Back to Journals » OncoTargets and Therapy » Volume 13
Comprehensive Analysis of CDC27 Related to Peritoneal Metastasis by Whole Exome Sequencing in Gastric Cancer
Authors Wu R, Li Q, Wu F, Shi C, Chen Q
Received 31 December 2019
Accepted for publication 8 March 2020
Published 21 April 2020 Volume 2020:13 Pages 3335—3346
DOI https://doi.org/10.2147/OTT.S244351
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Leo Jen-Liang Su
Riping Wu,1,* Qiaolian Li,1,* Fan Wu,2 Chunmei Shi,1 Qiang Chen3
1Fujian Medical University Union Hospital, Fuzhou, Fujian Province, People’s Republic of China; 2Fujian Medical University, Fuzhou, Fujian Province, People’s Republic of China; 3Fujian Medical University Union Hospital, Fuzhou, Fujian Province, People’s Republic of China, Stem Cell Research Institute, Fujian Medical University, Fuzhou, Fujian Province, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Qiang Chen; Chunmei Shi
Department of Medical Oncology, Fujian Medical University Union Hospital, Fuzhou, Fujian Province, People’s Republic of China
Email [email protected]; [email protected]
Introduction: The peritoneum is the most common metastatic site of gastric cancer and is associated with a dismal prognosis. However, there is no reliable biomarker for predicting peritoneal metastasis (PM).
Materials and Methods: Whole-exome sequencing (WES) was performed on formalin-fixed, paraffin-embedded (FFPE) samples from 63 patients with stage I–III gastric cancer and circulating tumor DNA (ctDNA) samples from 10 patients with stage IV gastric cancer. Differentially expressed genes (DEGs) were identified between the PM and non-PM groups and analyzed by multiple bioinformatics analyses. Univariate and multivariate Cox regression analyses were used to identify the risk factors for PM and a risk score model was developed.
Results: The number of mutant genes and the tumor mutation burden (TMB) in the PM group were higher than those in the non-PM group (p < 0.05). There was a significant positive correlation between the number of mutant genes and the TMB (R2 = 0.9997). The risk of PM was significantly higher in the high TMB group than in the low TMB group (p = 0.045). Forty-nine DEGs were identified as associated with PM in gastric cancer. CDC27 mutations were associated with a higher risk for PM and poor survival. The CDC27 mutations were located in the Apc3 region, the TPR region, and the phosphorylation region, and new mutation sites were not included in the TCGA database. Multivariable Cox regression analysis demonstrated that pathological T stage, poor tumor differentiation, Borrmann type, and CDC27 mutations were independent predictive factors of PM. A risk score model was constructed that demonstrated good performance.
Conclusion: Through WES, we identified 49 DEGs relevant to PM in gastric cancer. CDC27 mutations were independently associated with PM by statistical and bioinformatics analyses. A risk score model was built and was demonstrated to effectively discriminate gastric cancer patients with and without PM.
Keywords: gastric cancer, peritoneal metastasis, whole-exome sequencing, CDC27
Introduction
Gastric cancer is one of the most common malignancies around the world.1 Peritoneal metastasis (PM), mainly induced by the dissemination of free tumor cells into the peritoneal cavity, is the most metastatic pattern of gastric cancer.2 PM from gastric cancer results in a dismal prognosis and contributes to the major cause of gastric cancer-related deaths.3 Currently, many management strategies, such as neoadjuvant intraoperative chemotherapy, cytoreductive surgery (CRS), hyperthermic intraperitoneal chemoperfusion (HIPEC), and postoperative chemotherapy, have been developed to improve the prognosis of gastric cancer patients with PM.3,4 Given that any delay in diagnosis is detrimental for patients with peritoneal metastatic gastric cancer, early prediction of gastric cancer with PM has become imperative for treatment decision-making and improving patient prognosis.
Currently, early detection of gastric cancer with PM is difficult owing to a high rate of false-negative assessment by conventional imaging examinations.5,6 Serum tumor markers, such as carcinoembryonic antigen (CEA), carbohydrate antigen 19–9 (CA19-9), and carbohydrate antigen 125 (CA125), have been demonstrated to correlate with PM from gastric cancer. However, the clinical utility of these markers in PM from gastric cancer is ambiguous due to the relatively low accuracy.7,8 Currently, early prediction or diagnosis of PM from gastric cancer is difficult before an invasive laparotomy.9 Therefore, it is imperative to identify a non-invasive and sensitive predictive biomarker of gastric cancer with PM.
Recently, the next-generation sequencing (NGS) technology has revolutionized cancer research. In the era of precision medicine, NGS provides a high-throughput tool to systematically identify genetic alterations and potential therapeutic targets.10 Compared with the costly whole-genome sequencing (WGS), the cost-effective whole-exome sequencing (WES) directly sequences all the coding regions (exons) to evaluate the relationship between mutations and phenotypes in tumors, including gastric cancer.11 By identifying driver genes involved in tumorigenesis, WES analyses could enable the identification of novel predictive biomarkers and potential therapeutic targets in metastatic cancer.12 However, studies regarding the utility of WES in PM from gastric cancer are limited.13,14
In this study, we performed WES using tissue and blood samples from gastric cancer patients. Using a combination of bioinformatics analysis results and clinical data, a predictive model for postoperative PM of gastric cancer was constructed. The key gene, CDC27, predicting PM was also screened.
Materials and Methods
Sample Collection
Tissue and blood samples were collected from pathological proven gastric adenocarcinoma in the Fujian Medical University Union Hospital between March 2008 and December 2012. All patients who provided tissue samples were clinically staged as TNM stage I–III and did not receive any anticancer treatments such as chemotherapy before surgery. Patients were divided into two groups according to whether postoperative PM was developed, including PM (n=26) and non-PM (n=37) groups. Tissue samples including gastric cancer and adjacent non-cancerous tissues were harvested from surgical specimens after surgical resection. Each piece was formalin-fixed, paraffin-embedded (FFPE) and then sliced into 10μm sections. Blood samples were collected from 10 stage IV gastric cancer patients (PM group n=4, and non-PM group n=6). Plasma was separated within 2 hours of collection and stored at −80°C for future research. This study was approved by the Ethical Committee of Fujian Medical University Union Hospital and was conducted according to the approved guidelines. All patients signed an informed consent prior to their enrollment.
