Back to Journals » Journal of Blood Medicine » Volume 12
Complement in Sickle Cell Disease: Are We Ready for Prime Time?
Authors Varelas C, Tampaki A ![]() , Sakellari I, Anagnostopoulos A, Gavriilaki E, Vlachaki E
, Sakellari I, Anagnostopoulos A, Gavriilaki E, Vlachaki E ![]()
Received 15 February 2021
Accepted for publication 11 March 2021
Published 23 March 2021 Volume 2021:12 Pages 177—187
DOI https://doi.org/10.2147/JBM.S287301
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Christos Varelas,1 Athina Tampaki,2 Ioanna Sakellari,1 &Agr;chilles Anagnostopoulos,1 Eleni Gavriilaki,1,* Efthymia Vlachaki2,*
1Hematology Department – BMT Unit, G. Papanicolaou Hospital, Thessaloniki, Greece; 2Adults Thalassemia Unit, 2nd Department of Internal Medicine, Hippokration Hospital, Thessaloniki, Greece
*These authors contributed equally to this work
Correspondence: Eleni Gavriilaki
Hematology Department - BMT Unit, G. Papanicolaou Hospital, Exochi, Thessaloniki, 57010, Greece
Email [email protected]
Abstract: Sickle cell disease (SCD) is a widely spread inherited hemoglobinopathy that includes a group of congenital hemolytic anemias, all characterized by the predominance of sickle hemoglobin (HbS). Its features are anemia, predisposal to bacterial infections and complications such as vaso-occlusive crisis (VOC) or delayed hemolytic transfusion reaction (DHTR), which lead to increased rate of morbidity and mortality even in the era of hydroxyurea. The interaction between sickle cells, neutrophils, platelets or endothelial cells in small vessels results in hemolysis and has been considered the disease’s main pathophysiological mechanism. Complement activation has been reported in small cohorts of SCD patients, but the governing mechanism has not been fully elucidated. This will be important to predict the patient group that would benefit from complement inhibition. Until now, eculizumab-mediated complement inhibition has shown beneficial effects in DHTR, with limited reports in patients with VOC. In the meantime, several innovative agents are under clinical development Our state-of-the-art review summarizes current data on 1) complement activation in SCD both in steady state and crisis, 2) underlying mechanisms of complement over-activation for the clinician in the context of SCD, 3) actions of hydroxyurea and new therapeutic approaches including indirect involvement in complement activation, and 4) novel paradigms in complement inhibition.
Keywords: sickle cell disease, complement system, eculizumab, complement inhibition
Introduction
Sickle cell disease (SCD) still remains a devastating and dire condition with subsequent increased rates of morbidity and mortality in the era of hydroxyurea.1 It is a genetic, autosomal recessive condition caused by a single β-globin gene mutation on chromosome 11, leading to an amino-acid substitution (Glutamine -> Valine, βΑ -> βs), thus resulting in the formation of the abnormal hemoglobin S (HbS) tetramer.2 HbS is a tetramer with abnormal physicochemical properties that will polymerize under hypoxic stress, leading into the sickling of circulating red blood cells (RBCs).3
Our current understanding of the disease’s pathophysiology has mostly focused on the interaction between red blood cells and neutrophils, platelets or endothelial cells in small blood vessels.4 More recently, the effects of red blood cell adhesion and hemolysis that result in vaso-occlusive crisis (VOC) have also been investigated.5 A rather neglected entity in SCD that seems to be a key component of this pathophysiological mechanism may be complement activation. In this context, increased interest has been shown in the identification of the innate immune system’s pivotal role in the promotion of inflammation in SCD.6 The activation of the complement cascade is one of the hallmarks in this inflammatory process.7 In general, systemic complement dysregulation induces host tissue damage.8 Biomarkers of complement activation in the serum of SCD patients were revealed in various clinical studies, along with increased levels of C5b-9 – which is the definitive marker of complement activation - and other surface-bound C3 fragments not only on patients’ erythrocytes, but also in skin and kidney biopsies.10–14 Additionally, further experience about the role of complement activation and inhibition has been gained in the context of other disorders. Findings of complement activation in β-thalassemia major, thrombotic microangiopathies (TMAs), antiphospholipid antibody syndrome, HELLP syndrome and malaria enhance our efforts in understanding complement activation and its role in the pathophysiology of SCD.15–21
Our review originated from the aspiration to provide further evidence in the investigation of complement activation in SCD. This regards a summary of current data on complement activation both in steady state and crisis, probable elemental mechanisms of complement activation in the frame of SCD, actions of hydroxyurea, novel therapeutic approaches including indirect involvement in complement activation and novel endeavours of complement inhibition; all under the prism of a clinician’s point of view.
Complement Activation in SCD
Scientific effort to explain increased rates of bacterial infections and mortality in SCD patients1,22,23 led to the investigation of a possible involvement of the complement system in the disease’s pathophysiology, with the hypothesis that this excessive sensitivity to infections was a result of defective opsonization.9,24,25 Complement components, such as C3b and iC3b, are key ingredients of innate immune system and not only opsonize pathogens but also generate sequential adaptive immune responses. As a result, the complement system was placed at the center of scientific community’s research.
Francis and Womack were the first investigators who reported remarkable complement activity in the serum of SCD patients in 1967.26 Six years later, Johnston et al followed the same hypothesis and confirmed complement over-activation through the AP pathway in SCD.27 In 1976, Wilson et al revealed total AP function (AP50) in abnormally low levels, along with significantly depressed levels of factor B in serum of SCD patients compared to normal controls.28 Generally, initial surveys’ results with the depiction of depressed levels of Factor B, functional deficiency, and C3 inferred to the assumption that defective AP activation was responsible for defective opsonization. However, the possible underlying mechanisms were not further investigated.27–31
Later studies managed to measure Bb fragments and C3, P (properdin) complexes in serum from SCD patients in crisis.9 This study revealed elevated concentrations of both factors and was consistent with the hypothesis that irreversibly sickled red cells, or membrane spicules and vesicles are the site of increased AP activation. Furthermore, complement over-activation markers such as Bb, C5a, soluble sC5b9 were measured in patients’ serum or plasma and associated with SCD in steady state or in crisis.10,12–14,32 Additional results reported over-activation of the AP pathway in painful crisis periods, in contrast with asymptomatic circumstances.
