Back to Journals » Neuropsychiatric Disease and Treatment » Volume 13
Compound porcine cerebroside and ganglioside injection attenuates cerebral ischemia–reperfusion injury in rats by targeting multiple cellular processes
Authors Wang M, Zhang Y, Feng L, Zheng J, Fan S, Liu J, Yang N, Liu Y, Zuo P
Received 5 December 2016
Accepted for publication 21 February 2017
Published 27 March 2017 Volume 2017:13 Pages 927—935
DOI https://doi.org/10.2147/NDT.S129522
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Wai Kwong Tang
Mingyang Wang,* Yi Zhang,* Lu Feng, Ji Zheng, Shujie Fan, Junya Liu, Nan Yang, Yanyong Liu, Pingping Zuo
Department of Pharmacology, Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, China
*These authors contributed equally to this work
Background: Compound porcine cerebroside and ganglioside injection (CPCGI) is a neurotrophic drug used clinically to treat certain functional disorders of brain. Despite its extensive usage throughout China, the exact mechanistic targets of CPCGI are unknown. This study was carried out to investigate the protective effect of CPCGI against ischemic neuronal damage in rats with middle cerebral artery occlusion (MCAO) reperfusion injury and to investigate the neuroprotective mechanisms of CPCGI.
Materials and methods: Adult male Sprague-Dawley rats were subjected to MCAO surgery for 2 hours followed by reperfusion. The rats were administered CPCGI once a day for 14 days after reperfusion, and behavioral tests were performed 1, 3, 7, and 14 days post MCAO. Hematoxylin–eosin staining was used to measure infarct volume, and immunohistochemical analysis was performed to determine the number of NeuN-positive neurons in the ischemic cortex penumbra. Finally, the relative expression levels of proteins associated with apoptosis (Bcl-2, Bax, and GADD45α), synaptic function (Synaptophysin, SNAP25, Syntaxin, and Complexin-1/2), and mitochondrial function (KIFC2 and UCP3) were determined by Western blot.
Results: CPCGI treatment reduced infarct size, decreased neurological deficit scores, and accelerated the recovery of somatosensory function 14 days after MCAO. In addition, CPCGI reduced the loss of NeuN-positive cells in the ischemic cortex penumbra. In the ischemic cortex, CPCGI treatment decreased GADD45α expression, increased the Bcl-2/Bax ratio, augmented Synaptophysin, SNAP25, and Complexin-1/2 expression, and increased the expression of KIFC2 and UCP3 compared with sham rats 14 days after MCAO reperfusion injury.
Conclusion: CPCGI displays neuroprotective properties in rats subjected to MCAO injury by inhibiting apoptosis and improving synaptic and mitochondrial function.
Keywords: compound porcine cerebroside and ganglioside injection, cerebral ischemia reperfusion, multitarget, neuroprotection
Introduction
Tissue plasminogen activator is the only clinically approved therapy to treat the early onset of ischemic stroke;1–3 hence, many researchers have attempted to develop novel neuroprotective therapeutics. Despite encouraging preclinical testing, currently there remain no drugs that result in consistent clinical improvements,4,5 possibly due to the highly complex pathophysiologic responses that occur in brain during stroke. It is well known that ischemia is a complex process that finally leads to neuronal death via multiple pathways including apoptosis, necrosis, inflammation, immune modulation, oxidative stress, excitotoxity, and mitochondrial dysfunction.6–8 Therefore, a paradigm has emerged whereby the development of novel therapeutics now includes the identification of multifunctional drugs that can target a variety of biological pathways.9
Compound porcine cerebroside and ganglioside injection (CPCGI, drug approval H22026472; Buchang Pharmaceutical Group Ltd., Jilin, China), which was approved by the China Food and Drug Administration in 2010, is widely used in China for the treatment of stroke, Alzheimer’s disease, central and peripheral nerve injuries, and functional disorders caused by diseases related to the brain. Each milliliter of CPCGI is estimated to contain ~50 μg of gangliosides (GM-1), 3.2 mg of polypeptides, and 0.25 mg of hypoxanthine. The active ingredients of CPCGI, GM-1 and carnosine, have been reported to possess significant therapeutic potency with respect to the treatment of ischemic stroke.10,11 GM-1 acts as a neurotrophic factor in the central nervous system,12 promoting survival, differentiation, neurodegeneration, axon stability, and regeneration in both injured and aging brains.13–16 In addition, carnosine, a dipeptide composed of β-alanine and histidine, protects against neurological damage in both permanent and transient cerebral ischemia models by modulating multiple cellular processes including oxidative stress, apoptosis, excitotoxity, and autophagy with no apparent toxicity or side effects.17–21 However, the role of CPCGI in cerebral ischemia–reperfusion has not yet been described.
In the present study, the neuroprotective effects of CPCGI were assessed in a rat model of middle cerebral artery occlusion (MCAO). Furthermore, the study attempted to discern the molecular mechanism, whereby CPCGI treatment exhibits a multitargeted effect on ischemia–reperfusion.