WES for FFPE Samples
The genomic DNAs of FFPE samples were isolated and purified using QIAamp DNA Micro kits (Qiagen, Hilden, Germany) following the manufacturer’s instructions. Whole exomes were captured using the BGI Human All Exon V4 kit. DNA Nanoballs (DNBs) were produced by rolling circle amplification (RCA). All qualified capture libraries were loaded onto the BGISEQ-500 platform and each captured library was subjected to pair-ended 50 or 100 bp sequencing on an HiSeq 3000 platform (Illumina Inc., San Diego, CA, USA). After quality filtering and repeat removal of the reads, the average sequencing of each sample was around 100 million reads. The average sequencing depths of FFPE tumors and corresponding normal tissues were 117X and 92X, respectively. The raw data was filtered using the software SOAPnuke-1.5.6, and the filtered clean reads were mapped to the human HG19 reference genome and the duplicate reads were deleted using Edico software (http://edicogenome.com/dragen-bioit-platform). The remaining reads were calibrated and realigned using Genome Analysis Toolkit (GATK).15 Single nucleotide variations (SNVs) and insertions/deletions (InDels) were detected using MuTect16 and Varscan2 software,17 respectively. The detected mutations were annotated with ANNOVAR,18 and then through the screening steps to obtain reliable results. Mutation sites were randomly selected in tumor tissues and adjacent normal tissues, and verified by MassARRAY platform. Tumor mutation burden (TMB) was defined as the number of non-synonymous SNVs and InDels occurring per Mb base.
WES for Circulating Tumor DNA (ctDNA) Samples
Cell-free ctDNA was isolated from 1.7–3.3 mL plasma sample using QIAsymphony Circulating DNA Kit (Qiagen, Hilden, Germany), and peripheral blood lymphocytes (PBL) DNA was extracted using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. DNA concentration was measured using the Qubit 3.0 Fluorometer (Thermo Fisher Scientific, USA) and the Qubit dsDNA HS (High Sensitivity) Assay Kit (Thermo Fisher Scientific, USA). Every cfDNA sample was analyzed on the Agilent 2100 BioAnalyzer using the Agilent High Sensitivity DNA Kit, and cfDNA fragments were distributed with a dominant peak at approximately 170 bp. Sequencing libraries of both cfDNA and PBL DNA were constructed using the KAPA DNA Library Preparation Kit, and measured using an Agilent 2100 Bioanalyzer and an Applied Biosystems 7500 real-time PCR system. DNA sequencing was performed on the HiSeq3000 Sequencing System with 2×75 bp paired-end reads. The processed clean reads were aligned to the human genome build GRCh37 using BWA (a Burrows-Wheeler aligner).19 Picard tools (http://broadinstitute.github.io/picard) were used to mark PCR duplicates. SNVs and small Indels were detected using MuTect and GATK, respectively. All candidate somatic mutations identified by the bioinformatics pipeline were manually reviewed in the Integrative Genomics Viewer (IGV)20 through assessing the quality of base calls, the mapping quality of the reads, and the overall read depth at each mutation site. Mutations were annotated to genes by ANNOVAR software to identify the mutated protein-coding position and filtered intronic and silent changes. CONTRA21 was used to detect copy number variants (CNVs).
Statistical Analysis
All statistical analyses were performed using IBM SPSS software version 23.0 (SPSS Inc, Chicago, IL) and R version 3.4.3. Comparison of categorized variables was evaluated using the chi-square test, and comparison of numerical variables were evaluated using the Wilcoxon test. Survival analysis was performed using the Kaplan-Meier method and the Log rank test. Least absolute shrinkage and selection operator (LASSO) model was applied to determine the ideal coefficient for each prognostic feature and estimate the likelihood deviance with the “glmnet” package in R software.22 All variables were entered into the Cox regression model to identify predictors of PM. The corresponding risk scores for the samples from validation datasets were calculated using the risk score system. Patients were divided into high-risk and low-risk groups based on the median values of the risk score. The entire patient cohort was divided into two subgroups according to patient outcomes (dead or alive). Then, Receiver Operator Characteristic (ROC) curves were plotted based on the risk scores and survival status. The risk score was selected as the cut-off value when the AUC reached its maximum. Kaplan-Meier curves and Cox regression analysis were performed to compare PM risk between high-risk and low-risk groups. The performance of the model was evaluated by time-dependent ROC analysis. A two-sided p < 0.05 was considered statistically significant.
Results
WES Analysis of FFPE Samples from 63 Gastric Cancer Patients
WES was performed on FFPE samples from 63 gastric cancer patients (PM group n = 26, non-PM group n = 37). As shown in Figure 1A, 5014 mutated genes and 9559 mutation sites were detected. The heatmap showed that the number of mutant genes and the TMB were significantly higher in the PM group than in the non-PM group. The median number of mutation sites was 66 (range 2–1180), including 94.9% SNVs and 5.1% InDels (Figures 1C–F). As shown in Figure 1B, missense mutations (61.9%) were the most common mutation type, followed by synonymous mutations (27%) and frameshift insertion/deletion mutations (4.1%). Point mutations were characterized by a predominance of C > T, followed by C > A and T > C (Figure 1D). The frequency distribution of the mutant genes associated with PM was as follows (in order of greatest to least): TTN, TP53, MUC16, CDC27, and SYNE1, as shown in Figure 1G.