On the other hand, more measurable molecules of the complement system such as factors D, I and H were found to be within normal range or even reduced but not statistically significantly in SCD patients who exhibit complement activation.28,32,33 Moreover, a defective regulation mechanism of soluble C5b-9 has been correlated to a major increase in lysis of sickled cells.34 In addition, elevated soluble c5b-9 and sC5b-9 vascular deposition were revealed.11,12,14
Other studies showed that regulation of C5b-9 (membrane attack complex/MAC) formation in sickle erythrocytes was defective, especially in the densest cells. The defect is characterized by increased binding of C5b-7 and of C9 to more dense sickle cells, and as a result, sickle cells are more susceptible to C5b-9-mediated (reactive) lysis. Among the dense sickle cells, irreversibly sickled cells (ISC) are those that are more sensitive to lysis.34 Interestingly, dense ISC have the ability to directly activate the AP.9 Their density propagates an increased AP activation when compared to sickled RBCs or control RBCs. Defective modulation from surface-anchored complement regulators on dense sickled RBCs, along with an increased amount of C3 are potent complement activators.10,12
Since C3a and C3b fragments can be produced by either pathway, whereas Bb is specific to the AP and C4b both to the classical pathway (CP) and the lectin pathway (LP), several surveys used additional methods to block CP activation, in order to achieve only AP to be activated.9,27,32 Collectively, these data are conclusive in that complement activation is a major characteristic of SCD and sickled RBC’s death in vivo or in vitro is AP mediated. This mechanism of complement over-activation needs further exploration. However, numerous observational studies, mouse model data and experiments have attempted to shed a light in this obscure area of SCD pathophysiology.
Underlying Mechanisms of Complement Activation in SCD for the Clinician
Complement system and its role in the pathophysiology of SCD is considered a complex and vast entity. The key aspects that bind complement and SCD are cell-free heme and hemolysis-derived molecules, C5a together with P-selectin, the surface complement regulatory protein, the hypercoagulant state in SCD, the amendment of the bilayer membrane structure in sickle RBCs and the very essence of SCD pathobiology: the vicious cycle of ischemia/reperfusion (I/R). These mechanisms can forward complement activation through different, distinct pathways.32,35,36 Circulating microvesicles (MVs) of damaged SCD erythrocytes during oxygenation–deoxygenation sickling cycles have been identified with the ability to activate the complement system, and as a result, they can be used as biomarkers.35,37,38
AP activation due to alterations in the organization of membrane phospholipids of sickled RBCs is a speculated adjunctive mechanism. Early studies made reports of in vivo AP activation in the circulations of SCD patients during painful crisis32 and subsequent studies further evidenced an abnormal translocation of the sickled RBC membrane phospholipids, which transpires as a potential locale of AP activation in SCD.14,39
Chronic intravascular hemolysis is identified as an essential characteristic of SCD.40 Free extracellular heme interacts with toll-like receptor 4 (TLR-4) leading to the production of cytokines and adhesion molecules that promote inflammatory response and ultimately VOC. Endothelial TLR-4 signaling is rather critical for SCD VOC and recent data suggest that targeted inhibition of heme-induced TLR-4 signaling can actually reduce VOC.41 Moreover, free heme is released in circulation due to hemoglobin degradation during hemolysis. Counter protective mechanisms are not sufficient to prevent hemolysis from accumulating free heme in plasma.42 Recent studies exhibit increased plasma free heme (PFH), along with MVs, in many SCD patients and recognized their devastating effects – most notably intense kidney VOC, acute chest syndrome (ACS) not only in mouse models but also pediatric population.43–45 However, a recent single-cell RNA study reported that liver macrophages, are turned into “erythrophagocytes” that dominate the macrophage population and provide an on-demand adaptation to hemolytic driven oxidative stress.46 Recent studies report that loss.
C5a anaphylatoxin, an inflammatory molecule, has the ability to propagate P-selectin-mediated vaso-occlusion. A recent study by Vercelloti et al demonstrated that C5a alone when given to SS mice promotes VOC. Another finding was that C5a up-regulates P-selectin and vWF in vitro and in vivo in the same population, inducing P-selectin mediated VOC.36 In line with these findings, another study established that AP opsonins, anchored on sickled RBCs, serve actually as adhesion sites on vascular endothelium, resulting in the irregular trajectory of these cells by involvement of P-selectin/Mac-1.14 Other studies revealed the capacity of P-selectin to bind C3b and C3(H2O)-like, promoting complement activation.47,48
Early evidence suggests that abnormal expression of surface complement regulatory proteins is not just a key element of paroxysmal nocturnal hemoglobinuria (PNH). These proteins propagate complement-mediated cell lysis and their deficiency or dysfunction has been associated with the pathophysiology of other diseases as well.49–53 Most studies have focused on surface-bound complement regulators – CD55, CD59, CD46 and CR1 -. In the frame of SCD, some studies revealed decreased CD55 and CD59 on sickled erythrocytes, in comparison with control subjects.34,54 Other surveys showed difference in the expression of these regulators on dense vs non-dense RBC but not in the total population of RBC between controls and SCD patients.12
A growing consensus has been achieved between the complement and coagulation/fibrinolysis pathways in the context of clinically significant tissue damage and activation of inflammatory process. Thrombin effectively catalyzes the cleavage of both C3 and C5 acting as convertase, producing active anaphylatoxins and other complement sub-products.55 Interestingly, MVs propagate complement activation through a thrombin-mediated signal route both in vitro and in vivo, independently of CP or AP activation.56 Another noteworthy finding was that thrombin acts as an alternative molecule for C5-convertase in the absence of C3.57
Last but not least, I/R underlies the pathophysiology of SCD. The paradox of this cycle is that tissue damage is caused not only by ischemia but also by the following reperfusion-mediated reentry of oxygen58 I/R results in complement activation and neutrophilic stimulation, along with rapid oxygen production and ultimately contributes to tissue damage in various organs.59 Significant interest has been risen for the role of the LP and its role in inflammatory responses during I/R injury. There have been reports of “bypass” activation events that lead to lectin-mediated cleavage of C3, and as a result, I/R tissue damage is caused. Moreover, certain deficiencies of mannose-binding lectin (MBL) have been positively associated with better outcomes in experimental models of renal reperfusion injury after transplantation.60 More recent observations report that inhibition of mannose‐binding lectin-associated serine protease (MASP2), the effector enzyme of LP, may be beneficial in conditions such as thrombotic microangiopathy (TMA) post allogeneic hematopoietic stem cell transplantation (alloHSCT).61
In general, all these proposed mechanisms share in common the dominant role of complement activation – especially through the AP pathway – in the pathophysiology of SCD. Some studies revealed elevated biomarkers of complement activation in both steady state and crisis of SCD patients or mouse models. In other cases, despite preliminary data, no complement activation was reported in association with the factors studied. Nevertheless, there are still many elements of complement deregulation that have yet to be explored. Importantly, new therapeutic perspective is needed, since despite the fact that SCD is the first genetic disease described, treatment evolution seems to be stuck in the era of hydroxyurea.
