Back to Journals » International Journal of Nanomedicine » Volume 12
Complement C3-dependent uptake of targeted liposomes into human macrophages, B cells, dendritic cells, neutrophils, and MDSCs
Authors Francian A, Mann K ![]() , Kullberg M
, Kullberg M
Received 4 April 2017
Accepted for publication 2 June 2017
Published 19 July 2017 Volume 2017:12 Pages 5149—5161
DOI https://doi.org/10.2147/IJN.S138787
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Alexandra Francian,1 Kristine Mann,1,2 Max Kullberg1
1WWAMI Medical Education Program, 2Department of Biological Sciences, University of Alaska Anchorage, Anchorage, AK, USA
Abstract: Antitumor immunity in cancer patients is heavily modulated by cells of the innate immune system. Antigen-presenting cells, including dendritic cells, macrophages, and B cells, initiate immune recognition of tumor antigen by displaying antigen to effector cells. Countering this immune stimulation are immunosuppressive cells which include M2 macrophages, N2 neutrophils, and myeloid-derived suppressor cells (MDSCs). To create effective cancer immunotherapies, it is critical that we can target these important cell types of the immune system with immunostimulatory compounds. A commonality of these cell types is the complement receptor, which recognizes pathogens that are bound to activated complement C3 in human blood. To target the complement receptor, we have created a liposome that has a small molecule, orthopyridyl disulfide (OPSS), conjugated to its surface. OPSS forms a disulfide bond with activated complement C3, which then targets liposomes for uptake by dendritic cells, macrophages, B cells, MDSCs, and neutrophils in human blood. Internalization is efficient and specific to cells that display the complement receptor. Liposomes are a versatile drug delivery device. Possible applications for this system include delivery of toll-receptor agonists or tumor antigen to antigen-presenting cells and delivery of immunostimulatory drugs to M2, N2, and MDSC immunosuppressive cells.
Keywords: cancer immuno therapy, antigen-presenting cells, complement C3, nanoparticle, targeted delivery
Introduction
Dendritic cells, B cells, macrophages, neutrophils, and myeloid-derived suppressor cells (MDSCs) are all involved in regulation of the immune response against cancer.1,2 The first step in an adaptive immune response against a tumor is carried out by antigen-presenting cells (APCs), which include the dendritic cells, B cells, and macrophages.1,3 After engulfing tumor cells, endocytic processing in APCs results in antigen presentation by major histocompatibility complexes to helper and cytotoxic T-cells. Opposing this immunostimulatory action are immunosuppressive cells. The tumor microenvironment recruits and promotes the production of numerous suppressive cell types, including protumor M2 macrophages, N2 neutrophils, and MDSCs, which produce suppressive cytokines such as IL-10 and TGF-β, reactive oxygen species, nitric oxide synthetase, and arginase to inhibit cytotoxic T-cells.4,5 Whether targeting antigen to APCs or delivering drugs to relieve immunosuppression, cancer immunotherapies could benefit from a nanoparticle delivery system to both cell types. We have therefore developed a system that targets the receptor for complement C3, which is a commonality among dendritic cells, B cells, macrophages, neutrophils, and MDSCs.6,7
Various strategies have been employed in the nanoparticle field to target macrophages and dendritic cells including cationic, mannose, Fc-targeted, CD11c-targeted, and DC-SIGN-targeted liposome carriers.8,9 These have had various degrees of success, but often have the drawback of requiring complex targeting molecules or antibodies that present challenges to large-scale production and storage. An exception is the mannose targeting system that utilizes a mannose sugar to target the macrophage mannose receptor and is a robust and simple system.10 Cationic liposomes appear attractive for targeting cells, but when injected systemically, they aggregate and accumulate almost entirely in the lung and liver.11 While many systems have been developed to target macrophages and dendritic cells, there are few available options for the targeting of MDSCs, neutrophils, and B cells.9 To overcome these shortcomings and challenges, we have designed a system that utilizes the patient’s own endogenous complement C3.7 The liposomes would bind to C3 after injection, resulting in targeting to cell types that have the receptor for C3. The system utilizes small molecules that would allow for scaling up and storage, binds to endogenous C3 which would cut down on toxicities, and targets a wide variety of immune cells that regulate the antitumor immune response.
Complement C3 is a protein that is present in normal human blood and is activated in the presence of a pathogen or foreign molecule.6 Cleavage of C3 protein results in active C3b with a thioester domain that can react with antigens or with water to yield a free sulfhydryl group. The activated fragments bind to pathogen surfaces that are then recognized by APCs for phagocytosis, destruction, and antigen presentation.12,13 The liposome system we have developed contains lipid bound to an orthopyridyl disulfide (OPSS) moiety, which forms a disulfide bond with the exposed sulfhydryl group of activated C3 protein present in normal serum. OPSS-liposomes coated in C3 proteins are targeted for phagocytosis by the innate immune system. In a previous study, we have shown that liposomes containing OPSS bind to complement C3 in mouse serum, resulting in uptake by immune cells that have the receptor for complement.7 We went on to show that upon systemic administration into tumor-bearing mice, liposomes were taken up by MDSCs which infiltrated the spleen and tumor.7
The goal of the current study was to establish if the liposomes also bound to human complement C3, and if so to fully characterize the cell types in human blood that engulf C3 bound liposomes using flow cytometry analysis. Possible uses of complement C3-bound liposomes include delivery of tumor antigen or immunostimulating molecules to APCs, and delivery of drugs that can reprogram immunosuppressive MDSCs, macrophages, and neutrophils to an immunostimulatory phenotype.
Materials and methods
Reagents
1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[poly(ethylene glycol)-2000] (DSPE-PEG(2000)), and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[PDP-poly(ethylene glycol)-2000] (DSPE-PEG(2000)-PDP) used for liposome preparation were purchased from Avanti Polar Lipids (Alabaster, AL, USA). Fluorescently tagged lipid, Lissamine rhodamine B 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (RhodaminePE), was purchased from Life Technologies (Grand Island, NY, USA). Size exclusion chromatography utilized CL-4B Sepharose gel, purchased from Sigma-Aldrich (St Louis, MO, USA). Red blood cell lysis buffer was purchased from eBioscience (San Diego, CA, USA). Goat anti-human complement C3 was obtained from MP Biomedicals (Solon, OH, USA). Secondary donkey anti-goat 800 IgG was purchased from Li-Cor Bioscience (Lincoln, NE, USA). Normal human serum complement and C3-depleted human serum were obtained from Quidel Corporation (Athens, OH, USA). Flow cytometry antibodies, PE/Cy7 anti-human CD16, Brilliant Violet 605 anti-human CD33, Brilliant Violet 650 anti-human CD20, and Brilliant Violet 785 anti-human CD56 (NCAM), were purchased from BioLegend (San Diego, CA, USA). Flow cytometry antibodies, APC-Alexa Fluor 700 anti-human CD11c, APC-Alexa Fluor 750 anti-human CD11b, PC5.5 anti-human HLA-DR, FITC anti-human CD45, ECD anti-human CD3, Pacific Blue anti-human CD15, and APC anti-human CD14, were purchased from Beckman Coulter (Brea, CA, USA). All other chemicals and reagents were purchased from Thermo Fisher Scientific (Pittsburgh, PA, USA).