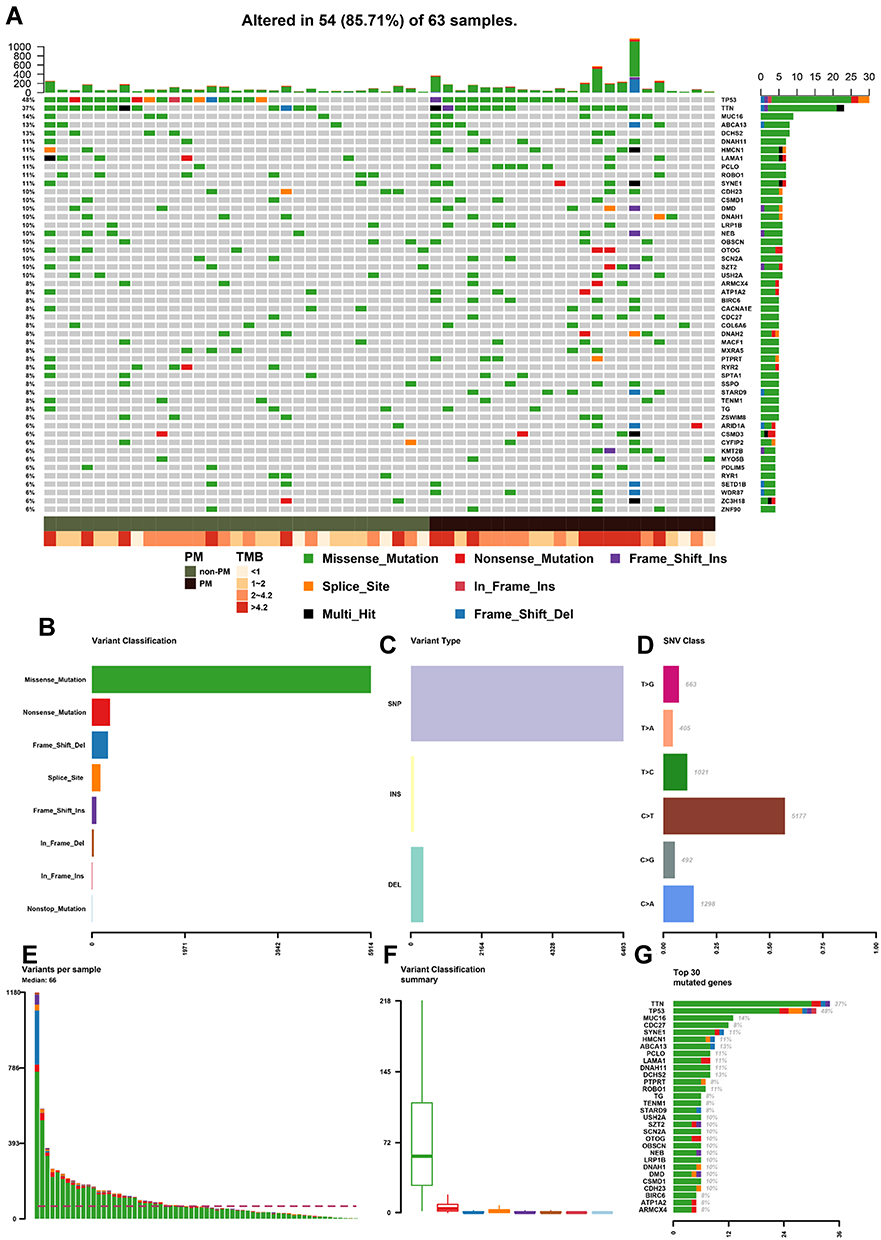 |
Figure 1 (A) Heatmap of mutated genes in stage I–III GC patients by WES. (B) Number of each type of mutation. (C) Number of SNV and Indels in all mutations. (D) Distribution of point mutation types in SNV. E. Number of mutation sites in each sample. (F) Median and interquartile range of each mutation type. (G) Top 30 genes with the most mutation sites. Abbreviations: GC, gastric cancer; WES, whole exome sequencing; SNV, single nucleotide variation. |
TMB Analysis
The number of mutant genes in the PM group was significantly higher than that in the non-PM group (91/person vs. 53/person, p = 0.04, Figure 2A). The TMB of the PM group was also higher than that of the non-PM group (median TMB2.84/Mb vs. 1.84/Mb, p = 0.064, Figure 2B). A significant positive correlation was found between mutant genes and TMB (R2 = 0.9997, Figure 2C). The PM group was associated with a significantly worse overall survival (OS) compared to the non-PM group (p < 0.01, Figure 2E). The patients were divided into a high TMB group and a low TMB group according to the cutoff value determined by the ROC curve (TMB = 2.13/Mb, sensitivity 61.5%, specificity 64.9%, and Youden index 0.264). The Kaplan-Meier curve demonstrated that the high TMB group was associated with a higher risk of PM than the low TMB group (p = 0.045, Figure 2D).