Hydroxyurea and New Therapeutic Approaches
Hydroxyurea was the first drug approved by the Food and Drug Administration (FDA) to be administered in adult SCD patients and it remains the main therapeutic choice for more than 30 years.62 It has many features that make it an ideal drug for SCD and can provide multiple benefits through several mechanisms of action. Over the past 30 years, substantial experience has been earned in matters of safety and efficacy for SCD patients.
Its main effect – through a mechanism still not fully understood – is the induction of fetal hemoglobin (HbF), high levels of which reduce significantly SCD severity. Hydroxyurea improves erythrocyte deformation, decreases the population of circulating neutrophils and reticulocytes and reduces the expression of adhesion receptors.63 Early findings of principle studies were followed by prospective Phase 1/2 trials that demonstrated efficacy first in affected adults, adolescents and children, and more recently even in infants and toddlers.64
In a double-blind, randomized clinical trial, hydroxyurea was proved to be effective and efficient in reducing the frequency of painful crises in adults who had a history of three or more such crises per year. A mean follow-up of 21 months was reported at the end of the trial.65 This study reported that patients assigned to hydroxyurea had lower annual rates of crises, and fewer of them suffered from acute chest syndrome or underwent transfusion in comparison with patients that received the placebo.65 Hydroxyurea treatment did not cause any severe adverse effects.
Hydroxyurea has proven its clinical efficacy and become acknowledged as the main therapeutic option for many SCD patients. It reduces number of painful crises and hospitalizations66 and is useful to treat acute VOCs both in adults and children. Infants receiving hydroxyurea preserve their splenic function.67 Furthermore, hydroxyurea has been correlated to fine growth and development,68 along with no delays on sexual maturation, including menarche.69
Interestingly, a recent study by Roumenina et al revealed novel evidence of an indirect effect of hydroxyurea in complement activation. This study revealed that although complement activation is a rather common event in the pathophysiology of SCD that is associated with the development of dense RBCs and hemolysis, treatment with hydroxyurea may partly alleviate this effect.12 More specifically, complement activation was shown by sC5b-9 concentration and upregulation of CD46 and it was reported substantially reduced in SCD patients treated with hydroxyurea.
However, hydroxyurea has many contraindications as well. Pregnancy or unwillingness to use contraception, history of severe hydroxyurea toxicity or hypersensitivity and history of significant non-compliance with recommended medical care, deprive patients of the benefits of this myelosuppressive agent.70 All in all, despite its multiple benefits, an important number of patients do not achieve satisfactory clinical response.
Due to the fact that hydroxyurea along with anticoagulants remain the sole treatment for SCD for over 30 years, several novel treatments are currently under advanced clinical development in SCD.
Another novel agent is voxelotor. It is an orally administered drug that increases Hb’s affinity to oxygen and inhibits the sickling of RBCs. Several clinical trials have reported its benefits.73 Crizanlizumab is a new, FDA-approved drug for the prevention of VOC. It is a humanized IgG2 kappa monoclonal antibody that binds to P-selectin and blocks interactions with its ligands including P-selectin glycoprotein ligand 1. Binding P-selectin on the surface of the activated endothelium and platelets blocks interactions between endothelial cells, platelets, RBCs, and leukocytes.74 Baseline analysis from systematic literature review and network meta-analysis proved that this monoclonal antibody can reduce crises and hospitalization days in comparison with placebo or other therapeutic agents with an acceptable adverse event profile in adult and adolescent SCD patients.75 Furthermore, binding of P-selectin by crizanlizumab also inhibits C5-a, which may be beneficial for SCD patients.36
Hematopoietic cell transplantation (HCT) is a potential definitive cure for SCD. The goal is to eliminate the sickle erythrocyte and its cellular progenitors and replace them with donor hematopoietic pluripotent stem cells that will produce mature erythrocytes which will not express sickle hemoglobin (Hb S), thereby reducing Hb S levels to those associated with the trait condition.76 However, the risk of post-transplantation severe adverse events must be balanced against SCD’s own serious complications which are known for causing morbidity and even death.77
Genetic modulation of phenotype may also have curative potential for SCD patients. SCD has different phenotypes due to differences in the genetic makeup of the affected patient. Although the exact genes are still under extensive study, further advances in our understanding of the pathophysiology imply that genes involved in numerous mechanisms might have epistatic potential in SCD.78 This may actually lead to gene therapy with the aim to replace the defective gene with a normal one.
Additional novel agents are being tested that target abnormal interactions between RBCs and their surrounding micro-environment (specifically the endothelium, neutrophils and platelets), as well as the inflammatory and prothrombotic setting. Some of these drugs may indirectly interact with the complement system. A recent review reported 20 Phase 1 studies, 10 Phase 2 and 3 Phase 3.79 Of all these studies, eight investigate drugs which are applied during an acute VOC, while the others are aimed at the reduction of VOC or adjustment of biomarkers. Table 1 summarizes novel approved and under development therapies in SCD.