Liposome preparation
Liposomes were prepared using the film hydration–extrusion method as previously described.7,14 Liposomes containing DSPE-PEG(2000)-PDP are referred to as OPSS-liposomes; liposomes containing DSPE-PEG(2000) are referred to as control-liposomes. To produce OPSS-liposomes, DPPC/DSPC/DSPE-PEG(2000)-PDP/DSPE-PEG(2000)/RhodaminePE in chloroform were briefly mixed at a molar ratio of 83:12:1:3:1). For control-liposomes, DSPE-PEG(2000) was substituted for DSPE-PEG(2000)-PDP to maintain the same ratio of DSPE-PEG. Lipids were dried under a nitrogen stream for 1 hour to remove any chloroform residue. The lipid film was rehydrated in 0.7 mL of filtered water and extruded 9 times through a 200-nm polycarbonate membrane filter at 47°C. Liposomes were column purified using a CL-4B Sepharose column hydrated in 1× PBS, pH 7.4. Liposome fraction was diluted to a concentration of 0.875 mg lipid/mL. The amount of OPSS-liposome and control-liposome in each sample was normalized using a NanoDrop 2000 UV-Vis Spectrophotometer. Liposome size was obtained using a Malvern Zetasizer Nano-S (Malvern Instruments, Malvern, UK). Control-liposome diameter was measured as 141.8±47.29 nm, and OPSS-liposome diameter was 140.4±43.76 nm. The diameter of OPSS-liposomes incubated in serum for 1 hour was determined to be 165±89 nm.
Liposome binding of activated C3
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot techniques were used to determine if liposomes bind activated C3 when exposed to complete human serum. A 1:1 sample of OPSS-liposomes or control-liposomes with human serum was incubated for 1 hour at 37°C. Liposomes were isolated from the serum by centrifuging in Beckman 5×41 mm ultraclear tubes in a SW50.1 rotor at 200,000× g for 10 minutes at 4°C in a Beckman L8-70 ultracentrifuge. Liposomes were centrifuged and rinsed three times in 1× PBS before being rehydrated in 1× PBS. Samples were mixed 1:1 with a 2× reducing sample loading buffer and heated at 95°C for 4 minutes. Samples were then run on a precast 10% SDS-PAGE gel (Bio-Rad Laboratories, Hercules, CA, USA) for 1 hour at 120 volts. After electrophoresis was complete, the gel was soaked in transfer buffer (25 mM Tris-base, 192 mM glycine) for 15–20 minutes to equilibrate before transfer. The proteins were then electroblotted onto Immobilon PVDF membrane (Sigma-Aldrich) at 12 volts overnight. Total proteins associated with the liposomes were identified by colloidal gold staining of the blot. C3 proteins associated with the liposomes were detected with goat anti-human complement C3 at a 1:1,000 dilution and secondary donkey anti-goat 800 IgG at a 1:10,000 dilution and visualized with a Li-Cor infrared scanner with Odyssey software (West Henrietta, NY, USA).
In vitro uptake of liposomes
An in vitro analysis of liposome uptake was performed to determine which cell types take up liposomes in human blood. Peripheral blood mononuclear cells were isolated from whole blood in heparinized tubes obtained from five healthy human volunteers. The protocol for blood draw was approved by the University of Alaska Anchorage Institutional Review Board, in accordance with the U.S. Department of Health and Human Services requirements for the protection of human research subjects (45 CFR 46 as amended/revised), and all volunteer donors provided written informed consent. Immediately after drawing, the blood was incubated in red blood cell lysis buffer for 10–15 minutes. The samples were then centrifuged at 500× g for 5 minutes in an Eppendorf 5804 centrifuge. Samples were rinsed in 1× PBS and resuspended in Roswell Park Memorial Institute media. Cells were aliquoted into a 96-well V-bottom plate with 80 μL per well to achieve a concentration of approximately 160,000 cells per well (2×106 per mL). OPSS-liposomes and control-liposomes were incubated for 1 hour at 37°C with an equal volume of normal human serum or serum that had been depleted of complement C3. Twenty microliters of the liposomes + serum sample was added to the 80 μL of cells in each well to bring the final volume in each well up to 100 μL with a concentration of 10% serum. Cells were exposed to liposomes for 2 hours before collection and analysis by flow cytometry.
Flow cytometry analysis
Cells were analyzed by flow cytometry to determine the populations of cells that were positive for rhodamine-labeled liposomes. Collected cells were centrifuged in a 96-well V-bottom polystyrene microplate at 2,000 rpm in a Sorvall T6000D centrifuge for 3 minutes and resuspended in 100 μL FACS buffer (1× PBS +1% bovine serum albumin) containing 1 μL each of anti-human antibodies against CD45, CD3, HLA-DR, CD16, CD14, CD11c, CD11b, CD15, CD33, CD20, and CD56. Cells were incubated in the dark with the staining buffer at 4°C for 20 minutes. After staining, cells were centrifuged (as mentioned earlier) and resuspended in 200 μL of FACS buffer and analyzed using a Beckman Coulter CytoFLEX flow cytometer with CytExpert software (Beckman Coulter, Brea, CA, USA). After gating to find cell populations, the percentage of rhodamine-liposome positive cells was determined, averaged for the five patients, and presented as mean ± standard error (n=5).
Fluorescent microscopy
Cells were treated for 2 hours with OPSS- or control-liposomes that had been incubated in complement C3-containing or depleted human serum, as described earlier. Cells were centrifuged at 500× g for 5 minutes and rinsed twice with PBS before resuspension and transfer to a flat bottom Falcon microtest 96-well assay plate, black/clear bottom (Becton Dickinson Labware, Franklin Lakes, NJ, USA). Cells were imaged with a Leica DMI6000B inverted fluorescence microscope (Leica Microsystems, Buffalo Grove, IL, USA), and photos were taken using a 10× objective utilizing Leica Application Suite, version 3.7.0 software (Leica microsystems Inc., Wetzlar, Germany).
Results
OPSS-liposomes bind complement C3 in normal human serum
The ability of OPSS-liposomes to bind complement protein C3 was determined by SDS-PAGE and Western blot analysis (Figure 1). When complement C3 protein in normal serum is cleaved, C3a is released and C3b undergoes a conformational change resulting in an exposed sulfhydryl group on the activated C3b fragment. The OPSS group on the PEGylated lipid binds to these activated fragments and forms the C3 bound liposomes (C3-liposomes), which are taken up by cells that have receptors for activated complement C3. OPSS-liposomes and liposomes lacking the OPSS group (control-liposomes) were incubated in normal human serum containing all the complement proteins to test the specificity toward complement C3, one of the most abundant complement proteins in serum. After incubation with serum, the liposomes were pelleted by ultracentrifugation and rinsed thoroughly to remove the serum before analysis by SDS-PAGE gel electrophoresis and Western blot. As shown in the anti-C3 immunoblot, control-liposomes lacking the OPSS group did not bind complement C3, while the OPSS-liposomes bound complement C3 and its activated fragments (Figure 1).15 A duplicate Western blot was stained with colloidal gold to detect all proteins that were associated with the liposomes. The overlap in bands between the colloidal gold stain and the anti-C3 blot shows that binding of C3 to the liposomes with the OPSS group is specific and does not occur in control-liposomes without the OPSS group. It is likely that the interaction of C3b with liposomes will lead to the formation of larger complement complexes including C3bBb, C3bBbC3b, and C3bBbP.16,17 These higher molecular weight bands that are apparent on the Western blot are therefore labeled as complement C3 complexes.
