao S, Wang Y, Li Y ![]() , Wang Z
, Wang Z
Received 21 February 2017
Accepted for publication 19 April 2017
Published 21 June 2017 Volume 2017:10 Pages 3095—3105
DOI https://doi.org/10.2147/OTT.S135312
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yao Dai
Zhen Li,1 Qianlan Yao,1 Songjian Zhao,1 Yin Wang,2,3 Yixue Li,1,4 Zhen Wang4
1School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, 2Shanghai Center for Bioinformation Technology, Shanghai Academy of Science and Technology, 3Collaborative Innovation Center for Genetics and Development, Fudan University, 4Key Laboratory of Computational Biology, CAS-MPG Partner Institute for Computational Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai, People’s Republic of China
Abstract: Esophageal squamous cell carcinoma (ESCC) is one of the most common malignancies worldwide and occurs at a relatively high frequency in People’s Republic of China. However, the molecular mechanism underlying ESCC is still unclear. In this study, the mRNA and long non-coding RNA (lncRNA) expression profiles of ESCC were downloaded from the Gene Expression Omnibus database, and then differential co-expression analysis was used to reveal the altered co-expression relationship of gene pairs in ESCC tumors. A total of 3,709 mRNAs and 923 lncRNAs were differentially co-expressed between normal and tumor tissues, and we found that most of the gene pairs lost associations in the tumor tissues. The differential regulatory networking approach deciphered that transcriptional dysregulation was ubiquitous in ESCC, and most of the differentially regulated links were modulated by 37 TFs. Our study also found that two novel lncRNAs (ADAMTS9-AS1 and AP000696.2) might be essential in the development of ectoderm and epithelial cells, which could significantly stratify ESCC patients into high-risk and low-risk groups, and were much better than traditional clinical tumor markers. Further inspection of two risk groups showed that the changes in TF-target regulation in the high-risk patients were significantly higher than those in the low-risk patients. In addition, four signal transduction-related DCmRNAs (ERBB3, ENSA, KCNK7, MFSD5), which were differentially co-expressed with the two lncRNAs, might also have the predictive capacity. Our findings will enhance the understanding of ESCC transcriptional dysregulation from a view of cross-link of lncRNA and mRNA, and the two-lncRNA combination may serve as a novel prognostic biomarker for clinical applications of ESCC.
Keywords: ESCC, differential co-expression analysis, differential regulation analysis, dysregulation, lncRNA, prognostic biomarker
Introduction
Esophageal cancer (EC) is one of the most lethal types of digestive tract malignancy in the world.1,2 Esophageal squamous cell carcinoma (ESCC) is the predominant subtype of EC, with the highest incidence in People’s Republic of China.3 Although the diagnosis and treatment have been advanced during recent years, ESCC still ranks among the fourth leading cause of cancer-related death.4 Because it is hard to detect at the early stage, and the tumor recurrence and metastasis after surgery are also very intractable, the long-term outcome of this malignancy is dismal, with an overall 5-year survival rate less than 10%.5–7 A poor understanding of carcinogenic mechanism and a lack of biomarkers with desired sensitivity and specificity pose a major challenge in diagnosing ESCC. Therefore, comprehensive surveying of the molecular mechanism will facilitate parsing of the esophageal tumorigenesis progress and detection of efficient prognostic biomarkers.
Protein coding genes are usually regarded as tumor markers in ESCC pathology,8 but only ~1.2% of the human genome encodes for protein-coding genes, and the large majority is transcribed into non-coding RNAs (ncRNAs).9–11 Long non-coding RNAs (lncRNAs), which are longer than 200 nucleotides without protein coding capability, can regulate the expression of genes in various biological processes.12,13 Aberrant lncRNA expression participates in carcinogenesis by disrupting the major biological processes,14 and could also serve as a potential diagnostic or prognostic biomarker for diverse human malignancies.15–17 For example, HOTAIR is a well-known lncRNA, whose expression levels powerfully predict metastases and survival in different cancer types,13,18 including ESCC.19,20 However, the understanding of these lncRNAs is insufficient, and the functional implications of most lncRNAs remain to be explored.
Genome-wide gene expression profile has become an instrumental resource and been used pervasively in cancer research. Many of prognostic and diagnostic mRNAs and lncRNAs have been identified by the traditional differential expression approach. However, this method focuses on the differentially expressed individual genes and cannot present the changes in gene interconnection in response to different conditions.21 Fortunately, differential co-expression analysis is not only emerging to complement the shortage, but also can hint the altered regulatory relationships and cancer-specific dysregulations.22–24 Construction of differential co-expression models, which integrate mRNAs and lncRNAs, could uncover the underlying functional roles of lncRNAs in tumorigenesis progress. Therefore, it is reliable to study the molecular biological mechanism of cancerogenesis and identify cancer-related prognostic lncRNAs by using differential co-expression analysis.
Here, we used a differentially co-expressed method by integrating information of mRNAs and lncRNAs from 119 ESCC tissues and matched adjacent normal tissues to identify a set of differentially co-expressed genes (DCGs; mRNAs and lncRNAs) and links. To systemically explain the mechanism of transcriptome alteration in ESCC, we developed a differential regulatory network by harnessing these differentially co-expressed mRNAs (DCmRNAs). Furthermore, based on the differentially co-expressed lncRNAs (DClncRNAs), we found two novel lncRNAs, ADAMTS9-AS1 and AP000696.2, that could predict the survival of patients with ESCC and might be essential in the development of the ectoderm and epithelial cells. Our study demonstrates that the transcriptional dysregulation is the critical cause of ESCC tumorigenesis, and the two-lncRNA signature may be regarded as a novel prognostic molecular marker, which is better than traditional biomarkers. Furthermore, it will be helpful to further experimental studies on lncRNAs in ESCC.
Materials and methods
Expression profile dataset
The normalized gene expression datasets of ESCC (GSE5362425 and GSE5362225) were downloaded from National Center for Biotechnology Information Gene Expression Omnibus database. The training dataset (GSE53624) included 119 ESCC and matched adjacent normal tissue samples. The independent testing dataset (GSE53622) contained 60 ESCC and matched adjacent normal tissue samples. All of patients in GSE53624 and GSE53622 were followed up for 5 years at least. The median follow-up time of patients in GSE53624 and GSE53622 was 32.2 and 39.5 months, respectively. The clinical characteristics of the two dataset populations are presented in Supplementary materials, Table S1. The expression profile of ESCC was performed using Agilent-038314 CBC Homo sapiens lncRNA+mRNA array V2.0 platform. (Agilent Technologies, Santa Clara, CA, USA). Each array contained probes interrogating 32,000 human mRNAs and 39,000 human lncRNAs. The probes with the same sequence were merged, resulting in 35,025 unique probes. GENCODE database was taken as reference to annotate mRNAs and lncRNAs. Then, we employed the Basic Local Alignment Search Tool (BLAST) program to map the unique probes to the reference; 17,245 mRNAs and 5,760 lncRNAs with at least one unique probe were retrieved.