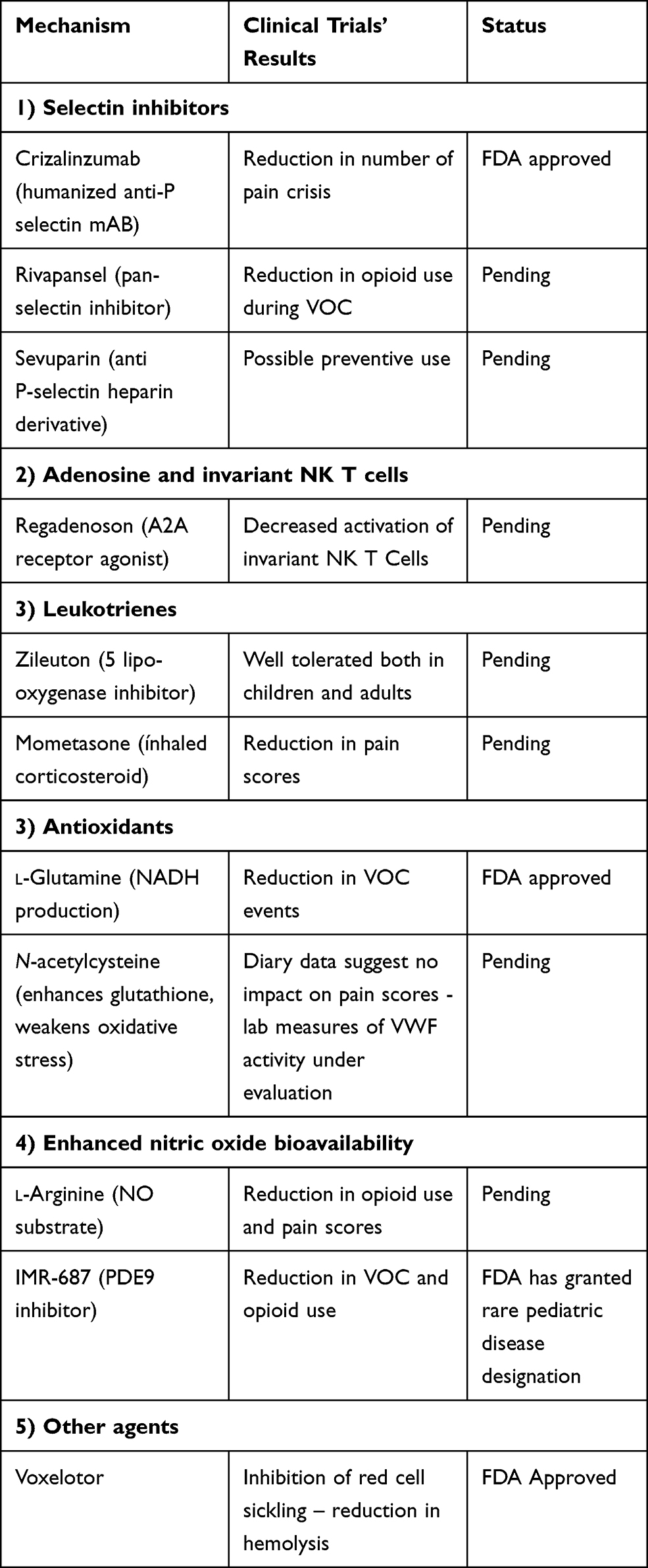 |
Table 1 Summary of Novel Approved and Under Development Therapies in SCD |
As our understanding in the pathophysiology of sickling is constantly improving, many potential novel therapeutic agents are being in development. This is indeed an exciting and optimistic time in SCD research. Nevertheless, the multifactorial and vast nature of the disease is more likely to require combination therapies, and some single agents are doomed to be unsuccessful. More studies are needed in this effort to unravel the mystery of SCD and its pathophysiology.
Novel Paradigms in Complement Inhibition
Complement inhibition has already been tested in Paroxysmal Nocturnal Hemoglobinuria (PNH) with two FDA-approved drugs: eculizumab since 2007 and ravulizumab since 2019. They are both monoclonal antibodies that block terminal complement activation.80,81 Ravulizumab has a longer half-life, thus providing sustained C5 inhibition,82,83 while eculizumab has already been used in other diseases and was found to be safe and have long-term efficacy.84 In order to overcome eculizumab’s own limitations, several other novel inhibitors are currently in advanced clinical development.49 Novel proximal inhibitors are being tested aiming at proteins within the early stages of the complement cascade, such as C3, factor B and factor D.85,86 Figure 1 summarizes the complement cascade, and inhibitors’ target points which could provide potential benefits in SCD.
 |
Figure 1 Complement activation and inhibition. The complement cascade has been traditionally considered to be activated by the classical, alternative and lectin pathway. The alternative pathway serves as an amplification loop for the lectin and classical pathway accounting for almost 80% of complement activation products. Spontaneous hydrolysis of C3 resulting from different triggers allows propagation of C3 convertase (C3bBb) and as a result the alternative pathway is constantly “on”. C3 convertase sustains the amplification loop together with factors B and D. Terminal complement pathway begins with propagation of C5 convertase which in turn cleaves C5 into C5a, a potent inflammatory mediator, and C5b; C5b together with C6-9 form C5b-9 (membrane attack complex/MAC), a cytolytic complex. Eculizumab, ravulizumab, ABP959, SKY59/RO7112650, tesidolumab, REGN3918, mubodina, coversin, RA101495, cemdisiran and zimura inhibit C5; AMY-101 and APL-2 inhibit C3 and C3 convertase activity; mini-FH/AMY-201 inhibits alternative pathway C3 convertase; LPN023 and IONIS-FB-LRx inhibit factor B; danicopan and lampalizumab inhibit factor D; mirococept inhibits C3 and C5 convertases; avacopan inhibits C5a receptor and IFX-1 C5a. |
Despite our better knowledge about complement’s role in SCD, advanced options for new treatment plans are not extensively explored. Eculizumab has been used with success as a salvage therapy in SCD patients presenting with delayed hemolytic transfusion reaction (DHTR), as it has been documented in some case reports.87–91 Patients received the monoclonal antibody after multiple lines of treatments and after confirmation of complement activation by analyses. It managed to overcome and reverse complement-mediated hemolysis and its devastating effects.87,89 It has also been found safe to be used during pregnancy as revealed in at least one case report.92 More recently, a study summarized the results of 18 SCD patients with DHTR receiving eculizumab.93 Most recent guidelines by the American Society of Hematology (ASH) recommend complement inhibition in patients with DHTR and ongoing hyperhemolysis.94
In addition, eculizumab has also been used in patients suffering with SCD crisis. These patients were considerably benefited by the reduction of heme-induced thrombo-inflammation through C5 inhibition.95 Importantly, eculizumab was found to be effective even in one case of bone marrow necrosis of a young adult SCD patient, where other lines of treatment failed.96 It has also found further use in patients with hemoglobinopathies who develop TMA after alloHSCT.97 Stress factors during transplantation process along with their constant underlying hemolytic condition puts these patients at high risk for complement-mediated hemolysis. These patients seem to benefit from inhibition of complement activation, thus keeping TMA activity biomarkers under careful monitoring can help establish the right moment for applying treatment with eculizumab.98 Moreover, inhibition of proximal complement activators, such as C3 inhibition with Compstatin or its analog AMY-101, is currently under investigation. Compstatin analogs have been used in preclinical and clinical studies for a plethora of disease models, such as age-related macular degeneration, sepsis, PNH, hemodialysis-induced inflammation and transplantation.99
Microvascular thrombosis and endothelial dysfunction are major characteristics of SCD and its complications. Our experience from severe coronavirus/COVID-19 infection may prove to be useful in SCD management.100 Undeniably, early reports indicate that severe COVID-19 infection bears many similarities to complement-mediated TMA.101 We have the knowledge from previous studies of other coronaviruses that inhibition of C3 activation results in weakening the lung-directed proinflammatory conditions. The genetic absence of C3 and the blockade of downstream complement effectors as well, have given rise to significant optimism.102,103 In line with these findings, AMY-101 – a C3 inhibitor – has been successfully administrated in a patient suffering from severe COVID-19 infection.100 Eculizumab was proven to be successful in such cases.104 As a matter of fact, such encouraging results led to ongoing clinical trials aiming at results that could not only shape future COVID-19 management but also affect the rest of complement-mediated diseases.