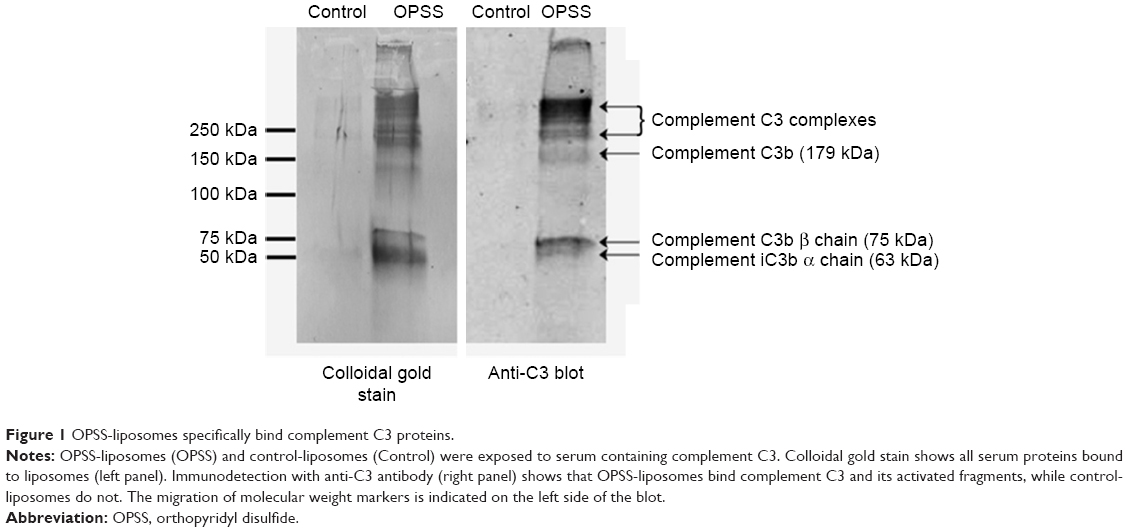 | Figure 1 OPSS-liposomes specifically bind complement C3 proteins. |
White blood cells internalize OPSS-liposomes
OPSS-liposomes were incubated in human serum that had functional complement C3 to create C3-bound liposomes (C3-liposomes). Controls included OPSS-liposomes incubated in C3-depleted serum as well as control-liposomes incubated in both C3+ serum and C3-depleted serum. These liposomes were then administered to white blood cells isolated from human blood, and uptake of liposomes was observed via a fluorescent rhodamine probe incorporated into the liposomal membranes. Members of the complement receptor family that are found on white blood cells include complement receptors 1, 2, and 3 (CR1, CR2, and CR3). CR3 can be identified by the surface marker CD11b and is the major complement receptor of the myeloid cell populations. These receptors are expressed on the surface of cells and bind and internalize particles attached to complement proteins. Fluorescent microscopy and flow cytometry analysis showed that OPSS-liposomes incubated in C3-containing serum were readily taken up by CD11b+ cells (71.87%±2.5%), while control-liposomes showed very little uptake (5.06%±1.9% of cells) (Figure 2). OPSS-liposomes and control-liposomes incubated in serum depleted of complement C3 (C3−) also displayed little internalization into CD11b+ cells (OPSS: 1.98%±0.7% of cells, Control: 1.74%±0.9% of cells) demonstrating the importance of both the OPSS group and the bound activated-C3 protein in the targeting mechanism (Figure 2). The CD11b-negative cells that had taken up OPSS-liposomes in C3-positive serum (lower right quadrant of left panel, Figure 2) were identified as B cells (data not shown), known to contain CR2 receptors (CD21) that can bind activated complement C3 fragments.18
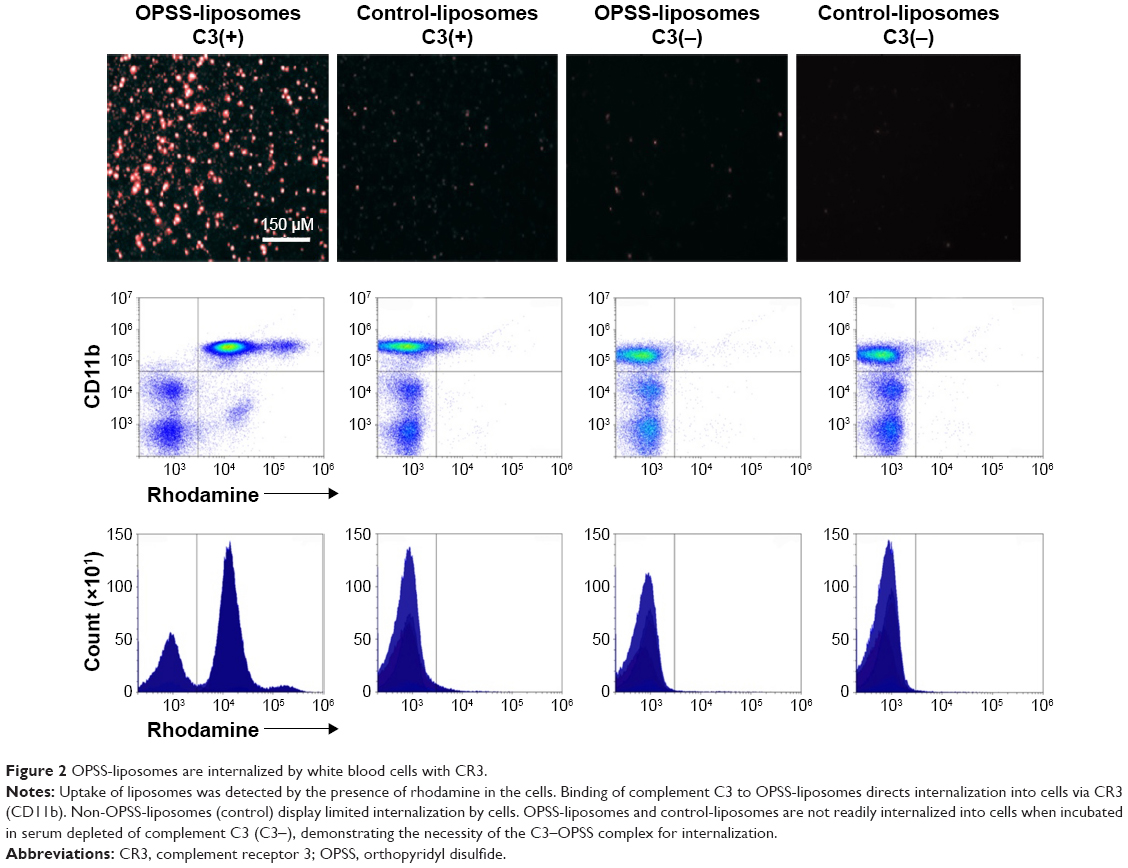 | Figure 2 OPSS-liposomes are internalized by white blood cells with CR3. |
Liposome uptake by MDSCs and neutrophils
MDSCs are a heterogeneous population of cells that express complement receptor CR3 (CD11b+), enabling C3-bound OPSS-liposomes to target both monocytic MDSC (M-MDSC) and granulocytic MDSC (G-MDSC). Normal human white blood cells were stained with antibodies against several cell surface markers to identify the MDSCs by flow cytometry. M-MDSCs were identified according to their cell surface marker phenotype: CD33+/hi, CD11b+, HLA-DR−/low, and CD14+/hi. G-MDSCs were characterized by: CD33+/low, CD11b+, HLA-DR−/low, CD14−, and CD15+ (Figure 3). The gating strategy used to distinguish M-MDSCs and G-MDSCs can be seen in Figure 3. Using the same gating strategy, neutrophils were identified as CD33mid/low, CD15+ (Figure 3, lower right panel).