Materials and methods
Animals
All in vivo experiments were conducted in accordance with the guidelines established by the National Institutes of Health for the care and the use of laboratory animals and approved by the Animal Care Committee of the Peking Union Medical College and Chinese Academy of Medical Sciences. Adult male Sprague-Dawley rats weighing 250–270 g were purchased from the Academy of Military Medical Sciences (Beijing, China). Before surgery, the rats were acclimated for 1 week and housed in a temperature-controlled environment with a 12-hour light/dark cycle and ad libitum access to food and water.
Cerebral ischemia–reperfusion model
The MCAO surgery was conducted as previously described with several modifications.22,23 All rats were anesthetized with 1% pentobarbital sodium (50 mg/kg) by intraperitoneal injection followed by the insertion of an intraluminal suture from the external carotid artery stump into the internal carotid artery for 2 hours. The suture was then withdrawn to recover blood circulation. After the surgery, all animals were kept warm before regaining consciousness. The rats of the sham group underwent the same procedure without the ligation of arteries. Animals were then randomly assigned into 5 groups (n=10): sham, model, CPCGI-low (L) (0.5 mL/kg/d), CPCGI-high (H) (1 mL/kg/d), and Ginaton (50 mg/kg/d, as positive control) groups.24,25 CPCGI was obtained from the Jilin Buchang Pharmaceutical Group Co., Ltd (Jilin, People’s Republic of China). Ginaton injection solution was purchased from the Chi Sheng Chemical Corporation (Taiwan, China). Vehicle, CPCGI, or Ginaton were injected intraperitoneally from the start of reperfusion once a day for 14 days until the animals were killed. Sham and model groups received an equivalent volume of saline.
Neurological function assessment
For all animals, a blind neurological function assessment was performed 1, 3, 7, and 14 days after MCAO. Neurological deficits were evaluated, as previously described, by a set of modified Neurological Severity Scores (NSS) tests, involving a series of motor, sensory, reflex, and balance measurements.26,27 Using the NSS test, neurological function was graded from 0 to 18 (normal score =0; maximal deficit score =18).
Adhesive removal test
Adhesive removal tests were carried out 14 days after MCAO, as described previously, with several modifications.26 In brief, two small pieces of adhesive backed paper dots of equal size (100 mm2) were used as bilateral tactile stimuli occupying the distal–radial region on the wrist of each forelimb. After adhesion of the paper dots, the rats were then placed into hyaline cages for observation. The time taken by the rats to remove the stimulus from the forelimbs was recorded. Before the adhesive removal test, the rats were trained to familiarize themselves with their environment. The maximal time allowed for each test was 2 minutes.
Tissue preparation
Fourteen days after MCAO, rats (n=6 for each group) were anesthetized with 1% pentobarbital sodium (50 mg/kg) intraperitoneally and perfused first with 0.1 mol/L phosphate-buffered saline (PBS) and then 4% paraformaldehyde (pH 7.4) through the left ventricle. The brains were harvested and fixed in 4% paraformaldehyde solution at 4°C, followed by achievement of equilibrium with 0.1 mol/L PBS containing 15%, 20%, and 30% sucrose at 4°C, respectively. Each brain was coronally sectioned (2-mm thick), from frontal to occipital pole, into five segments. Coronal sections were then prepared for hematoxylin–eosin (HE) staining (10-μm thick) and immunohistochemical analysis (35-μm thick) using a cryostat.
Infarct volume assessment
One coronal section from each segment described earlier was stained with HE to measure the infarct volume.28 The sections were photographed by using a digital camera and analyzed by Image-Pro Plus 6.0. The infarct volumes of the lesion structures were expressed as a percentage of the volume of the structures from the control hemispheres by using the formula [(VC − VL)/VC] ×100%, where VC is the volume of control hemisphere and VL is the volume of noninfarcted tissue in the lesion hemisphere.29 The total infarct volume of each brain was calculated as the sum of the infarct volumes of the five brain slides.
Immunohistochemical analysis
Immunohistochemical analysis was performed as previously described.23 Briefly, brain slices were taken 0.20–1.00 mm rostral from the bregma (motor cortex and primary somatosensory cortex, forelimb region) followed by three washes with PBS and incubation in 3% H2O2 solution for 10 minutes to reduce endogenous peroxidase activity. After another three washes with phosphate-buffered saline-Triton 100 (PBST), the slices were blocked in 10% bovine serum for 1 hour and incubated overnight with a mouse anti-NeuN antibody (1:400; Merck Millipore, Darmstadt, Germany) at 4°C. The slides were washed another three times with PBS, then incubated with a horseradish peroxidase (HRP) goat anti-mouse secondary antibody per the manufacturer’s instructions (Beijing Zhong Shan Biotechnology Co., Beijing, China). The slices were then visualized using a 3′,3′-diaminobenzidine (DAB) substrate kit (Xi Ya Jin Qiao Biological Technology, Beijing, People’s republic of China), and images were captured using a charge coupled device (CCD) camera (Olympus, Tokyo, Japan). The number of NeuN-positive cells was blindly counted for three fields of view evenly distributed throughout the areas of interest by using Image-Pro Plus 6.0 software.