Differential co-expression analysis
Differential co-expression analysis for the expression profile of the training dataset was conducted in R environment (V3.2.3) using differentially co-expressed genes and links (DCGL) package (V2.0).22–24 First, the genes were filtered by the method of gene variance with default options, which resulted in a total of 12,426 genes preserved. Next, differential co-expression profile and differential co-expression enrichment (DCe) methods were adopted to identify DCGs, while differentially co-expressed links (DCLs) were identified by the modified limit fold change model incorporated in the DCe method. In summary, 4,632 DCGs and about 6,384,526 DCLs were obtained, with each DCL containing at least one DCG. The DCGs consisted of 3,709 DCmRNAs and 923 DClncRNAs. In order to remove the low correlation DCLs, the correlation coefficients of gene pairs less than 0.2, 0.3 and 0.4 in both normal and tumor conditions were used as cutoffs. As similar results could be observed under the three criteria (see “Results” section), we selected 0.3 to be the final cutoff for removing low correlation DCLs, and the remaining DCLs were used for further analysis.
Functional analysis of ESCC-associated DCGs
The gene ontology (GO) and pathway enrichment analysis were performed using DAVID software26 to investigate the functional roles of DCmRNAs in the development of ESCC (Benjamin adjust P-value <0.01). In addition, priori knowledge was incorporated to verify the association of DCGs with the disease. Gene enrichment analysis was also performed for the DCmRNAs based on 572 cancer genes from Cancer Gene Census,27 1,601 human drug targets from DrugBank,28 as well as 363 ESCC-related genes from seven high-quality literatures (Fisher test, P-value <0.05).29–35 Meanwhile, gene enrichment analysis was performed for the DClncRNAs based on 253 cancer-related lncRNAs from Lnc2Cancer (Fisher test, P-value <0.05).36
DCmRNAs: construction of regulatory network
The regulatory networks were constructed to reveal the molecular mechanism of transcriptional alteration in ESCC. The transcription factor (TF)-target relationships predicted in our previous study were treated as the reference network of transcriptional regulation.37 The gene set of 3,709 DCmRNAs was used for the construction of ESCC-related TF-target networks in the normal and tumor conditions by using linear regression model.37 The important differentially regulated genes (DRGs) were identified by calculating the differential regulation (DR) value of genes between the two regulatory networks.37 The DR value could measure whether a DRG was highly relevant to the tumorigenesis. According to the changes of the regulation efficacy between a TF and its target, the differentially regulated links (DRLs) were identified, which were equal or greater than the average value across all TF-target pairs.
DClncRNAs: the identification of novel prognostic lncRNAs
The 923 DClncRNAs were taken as the initial gene set for screening prognostic biomarkers. To choose the cancer-related candidates, only the lncRNAs differentially co-expressed with cancer genes, drug targets or ESCC genes were considered. To shrink the DClncRNAs, we considered the differential expression between the normal and tumor tissues (Paired Student’s t-test, Benjamin-Hochberg adjust P-value <0.05), and the top 10 lncRNAs with the highest fold changes were selected as the prognostic candidates.
To obtain an optimal combination from the top 10 lncRNAs, 5-fold cross-validation was used. There are 1,023 combinations (210-1) for the 10 lncRNAs. For each combination, prognostic accuracy was calculated by the K-means algorithm based on their expression profile to stratify ESCC samples into high-risk and low-risk groups. Kaplan–Meier survival analysis was performed for the two groups, and statistical significance was assessed using the log-rank test by using the R survival package.38 The optimum lncRNA combination was determined by the most significant average P-value in the 5-fold cross-validation.
To explore the function of the identified prognostic lncRNAs, functional enrichment analysis was performed for mRNAs, which were differentially co-expressed with them. By doing this, 675 DCmRNAs were used to predict their prognostic capacities. The hazard ratio (HR) of genes were evaluated by using the SurvComp package in R, and a univariate Cox regression model was implemented to analyze the relationship between the gene expression and survival time. The genes with P-value <0.05 were selected for the survival analysis39,40 as we did for lncRNAs.
Results
Identified DCGs and DCLs
A total of 17,245 mRNAs and 5,760 lncRNAs across 119 ESCC and matched adjacent healthy tissue samples were retrieved from the training dataset. After being filtered by variance, 9,078 mRNAs and 3,348 lncRNAs were preserved for differential co-expression analysis by the DCGL package (see “Materials and methods” section). As a result, DCLs containing 4,632 DCGs were yielded for further analysis, including 3,709 DCmRNAs and 923 DClncRNAs.
To investigate the functions of the ESCC DCGs, functional enrichment analysis was carried out for the DCmRNAs, and significant GO terms are listed in Supplementary materials, Figure S1. These DCmRNAs might take part in ESCC cell growth (such as development, differentiation and proliferation of cells), in agreement with the fact that cancer is a disease involving dysregulation of multiple fundamental cell processes such as development, proliferation, differentiation, migration and apoptosis.41 The DCmRNAs were also enriched in two Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways of extracellular matrix (ECM)-receptor interaction and arachidonic acid metabolism, which were reported to contribute to esophageal squamous cell carcinogenesis.42–44 Gene enrichment analysis proved that the DCGs were also significantly enriched in drug targets (P-value =9.85E-6), ESCC genes (P-value =1.15E-14) and cancer related lncRNAs (P-value =3.04E-5), except cancer genes (P-value =0.34). We purposed that there would be some novel and important cancer-related genes in the DCGs.
After removing the DCLs with the absolute correlation coefficient less than 0.3 in both normal and tumor conditions, the rest of DCLs included 939,936 mRNA-lncRNA associations; 2,192,442 mRNA-mRNA associations and 112,005 lncRNA-lncRNA associations. We divided the DCLs into three types, “loss-of-association”, “gain-of-association” and “reverse-of-association”. If the absolute correlation coefficient of a DCL was equal or greater than 0.3 in the normal, but smaller than 0.3 in the ESCC, it would be grouped into the “loss-of-association” type. On the contrary, it belonged to the “gain-of-association” type. The “reverse-of-association” was the case wherein the absolute correlation coefficient was larger than 0.3 in both normal and tumor conditions, but the direction of association was reversed during tumorigenesis (Figure 1A). Our results showed that almost 80% DCLs lost associations, 20% gained new associations and only few of DCLs reversed associations (Figure 1B). We also tested other cutoffs of 0.2 and 0.4, and similar results were obtained (Supplementary materials, Figure S2). When DCLs were classified into three types, mRNA-lncRNA, mRNA-mRNA and lncRNA-lncRNA, this result still held, suggesting a widespread alteration of gene relationships in the transcriptome of ESCC.