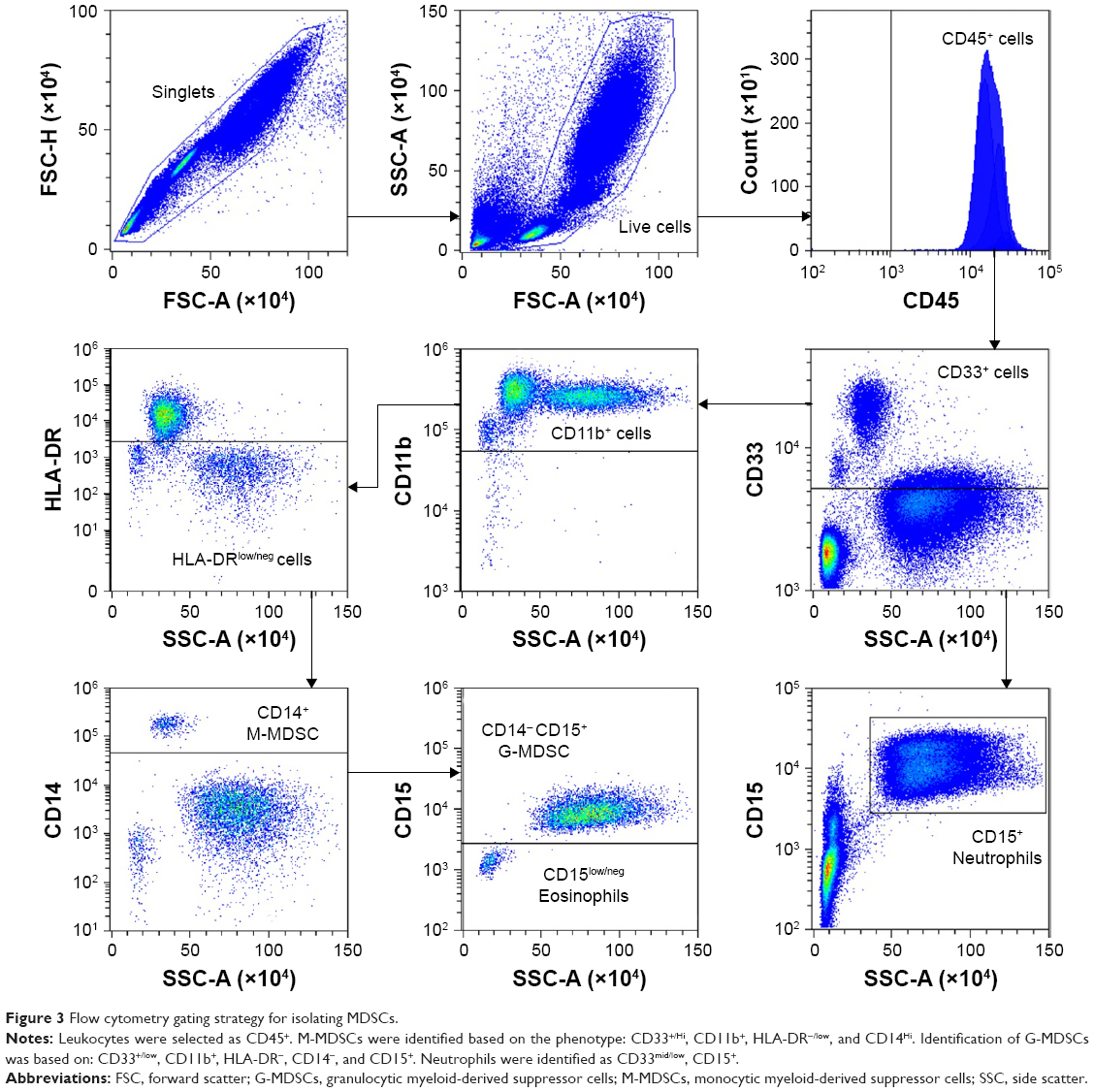 | Figure 3 Flow cytometry gating strategy for isolating MDSCs. |
Human white blood cells were exposed to rhodamine-labeled liposomes that had been incubated in C3+ vs C3-depleted serum to determine if bound C3 led to internalization of OPSS-liposomes. Both M-MDSC and G-MDSC showed high internalization of C3-liposomes, with 99.8%±0.1% and 96.7%±0.9% of cells taking up liposomes, respectively. Control-liposomes incubated in C3+ serum as well as control- and OPSS-liposomes incubated in serum depleted of complement C3 had significantly reduced uptake, with G-MDSC showing less than 27% uptake and M-MDSC showing less than 12% uptake in all three control conditions (Figure 4). Neutrophils that were CD15+ and CD16+, but did not display high levels of the immature myeloid marker CD33, also took up C3-bound OPSS-liposomes (77%±7%), with less than 3% of cells taking up liposomes if serum was depleted of C3 or if OPSS was not present in the liposomes. Eosinophils, separated from neutrophils by low CD16 expression (dot plot not shown), showed a similar pattern of uptake, with 71%±9% internalizing C3-bound OPSS-liposomes and less than 8% of cells taking up liposomes in C3-depleted serum or liposomes lacking the OPSS group (Figure 4).
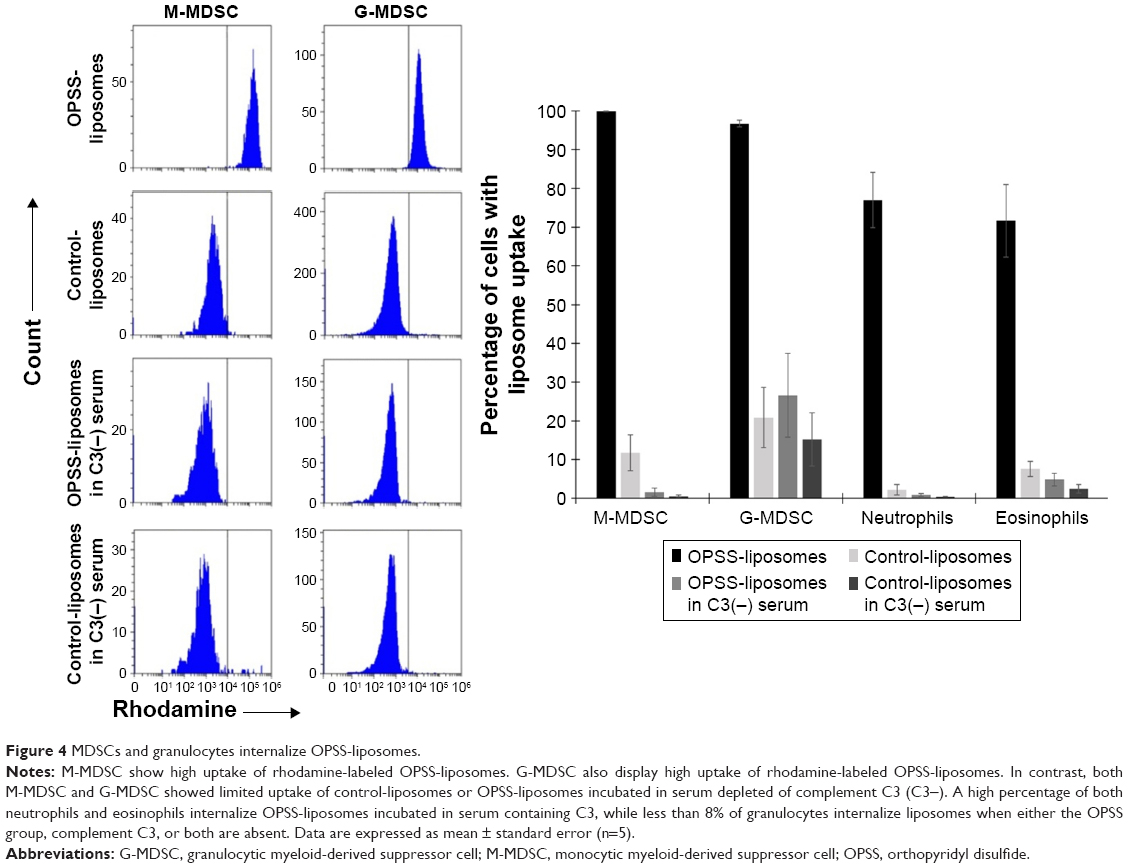 | Figure 4 MDSCs and granulocytes internalize OPSS-liposomes. |
Liposome uptake by APCs
APCs (macrophages, dendritic cells, and B cells) were also identified by flow cytometry and analyzed for uptake of rhodamine-labeled liposomes. Singlets were first selected that were positive for the common leukocyte antigen, CD45. These cells were then selected by size and internal complexity (side scatter vs forward scatter) to separate the monocyte/granulocyte population from the lymphocytes (Figure 5). B cells were identified from the lymphocyte population by the surface markers HLA-DR and CD20. Macrophages were identified from the monocyte/granulocyte population by the presence of HLA-DR and CD14 surface marker expression. Myeloid dendritic cells were isolated from the same population by expression of HLA-DR, low expression of CD14, and high expression of CD11c (Figure 5).