Western blotting
Total proteins were extracted from the ischemic cortex 14 days after MCAO (n=3 for each group). Western blots were performed as previously described.23 Equal amounts of sample from each group were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes. The membranes were blocked in 5% nonfat milk for 1 hour at room temperature and then incubated overnight at 4°C with the desired primary antibodies: rabbit anti-GADD45α (1:1,000; CST, Danvers, MA, USA), rabbit anti-Bcl-2 (1:1,000; CST), rabbit anti-Bax (1:1,000; CST), rabbit anti-KIFC2 (1:200; Santa Cruz Biotechnology Inc., Dallas, TX, USA), rabbit anti-SNAP25 (1:1,000; CST), rabbit anti-Syntaxin (1:5,000; Abcam, Cambridge, UK), rabbit anti-Complexin-1/2 (1:1,000; CST), rabbit anti-UCP3 (1:200; Novus, San Jose, CA, USA), and mouse anti-β-actin (1:1,000; Applygen, Beijing, People’s republic of China). After washing three times with tris-buffered saline-Tween 20 (TBST), the membranes were incubated with secondary antibodies conjugated to HRP for 1 hour. The immunoblots were visualized with enhanced chemiluminescence and analyzed using GelPro software. For each blot, β-actin served as an internal loading control.
Statistical analysis
All data were analyzed by one-way analysis of variance using Statistical Package for the Social Sciences (SPSS) 17.0 software (SPSS Inc., Chicago, IL, USA). Multiple comparison post hoc tests among groups was performed with the Least-Significant Difference test or Dunnett’s post hoc test depending on the homogeneity of variance test. Data were presented as mean values ± standard deviation (SD), and differences between groups were considered significant at P<0.05.
Results
CPCGI protects the rat brain from ischemic damage
We first investigated the neuroprotective effect of CPCGI in rat focal cerebral ischemia. Fourteen days after reperfusion, no cerebral injury was detected in the sham group, whereas the model group showed a significant area of infarct. Treatment with a high dose of CPCGI significantly decreased infarct volumes from 50.5% to 32.9%, and a