 | Figure 1 The presentation of the DCLs. The expression profile was used for differential co-expression analysis to identify DCLs. (A) The cartoon sketch presented the association types between genes, and the DCLs were grouped into three different types. (B) The percentages of three types of DCLs. The loss-of-association DCLs was the predominant type. |
Comparison of regulatory network
We hypothesized that the dysfunctional regulation of TFs may be one of the important causes for transcriptome alteration. To prove the hypothesis, we used the 3,709 DCmRNAs to build normal and ESCC regulatory networks (see “Materials and methods” section). The regulated links could represent the causal influences in the network. By comparing the difference of the two regulatory networks, 4,442 and 3,492 regulated links were observed in the normal and ESCC conditions. The changes of the regulation efficacy were employed to screen the TF-target pairs, and 2,208 DRLs contributed by 37 TFs were identified. We found that seven of top 10 DRmRNAs that had the largest DR values (Supplementary materials, Table S2) were TFs, and these TFs regulated nearly half of DRLs. Among them, TCF3, TP53, MYB and JUN were cancer-related genes in the database of Cancer Gene Census.27 Next, DRLs were divided into five types according to the changes of the regulation efficacy in the tumor compared with the normal cells, “loss-of-regulation” (42.66%), “gain-of-regulation” (24%), “reverse-of-regulation” (20.11%), “weaken-of-regulation” (9.15%) and “strengthen-of-regulation” (4.08%) (Figure 2A). This result was consistent with our differential co-expression analysis that most DCLs belonged to the loss-of-association type.
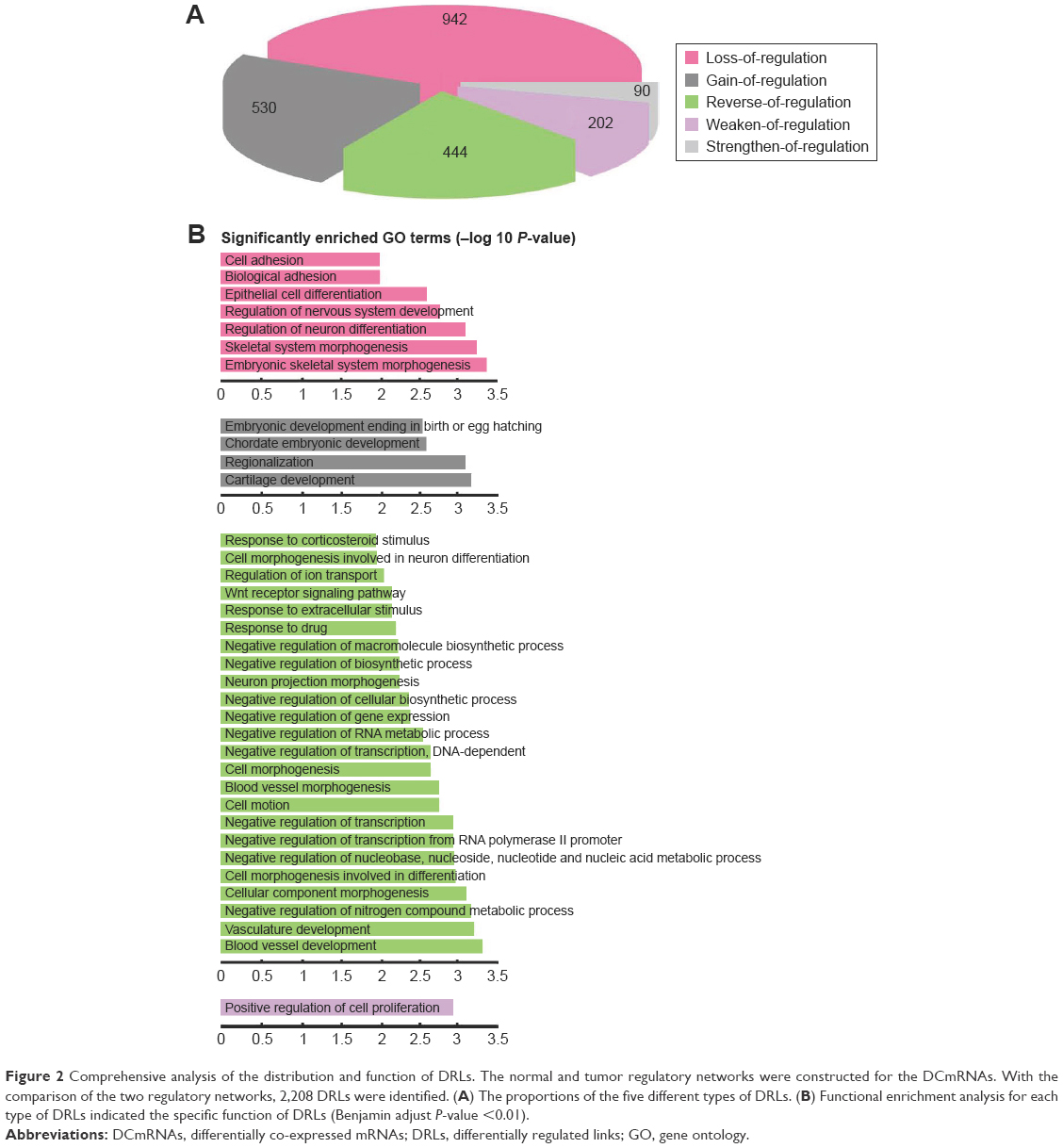 | Figure 2 Comprehensive analysis of the distribution and function of DRLs. The normal and tumor regulatory networks were constructed for the DCmRNAs. With the comparison of the two regulatory networks, 2,208 DRLs were identified. (A) The proportions of the five different types of DRLs. (B) Functional enrichment analysis for each type of DRLs indicated the specific function of DRLs (Benjamin adjust P-value <0.01). |
Then, the GO term functional enrichment analysis was separately performed for TF-target pairs in the five types of DRLs. We found that the five types were all significantly enriched in positive regulation of biosynthetic and metabolic processes and transcription (Supplementary materials, Table S3), and some biological functions were specific to each type (Figure 2B). Interestingly, 20.11% of reverse-of-regulation DRLs had the most significant GO terms, and they were enriched in both positive and negative regulation of biosynthetic and metabolic process and transcription, suggesting their capacity of bi-directional regulation. The KEGG pathway enrichment analysis revealed that the reverse-of-regulation DRLs significantly enriched in Wnt signaling pathway, which was proved to be associated with the ESCC progression and metastasis.33,45–47 Our results demonstrated that the alterations of gene-to-gene relationships in the ESCC may be mediated by the dysregulation of TFs to some extent.
Identified novel lncRNAs associated with the overall survival of ESCC patients
lncRNAs carry out the regulatory functions base on their complex structures which are convenient for binding proteins, RNA, DNA, and closely associate with the progression of disease that have been widely regarded as biomarkers. In order to investigate whether the DClncRNAs could become prognostic indicators for the survival of ESCC patients, differential co-expression method identified 923 DClncRNAs, which served as the initial prognostic candidates. In order to narrow down the DClncRNAs, a total of 820 cancer-related lncRNAs were found through differential co-expression with cancer genes, drug targets and ESCC genes, simultaneously (Figure 3). The differential expression analysis was adopted to narrow down the candidates (see “Materials and methods” section). The top 10 lncRNAs in terms of their fold change were selected for the cross-validation analysis. A two-lncRNA combination, ADAMTS9-AS1 (ENSG00000241158.5) and AP000696.2 (ENSG00000231324.1), was identified as the optimal combination for predicting the survival of ESCC patients (Figure 3).