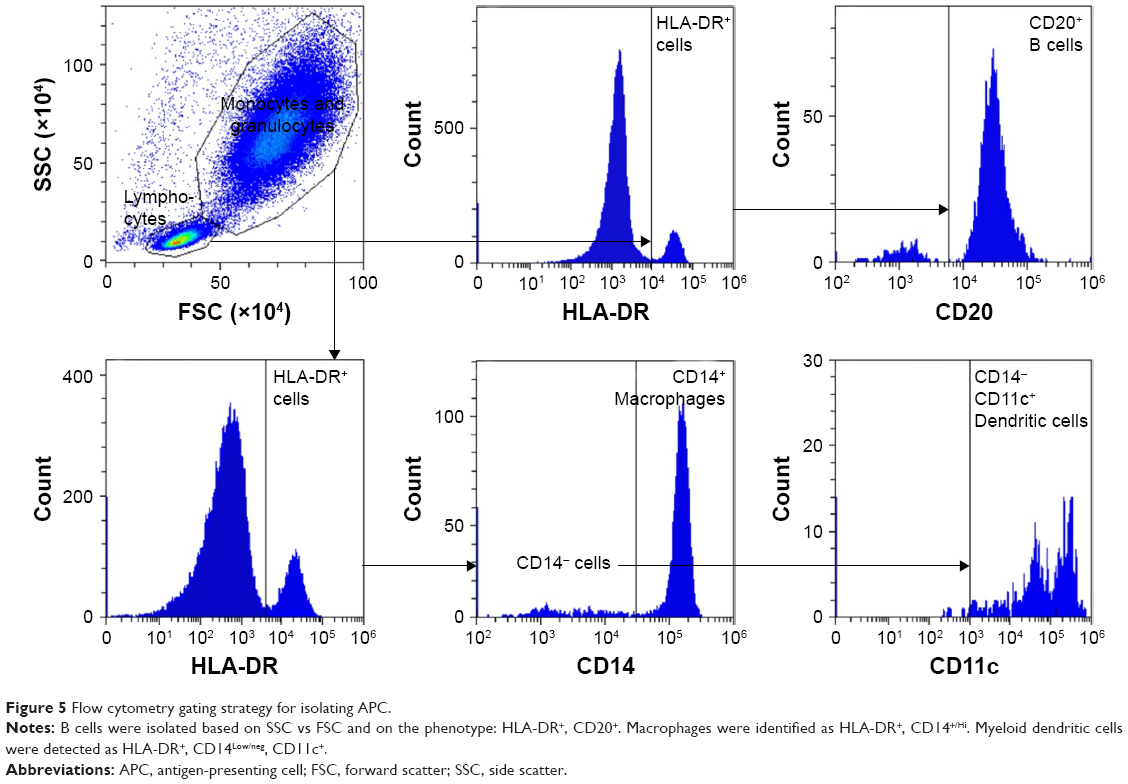 | Figure 5 Flow cytometry gating strategy for isolating APC. |
All three types of APCs displayed selective uptake of rhodamine-labeled C3-liposomes. OPSS-liposomes incubated in C3-depleted serum and control-liposomes incubated in C3-positive or C3-depleted serum showed little uptake by APCs (Figure 6). OPSS-liposomes and control-liposomes incubated in C3-positive serum showed internalization into 99.99%±0.01% and 8%±1% of macrophages, respectively. This demonstrates the ability of the C3-bound liposomes to enhance the natural liposomal clearance by phagocytic macrophages and specifically target the complement receptors for uptake. 66%±7% of myeloid dendritic cells internalized C3-liposomes, while only 10%±3% internalized control-liposomes after incubation in C3-positive serum. B cells displayed a similar pattern with 90%±2% uptake of C3-liposomes compared to 0.8%±0.2% uptake of control-liposomes. When liposomes were incubated in C3 depleted serum, uptake of either OPSS- or control-liposomes was seen in less than 4% of dendritic cells, macrophages, or B cells (Figure 6). These results show that both the OPSS group and the presence of complement C3 drive the uptake of liposomes into the APCs.