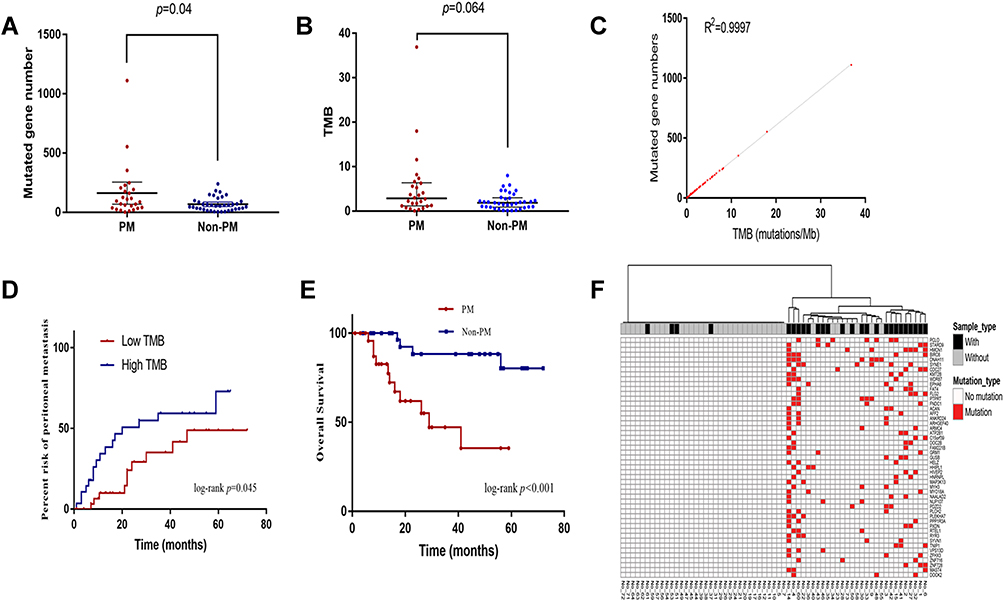 |
Figure 2 (A) The number of mutant genes in PM patients was higher than that of non-PM patients. (B) TMB in PM patients was higher than that of non-PM patients. (C) The positive correlation between TMB and the number of mutant genes. (D) Patients with high TMB have a higher risk of PM than low TMB patients. (E) The PM group was associated with a worse OS compared to the non-PM group. (F) The heatmap demonstrated that these 49 genes could discriminate patients with and without PM. Abbreviations: PM, peritoneal metastasis; TMB, tumor mutation burden; OS, overall survival. |
Identification of PM-Associated DEGs
We used Chi-squared tests (one-sided test, p < 0.1) to analyze the differential distribution of the mutant genes between the two groups. As listed in Table 1, 49 genes associated with PM were identified and defined as PM-related DEGs. A supervised hierarchical cluster analysis of 49 PM-related DEGs was performed based on whether the 63 gastric cancer patients had mutations. As shown in Figure 2F, the heatmap demonstrated that these 49 genes could discriminate patients with and without PM. Among them, four genes were classified into the non-PM group and did not contain differential genes.
 |
Table 1 Differential Genes with Higher Mutation Rate in PM Patients |
WES Analysis of Plasma Samples from 10 Stage IV Gastric Cancer Patients
WES analysis was performed on plasma samples from 10 stage IV gastric cancer patients (PM group n = 4, non-PM group n = 6). A total of 51 mutated genes were detected (PM n = 21, non-PM n = 32). The intersection of the two groups was MSH3 and TP53 mutations. A total of five genes showed copy number variations (CNVs). The three genes with the highest variant allele frequency (VAF) were TP53 (45.82%), MAP3K13 (31.71%), and PIK3CA (28.08%). Among the 51 mutant genes, the highest proportion of TP53 was 50% (5/10), as shown in Figure 3A. EPHB6 was only found in the PM group and KCNH2 was only found in the non-PM group. In copy number variation, HER2 and MYC amplifications occurred only in the PM group, whereas CCNE1, FGFR2, and MET amplifications were found only in the non-PM group. All variant genes in ctDNA were detected as heatmaps shown in Figure 3A. Gene Ontology (GO) functional analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of all variant genes were performed using the DAVID database,23 as shown in Figure 3B and C. Variant gene analysis was performed in the PM group. The results demonstrated that PM was associated with an abnormal Wnt signaling pathway, particularly related to abnormal proteins involved in the cell cycle, apoptosis regulation, and DNA damage repair.
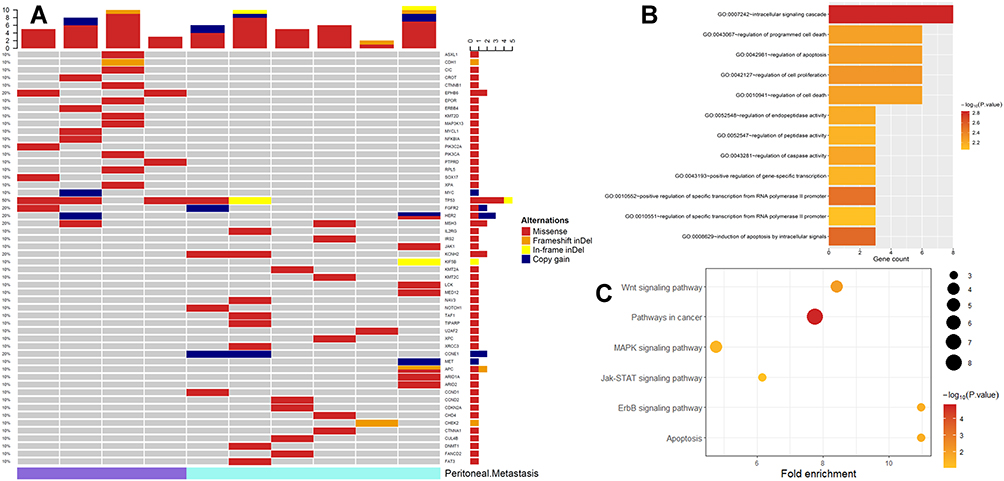 |
Figure 3 (A) Heatmap of all variant genes detected in ctDNA; (B) Bubble map of KEGG analysis in ctDNA samples; (C) Display of CDH1, CTNNB1 and TP53 mutated only in PM patients in the Wnt pathway and downstream abnormal cell function in ctDNA samples. Abbreviations: GO, gene ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; PM, peritoneal metastasis. |
Comprehensive Analysis of Biological Information from FFPE and Plasma Samples
GO enrichment analysis was performed in the FFPE samples to investigate the molecular mechanism of 49 DEGs involved in PM in gastric cancer patients. The results demonstrated that the function of these genes was mostly related to apoptosis and cell cycle and the gene functional protein cells were located in the cytoskeletal structure (p < 0.05, Figure 4A). GO functional analysis revealed that these genes are mainly involved in the negative regulation of cell death (p < 0.05, Figure 4B). KEGG pathway analysis revealed that these genes are mainly involved in ubiquitin-mediated proteolysis (p < 0.05, Figure 4C). Protein-protein interaction (PPI) analysis was performed on 49 DEGs in the STRING database. The results demonstrated that CDC27 and BIRC6 were at the core of the network (Figure 4D). CDC27 was the most abundant in the above GO and KEGG analyses. Thus, CDC27 was selected for further analysis.