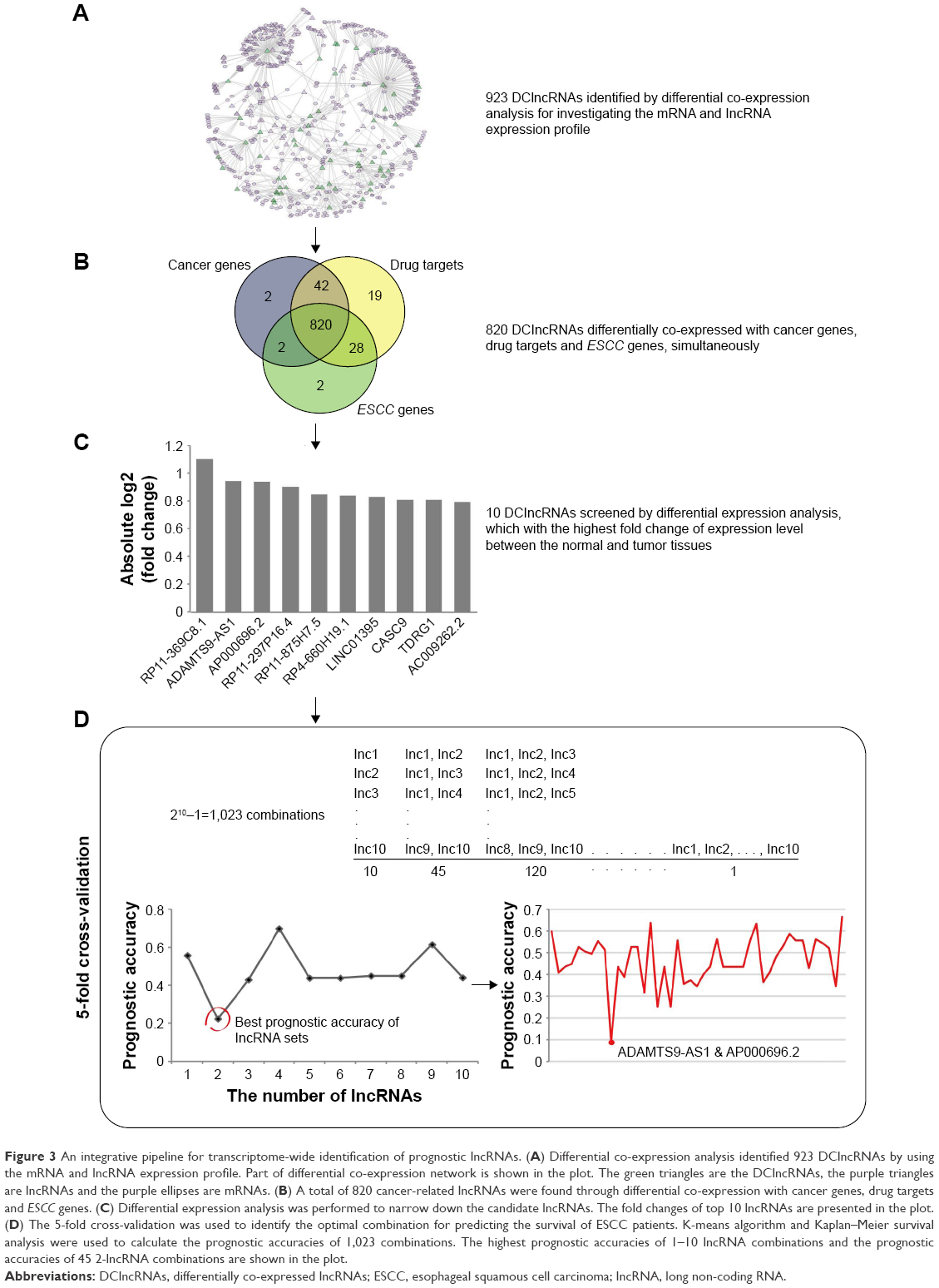 | Figure 3 An integrative pipeline for transcriptome-wide identification of prognostic lncRNAs. (A) Differential co-expression analysis identified 923 DClncRNAs by using the mRNA and lncRNA expression profile. Part of differential co-expression network is shown in the plot. The green triangles are the DClncRNAs, the purple triangles are lncRNAs and the purple ellipses are mRNAs. (B) A total of 820 cancer-related lncRNAs were found through differential co-expression with cancer genes, drug targets and ESCC genes. (C) Differential expression analysis was performed to narrow down the candidate lncRNAs. The fold changes of top 10 lncRNAs are presented in the plot. (D) The 5-fold cross-validation was used to identify the optimal combination for predicting the survival of ESCC patients. K-means algorithm and Kaplan–Meier survival analysis were used to calculate the prognostic accuracies of 1,023 combinations. The highest prognostic accuracies of 1–10 lncRNA combinations and the prognostic accuracies of 45 2-lncRNA combinations are shown in the plot. |
The expression of the two lncRNAs categorized 119 ESCC patients into high-risk and low-risk groups (log rank test, P-value =1.28E-2) (Figure 4A). Among them, 42 patients were identified as belonging to the “high-risk” group, in which less than 24% living people were observed and the median of overall survival time was 22.92 months. By contrast, 77 patients were grouped as belonging to the “low-risk” group, in which 47% living people were observed and the median of overall survival time was 48.77 months. Next, we verified the prediction power of the two lncRNAs in an independent dataset of 60 patients. The patients were classified into the high-risk (19) and low-risk groups (41) with significantly different survival time (log rank test, P-value =2.56E-2) (Figure 4B). With a similar result, there were 26% living patients in the high-risk group and the median survival time was 16 months, and 54% living patients in the low-risk group and the median survival time was 48 months. CYFRA21-1 and CEA, two traditional tumor markers, have been used for diagnosis of ESCC.48 High CYFRA21-1 level in patients is associated with poor prognosis. CEA is significantly associated with overall 5-year survival in ESCC.49–51 Then, we compared our lncRNA biomarkers with the traditional tumor markers CYFRA21-1 and CEA. The result showed that our lncRNA markers had a higher power to predict the survival of human ESCC patients (Figure 5).