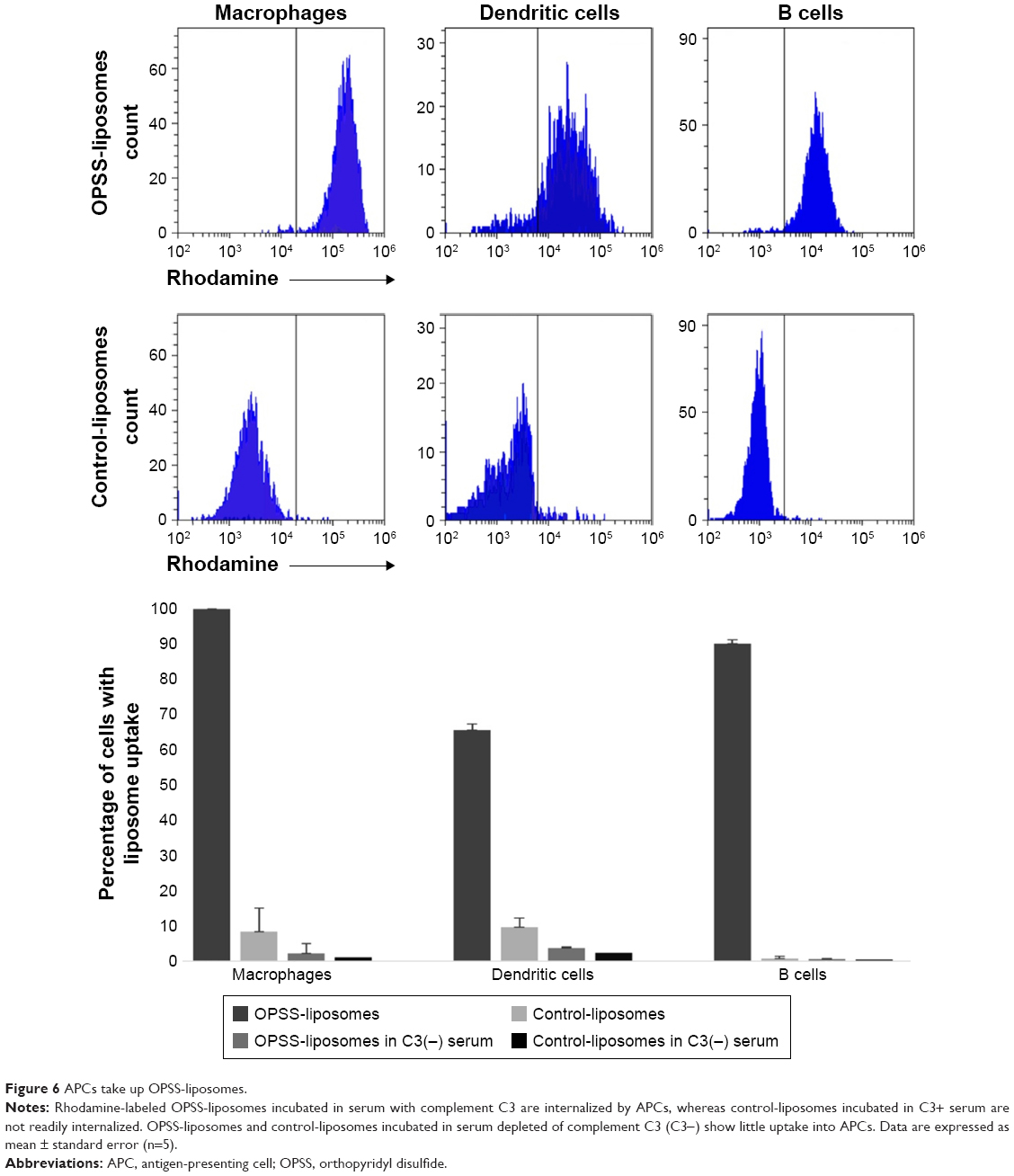 | Figure 6 APCs take up OPSS-liposomes. |
Liposome uptake by lymphocytes
T-cell, natural killer (NK) cell, and B cell populations were analyzed for their uptake of rhodamine-labeled liposomes. The lymphocyte population was selected by CD45 and by size and internal complexity (side scatter vs forward scatter). This population was further divided to identify CD20+ B cells, CD3+ T-cells, and CD56+ NK cells. The T-cell, and NK cell populations showed minimal uptake of OPSS-liposomes and control-liposomes incubated in either C3-positive or C3-depleted serum with less than 2% of T- and NK cells positive in all conditions. In contrast, 90%±2% of B cells took up OPSS-liposomes incubated in C3-positive serum, while less than 3% took up OPSS-liposomes incubated in C3-depleted serum or control-liposomes incubated in either C3-positive or C3-depleted serum (Figure 7). These data show that among the different leukocyte populations, B cells are the only population targeted by C3-bound liposomes.
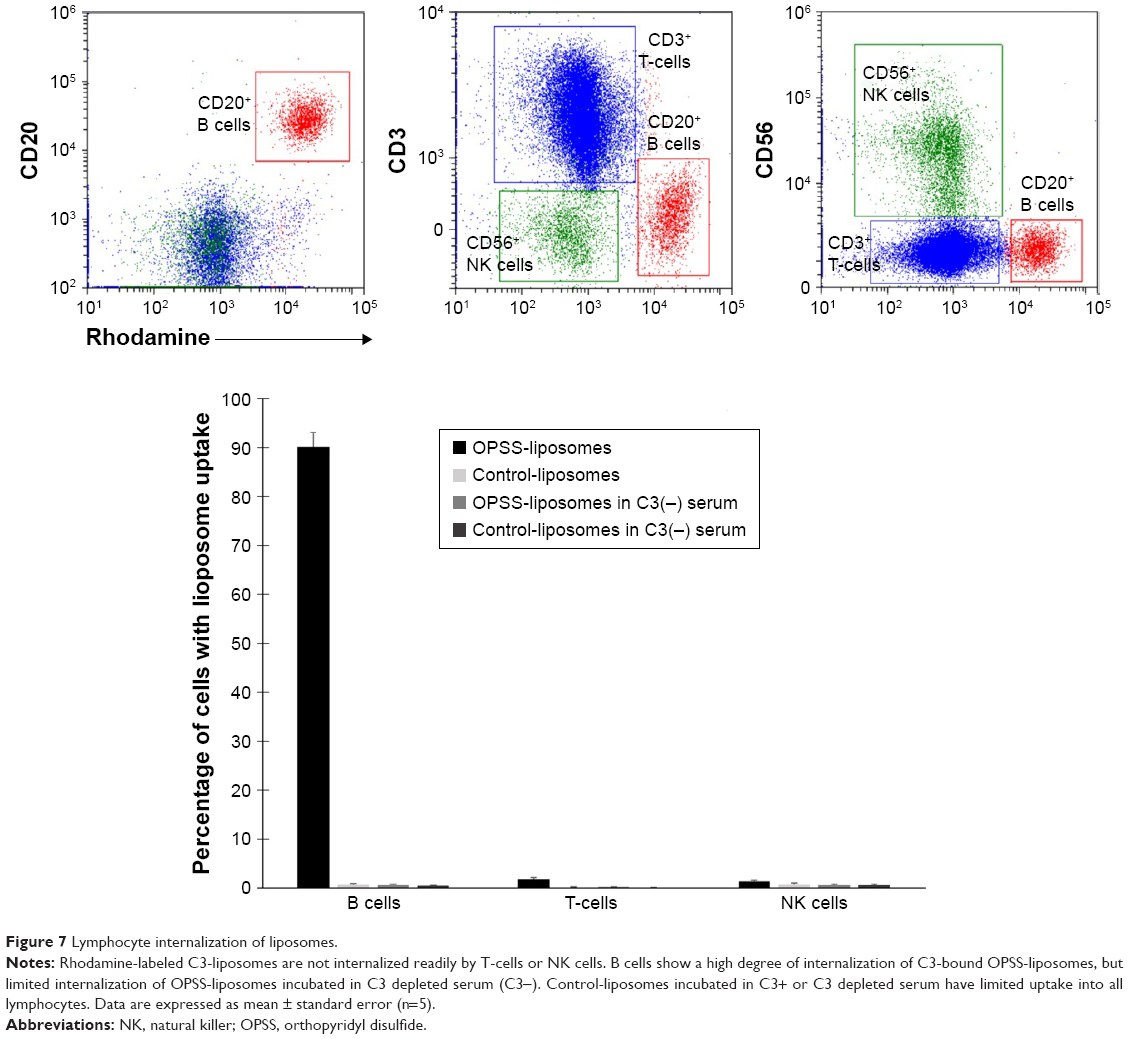 | Figure 7 Lymphocyte internalization of liposomes. |
C3-liposome uptake varies between white blood cell types
Isolated white blood cells were exposed to rhodamine-labeled C3-liposomes and analyzed for levels of uptake via flow cytometry. Relative fluorescence intensity was determined for each cell population. Macrophages and M-MDSC showed the highest levels of rhodamine-labeled liposomes, followed by the rest of the myeloid cell populations displaying similar but lower levels of liposome uptake. T-cells and NK cells showed the lowest levels of rhodamine fluorescence (Figure 8).