similar effect was observed with Ginaton treatment in rats subjected to MCAO (Figure 1A and B). It was found that CPCGI improved neurological performance post-MCAO, as measured using the NSS and adhesive removal tests. The rats in the model group showed the highest neurological deficit score, whereas the neurological deficit scores of rats administered CPCGI were significantly lower 14 days after reperfusion compared with the model group (Figure 1C). Rats exposed to a high dose of CPCGI also took a significantly shorter time (48.2 seconds) to remove an adhesive stimulus 14 days after reperfusion compared with the model group (93.5 seconds) (Figure 1D). In addition, treatment with CPCGI prevented the loss of NeuN-positive neurons in the ischemic cortex penumbra after reperfusion (Figure 2). Therefore, CPCGI treatment seemed to protect the brain against ischemic damage post-MCAO.
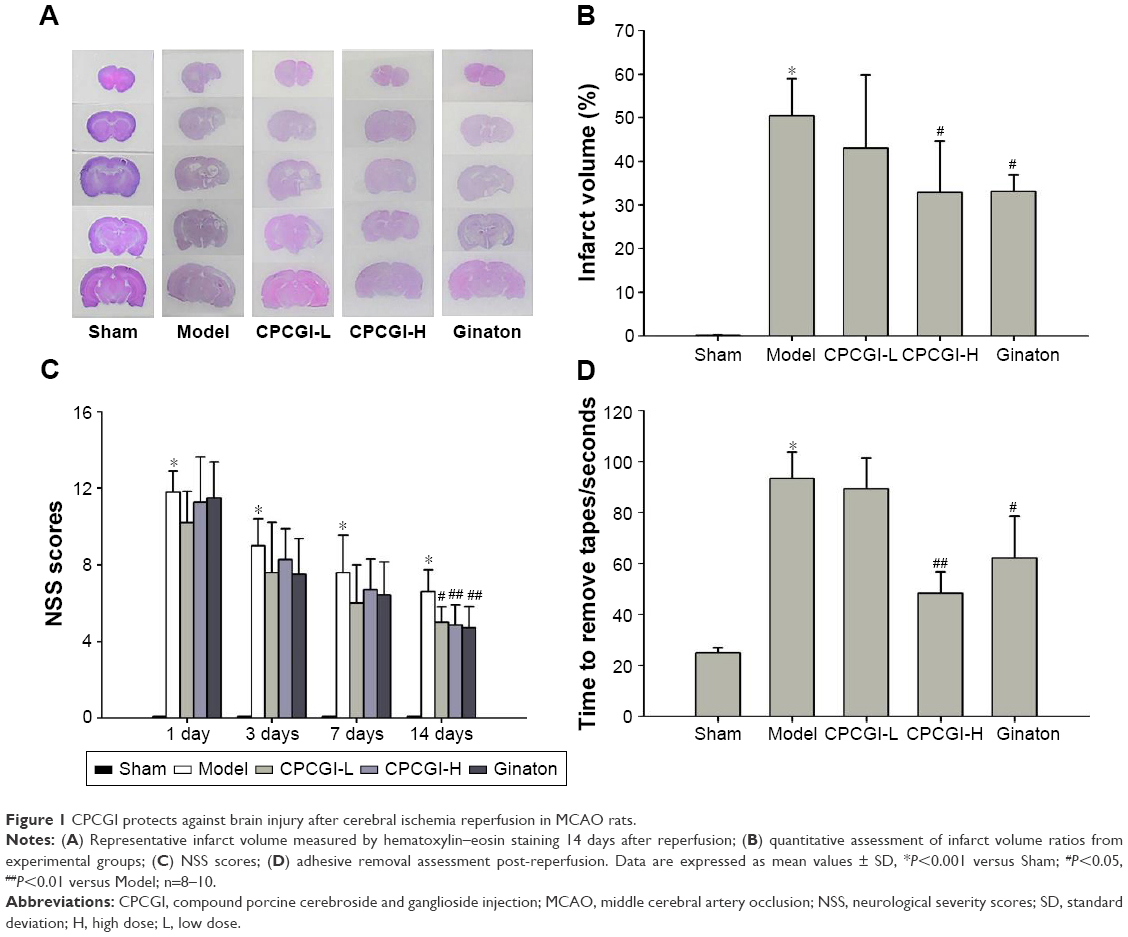 | Figure 1 CPCGI protects against brain injury after cerebral ischemia reperfusion in MCAO rats. |
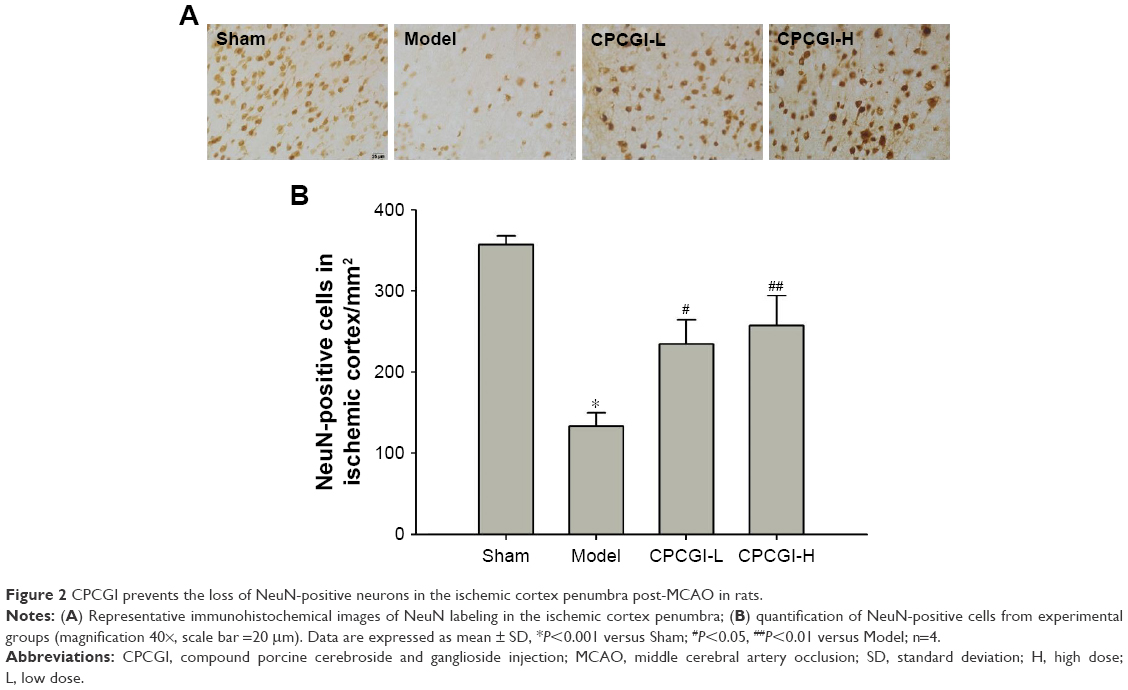 | Figure 2 CPCGI prevents the loss of NeuN-positive neurons in the ischemic cortex penumbra post-MCAO in rats. |
CPCGI abrogates MCAO-induced apoptosis in the ischemic cortex
Apoptosis is the primary biological process that drives neuronal cell death after ischemia.30 It has previously been shown that carnosine, one of the main components of CPCGI, plays an important role in inhibiting neuronal apoptosis after acute cerebral ischemia.20,31 To determine the role of apoptosis in CPCGI-mediated neuroprotection, the levels of apoptosis-related proteins (Bcl-2, Bax, and GADD45α) were measured in the ischemic cortex by Western blot. After reperfusion, it was found that the ratio of Bcl-2/Bax significantly decreased from 1.43 to 0.65 and the relative expression of GADD45α was 5.21 times higher (Figure 3). Treatment with a high dose of CPCGI reversed the effect of MCAO on Bcl-2/Bax ratio and GADD45α induction, suggesting CPCGI possesses antiapoptotic properties within the context of MCAO reperfusion injury.