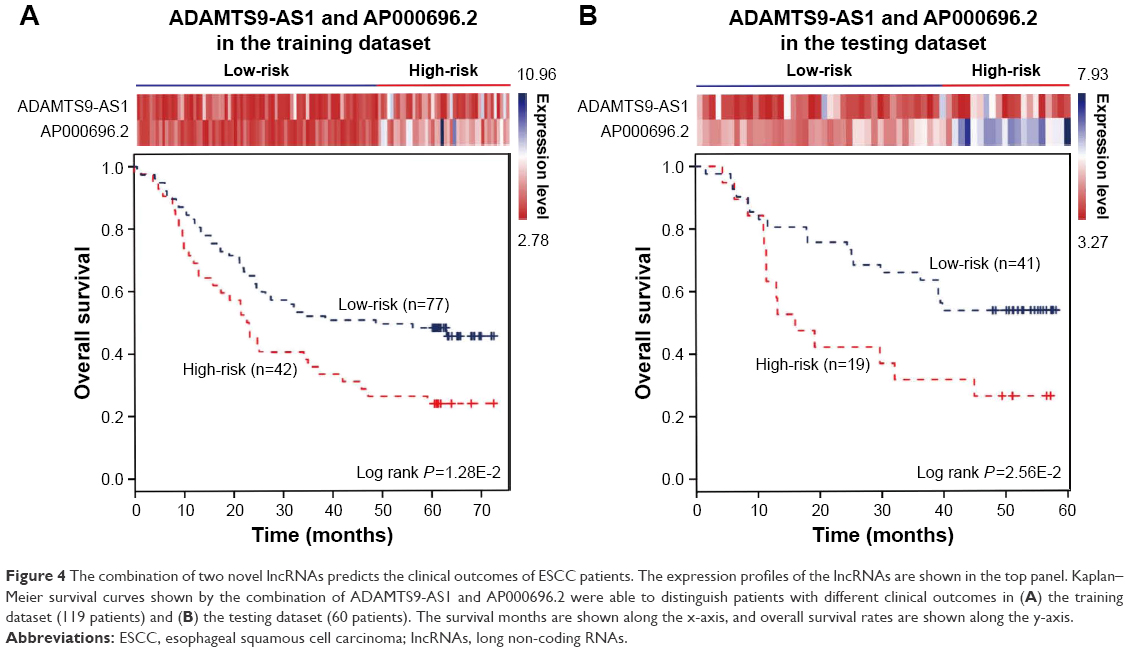 | Figure 4 The combination of two novel lncRNAs predicts the clinical outcomes of ESCC patients. The expression profiles of the lncRNAs are shown in the top panel. Kaplan–Meier survival curves shown by the combination of ADAMTS9-AS1 and AP000696.2 were able to distinguish patients with different clinical outcomes in (A) the training dataset (119 patients) and (B) the testing dataset (60 patients). The survival months are shown along the x-axis, and overall survival rates are shown along the y-axis. |
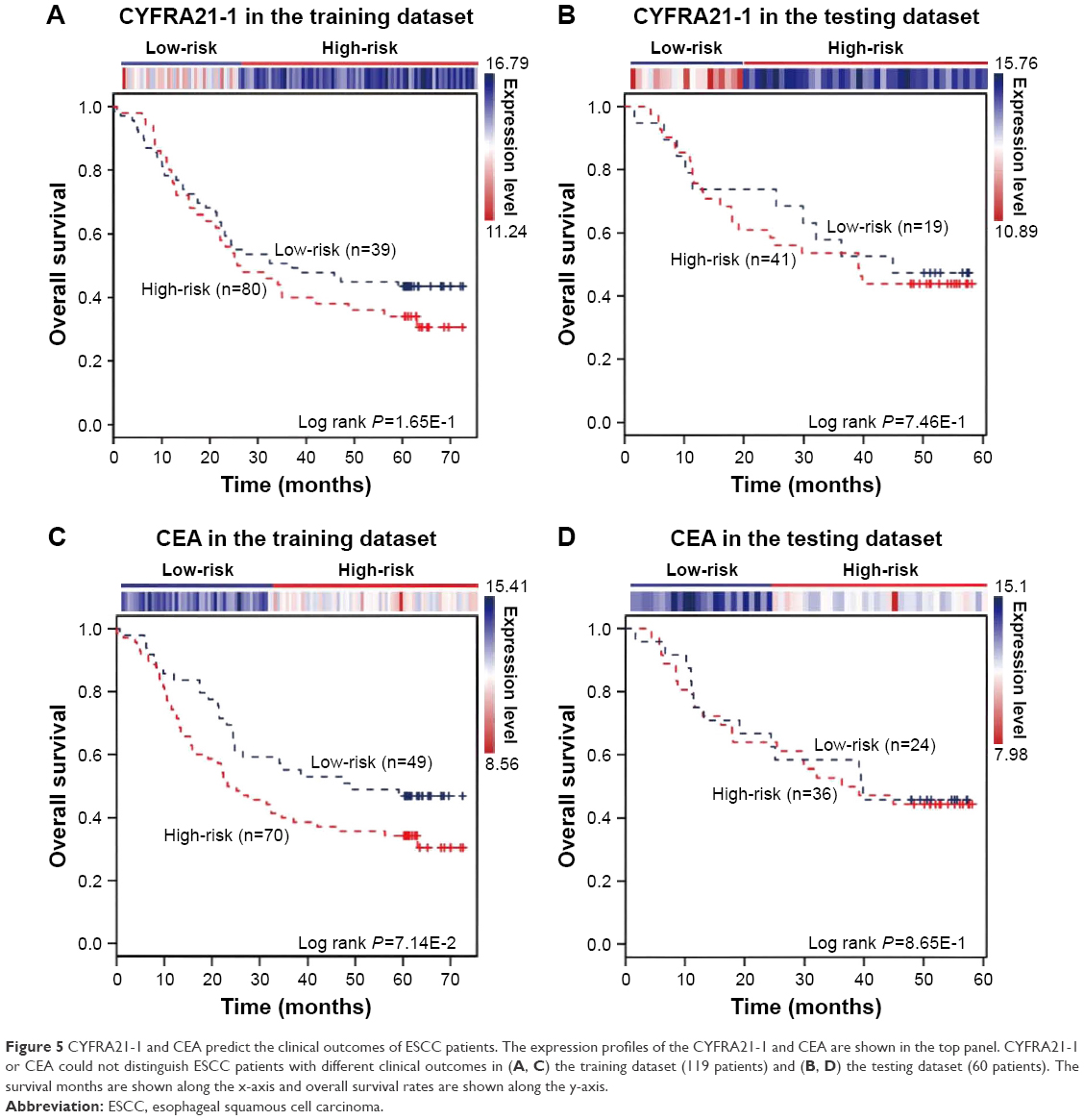 | Figure 5 CYFRA21-1 and CEA predict the clinical outcomes of ESCC patients. The expression profiles of the CYFRA21-1 and CEA are shown in the top panel. CYFRA21-1 or CEA could not distinguish ESCC patients with different clinical outcomes in (A, C) the training dataset (119 patients) and (B, D) the testing dataset (60 patients). The survival months are shown along the x-axis and overall survival rates are shown along the y-axis. |
mRNAs differentially co-expressed with the two-lncRNA biomarker
ADAMTS9-AS1 and AP000696.2 were two novel lncRNAs without any functional annotations, except another ADAMTS9 antisense transcript. ADAMTS9-AS2 could regulate the expression level of tumor suppressor ADAMTS9, and its overexpression resulted in significant inhibition of glioma cell migration.52 Furthermore, the starBase53 showed that ADAMTS9-AS1 interacted with two RNA-binding protein genes, DCGR8 and FUS. lncRNAs exert regulatory function in cancer biology mainly through their relationships with RNA-binding proteins. H19, HOTAIR, MALAT1 and HOTTIP are essential in many biological events for cell proliferation and differentiation, apoptosis and tumorigenesis via their impact on RNA-binding protein in HCC.54 Therefore, we proposed that ADAMTS9-AS1 might be functional in the development of cancer. To investigate the potential function of ADAMTS9-AS1 and AP000696.2 in ESCC, GO enrichment was performed to analyze the mRNAs that were differentially co-expressed with them. We got 1,139 mRNAs that were differentially co-expressed with ADAMTS9-AS1 or AP000696.2, in which 1,056 mRNAs lost associations and 86 mRNAs gained associations with the two lncRNAs. First, we performed the GO enrichment analysis for these mRNAs. The result indicated that they were significantly enriched in the biological process of ectoderm development and epidermis development (adjust P-value <0.01). The epidermis, which is formed by ectoderm, is the outermost layer of the skin, and protects the body from environmental insults.55 The development of the ectoderm and epidermis are two crucial functions in the progression of ESCC and hence the two novel lncRNAs might be very important in the development of the ESCC.