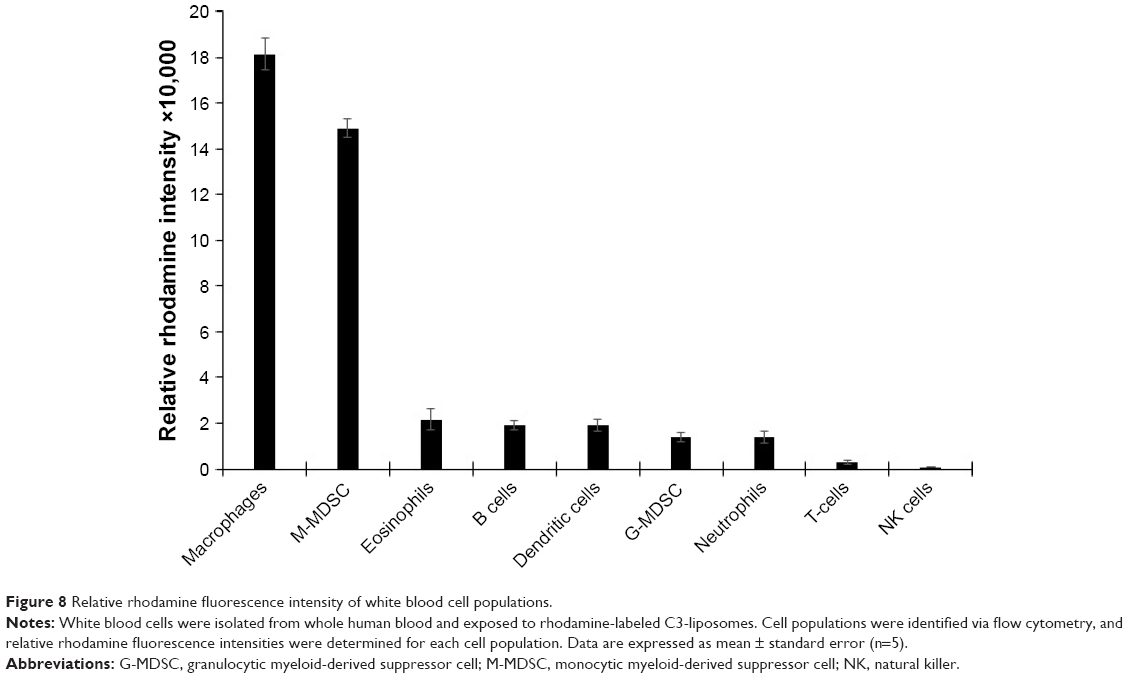 | Figure 8 Relative rhodamine fluorescence intensity of white blood cell populations. |
Discussion
APCs and immunosuppressive cells play an important role in cancer progression and immune evasion.1,18 Strategies for promoting an antitumor immune response would benefit from a nanoparticle system that can target these cells, all of which display the receptor for complement C3.3,9,19 Liposomes were therefore formulated with a lipid-attached OPSS group, which can form a disulfide bond with activated complement C3. After binding complement, we show that these liposomes are taken up by human macrophages, M-MDSCs, G-MDSCs, neutrophils, dendritic cells, and B cells. By utilizing this targeting mechanism, the C3-bound OPSS-liposomes should allow the delivery of tumor antigen or immunostimulatory drugs to these various cell types.
Complement C3 is a major component of our blood that is inert but can be activated to C3b, exposing a thioester group capable of forming a disulfide bond with OPSS.6,7 Western blot analysis reveals that incubation of OPSS-liposomes in serum for 1 hour allows conjugation of C3b to the liposomes and that this binding is relatively specific with little other protein attached. C3b targets the complement CR1 receptor, but can be further metabolized to iC3b and C3dg, which can target CR2 (iC3b, C3dg), CR3 (iC3b), CRIg (iC3b), and CR4 (iC3b) receptors.6 Most of the cells targeted by C3-bound OPSS-liposomes have the CR3 receptor (CD11b), including macrophages, neutrophils, dendritic cells, and MDSCs. However, B cells, which express the CR2 receptor also readily engulf liposomes, implying that the liposomes can target through the iC3b or C3dg breakdown products. Indeed, the Western blot shows that, on the basis of molecular weight, iC3b is part of the complex that is conjugated to our liposomes. While all cells of myeloid lineage internalize C3-bound OPSS-liposomes, presumably due to the presence of the myeloid CR3 receptor, the lymphocyte population of cells shows limited uptake of liposomes, with the exception of B cells.
MDSCs show a high level of uptake of activated C3-bound OPSS-liposomes. MDSCs do not take up liposomes readily when liposomes are formulated without OPSS or when serum is depleted of complement C3, demonstrating the importance of both the liposomal OPSS group and complement C3 for targeting. MDSCs are a heterogeneous population of immature cells that include granulocytic and monocytic subtypes.2,20 Both types are targeted by C3-liposomes in human blood. In cancer patients, the population of MDSCs expands in number in response to cytokines, such as GM-CSF, released from the tumor and are critical to creating immunosuppressive conditions.2,21,22 The overall number of MDSCs correlates directly with cancer stage and level of metastasis. Being able to target this cell population and reverse the suppression would significantly improve treatments and therapies.23–25 Reprogramming of MDSCs has been shown using all-trans retinoic acid, vitamin D, and CpG oligonucleotides, but techniques for specific delivery of these compounds are still lacking.23,24 Our targeted liposomal system provides a means to target MDSCs and test different treatments, which could relieve the suppression and possibly revert MDSCs toward their non-suppressive phenotype.
C3-liposomes are also taken up in a complement-dependent pathway by all three types of APCs: dendritic cells, macrophages, and B cells. The first step in creating a robust adaptive immune response against cancer cells requires efficient presentation of tumor antigen by APCs to the effector cells of the immune system.1,3 APCs present antigen to T helper cells via MHCII molecules. Additionally, dendritic cells and B cells have been shown to cross-present antigen via MHCI molecules, allowing for the stimulation of cytotoxic T-cells.26,27 Techniques to improve antigen presentation include ex vivo strategies such as adoptive T-cell transfer and in vivo strategies such as nanoparticle antigen delivery.8,9,19,28 Drawbacks of ex vivo techniques include high cost and lack of a memory T-cell population after inoculation into the patient.29,30 Nanoparticle delivery systems have had some success, but often they are targeted only to macrophages and dendritic cells, and most targeted systems require costly antibodies or peptides that are difficult to store and scale up to pharmaceutical quantities.8,9 The advantage of using OPSS-liposomes is that OPSS is a small low-cost molecule that binds endogenous complement C3 and targets all three APCs, including B cells. OPSS-liposomes could encapsulate tumor antigen or activating oligonucleotides to improve antigen presentation to effector cells. In addition, to provoke a B cell antibody response, it is critical that B cells are stimulated through their complement receptor, thereby lowering the stimulation threshold at which they produce antibody by approximately 1,000-fold.18,31,32 By targeting antigen to all three APC cell types via the complement system, C3-liposomes could activate T-cells and increase antibody production by B cells, leading to a robust and enduring antitumor immune response.
C3-liposome delivery may activate the complement system, leading to initiation of the complement cascade, possibly leading to systemic cytokine release or the formation of the membrane attack complex.33,34 While overactivation of the complement system is a concern, preliminary studies in mice with systemic administration of C3-liposomes did not appear to cause any signs of distress.7 The tumor microenvironment causes an overall suppression of the immune system, so there is a possibility that activation of immune cells upon binding of the complement receptor could be beneficial.18,35 More experimentation will be done to characterize the response of immune cells to C3-liposomes.