However, special attention should be paid in complement inhibition in the context of SCD. The biggest barrier in complement inhibition seems to be the increased risk of infections. These patients are prone to infection for a number of reasons including splenic dysfunction, defective opsonization and impaired adaptive immunity.105 Atypical viruses and encapsulated bacteria are at the front line. Use of eculizumab is associated with a 1000-fold to 2000-fold increased incidence of meningococcal disease despite preventive vaccination.106 Therefore, SCD patients are even at a higher risk for invasive infections because of probable complement inhibition.
Conclusions and Future Considerations
Our comprehension of complement activation and its role in the pathophysiology of SCD has expanded over the course of the past few decades. Of note, complement regulation and its mechanisms require further interpretation in the context of the disease. Current results are more than encouraging. However, improvements need to be made in complement biomarkers monitoring, in order to achieve a sufficient, modern approach to SCD management.
This review presents data that underline the expanding need for supplementary prospective studies – with more sensitive biomarkers and clinical assays included – so as to evaluate complement activation in SCD and determine selection of patients with clinical characteristics that are potentially predisposed to complement activation. Strong criteria are needed for early recognition of patients that may be benefited by complement inhibition. Additionally, should complement inhibition utilize in SCD other questions arise about the way they would be administered. These inhibitors will probably be able to stop a crisis or pain even in VOC; however, more clinical studies are needed to determine their place in treatment plans. It should also not be forgotten that hydroxyurea and other novel treatment options may also have beneficial effects on the vicious cycle that attenuates complement activation.
Acknowledgments
EG is supported by the ASH Global Research Award.
Disclosure
Eleni Gavriilaki reports serving on Alexion and Omeros advisory boards, outside the submitted work, and has consulted for Alexion Pharmaceuticals, Inc and Omeros Pharmaceuticals, Inc. The authors report no other potential conflicts of interest for this work.
References
1. Maitra P, Caughey M, Robinson L, et al. Risk factors for mortality in adult patients with sickle cell disease: a meta-analysis of studies in North America and Europe. Haematologica. 2017;102(4):626–636. doi:10.3324/haematol.2016.153791
2. Sundd P, Gladwin MT, Novelli EM. Pathophysiology of sickle cell disease. Annu Rev Pathol. 2019;14(1):263–292. doi:10.1146/annurev-pathmechdis-012418-012838
3. Stuart MJ, Nagel RL. Sickle-cell disease. Lancet. 2004;364(9442):1343–1360. doi:10.1016/S0140-6736(04)17192-4
4. Kim K, Li J, Barazia A, et al. ARQ 092, an orally-available, selective AKT inhibitor, attenuates neutrophil-platelet interactions in sickle cell disease. Haematologica. 2017;102:246–259. doi:10.3324/haematol.2016.151159
5. Lamarre Y, Romana M, Waltz X, et al. Hemorheological risk factors of acute chest syndrome and painful vaso-occlusive crisis in children with sickle cell disease. Haematologica. 2012;97:1641–1647. doi:10.3324/haematol.2012.066670
6. Allali S, Maciel TT, Hermine O, de Montalembert M. Innate immune cells, major protagonists of sickle cell disease pathophysiology. Haematologica. 2020;105(2):273–283. doi:10.3324/haematol.2019.229989
7. Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I - molecular mechanisms of activation and regulation. Front Immunol. 2015;6:262.
8. Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT. Complement system part II: role in immunity. Front Immunol. 2015;6:257.
9. Chudwin DS, Papierniak C, Lint TF, Korenblit AD. Activation of the alternative complement pathway by red blood cells from patients with sickle cell disease. Clin Immunol. 1994;71(2):199–202.
10. Mold C, Tamerius JD, Phillips G. Complement activation during painful crisis in sickle cell anemia. Clin Immunol Immunopathol. 1995;76(3):314–320. doi:10.1006/clin.1995.1131
11. Gavriilaki E, Mainou M, Christodoulou I, et al. In vitro evidence of complement activation in patients with sickle cell disease. Haematologica. 2017;102(12):e481. doi:10.3324/haematol.2017.174201
12. Roumenina LT, Chadebech P, Bodivit G, et al. Complement activation in sickle cell disease: dependence on cell density, hemolysis and modulation by hydroxyurea therapy. Am J Hematol. 2020;95(5):456–464. doi:10.1002/ajh.25742
13. Chudwin DS, Korenblit AD, Kingzette M, Artrip S, Rao S. Increased activation of the alternative complement pathway in sickle cell disease. Clin Immunol Immunopathol. 1985;37(1):93–97. doi:10.1016/0090-1229(85)90139-4
14. Lombardi E, Matte A, Risitano AM, et al. Factor H interferes with the adhesion of sickle red cells to vascular endothelium: a novel disease modulating molecule. Haematologica. 2019;104:919–928. doi:10.3324/haematol.2018.198622
15. Vaught AJ, Gavriilaki E, Hueppchen N, et al. Direct evidence of complement activation in HELLP syndrome: a link to atypical hemolytic uremic syndrome. Exp Hematol. 2016;44:390–398. doi:10.1016/j.exphem.2016.01.005
16. Brady TM, Pruette C, Loeffler LF, et al. Typical Hus: evidence of acute phase complement activation from a daycare outbreak. J Clin Exp Nephrol. 2016;1(2). doi:10.21767/2472-5056.100011
17. Gavriilaki E, Chrysanthopoulou A, Sakellari I, et al. Linking complement activation, coagulation, and neutrophils in transplant-associated thrombotic microangiopathy. Thromb Haemost. 2019;119(09):1433–1440. doi:10.1055/s-0039-1692721
18. Chaturvedi S, Braunstein EM, Yuan X, et al. Complement activity and complement regulatory gene mutations are associated with thrombosis in APS and CAPS. Blood. 2020;135(4):239–251. doi:10.1182/blood.2019003863
19. Gavriilaki E, Gkaliagkousi E, Grigoriadis S, Anyfanti P, Douma S, Anagnostopoulos A. Hypertension in hematologic malignancies and hematopoietic cell transplantation: an emerging issue with the introduction of novel treatments. Blood Rev. 2019;35:51–58. doi:10.1016/j.blre.2019.03.003
20. Gavriilaki E, Touloumenidou T, Sakellari I, et al. Pretransplant genetic susceptibility: clinical relevance in transplant-associated thrombotic microangiopathy. Thromb Haemost. 2020;120(04):638–646. doi:10.1055/s-0040-1702225
21. Gavriilaki E, Christodoulou I, Koravou EE, et al. Pre- and post-transfusion complement activation in transfusion-dependent beta-thalassaemia. Hemasphere. 2018;2:e58. doi:10.1097/HS9.0000000000000058
22. Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330(23):1639–1644. doi:10.1056/NEJM199406093302303
23. Manci EA, Culberson DE, Yang YM, et al. Causes of death in sickle cell disease: an autopsy study. Br J Haematol. 2003;123:359–365. doi:10.1046/j.1365-2141.2003.04594.x
24. Mold C, Gewurz H, Du Clos TW. Regulation of complement activation by C-reactive protein. Immunopharmacology. 1999;42(1–3):23–30. doi:10.1016/S0162-3109(99)00007-7
25. Neth O, Jack DL, Dodds AW, Holzel H, Klein NJ, Turner MW. Mannose-binding lectin binds to a range of clinically relevant microorganisms and promotes complement deposition. Infect Immun. 2000;68(2):688–693. doi:10.1128/IAI.68.2.688-693.2000
26. Francis WG, Womack CR. Serum complement activity in normal individuals and patients with sickle cell hemoglobin abnormalities. Am J Med Technol. 1967;33:77–86.