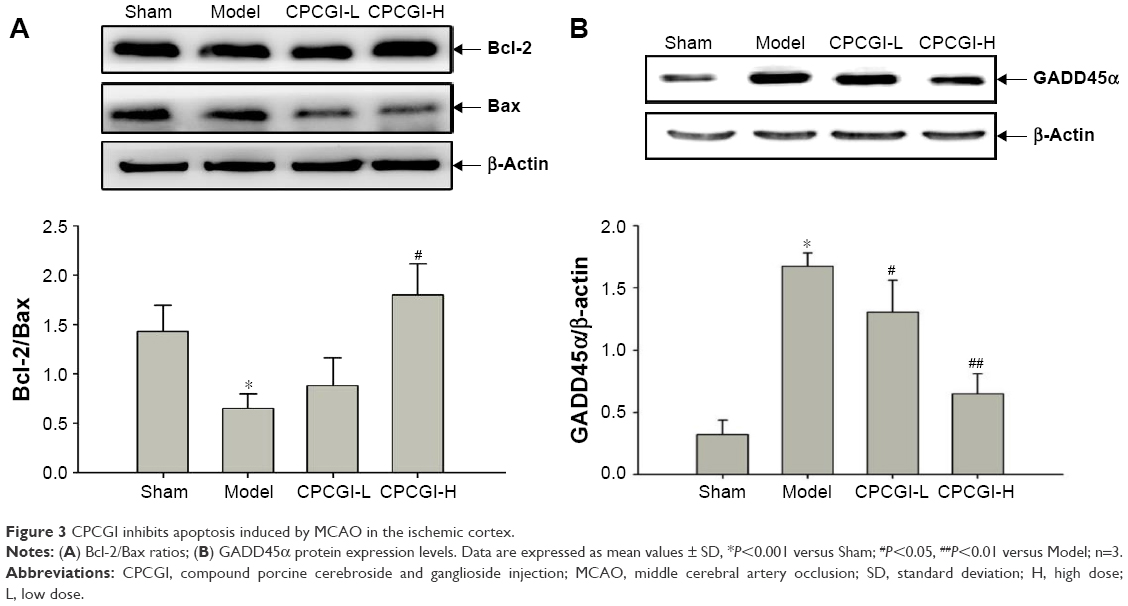 | Figure 3 CPCGI inhibits apoptosis induced by MCAO in the ischemic cortex. |
CPCGI improves synaptic function in the ischemic cortex
Functional synaptic activity is generally required to maintain the normal physiological functions of neurons. Because GM-1 can act as a neurotrophic factor in the central nervous system by modulating axon stability,12 the effect of CPCGI administration on synaptic function post-MCAO reperfusion was investigated. In the ischemic cortex, the expression levels of Synaptophysin, a protein necessary for the proper structure and function of synapses,32 were found to be markedly downregulated by ischemia–reperfusion injury 14 days post-MCAO, and this effect was partially reversed by treatment with CPCGI although not to a significant extent (Figure 4A and B).
 | Figure 4 CPCGI improves synaptic function in the ischemic cortex. |
Functional synaptic activity requires sufficient energy and proper synaptic protein expression. The expression levels of KIFC2, SNARE complex, and Complexin-1/2 were measured, which involved in the modulation of mitochondria retrograde axonal transport and the neurotransmitter release.33–35 It was found that KIFC2 protein levels in the ischemic cortex of MCAO-treated rats were significantly lower compared with the sham group (Figure 4A and F). Furthermore, MCAO also decreased the protein levels of SNAP25 and Complexin-1/2 (Figure 4A, C, and E). While CPCGI treatment partially reversed, these pathological changes and all above proteins showed a certain level of recovery. In addition, MCAO reperfusion had no effect on the expression levels of Syntaxin (Figure 4A and D).
CPCGI induces upregulation of UCP3 in the ischemic cortex after MCAO reperfusion
During ischemic stroke, GM-1 and carnosine exert their neuroprotective effects by inhibiting macromolecular oxidation.36,37 UCP3 is an uncoupling protein (UCP) family member and regulates mitochondrial adenosine-triphosphate (ATP) and reactive oxygen species (ROS) production.38 It has been shown that fluctuations in UCP3 expression mediate oxidative stress induced by ischemia–reperfusion.39 In our study, 14 days after MCAO reperfusion, UCP3 protein levels were ~4.95 times lower in the model group compared with the sham group, and treatment with a high dosage of CPCGI partially reversed this effect (Figure 5).