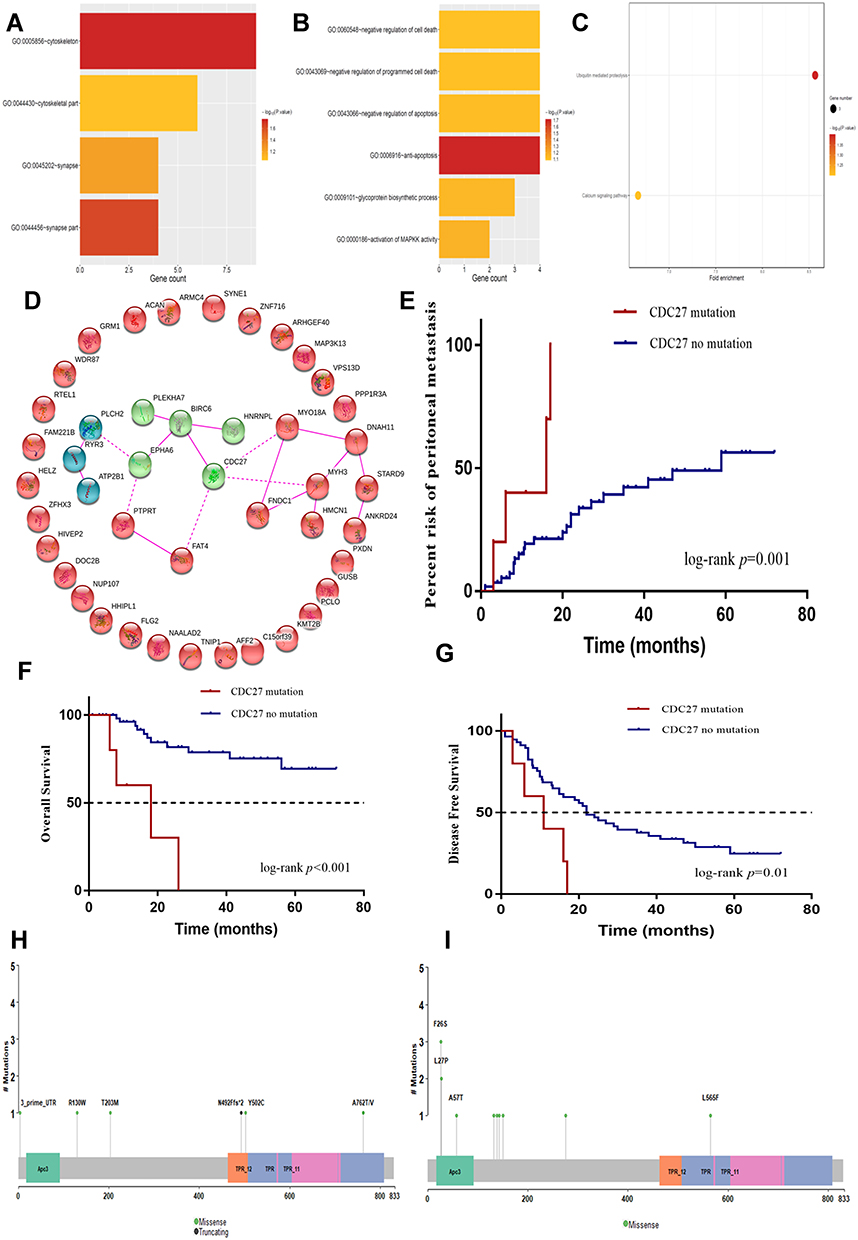 |
Figure 4 (A) Biological process GO analysis of differentially mutated genes in FFPE samples. (B) Cellular component GO analysis of differentially mutated genes in FFPE samples. (C) KEGG analysis of differentially mutated genes associated with peritoneal metastasis in gastric cancer patients in FFPE samples. (D) Protein-protein interaction network analysis of 49 differential genes (The line between the genes indicates that there is interaction between the two proteins, green and blue are two clusters respectively). (E) CDC27 mutation was associated with a significantly higher risk of postoperative PM. (F) CDC27 mutation was associated with a significantly lower overall survival. (G) CDC27 mutation was associated with a significantly lower disease-free survival. (H) Lollipop plot of CDC27 amino acid mutation in our study. (I) Lollipop plot of CDC27 amino acid mutation in TCGA database of gastric cancer patients. Abbreviations: GO, gene ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; PM, peritoneal metastasis; FFPE, formalin-fixed, paraffin-embedded; TCGA, The Cancer Genome Atlas. |
Relationship Between CDC27 Mutations and Clinicopathological Features
We next investigated whether CDC27 mutations were independent of other clinical factors. As demonstrated in Table 2, CDC27 mutations were not associated with gender, age, tumor location, T stage, N stage, differentiation, vascular tumor thrombus, neurological invasion, or postoperative PM (all p > 0.05). Patients with CDC27 mutations had a significantly higher risk of postoperative PM (1 year: 40% vs. 21.3%, 2 year: 70% vs. 33.7%, p = 0.001, Figure 4E). We then used Kaplan-Meier survival curves to further analyze the relationship between CDC27 mutations and patient survival. The results demonstrated that CDC27 mutations were associated with significantly lower OS and disease-free survival (DFS) (p < 0.001, p = 0.01, respectively), as shown in Figure 4F and G.
 |
Table 2 Relationship Between CDC27 Mutation in FFPE Samples and Clinicopathological Features |
Comparison of FFPE Sample Sequencing Results with the The Cancer Genome Atlas Database
The proportion of the 63 stage I–III gastric cancer patients with CDC27 mutations was 7.94% (5/63), compared to 1.59% (7/440) in the The Cancer Genome Atlas (TCGA) database. The lollipop chart of the mutation sites demonstrated the amino acid changes caused by the mutation site on the CDC27 protein structure, as shown in Figure 4H and I. Four new functional region mutation sites of CDC27 were identified, including three in the Apc3 region (F26S, L27P, and A57T), one in the TPR region (L565F), and five phosphorylation region mutations (A132T, Y138D, S143R, S150C, and L275V).