Second, we constructed the regulatory network for the high-risk and low-risk groups, which were classified by our two identified novel prognostic lncRNAs, respectively, and then compared with the normal regulatory network one by one. The results revealed that the changes of TF-target pairs between the normal and the low-risk group were smaller than those between the normal and the high-risk group (Wilcoxon test, P-value =2.15E-2) (Supplementary materials, Figure S3). This large number of dysregulation might contribute to the poor prognosis of the high-risk group.
Third, we wondered that if the mRNAs differentially co-expressed with the two prognostic lncRNAs also had the capacity of predicting the survival outcome of ESCC patients. To test this, we used the HR and survival analysis to screen the DCmRNAs from the 1,139 mRNAs (see “Materials and methods” section). Finally we identified four mRNAs, ERBB3, ENSA, KCNK7 and MFSD5, that could significantly divide the patients into high-risk and low-risk groups both in the training and testing datasets (Supplementary materials, Figure S4 and Supplementary materials, Table S4). These four mRNAs were all signal transduction-related genes.
Discussion
In this study, differential co-expression analysis of ESCC expression profiles was applied to analyze the mechanism of transcriptome alteration and identify the novel prognostic lncRNAs. We firstly proposed three different types of DCLs among the DCGs, “loss-of-association”, “gain-of-association” and “reverse-of-association”, and found that most DCLs belonged to the loss-of-association type. Considering the importance of DCmRNAs in ESCC, we constructed the normal and ESCC regulatory networks for the DCmRNAs to explore the alteration of gene co-expression caused by dysfunctional regulation of TFs. By comparison of two networks, we found that 37 TFs contributed to the all DRLs, in which the predominant type was the “loss-of-regulation”.
Nearly half of the DRLs were concerned by seven TFs in the top 10 DRGs, in which TCF3 and TP53 were very meaningful and worthy to notice as they contributed to more than a quarter of loss-of-regulation DRLs. TCF3 was the TF that had the highest DR value and the most DRLs in all DCmRNAs, and most of the DRLs regulated by TCF3 were loss-of-regulation type. It has been reported that TCF3 is a transcriptional repressor and plays the key role in cell fate, and its overexpression could block epithelial differentiation.56,57 TP53 was ranked the third one among the DRGs. In all, 76.92% of TP53-DRLs belonged to the loss-of-regulation, and none of the other TFs showed such obvious preference. The multifunctional TF TP53 was involved in the carcinogenesis of various malignancies, and is frequently mutated in >50% of human cancers.58–60 A lot of TP53 mutants can lead to the loss of its DNA-binding activity and affect its role as a TF.58–61
lncRNAs can be dysregulated in tumor progression62,63 and involved in tumorigenesis, invasion and metastasis.12,14 However, their potential as diagnostic and prognostic markers is less explored. In our study, we found and validated that a two-lncRNA combination (ADAMTS9-AS1 or AP000696.2) was the most optimal predictor of survival in ESCC, which significantly classified 42 and 77 patients into high-risk and low-risk groups with totally different survival times. The variance of TF-target regulation between the high- and low-risk groups indicated that transcriptional regulation might be altered with the deterioration of cancer. In addition, we found that our two-lncRNA combination had stronger predictive power than the known clinical markers, CYFRA21-1 and CEA, suggesting that it might be a potential clinical indicator. Functional enrichment analysis of the mRNAs differentially co-expressed with them suggested that they might be associated with the biological process of the development of the ectoderm and epithelial cells. Moreover, the four mRNAs (ERBB3,64,65 ENSA,66 KCNK7,67 MFSD568,69), involved in signal transductions and differentially co-expressed with ADAMTS9-AS1 or AP000696.2, could be regarded as predictors of survival outcomes in ESCC. This result provided another evidence for the rationality of the two novel lncRNAs. The original study of GSE53624 revealed a prognosis-related three-lncRNA signature, which classified the patients into two groups with significantly different overall survival. However, the machine learning method they used could not fully explain the underlying biological regulation mechanism, whereas our differential co-expression analysis method not only identified a more economical biomarker (a two-lncRNA combination) with similar prognostic capacity, but also provided an opportunity to investigate the possible functional role of the identified lncRNAs through the DCmRNAs.
Limitations
In summary, our study comprehensively analyzed the transcriptomes of ESCC. By using the differential co-expression method, we investigated the mechanism of abnormal regulation of mRNAs and lncRNAs, and identified a novel combination of two lncRNAs for predicting the survival of ESCC patients. There are limitations in our study. First, the functions of the two lncRNAs in tumorigenesis were still unknown, even though the potential biological processes had been inferred. Second, although the predictive ability of the two-lncRNAs signature was verified in the independent dataset, the sample size was limited. And it still needs experimental studies like RT-PCR, clinical trials and functional verification in the future. But computational investigation of functional lncRNAs is helpful to guide further experimental studies on lncRNAs. Our results may provide important resources for the future ESCC researches.
Acknowledgments
This work was supported by the National High Technology Research and Development Program of China (2015AA020104, 2015AA020108), the National Key Research and Development Program on Precision Medicine (2016YFC0901700, 2016YFC0901900, 2016YFC0901600), the National Key Technology Support Program (2013BAI101B09), the National Grand Program on Key Infectious Diseases (2015ZX10004801) and the Youth Innovation Promotion Association CAS.