27. Johnston RB, Newman SL, Struth AG. An abnormality of the alternate pathway of complement activation in sickle-cell disease. N Engl J Med. 1973;288(16):803–808. doi:10.1056/NEJM197304192881601
28. Wilson WA, Hughes GR, Lachmann PJ. Deficiency of factor B of the complement system in sickle cell anaemia. Br Med J. 1976;1:367–369. doi:10.1136/bmj.1.6006.367
29. deCiutiis AC, Peterson CM, Polley MJ, Metakis LJ. Alternative pathway activation in sickle cell disease and beta-thalassemia major. J Natl Med Assoc. 1978;70:503–506.
30. Larcher VF, Wyke RJ, Davis LR, Stroud CE, Williams R. Defective yeast opsonisation and functional deficiency of complement in sickle cell disease. Arch Dis Child. 1982;57:343–346. doi:10.1136/adc.57.5.343
31. Koethe SM, Casper JT, Rodey GE. Alternative complement pathway activity in sera from patients with sickle cell disease. Clin Exp Immunol. 1976;23:56–60.
32. Wang RH, Phillips G
33. Wilson WA, Thomas EJ, Sissons JG. Complement activation in asymptomatic patients with sickle cell anaemia. Clin Exp Immunol. 1979;36:130–139.
34. Test ST, Woolworth VS. Defective regulation of complement by the sickle erythrocyte: evidence for a defect in control of membrane attack complex formation. Blood. 1994;83(3):842. doi:10.1182/blood.V83.3.842.842
35. Merle NS, Grunenwald A, Rajaratnam H, et al. Intravascular hemolysis activates complement via cell-free heme and heme-loaded microvesicles. JCI Insight. 2018;3. doi:10.1172/jci.insight.96910
36. Vercellotti GM, Dalmasso AP, Schaid TR
37. Gkaliagkousi E, Gavriilaki E, Vasileiadis I, et al. Endothelial microvesicles circulating in peripheral and coronary circulation are associated with central blood pressure in coronary artery disease. Am J Hypertens. 2019;32(12):1199–1205. doi:10.1093/ajh/hpz116
38. Gkaliagkousi E, Nikolaidou B, Gavriilaki E, et al. Increased erythrocyte- and platelet-derived microvesicles in newly diagnosed type 2 diabetes mellitus. Diabetes Vascular Dis Res. 2019;16(5):458–465. doi:10.1177/1479164119844691
39. de Jong K, Larkin SK, Styles LA, Bookchin RM, Kuypers FA. Characterization of the phosphatidylserine-exposing subpopulation of sickle cells. Blood. 2001;98(3):860–867. doi:10.1182/blood.V98.3.860
40. Kato GJ, Steinberg MH, Gladwin MT. Intravascular hemolysis and the pathophysiology of sickle cell disease. J Clin Invest. 2017;127(3):750–760. doi:10.1172/JCI89741
41. Beckman JD, Abdullah F, Chen C, et al. Endothelial TLR4 expression mediates vaso-occlusive crisis in sickle cell disease. Front Immunol. 2021;11:613278. doi:10.3389/fimmu.2020.613278
42. Reiter CD, Wang X, Tanus-Santos JE, et al. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat Med. 2002;8(12):1383–1389. doi:10.1038/nm1202-799
43. Camus SM, De Moraes JA, Bonnin P, et al. Circulating cell membrane microparticles transfer heme to endothelial cells and trigger vasoocclusions in sickle cell disease. Blood. 2015;125(24):3805–3814. doi:10.1182/blood-2014-07-589283
44. Ghosh S, Adisa OA, Chappa P, et al. Extracellular hemin crisis triggers acute chest syndrome in sickle mice. J Clin Invest. 2013;123(11):4809–4820. doi:10.1172/JCI64578
45. Adisa OA, Hu Y, Ghosh S, Aryee D, Osunkwo I, Ofori-Acquah SF. Association between plasma free haem and incidence of vaso-occlusive episodes and acute chest syndrome in children with sickle cell disease. Br J Haematol. 2013;162(5):702–705. doi:10.1111/bjh.12445
46. Pfefferle M, Ingoglia G, Schaer CA, et al. Hemolysis transforms liver macrophages into antiinflammatory erythrophagocytes. J Clin Invest. 2020;130(10):5576–5590. doi:10.1172/JCI137282
47. Del Conde I, Crúz MA, Zhang H, López JA, Afshar-Kharghan V. Platelet activation leads to activation and propagation of the complement system. J Exp Med. 2005;201(6):871–879. doi:10.1084/jem.20041497
48. Merle NS, Paule R, Leon J, et al. P-selectin drives complement attack on endothelium during intravascular hemolysis in TLR-4/heme-dependent manner. Proc Natl Acad Sci U S A. 2019;116(13):6280–6285. doi:10.1073/pnas.1814797116
49. Ruiz-Delgado GJ, Vazquez-Garza E, Mendez-Ramirez N, Gomez-Almaguer D. Abnormalities in the expression of CD55 and CD59 surface molecules on peripheral blood cells are not specific to paroxysmal nocturnal hemoglobinuria. Hematology. 2009;14(1):33–37. doi:10.1179/102453309X385089
50. Asimakopoulos JV, Terpos E, Papageorgiou L, et al. The presence of CD55- and/or CD59-deficient erythrocytic populations in patients with rheumatic diseases reflects an immune-mediated bone-marrow derived phenomenon. Med Sci Monit. 2014;20:123–139. doi:10.12659/MSM.889727
51. Sartain SE, Turner NA, Moake JL. TNF regulates essential alternative complement pathway components and impairs activation of protein C in human glomerular endothelial cells. J Immunol. 2016;196(2):832–845. doi:10.4049/jimmunol.1500960