Construction of a Predictive Model of PM
We used the LASSO method to estimate the prognostic relationship between the 49 DEGs and PM. As shown in Figures 5A and B, a total of eight genes (CDC27, SYNE1, HMCN1, DNAH11, PTPRT, PCLO, STARD9, and BIRC6) were found to be independently associated with PM. To further evaluate the predictive value of these DEGs on PM, we incorporated both clinicopathological parameters (e.g., analysis of surgical methods, pathological T stage, number of lymph node, tumor location, Borrmann classification, tumor differentiation, and vascular/neural invasion) and gene parameters into a Cox regression model. Multivariable Cox regression analysis demonstrated that pathological T stage, poor tumor differentiation, Borrmann type, and CDC27 mutations were independent predictors of PM. As depicted in Figure 5C, the ROC curve demonstrated that a combination of these factors had the most powerful PM predictive ability (AUC = 0.875).
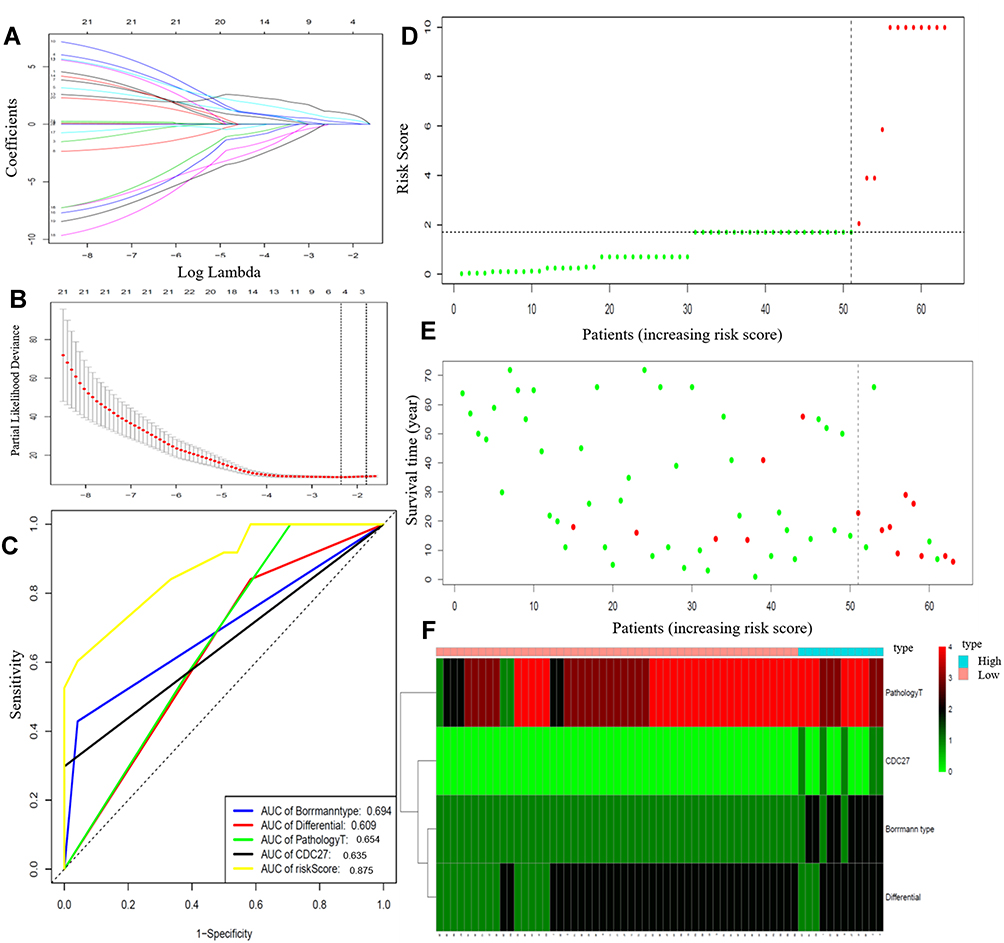 |
Figure 5 (A) Tuning parameter selection in the LASSO model. (B) LASSO coefficient profiles of 49 DEGs. (C) Time-dependent ROC analysis of the risk score model. (D) The risk score distribution of gastric patients; (E) The overall survival status of gastric patients; (F) The heatmap of four parameters in the low-risk and high-risk groups. Abbreviations: LASSO, least absolute shrinkage and selection operator; DEGs, differentially expressed genes. |
Risk scores were calculated based on the absolute value of the coefficients in the Cox regression model as follows: risk score = 2.759 × Borrmann type (ref. I/II) + 0.874 × pathological T stage + 1.924 × tumor differentiation (ref. high) + 2.115 × CDC27 mutation (yes vs. no). The risk scores were calculated for each patient and the distribution of risk scores and survival status of the patients are shown in Figure 5D and E. The patients were divided into high-risk and low-risk groups according to the median risk score. A risk heatmap demonstrated that the risk score could effectively discriminate gastric cancer patients with and without PM (Figure 5F).