Author contributions
ZL, ZW and YXL conceived the concept for this work; ZL performed the analyses and wrote the manuscript; ZL, QLY and ZW discussed the analyses; and ZW, SJZ and YW carried out the review. All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Lin Y, Totsuka Y, He Y, et al. Epidemiology of esophageal cancer in Japan and China. J Epidemiol. 2013;23(4):233–242. | ||
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Yang CS. Research on esophageal cancer in China: a review. Cancer Res. 1980;40(8 Pt 1):2633–2644. | ||
Holmes RS, Vaughan TL. Epidemiology and pathogenesis of esophageal cancer. Semin Radiat Oncol. 2007;17(1):2–9. | ||
Wang LS, Chow KC, Chi KH, et al. Prognosis of esophageal squamous cell carcinoma: analysis of clinicopathological and biological factors. Am J Gastroenterol. 1999;94(7):1933–1940. | ||
Liu J, Xie X, Zhou C, Peng S, Rao D, Fu J. Which factors are associated with actual 5-year survival of oesophageal squamous cell carcinoma? Eur J Cardiothorac Surg. 2012;41(3):e7–e11. | ||
Takeno S, Noguchi T, Takahashi Y, Fumoto S, Shibata T, Kawahara K. Assessment of clinical outcome in patients with esophageal squamous cell carcinoma using TNM classification score and molecular biological classification. Ann Surg Oncol. 2007;14(4):1431–1438. | ||
Lin DC, Du XL, Wang MR. Protein alterations in ESCC and clinical implications: a review. Dis Esophagus. 2009;22(1):9–20. | ||
Djebali S, Davis CA, Merkel A, et al. Landscape of transcription in human cells. Nature. 2012;489(7414):101–108. | ||
Derrien T, Johnson R, Bussotti G, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22(9):1775–1789. | ||
Hattori M. Finishing the euchromatic sequence of the human genome. Nature. 2005;50(2):162–168. | ||
Sugihara H, Ishimoto T, Miyake K, et al. Noncoding RNA expression aberration is associated with cancer progression and is a potential biomarker in esophageal squamous cell carcinoma. Int J Mol Sci. 2015;16(11):27824–27834. | ||
Gupta RA, Shah N, Wang KC, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464(7291):1071–1076. | ||
Qi P, Du X. The long non-coding RNAs, a new cancer diagnostic and therapeutic gold mine. Mod Pathol. 2013;26(2):155–165. | ||
Liu Q, Huang J, Zhou N, et al. LncRNA loc285194 is a p53-regulated tumor suppressor. Nucleic Acids Res. 2013;41(9):4976–4987. | ||
Gutschner T, Hämmerle M, Eissmann M, et al. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 2013;73(3):1180–1189. | ||
Yuan SX, Yang F, Yang Y, et al. Long noncoding RNA associated with microvascular invasion in hepatocellular carcinoma promotes angiogenesis and serves as a predictor for hepatocellular carcinoma patients’ poor recurrence-free survival after hepatectomy. Hepatology. 2012;56(6):2231–2241. | ||
Qiu JJ, Lin YY, Ye LC, et al. Overexpression of long non-coding RNA HOTAIR predicts poor patient prognosis and promotes tumor metastasis in epithelial ovarian cancer. Gynecol Oncol. 2014;134(1):121–128. | ||
Lv XB, Lian GY, Wang HR, Song E, Yao H, Wang MH. Long noncoding RNA HOTAIR is a prognostic marker for esophageal squamous cell carcinoma progression and survival. PLoS One. 2013;8(5):e63516. | ||
Li X, Wu Z, Mei Q, et al. Long non-coding RNA HOTAIR, a driver of malignancy, predicts negative prognosis and exhibits oncogenic activity in oesophageal squamous cell carcinoma. Br J Cancer. 2013;109(8):2266–2278. | ||
Cui X, Churchill GA. Statistical tests for differential expression in cDNA microarray experiments. Genome Biol. 2003;4(4):210. | ||
Yang J, Yu H, Liu BH, et al. DCGL v2.0: an R package for unveiling differential regulation from differential co-expression. PLoS One. 2013;8(11):e79729. | ||
Yu H, Liu BH, Ye ZQ, Li C, Li YX, Li YY. Link-based quantitative methods to identify differentially coexpressed genes and gene pairs. BMC Bioinformatics. 2011;12:315. | ||
Liu BH, Yu H, Tu K, Li C, Li YX, Li YY. DCGL: an R package for identifying differentially coexpressed genes and links from gene expression microarray data. Bioinformatics. 2010;26(20):2637–2638. | ||
Li J, Chen Z, Tian L, et al. LncRNA profile study reveals a three-lncRNA signature associated with the survival of patients with oesophageal squamous cell carcinoma. Gut. 2014;63(11):1700–1710. | ||
Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. | ||
Futreal PA, Coin L, Marshall M, et al. A census of human cancer genes. Nat Rev Cancer. 2004;4(3):177–183. | ||
Wishart DS, Knox C, Guo AC, et al. DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008;36(Database issue):D901–D906. | ||
Luo A, Kong J, Hu G, et al. Discovery of Ca2+-relevant and differentiation-associated genes downregulated in esophageal squamous cell carcinoma using cDNA microarray. Oncogene. 2004;23(6):1291–1299. | ||
Yamabuki T, Daigo Y, Kato T, et al. Genome-wide gene expression profile analysis of esophageal squamous cell carcinomas. Int J Oncol. 2006;28(6):1375–1384. | ||
Kashyap MK, Marimuthu A, Kishore CJH, et al. Genome-wide mRNA profiling of esophageal squamous cell carcinoma for identification of cancer biomarkers. Cancer Biol Ther. 2009;8(1):36–46. | ||
Lin DC, Hao JJ, Nagata Y, et al. Genomic and molecular characterization of esophageal squamous cell carcinoma. Nat Genet. 2014;46(5):467–473. | ||
Song Y, Li L, Ou Y, et al. Identification of genomic alterations in oesophageal squamous cell cancer. Nature. 2014;509(7498):91–95. | ||
Hyland PL, Zhang H, Yang Q, et al. Pathway, in silico and tissue-specific expression quantitative analyses of oesophageal squamous cell carcinoma genome-wide association studies data. Int J Epidemiol. 2015;45(1):206–220. | ||
Hu YC, Lam KY, Law S, Wong J, Srivastava G. Profiling of differentially expressed cancer-related genes in esophageal squamous cell carcinoma (ESCC) using human cancer cDNA arrays: overexpression of oncogene MET correlates with tumor differentiation in ESCC. Clin Cancer Res. 2001;7(11):3519–3525. | ||