These experiments show a new technique for targeting immune cells that play a key role in cancer progression, including MDSCs, neutrophils, macrophages, dendritic cells, and B cells. Since these data were obtained using the blood of healthy human volunteers, it is important to remember that the number and phenotypes of immune cells will presumably be different in cancer patients. It remains to be seen if the C3-bound liposomes can target M2 macrophages, N2 neutrophils, suppressive MDSCs, and APCs that have been exposed to immunosuppressive cytokines released by tumors. Future studies will evaluate the targeting potential of our liposomes using blood samples donated by cancer patients. In addition, in vivo studies will be done to determine the biodistribution of C3-bound liposomes in a tumor mouse model. If targeting to tumors is accomplished, these liposomes could be a potent tool for specific delivery of tumor antigens and immune-stimulating drugs, with the goal of creating a robust antitumor immune response.
Acknowledgments
We thank Dr Holly Martinson for her critical thinking and thoughtful discussion during the development of this targeting system. This research was supported by the WWAMI Medical Education Program, University of Alaska Anchorage; Alaska Run for Women Grants; and an Institutional Development Award (IDeA) from the National Institutes of Health/National Institute of General Medical Sciences under Grant P20GM103395.
Disclosure
The authors report no conflicts of interest in this work.
References
Cerezo-Wallis D, Soengas M. Understanding tumor-antigen presentation in the new era of cancer immunotherapy. Curr Pharm Des. 2016;22(41):6234–6250. | ||
Markowitz J, Wesolowski R, Papenfuss T, Brooks TR, Carson WE 3rd. Myeloid-derived suppressor cells in breast cancer. Breast Cancer Res Treat. 2013;140(1):13–21. | ||
Tagliamonte M, Petrizzo A, Tornesello ML, Buonaguro FM, Buonaguro L. Antigen-specific vaccines for cancer treatment. Hum Vaccin Immunother. 2014;10(11):3332–3346. | ||
Nefedova Y, Fishman M, Sherman S, Wang X, Beg AA, Gabrilovich DI. Mechanism of all-trans retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Res. 2007;67(22):11021–11028. | ||
Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008;181(8):5791–5802. | ||
Alcorlo M, Martinez-Barricarte R, Fernandez FJ, et al. Unique structure of iC3b resolved at a resolution of 24 A by 3D-electron microscopy. Proc Natl Acad Sci U S A. 2011;108(32):13236–13240. | ||
Kullberg M, Martinson H, Mann K, Anchordoquy TJ. Complement C3 mediated targeting of liposomes to granulocytic myeloid derived suppressor cells. Nanomedicine. 2015;11(6):1355–1363. | ||
Fan Y, Moon JJ. Nanoparticle drug delivery systems designed to improve cancer vaccines and immunotherapy. Vaccines (Basel). 2015;3(3):662–685. | ||
Joshi MD, Unger WJ, Storm G, van Kooyk Y, Mastrobattista E. Targeting tumor antigens to dendritic cells using particulate carriers. J Control Release. 2012;161(1):25–37. | ||
Irache JM, Salman HH, Gamazo C, Espuelas S. Mannose-targeted systems for the delivery of therapeutics. Expert Opin Drug Deliv. 2008;5(6):703–724. | ||
Xu L, Anchordoquy T. Drug delivery trends in clinical trials and translational medicine: challenges and opportunities in the delivery of nucleic acid-based therapeutics. J Pharm Sci. 2011;100(1):38–52. | ||
Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I – molecular mechanisms of activation and regulation. Front Immunol. 2015;6:262. | ||
Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT. Complement system part II: role in immunity. Front Immunol. 2015;6:257. | ||
Kullberg M, Mann K, Anchordoquy TJ. Targeting Her-2+ breast cancer cells with bleomycin immunoliposomes linked to LLO. Mol Pharm. 2012;9(7):2000–2008. | ||
Law SK, Dodds AW. The internal thioester and the covalent binding properties of the complement proteins C3 and C4. Protein Sci. 1997;6(2):263–274. | ||
Hourcade DE. The role of properdin in the assembly of the alternative pathway C3 convertases of complement. J Biol Chem. 2006;281(4):2128–2132. | ||
Turner N, Sartain S, Moake J. Ultralarge von Willebrand factor-induced platelet clumping and activation of the alternative complement pathway in thrombotic thrombocytopenic purpura and the hemolytic-uremic syndromes. Hematol Oncol Clin North Am. 2015;29(3):509–524. | ||
Hess MW, Schwendinger MG, Eskelinen EL, et al. Tracing uptake of C3dg-conjugated antigen into B cells via complement receptor type 2 (CR2, CD21). Blood. 2000;95(8):2617–2623. | ||
Geyer MB, Brentjens RJ. Review: current clinical applications of chimeric antigen receptor (CAR) modified T cells. Cytotherapy. 2016;18(11):1393–1409. | ||
Soliman H. Immunotherapy strategies in the treatment of breast cancer. Cancer Control. 2013;20(1):17–21. | ||
Almand B, Resser JR, Lindman B, et al. Clinical significance of defective dendritic cell differentiation in cancer. Clin Cancer Res. 2000;6(5):1755–1766. | ||
Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother. 2009;58(1):49–59. | ||
Kusmartsev S, Cheng FD, Yu B, et al. All-trans-retinoic acid eliminates immature myeloid cells from tumor-bearing mice and improves the effect of vaccination. Cancer Research. 2003;63(15):4441–4449. | ||
Mirza N, Fishman M, Fricke I, et al. All-trans-retinoic acid improves differentiation of myeloid cells and immune response in cancer patients. Cancer Res. 2006;66(18):9299–9307. | ||
Ugel S, Delpozzo F, Desantis G, et al. Therapeutic targeting of myeloid-derived suppressor cells. Curr Opin Pharmacol. 2009;9(4):470–481. | ||
Candolfi M, Curtin JF, Yagiz K, et al. B cells are critical to T-cell-mediated antitumor immunity induced by a combined immune-stimulatory/conditionally cytotoxic therapy for glioblastoma. Neoplasia. 2011;13(10):947–960. | ||
Schultze JL, Michalak S, Seamon MJ, et al. CD40-activated human B cells: an alternative source of highly efficient antigen presenting cells to generate autologous antigen-specific T cells for adoptive immunotherapy. J Clin Invest. 1997;100(11):2757–2765. | ||
Thomas S, Prendergast GC. Cancer vaccines: a brief overview. Methods Mol Biol. 2016;1403:755–761. | ||
Busch DH, Frassle SP, Sommermeyer D, Buchholz VR, Riddell SR. Role of memory T cell subsets for adoptive immunotherapy. Semin Immunol. 2016;28(1):28–34. | ||
Xu Y, Dotti G. Selection bias: maintaining less-differentiated T cells for adoptive immunotherapy. J Clin Invest. 2016;126(1):35–37. | ||
Barrington RA, Schneider TJ, Pitcher LA, et al. Uncoupling CD21 and CD19 of the B-cell coreceptor. Proc Natl Acad Sci U S A. 2009;106(34):14490–14495. | ||
Lyubchenko T, Dal Porto JM, Holers VM, Cambier JC. Cutting edge: complement (C3d)-linked antigens break B cell anergy. J Immunol. 2007;179(5):2695–2699. | ||
Nielsen CH, Marquart HV, Prodinger WM, Leslie RG. CR2-mediated activation of the complement alternative pathway results in formation of membrane attack complexes on human B lymphocytes. Immunology. 2001;104(4):418–422. | ||
Niyonzima N, Halvorsen B, Sporsheim B, et al. Complement activation by cholesterol crystals triggers a subsequent cytokine response. Mol Immunol. 2017;84:43–50. | ||
Lindau D, Gielen P, Kroesen M, Wesseling P, Adema GJ. The immunosuppressive tumour network: myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology. 2013;138(2):105–115. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.