Discussion
PM remains a major obstacle in the treatment of gastric cancer. Currently, there are no sensitive biomarkers or genetic signatures for PM from gastric cancer. Several studies have explored gene mutations associated with PM in gastric cancer by using WES. Liu et al13 performed WES to identify somatic mutations in peritoneal metastatic gastric adenocarcinoma, and revealed that nine differential genes are only mutated in metastatic lesions and may be therapeutic targets. Lim et al14 identified a unique mutational signature in malignant ascites from gastric cancer patients with PM. Nevertheless, these studies focused on gastric cancer patients who had already developed PM. Few studies have utilized WES to identify the predictive biomarkers of PM in gastric cancer. Using WES, our study identified a gene mutation relevant to postoperative PM in stage I–III gastric cancer patients and constructed a risk score model for predicting PM from gastric cancer for the first time.
PM from gastric cancer is triggered by a series of gene aberration. Advanced gastric cancer patients with high TMB were reported to have a better response to immunological checkpoint inhibitors.28 However, no study has reported the correlation between TMB and PM in gastric cancer. In this study, through WES conducted on the FFPE samples of 63 patients with stage I–III gastric cancer, we found that the number of mutant genes and the TMB in the PM group were higher than in the non-PM patients. Furthermore, most of the mutant genes were unique to PM. In addition, the risk of postoperative PM was significantly higher in patients with high TMB. Together, these findings suggest that TMB may be used as a biomarker to predict postoperative PM.
We further performed WES on plasma ctDNA from 10 stage IV gastric cancer patients with and without PM. It has been reported that ctDNA plays an important role in the recurrence and metastasis of gastric cancer.23 However, few studies have focused on the role of ctDNA in the PM of gastric cancer. In addition, the results demonstrated that genetic variants, such as TP53, EPHB6, MSH3, HER2, and MYC, may be associated with PM in gastric cancer. Due to the small sample size included in this study, it was difficult to draw reasonable conclusions and further studies with a larger sample size are needed.
In the next step, by using bioinformatic analysis of the FFPE and ctDNA sequencing results, we found that the protein dysfunction caused by mutations in gastric cancer PM was most likely related to cell cycle regulation, localization in the cytoskeletal structure, and ubiquitin-mediated proteolysis. The gene mutation related to PM affects the Wnt pathway function, which may lead to abnormal cell cycle regulation (cyclin), impaired function of the G1/G2 checkpoint, uncontrolled proliferation, and genomic instability. A high frequency of CDC27 gene mutations was the most abundant mutation in both GO and KEGG analyses and may be closely related to PM after gastric cancer surgery.
Cell division cycle 27 (CDC27) is an important subunit of the anaphase-promoting complex/cyclosome (APC/C). It has been reported that dysregulation of APC/C contribute to tumorigenesis in cancers.24 Emerging evidence has revealed the role of CDC27 in the invasion and metastasis in several tumors, including colorectal,25 gastric,26 and pancreatic27 cancer. The CDC27 protein contains two major functional regions, the Apc3 region and the TPR region. Apc3 is the major region encoding the function of the performing protein and the TPR region is a tetrapeptide repeat region. TPR is very important for protein-protein interaction between CDC27 and the other three members of the APC complex. This study identified five point-mutations in the phosphorylation region of the CDC27 protein. Kraft et al29 found that the phosphorylation activation binding site of CDC27 was located between the Apc3 region and the TPR region. A new missense mutation in the Apc3 and TPR regions of the CDC27 protein may affect the functional execution of CDC27 and mutations in the phosphorylation region may be associated with the abnormal activation of CDC27 phosphorylation. In addition, we demonstrated that CDC27 mutations were associated with postoperative PM and a worse DFS and OS in gastric cancer patients. These results suggested that CDC27 mutations might be used as a predictive factor for postoperative PM in gastric cancer.
According to the TCGA database, CDC27 exhibits somatic mutations in a variety of tumors, with a mutation rate of 1.59% in gastric cancer patients. The proportion of patients with CDC27 mutations in this study was significantly higher than that of the TCGA database, which may be related to the small proportion of an East Asian population in the TCGA database. Four new CDC27 functional region mutation sites were also found, and none of these mutation sites were reported in the TCGA database. The Apc3 region is the main functional structural region of CDC27, which is responsible for binding to APC/C and the TPR region is a region that binds to CDH1 and CDC20. The phosphorylation region is the phosphorylation site of CDC27 and is involved in its activation. Mutations in these three regions have the potential to affect the normal function of CDC27.
Several factors have been shown to be associated with PM in gastric cancer.29 The present study found that Borrmann type, tumor differentiation, and pathological T stage were independently associated with PM in gastric cancer patients. To further evaluate the predictive value of CDC27 for PM, we incorporated clinical parameters into a Cox regression model. Multivariable Cox regression analysis demonstrated that Borrmann type (III/IV), pathological T stage, poor tumor differentiation, and CDC27 mutations were independent predictive factors of PM. Finally, by incorporating the above significant parameters, we constructed a risk score model for predicting gastric cancer PM. Future studies are warranted to confirm the risk model in larger patient populations.
The present study had several limitations. First, the sample size was relatively small, especially for peripheral blood samples used for ctDNA detection. Second, the statistical and bioinformatic analyses in this study were carried out in silico. Therefore, the exact function and mechanism of the involvement of CDC27 in PM in gastric cancer need to be evaluated in further experiments. Given these limitations, our results provide the basis for screening for genes involved in PM after gastric cancer surgery.
In conclusion, through WES, we identified 49 DEGs relevant to PM in gastric cancer. CDC27 mutation was identified independently associated with PM by using statistical and bioinformatic analysis. A risk score model was built and might be help prediction of PM in gastric cancer.
Ethics and Consent Statement
Studies relative to human in this article were approved by the ethics committee of the The Fujian Medical University Union Hospital.