Ning S, Zhang J, Wang P, et al. Lnc2Cancer: a manually curated database of experimentally supported lncRNAs associated with various human cancers. Nucleic Acids Res. 2016;44(D1):D980–D985. | ||
Cao MS, Liu BY, Dai WT, Zhou WX, Li YX, Li YY. Differential network analysis reveals dysfunctional regulatory networks in gastric carcinogenesis. Am J Cancer Res. 2015;5(9):2605–2625. eCollection 2015. | ||
Kramar A, Com-Nougué C. [Estimate of adjusted survival curves]. Revue D’épidémiologie Et De Santé Publique. 1990;38(2):149–152. French [with English abstract]. | ||
Haibe-Kains B, Desmedt C, Sotiriou C, Bontempi G. A comparative study of survival models for breast cancer prognostication based on microarray data: does a single gene beat them all? Bioinformatics. 2008;24(19):2200–2208. | ||
Schröder MS, Culhane AC, Quackenbush J, Haibe-Kains B. Survcomp: an R/Bioconductor package for performance assessment and comparison of survival models. Bioinformatics. 2011;27(22):3206–3208. | ||
Kreeger PK, Lauffenburger DA. Cancer systems biology: a network modeling perspective. Carcinogenesis. 2010;31(1):2–8. | ||
Chen X, Li N, Wang S, et al. Aberrant arachidonic acid metabolism in esophageal adenocarcinogenesis, and the effects of sulindac, nordihydroguaiaretic acid, and alpha-difluoromethylornithine on tumorigenesis in a rat surgical model. Carcinogenesis. 2002;23(12):2095–2102. | ||
Zhi H, Zhang J, Hu G, et al. The deregulation of arachidonic acid metabolism-related genes in human esophageal squamous cell carcinoma. Int J Cancer. 2003;106(3):327–333. | ||
Li X, Jiang C, Wu X, et al. A systems biology approach to study the biology characteristics of esophageal squamous cell carcinoma by integrating microRNA and messenger RNA expression profiling. Cell Biochem Biophys. 2014;70(2):1369–1376. | ||
Moghbeli M, Abbaszadegan MR, Golmakani E, Forghanifard MM. Correlation of Wnt and NOTCH pathways in esophageal squamous cell carcinoma. J Cell Commun Signal. 2016;10(2):129–135. | ||
Wang X, Gao Z, Liao J, et al. lncRNA UCA1 inhibits esophageal squamous-cell carcinoma growth by regulating the Wnt signaling pathway. J Toxicol Environ Health Part A. 2016;79(9–10):407–418. | ||
Ai R, Sun Y, Guo Z, et al. NDRG1 overexpression promotes the progression of esophageal squamous cell carcinoma through modulating Wnt signaling pathway. Cancer Biol Ther. 2016;17(9):943–954. | ||
Mealy K, Feely J, Reid I, Mcsweeney J, Walsh T, Hennessy TPJ. Tumour marker detection in oesophageal carcinoma. Eur J Surg Oncol. 1996;22(5):505–507. | ||
Cao X, Zhang L, Feng GR, et al. Preoperative Cyfra21-1 and SCC-Ag serum titers predict survival in patients with stage II esophageal squamous cell carcinoma. J Transl Med. 2012;10:197. | ||
Dong J, Zeng BH, Xu LH, et al. Anti-CDC25B autoantibody predicts poor prognosis in patients with advanced esophageal squamous cell carcinoma. J Transl Med. 2010;8:81. | ||
Yi Y, Li B, Wang Z, Sun H, Gong H, Zhang Z. CYFRA21-1 and CEA are useful markers for predicting the sensitivity to chemoradiotherapy of esophageal squamous cell carcinoma. Biomarkers. 2009;14(7):480–485. | ||
Yao J, Zhou B, Zhang J, et al. A new tumor suppressor LncRNA ADAMTS9-AS2 is regulated by DNMT1 and inhibits migration of glioma cells. Tumor Biol. 2014;35(8):7935–7944. | ||
Li JH, Liu S, Zhou H, Qu LH, Yang JH. StarBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014;42(Database issue):D92–D97. | ||
Mohamadkhani A. Long noncoding RNAs in interaction with RNA binding proteins in hepatocellular carcinoma. Hepat Mon. 2014;14(5):e18794. | ||
Kern F, Niault T, Baccarini M. Ras and Raf pathways in epidermis development and carcinogenesis. Brit J Cancer. 2011;104(2):229–234. | ||
Howard JM, Nuguid JM, Ngole D, Nguyen H. Tcf3 expression marks both stem and progenitor cells in multiple epithelia. Development. 2014;141(16):3143–3152. | ||
Nguyen H, Rendl M, Fuchs E. Tcf3 governs stem cell features and represses cell fate determination in skin. Cell. 2006;127(1):171–183. | ||
Oijen MGCTV. Gain-of-function mutations in the tumor suppressor gene p53. Clin Cancer Res. 2000;6(6):2138–2145. | ||
Hainaut P, Hollstein M. p53 and human cancer: the first ten thousand mutations. Adv Cancer Res. 2000;77:81–137. | ||
Kropveld A, Rozemuller EH, Leppers FG, et al. Sequencing analysis of RNA and DNA of exons 1 through 11 shows p53 gene alterations to be present in almost 100% of head and neck squamous cell cancers. Lab Invest. 1999;79(3):347–353. | ||
Aranda M, Gonzalez-Nilo F, Riadi G, et al. Loss of TP53-DNA interaction induced by p.C135R in lung cancer. Oncol Rep. 2007;18(5):1213–1217. | ||
Prensner JR, Chinnaiyan AM. The emergence of lncRNAs in cancer biology. Cancer Discov. 2011;1(5):391–407. | ||
Spizzo R, Almeida MI, Colombatti A, Calin GA. Long non-coding RNAs and cancer: a new frontier of translational research? Oncogene. 2012;31(43):4577–4587. | ||
Choi BK, Fan X, Deng H, Zhang N, An Z. ERBB3 (HER3) is a key sensor in the regulation of ERBB-mediated signaling in both low and high ERBB2 (HER2) expressing cancer cells. Cancer Med. 2012;1(1):28–38. | ||
Yan X, Chen X, Liang H, et al. miR-143 and miR-145 synergistically regulate ERBB3 to suppress cell proliferation and invasion in breast cancer. Mol Cancer. 2014;13:220. | ||
Chen YL, Kuo MH, Lin PY, et al. ENSA expression correlates with attenuated tumor propagation in liver cancer. Biochem Biophys Res Commun. 2013;442(1–2):56–61. | ||
Yost CS, Oh I, Eger EI 2nd, Sonner JM. Knockout of the gene encoding the K(2P) channel KCNK7 does not alter volatile anesthetic sensitivity. Behav Brain Res. 2008;193(2):192–196. | ||
Tang T, Li L, Tang J, et al. A mouse knockout library for secreted and transmembrane proteins. Nat Biotechnol. 2010;28(7):749–755. | ||
Perland E, Lekholm E, Eriksson MM, Bagchi S, Arapi V, Fredriksson R. The putative SLC transporters Mfsd5 and Mfsd11 are abundantly expressed in the mouse brain and have a potential role in energy homeostasis. PLoS One. 2016;11(6):e0156912. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.