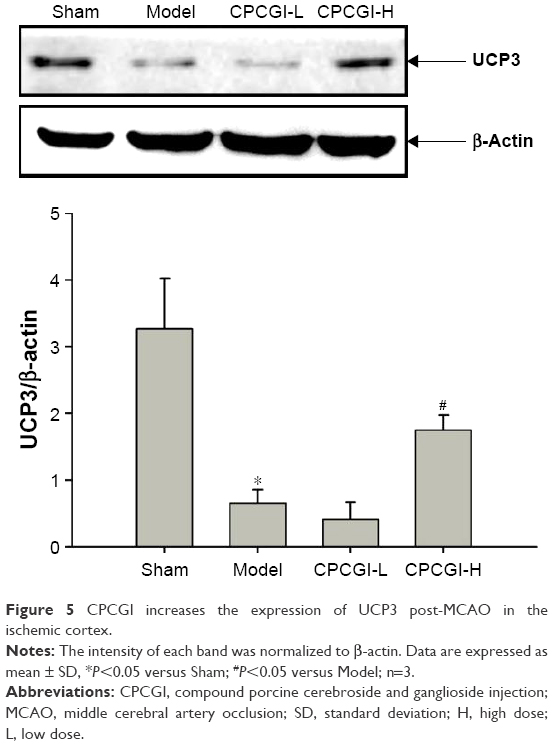 | Figure 5 CPCGI increases the expression of UCP3 post-MCAO in the ischemic cortex. |
Discussion
This study aimed to assess the protective effects of CPCGI post-cerebral ischemia–reperfusion injury in vivo. It was found that CPCGI reduced infarct size, improved neurological function, and accelerated the recovery of somatosensory function 14 days after MCAO reperfusion, as the positive control Ginaton did. Ginaton, so-called Ginkgo biloba extract (EGB), has been used in traditional medicines for centuries, and abundant evidences show that treatment with EGB has a significant protective effect on experimental animal models of stroke.25 Rats treated with a high dosage of CPCGI also showed an increase of NeuN-positive cells in ischemic cortex penumbra post-reperfusion. These results support the ability of CPCGI to protect against neurological damage caused by ischemia–reperfusion injury.
In a previous study, it was found that CPCGI protected against cerebral ischemia–reperfusion injury in rats via the activation of mitophagy,24 a process that maintains mitochondrial homeostasis by selectively removing damaged mitochondria. Accumulation of damaged mitochondria can result in the release of proapoptotic proteins (eg, cytochrome c) from the mitochondrial matrix into the cytoplasm, activating the intrinsic pathway of apoptosis and, eventually, inducing neuronal cell death. The present study further characterized the expression of apoptotic proteins (Bcl-2, Bax, and GADD45α) in the ischemic cortex using Western blot analysis. The ratio of Bcl-2/Bax, an important marker of intrinsic apoptosis,40 was significantly decreased by ischemia–reperfusion injury, and this effect was reversed by CPCGI treatment, suggesting that CPCGI inhibits apoptosis in the brain. CPCGI also suppressed the expression of GADD45α, a protein that has been shown to activate apoptosis by inducing the translocation of BCL2L11/Bim from the cytoskeleton to the mitochondria.41 Interestingly, GADD45α has also been shown to inhibit autophagy by directly interacting with Beclin1,42 suggesting that CPCGI might induce autophagy as a protective mechanism in the ischemic cortex. The results were in accordance with our previous data.
Most critical functions carried out by the nervous system require proper synaptic activity, including the release of neurotransmitters from presynaptic terminals to promote efficient communication between neurons. Some core proteins are involved in a complicated machinery that modulate the exquisite regulation of the release in varied presynaptic plasticity processes, underlying diverse forms of information processing in the brain.43 SNAREs and Complexins, through a series of complex interactions, facilitate neurotransmitter release by mediating the fusion of synaptic vesicles with the plasma membrane.44 Fourteen days post-MCAO reperfusion injury, a significant decrease in the expression of Complexin-1/2 and SNAP25 was observed. However, protein levels of Syntaxin (also a SNARE component) were unchanged. These data indicate that treatment with a high dosage of CPCGI prevented the MCAO-mediated downregulation of Complexin-1/2 and SNAP25. Moreover, CPCGI partially inhibited the effect of MCAO reperfusion on the expression levels of Synaptophysin, indicating the recovery of synaptic activity.
Proper synaptic activity requires a sufficient amount of ATP production from mitochondria that are transported away from the cell body to the location of the synapse. Therefore, the expression levels of KIFC2 in the ischemic cortex were assessed, which plays a crucial role in mitochondrial retrograde axonal transport. Remarkably, it was found that CPCGI attenuated the downregulation of KIFC2 induced by ischemia–reperfusion. Furthermore, CPCGI increased the expression of UCP3, a known regulator of mitochondrial ATP and ROS production,38 post-MCAO in the ischemic cortex. These data suggest that mitochondrial homeostasis is maintained in response to treatment with CPCGI, even after ischemia–reperfusion.