Acknowledgments
This study was supported by the Critical Patented Project of The Science & Technology Bureau of Fujian Province, China (grant number 2013YZ0002-2), and the Special Program for the Development of Strategic Emerging Industries of Fujian Province, China (grant number 2060404). Riping Wu and Qiaolian Li are co-first authors for this study.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that there are no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Kanda M, Kodera Y. Molecular mechanisms of peritoneal dissemination in gastric cancer. World J Gastroenterol. 2016;22(30):6829–6840. doi:10.3748/wjg.v22.i30.6829
3. Sugarbaker PH. Peritoneal metastases from gastrointestinal cancer. Curr Oncol Rep. 2018;20(8):62. doi:10.1007/s11912-018-0703-0
4. Glehen O, Gilly FN, Arvieux C, et al. Peritoneal carcinomatosis from gastric cancer: a multi-institutional study of 159 patients treated by cytoreductive surgery combined with perioperative intraperitoneal chemotherapy. Ann Surg Oncol. 2010;17(9):2370–2377. doi:10.1245/s10434-010-1039-7
5. Kwee RM, Kwee TC. Modern imaging techniques for preoperative detection of distant metastases in gastric cancer. World J Gastroenterol. 2015;21(37):10502–10509. doi:10.3748/wjg.v21.i37.10502
6. Kim SJ, Kim HH, Kim YH, et al. Peritoneal metastasis: detection with 16- or 64-detector row CT in patients undergoing surgery for gastric cancer. Radiology. 2009;253(2):407–415. doi:10.1148/radiol.2532082272
7. Emoto S, Ishigami H, Yamashita H, Yamaguchi H, Kaisaki S, Kitayama J. Clinical significance of CA125 and CA72-4 in gastric cancer with peritoneal dissemination. Gastric Cancer. 2012;15(2):154–161. doi:10.1007/s10120-011-0091-8
8. Huang C, Liu Z, Xiao L, et al. Clinical significance of serum CA125, CA19-9, CA72-4, and fibrinogen-to-lymphocyte ratio in gastric cancer with peritoneal dissemination. Front Oncol. 2019;9:1159. doi:10.3389/fonc.2019.01159
9. Ramos RF, Scalon FM, Scalon MM, Dias DI. Staging laparoscopy in gastric cancer to detect peritoneal metastases: a systematic review and meta-analysis. Eur J Surg Oncol. 2016;42(9):1315–1321. doi:10.1016/j.ejso.2016.06.401
10. Lin Y, Wu Z, Guo W, Li J. Gene mutations in gastric cancer: a review of recent next-generation sequencing studies. Tumour Biol. 2015;36(10):7385–7394. doi:10.1007/s13277-015-4002-1
11. Liu X, Wang J, Chen L. Whole-exome sequencing reveals recurrent somatic mutation networks in cancer. Cancer Lett. 2013;340(2):270–276. doi:10.1016/j.canlet.2012.11.002
12. Beltran H, Eng K, Mosquera JM, et al. Whole-exome sequencing of metastatic cancer and biomarkers of treatment response. JAMA Oncol. 2015;1(4):466–474. doi:10.1001/jamaoncol.2015.1313
13. Liu H, Li F, Zhu Y, et al. Whole-exome sequencing to identify somatic mutations in peritoneal metastatic gastric adenocarcinoma: a preliminary study. Oncotarget. 2016;7(28):43894–43906. doi:10.18632/oncotarget.9707
14. Lim B, Kim C, Kim JH, et al. Genetic alterations and their clinical implications in gastric cancer peritoneal carcinomatosis revealed by whole-exome sequencing of malignant ascites. Oncotarget. 2016;7(7):8055–8066. doi:10.18632/oncotarget.6977
15. Mckenna A, Hanna M, Banks E, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–1303. doi:10.1101/gr.107524.110
16. Cibulskis K, Lawrence MS, Carter SL, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31(3):213–219. doi:10.1038/nbt.2514
17. Koboldt DC, Zhang Q, Larson DE, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22(3):568–576. doi:10.1101/gr.129684.111
18. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi:10.1093/nar/gkq603
19. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi:10.1093/bioinformatics/btp324
20. Robinson JT, Thorvaldsdottir H, Winckler W, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24–26. doi:10.1038/nbt.1754
21. Li J, Lupat R, Amarasinghe KC, et al. CONTRA: copy number analysis for targeted resequencing. Bioinformatics. 2012;28(10):1307–1313. doi:10.1093/bioinformatics/bts146
22. Gao J, Kwan PW, Shi D. Sparse kernel learning with LASSO and Bayesian inference algorithm. Neural Netw. 2010;23(2):257–264. doi:10.1016/j.neunet.2009.07.001
23. Li TT, Liu H, Yu J, Shi GY, Zhao LY, Li GX. Prognostic and predictive blood biomarkers in gastric cancer and the potential application of circulating tumor cells. World J Gastroenterol. 2018;24(21):2236–2246. doi:10.3748/wjg.v24.i21.2236
24. Holland AJ, Cleveland DW. Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat Rev Mol Cell Biol. 2009;10(7):478–487. doi:10.1038/nrm2718
25. Qiu L, Tan X, Lin J, et al. CDC27 induces metastasis and invasion in colorectal cancer via the promotion of epithelial-to-mesenchymal transition. J Cancer. 2017;8(13):2626–2635. doi:10.7150/jca.19381
26. Xin Y, Ning S, Zhang L, Cui M. CDC27 facilitates gastric cancer cell proliferation, invasion and metastasis via twist-induced epithelial-mesenchymal transition. Cell Physiol Biochem. 2018;50(2):501–511. doi:10.1159/000494164
27. Yu M, Hong W, Ruan S, et al. Genome-wide profiling of prognostic alternative splicing pattern in pancreatic cancer. Front Oncol. 2019;9:773. doi:10.3389/fonc.2019.00773
28. Thomassen I, van Gestel YR, van Ramshorst B, et al. Peritoneal carcinomatosis of gastric origin: a population-based study on incidence, survival and risk factors. Int J Cancer. 2014;134(3):622–628. doi:10.1002/ijc.28373
29. Kraft C, Herzog F, Gieffers C, et al. Mitotic regulation of the human anaphase-promoting complex by phosphorylation. EMBO J. 2003;22(24 ):6598–6609.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.