52. Salama MA, Sadek NA, Hassab HM, Abadeer AF, Mikhael IL. Erythrocyte autoantibodies and expression of CD59 on the surface of red blood cells of polytransfused patients with beta-thalassaemia major. Br J Biomed Sci. 2004;61:88–92. doi:10.1080/09674845.2004.11732650
53. Takeda J, Miyata T, Kawagoe K, et al. Deficiency of the GPI anchor caused by a somatic mutation of the PIG-A gene in paroxysmal nocturnal hemoglobinuria. Cell. 1993;73(4):703–711. doi:10.1016/0092-8674(93)90250-T
54. Chapin J, Terry HS, Kleinert D, Laurence J. The role of complement activation in thrombosis and hemolytic anemias. Transfus Apher Sci. 2016;54(2):191–198. doi:10.1016/j.transci.2016.04.008
55. Amara U, Flierl MA, Rittirsch D, et al. Molecular intercommunication between the complement and coagulation systems. J Immunol. 2010;185(9):5628–5636. doi:10.4049/jimmunol.0903678
56. Zecher D, Cumpelik A, Schifferli JA. Erythrocyte-derived microvesicles amplify systemic inflammation by thrombin-dependent activation of complement. Arterioscler Thromb Vasc Biol. 2014;34(2):313–320. doi:10.1161/ATVBAHA.113.302378
57. Huber-Lang M, Sarma JV, Zetoune FS, et al. Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med. 2006;12(6):682–687. doi:10.1038/nm1419
58. Hebbel RP, Belcher JD, Vercellotti GM. The multifaceted role of ischemia/reperfusion in sickle cell anemia. J Clin Invest. 2020;130(3):1062–1072. doi:10.1172/JCI133639
59. Riedemann NC, Ward PA. Complement in ischemia reperfusion injury. Am J Pathol. 2003;162(2):363–367. doi:10.1016/S0002-9440(10)63830-8
60. Farrar CA, Asgari E, Schwaeble WJ, Sacks SH. Which pathways trigger the role of complement in ischaemia/reperfusion injury? Front Immunol. 2012;3:341. doi:10.3389/fimmu.2012.00341
61. Elhadad S, Chapin J, Copertino D, Van Besien K, Ahamed J, Laurence J. MASP2 levels are elevated in thrombotic microangiopathies: association with microvascular endothelial cell injury and suppression by anti-MASP2 antibody narsoplimab. Clin Exp Immunol. 2021;203(1):96–104. doi:10.1111/cei.13497
62. Rai P, Ataga K. Drug therapies for the management of sickle cell disease. F1000Research. 2020;9:592. doi:10.12688/f1000research.22433.1
63. Baliga B, Pace B, Chen H, Shah A, Yang Y. Mechanisms for fetal hemoglobin induction by hydroxyurea in sickle cell erythroid progenitors. AJH. 2000;65(3):227–233.
64. Agrawal RK, Patel RK, Shah V, et al. Hydroxyurea in Sickle Cell Disease: drug Review. Indian J Hematol Blood Transfus. 2014;30(2):91–96. doi:10.1007/s12288-013-0261-4
65. Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. investigators of the multicenter study of hydroxyurea in sickle cell anemia. N Engl J Med. 1995;332(20):1317–1322. doi:10.1056/NEJM199505183322001
66. Jayabose S, Tugal O, Sandoval C, et al. Clinical and hematologic effects of hydroxyurea in children with sickle cell anemia. J Pediatr. 1996;129(4):559–565. doi:10.1016/S0022-3476(96)70121-X
67. Wang WC, Wynn LW, Rogers ZR, Scott JP, Lane PA, Ware RE. A two-year pilot trial of hydroxyurea in very young children with sickle-cell anemia. J Pediatr. 2001;139(6):790–796. doi:10.1067/mpd.2001.119590
68. Wang WC, Helms RW, Lynn HS, et al. Effect of hydroxyurea on growth in children with sickle cell anemia: results of the HUG-KIDS study. J Pediatr. 2002;140(2):225–229. doi:10.1067/mpd.2002.121383
69. Hankins JS, Ware RE, Rogers ZR, et al. Longterm hydroxyurea therapy for infants with sickle cell anemia: the HUSOFT extension study. Blood. 2005;106(7):2269–2275. doi:10.1182/blood-2004-12-4973
70. Ryan K, Heeney M. New England pediatric sickle cell consortium. 2004.
71. Sadaf A, Quinn CT. L-glutamine for sickle cell disease: knight or pawn? Exp Biol Med. 2020;245(2):146–154. doi:10.1177/1535370219900637
72. Cox SE, Hart E, Kirkham FJ, Stotesbury H. L-Glutamine in sickle cell disease. Drugs Today. 2020;56(4):257–268. doi:10.1358/dot.2020.56.4.3110575
73. AlDallal SM. Voxelotor: a ray of hope for sickle disease. Cureus. 2020;12(2):e7105.
74. Prescribing information for Adakveo (crizanlizumab): US food and drug administration; 2019. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761128s000lbl.pdf.
75. Thom H, Jansen J, Shafrin J, et al. Crizanlizumab and comparators for adults with sickle cell disease: a systematic review and network meta-analysis. BMJ Open. 2020;10(9):e034147. doi:10.1136/bmjopen-2019-034147
76. Parkman R. The application of bone marrow transplantation to the treatment of genetic diseases. Science. 1986;232(4756):1373–1378. doi:10.1126/science.3520819
77. Brodsky RA, DeBaun MR. Are genetic approaches still needed to cure sickle cell disease? J Clin Invest. 2020;130(1):7–9. doi:10.1172/JCI133856
78. Ferrone F, Nagel RL, et al. Polymer structure and polymerization of deoxyhemoglobin S. In: Steinberg MH, Forget BG, Higgs DR, editors. Disorders of Hemoglobin: Genetics, Pathophysiology, Clinical Management. Cambridge, UK: Cambridge University Press; 2001.