Conclusion
In summary, this study confirms the neuroprotective effect of CPCGI in rats after MCAO reperfusion injury and validates its clinical potential as a potent treatment for ischemic stroke. It proposes that CPCGI acts as a multitarget drug, inhibiting apoptosis, improving synaptic function, and reducing mitochondrial dysfunction. However, because the observations are limited to an MCAO model of ischemia–reperfusion, additional investigation is required to characterize the nature of the protective effects of CPCGI.
Acknowledgments
This work was supported by Chinese Academy of Medical Sciences (CAMS) Innovation Fund for Medical Sciences (CIFMS) (2016-I2M-3-004), Peking Union Medical College (PUMC) Youth Fund and Fundamental Research Funds for the Central Universities (3332015113), and grants from Buchang Pharmaceutical Co. Ltd.
Disclosure
The authors report no conflicts of interest in this work.
References
Goldstein LB, Rothwell PM. Advances in prevention and health services delivery. Stroke. 2008;39(2):258–260. | ||
Xing C, Arai K, Lo EH, Hommel M. Pathophysiologic cascades in ischemic stroke. Int J Stroke. 2012;7(5):378–385. | ||
Broussalis E, Killer M, McCoy M, Harrer A, Trinka E, Kraus J. Current therapies in ischemic stroke. Part A. Recent developments in acute stroke treatment and in stroke prevention. Drug Discov Today. 2012; 17(7–8):296–309. | ||
Fisher M. New approaches to neuroprotective drug development. Stroke. 2011;42(1 Suppl):S24–S27. | ||
Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges, and opportunities in stroke. Nat Rev Neurosci. 2003;4(5):399–415. | ||
Pandya RS, Mao L, Zhou H, et al. Central nervous system agents for ischemic stroke: neuroprotection mechanisms. Cent Nerv Syst Agents Med Chem. 2011;11(2):81–97. | ||
Sims NR, Anderson MF. Mitochondrial contributions to tissue damage in stroke. Neurochem Int. 2002;40(6):511–526. | ||
Sims NR, Muyderman H. Mitochondria, oxidative metabolism and cell death in stroke. Biochim Biophys Acta. 2010;1802(1):80–91. | ||
Nuñez-Figueredo Y, Ramírez-Sánchez J, Hansel G, et al. A novel multi-target ligand (JM-20) protects mitochondrial integrity, inhibits brain excitatory amino acid release and reduces cerebral ischemia injury in vitro and in vivo. Neuropharmacology. 2014;85:517–527. | ||
Kwak DH, Kim SM, Lee DH, et al. Differential expression patterns of Gangliosides in the ischemic cerebral cortex produced by middle cerebral artery occlusion. Mol Cells. 2005;20(3):354–360. | ||
Davis CK, Laud PJ, Bahor Z, Rajanikant GK, Majid A. Systematic review and stratified meta-analysis of the efficacy of carnosine in animal models of ischemic stroke. J Cereb Blood Flow Metab. 2016;36(10):1686–1694. | ||
Zamfir AD. Neurological analyses: focus on gangliosides and mass spectrometry. Adv Exp Med Biol. 2014;806:153–204. | ||
Ohmi Y, Tajima O, Ohkawa Y, et al. Gangliosides play pivotal roles in the regulation of complement systems and in the maintenance of integrity in nerve tissues. Proc Natl Acad Sci U S A. 2009;106(52):22405–22410. | ||
Posse de Chaves E, Sipione S. Sphingolipids and gangliosides of the nervous system in membrane function and dysfunction. FEBS Lett. 2010;584(9):1748–1759. | ||
Li L, Tian J, Long MK, et al. Protection against experimental stroke by ganglioside GM1 is associated with the inhibition of autophagy. PLoS One. 2016;11(1):e0144219. | ||
Mo L, Ren Q, Duchemin AM, Neff NH, Hadjiconstantinou M. GM1 and ERK signaling in the aged brain. Brain Res. 2005;1054(2):125–134. | ||
Rajanikant GK, Zemke D, Senut MC, et al. Carnosine is neuroprotective against permanent focal cerebral ischemia in mice. Stroke. 2007;38(11):3023–3031. | ||
Bae ON, Serfozo K, Baek SH, et al. Safety and efficacy evaluation of carnosine, an endogenous neuroprotective agent for ischemic stroke. Stroke. 2013;44(1):205–212. | ||
Baek SH, Noh AR, Kim KA, et al. Modulation of mitochondrial function and autophagy mediates carnosine neuroprotection against ischemic brain damage. Stroke. 2014;45(8):2438–2443. | ||
Pekcetin C, Kiray M, Ergur BU, et al. Carnosine attenuates oxidative stress and apoptosis in transient cerebral ischemia in rats. Acta Biol Hung. 2009;60(2):137–148. | ||