79. Morrone K, Mitchell WB, Manwani D. Novel sickle cell disease therapies: targeting pathways downstream of sickling. Semin Hematol. 2018;55(2):68–75. doi:10.1053/j.seminhematol.2018.04.007
80. Hillmen P, Young NS, Schubert J, et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2006;355(12):1233–1243. doi:10.1056/NEJMoa061648
81. Brodsky RA, Young NS, Antonioli E, et al. Multicenter phase 3 study of the complement inhibitor eculizumab for the treatment of patients with paroxysmal nocturnal hemoglobinuria. Blood. 2008;111(4):1840–1847. doi:10.1182/blood-2007-06-094136
82. Kulasekararaj AG, Hill A, Rottinghaus ST, et al. Ravulizumab (ALXN1210) vs eculizumab in C5-inhibitor-experienced adult patients with PNH: the 302 study. Blood. 2019;133:540–549. doi:10.1182/blood-2018-09-876805
83. Lee JW, Sicre de Fontbrune F, Wong lee lee L, et al. Ravulizumab (ALXN1210) vs eculizumab in adult patients with PNH naive to complement inhibitors: the 301 study. Blood. 2019;133(6):530–539. doi:10.1182/blood-2018-09-876136
84. Legendre CM, Licht C, Muus P, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med. 2013;368:2169–2181. doi:10.1056/NEJMoa1208981
85. Gavriilaki E, Anagnostopoulos A, Mastellos DC. Complement in thrombotic microangiopathies: unraveling ariadne’s thread into the labyrinth of complement therapeutics. Front Immunol. 2019;10:337. doi:10.3389/fimmu.2019.00337
86. Yuan X, Gavriilaki E, Thanassi JA, et al. Small-molecule factor D inhibitors selectively block the alternative pathway of complement in paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome. Haematologica. 2017;102(3):466–475. doi:10.3324/haematol.2016.153312
87. Ricklin D, Mastellos DC, Reis ES, Lambris JD. The renaissance of complement therapeutics. Nat Rev Nephrol. 2018;14(1):26–47. doi:10.1038/nrneph.2017.156
88. Dumas G, Habibi A, Onimus T, et al. Eculizumab salvage therapy for delayed hemolysis transfusion reaction in sickle cell disease patients. Blood. 2016;127(8):1062–1064. doi:10.1182/blood-2015-09-669770
89. Vlachaki E, Gavriilaki E, Kafantari K, et al. Successful outcome of hyperhemolysis in sickle cell disease following multiple lines of treatment: the role of complement inhibition. Hemoglobin. 2018;42(5–6):339–341. doi:10.1080/03630269.2018.1540353
90. Boonyasampant M, Weitz IC, Kay B, Boonchalermvichian C, Liebman HA, Shulman IA. Life-threatening delayed hyperhemolytic transfusion reaction in a patient with sickle cell disease: effective treatment with eculizumab followed by rituximab. Transfusion. 2015;55(10):2398–2403. doi:10.1111/trf.13144
91. Chonat S, Quarmyne MO, Bennett CM, et al. Contribution of alternative complement pathway to delayed hemolytic transfusion reaction in sickle cell disease. Haematologica. 2018;103(10):e483–e485. doi:10.3324/haematol.2018.194670
92. Kirui Chebet L, Scully M, Mcqueen N, Porter J, Elftheriou P. Use of eculizumab for the treatment of hyperhaemolysis in pregnancy in sickle cell disease: a case report. Blood. 2018;132(Supplement 1):4922. doi:10.1182/blood-2018-99-118396
93. Floch A, Morel A, Zanchetta-Balint F, et al. Anti-C5 antibody treatment for delayed hemolytic transfusion reactions in sickle cell disease. Haematologica. 2020;105(11):2694–2697. doi:10.3324/haematol.2020.253856
94. Chou ST, Alsawas M, Fasano RM, et al. American Society of Hematology 2020 guidelines for sickle cell disease: transfusion support. Blood Adv. 2020;4(2):327–355. doi:10.1182/bloodadvances.2019001143
95. Chonat S, Graciaa S, Shin HS, et al. Eculizumab for complement mediated thrombotic microangiopathy in sickle cell disease. Haematologica. 2020;105(12).
96. Azul M, Shah S, Williams S, Vercellotti GM, Boucher AA. Evidence for complement-mediated bone marrow necrosis in a young adult with sickle cell disease. Blood Cells Mol Dis. 2021;86:102508. doi:10.1016/j.bcmd.2020.102508
97. Jodele S. Double trouble: complement-mediated thrombotic microangiopathy in patients with hemoglobinopathies after stem cell transplantation. Pediatr Blood Cancer. 2017;64(9):e26566. doi:10.1002/pbc.26566
98. Chonat S, Chandrakasan S, Kalinyak KA, et al. Atypical haemolytic uraemic syndrome in a patient with sickle cell disease, successfully treated with eculizumab. Br J Haematol. 2016;175(4):744–747. doi:10.1111/bjh.13884
99. Mastellos DC, Yancopoulou D, Kokkinos P, et al. Compstatin: a C3-targeted complement inhibitor reaching its prime for bedside intervention. Eur J Clin Invest. 2015;45(4):423–440. doi:10.1111/eci.12419
100. Mastaglio S, Ruggeri A, Risitano AM, et al. The first case of COVID-19 treated with the complement C3 inhibitor AMY-101. Clinical Immunology. 2020;215:108450.
101. Gavriilaki E, Brodsky RA. Severe COVID-19 infection and thrombotic microangiopathy: success does not come easily. Br J Haematol. 2020;189(6). doi:10.1111/bjh.16783
102. Gralinski LE, Sheahan TP, Morrison TE, et al. Complement activation contributes to severe acute respiratory syndrome coronavirus pathogenesis. mBio. 2018;9(5). doi:10.1128/mBio.01753-18
103. Jiang Y, Zhao G, Song N, et al. Blockade of the C5a–C5aR axis alleviates lung damage in hDPP4 transgenic mice infected with MERS-CoV. Emerg Microbes Infect. 2018;7(77):1–12. doi:10.1038/s41426-018-0063-8
104. Diurno F, Numis FG, Porta G, et al. Facchini, Eculizumab treatment in patients with COVID-19: preliminary results from real life ASL Napoli 2 Nord experience. Eur Rev Med Pharmacol Sci. 2020;24:4040–4047. doi:10.26355/eurrev_202004_20875
105. Ochocinski D, Dalal M, Black LV, et al. Life-threatening infectious complications in sickle cell disease: a concise narrative review. Front Pediatr. 2020;8:38. doi:10.3389/fped.2020.00038
106. McNamara LA, Topaz N, Wang X, Hariri S, Fox L, MacNeil JR. High risk for invasive meningococcal disease among patients receiving eculizumab (Soliris) despite receipt of meningococcal vaccine. MMWR Morb Mortal Wkly Rep. 2017;66(27):734–737. doi:10.15585/mmwr.mm6627e1
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.