Boldyrev AA, Stvolinsky SL, Fedorova TN, Suslina ZA. Carnosine as a natural antioxidant and geroprotector: from molecular mechanisms to clinical trials. Rejuvenation Res. 2010;13(2–3):156–158. | ||
Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20(1):84–91. | ||
Zhang R, Yang N, Ji C, et al. Neuroprotective effects of Aceglutamide on motor function in a rat model of cerebral ischemia and reperfusion. Restor Neurol Neurosci. 2015;33(5):741–759. | ||
Wang MY, Feng L, Fan SJ, et al. [Effect of Compound Porcine Cerebroside and Ganglioside Injection on cerebral ischemia-reperfusion injury in rats]. Chin J Rehabil Theory Pract. 2016;22(7):750–753. Chinese. | ||
Yin B, Xu Y, Wei R, Luo B. Ginkgo biloba on focal cerebral ischemia: a systematic review and meta-analysis. Am J Chin Med. 2014;42(4):769–783. | ||
Chen J, Li Y, Wang L, et al. Therapeutic benefit of intravenous administration of bone marrow stromal cells after cerebral ischemia in rats. Stroke. 2001;32(4):1005–1011. | ||
Germano AF, Dixon CE, d’Avella D, Hayes RL, Tomasello F. Behavioral deficits following experimental subarachnoid hemorrhage in the rat. J Neurotrauma. 1994;11(3):345–353. | ||
Dang S, Liu X, Fu P, et al. Neuroprotection by local intra-arterial infusion of erythropoietin after focal cerebral ischemia in rats. Neurol Res. 2011;33(5):520–528. | ||
Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab. 1990;10(2):290–293. | ||
Yu XQ, Xue CC, Zhou ZW, et al. In vitro and in vivo neuroprotective effect and mechanisms of glabridin, a major active isoflavan from Glycyrrhiza glabra (licorice). Life Sci. 2008;82(1–2):68–78. | ||
Wang JP, Yang ZT, Liu C, He YH, Zhao SS. L-carnosine inhibits neuronal cell apoptosis through signal transducer and activator of transcription 3 signaling pathway after acute focal cerebral ischemia. Brain Res. 2013;1507:125–133. | ||
Tarsa L, Goda Y. Synaptophysin regulates activity dependent synapse formation in cultured hippocampal neurons. Proc Natl Acad Sci U S A. 2002;99(2):1012–1016. | ||
Saito N, Okada Y, Noda Y, Kinoshita Y, Kondo S, Hirokawa N. KIFC2 is a novel neuron-specific C-terminal type kinesin superfamily motor for dendritic transport of multivesicular body-like organelles. Neuron. 1997;18(3):425–438. | ||
Antonucci F, Corradini I, Fossati G, Tomasoni R, Menna E, Matteoli M. SNAP-25, a known presynaptic protein with emerging postsynaptic functions. Front Synaptic Neurosci. 2016;8:7. | ||
Trimbuch T, Rosenmund C. Should I stop or should I go? The role of complexin in neurotransmitter release. Nat Rev Neurosci. 2016;17(2):118–125. | ||
Zhang J, Fang X, Zhou Y, et al. The possible damaged mechanism and the preventive effect of monosialotetrahexosylganglioside in a rat model of cerebral ischemia-reperfusion injury. J Stroke Cerebrovasc Dis. 2015;24(7):1471–1478. | ||
Park HS, Han KH, Shin JA, Park JH, Song KY, Kim DH. The neuroprotective effects of carnosine in early stage of focal ischemia rodent model. J Korean Neurosurg Soc. 2014;55(3):125–130. | ||
Ricquier D, Bouillaud F. The uncoupling protein homologues: UCP1, UCP2, UCP3, StUCP and AtUCP. Biochem J. 2000;345 Pt 2:161–179. | ||
Ozcan C, Palmeri M, Horvath TL, Russell KS, Russell RR. Role of uncoupling protein 3 in ischemia-reperfusion injury, arrhythmias, and preconditioning. Am J Physiol Heart Circ Physiol. 2013;304(9):H1192–H1200. | ||
Tamm I, Schriever F, Dörken B. Apoptosis: implications of basic research for clinical oncology. Lancet Oncol. 2001;2(1):33–42. | ||
Tong T, Ji J, Jin S, et al. Gadd45a expression induces Bim dissociation from the cytoskeleton and translocation to mitochondria. Mol Cell Biol. 2005;25(11):4488–4500. | ||
Zhang D, Zhang W, Li D, Fu M, Chen R, Zhan Q. GADD45A inhibits autophagy through regulating the interaction between BECN1 and PIK3C3. Autophagy. 2015;11(12):2247–2258. | ||
Regehr WG. Short-term presynaptic plasticity. Cold Spring Harb Perspect Biol. 2012;4(7):a005702. | ||
Rizo J, Xu JJ. The synaptic vesicle release machinery. Annu Rev Biophys. 2015;44:339–367. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.