Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Competitive inhibition of survivin using a cell-permeable recombinant protein induces cancer-specific apoptosis in colon cancer model
Authors Roy K, Kanwar R, Krishnakumar S, Cheung CHA ![]() , Kanwar J
, Kanwar J
Received 7 September 2014
Accepted for publication 16 October 2014
Published 2 February 2015 Volume 2015:10(1) Pages 1019—1043
DOI https://doi.org/10.2147/IJN.S73916
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Kislay Roy,1 Rupinder K Kanwar,1 Subramanian Krishnakumar,2,3 Chun Hei Antonio Cheung,4 Jagat R Kanwar1
1Nanomedicine-Laboratory of Immunology and Molecular Biomedical Research (NLIMBR), Molecular and Medical Research (MMR) Strategic Research Centre, School of Medicine (SoM), Faculty of Health, Deakin University, Waurn Ponds, VIC, Australia; 2Department of Nanobiotechnology, 3Larsen & Toubro (L&T) Ocular Pathology Department, Vision Research Foundation, Kamalnayan Bajaj Institute for Research in Vision and Ophthalmology, Chennai, India; 4Department of Pharmacology, College of Medicine, National Cheng Kung University, Tainan, Taiwan, Republic of China
Abstract: Endogenous survivin expression has been related with cancer survival, drug resistance, and metastasis. Therapies targeting survivin have been shown to significantly inhibit tumor growth and recurrence. We found out that a cell-permeable dominant negative survivin (SurR9-C84A, referred to as SR9) competitively inhibited endogenous survivin and blocked the cell cycle at the G1/S phase. Nanoencapsulation in mucoadhesive chitosan nanoparticles (CHNP) substantially increased the bioavailability and serum stability of SR9. The mechanism of nanoparticle uptake was studied extensively in vitro and in ex vivo models. Our results confirmed that CHNP–SR9 protected primary cells from autophagy and successfully induced tumor-specific apoptosis via both extrinsic and intrinsic apoptotic pathways. CHNP–SR9 significantly reduced the tumor spheroid size (three-dimensional model) by nearly 7-fold. Effects of SR9 and CHNP–SR9 were studied on 35 key molecules involved in the apoptotic pathway. Highly significant (4.26-fold, P≤0.005) reduction in tumor volume was observed using an in vivo mouse xenograft colon cancer model. It was also observed that net apoptotic (6.25-fold, P≤0.005) and necrotic indexes (3.5-fold, P≤0.05) were comparatively higher in CHNP–SR9 when compared to void CHNP and CHNP–SR9 internalized more in cancer stem cells (4.5-fold, P≤0.005). We concluded that nanoformulation of SR9 did not reduce its therapeutic potential; however, nanoformulation provided SR9 with enhanced stability and better bioavailability. Our study presents a highly tumor-specific protein-based cancer therapy that has several advantages over the normally used chemotherapeutics.
Keywords: nanoparticle, chitosan, mucoadhesive, cytotoxicity, xenograft
Introduction
Bowel cancer, also known as colorectal cancer (CRC), is the third largest cause of cancer deaths. About 1.23 million people worldwide are estimated to be suffering from colon cancer. If detected at an early stage, most patients have a chance to recover from colon cancer; however, most cases are detected at a later stage and in Australia, out of the 274 new cases diagnosed every week, 78 people die from the disease.1 The treatment of CRC has not been very successful, especially in patients with metastatic tumors, despite understanding the CRC biology and availability of modern targeted therapeutics.2
Survivin belongs to the inhibitor of apoptosis (IAP) family of proteins encoded by the baculovirus IAP repeat (BIR) C5 gene that plays a key role in cell division and apoptosis.3,4 It is expressed during human embryonic and fetal development but is absent in most adult tissues.5 Survivin is present on β-tubulin during cell division and causes a mitotic progression once overexpressed in tumors of adults.6 Survivin expression has been observed to be 10-fold higher in the G2/M phase when compared to the G2 or S phase.7 Apoptosis is inhibited by the BIR domain of survivin by interfering with caspase-3 and caspase-7.8 It inhibits caspase-3, which is essential for suppression of apoptosis, and survivin’s affinity to associate with caspase-9 and second mitochondria-derived activator of caspases (Smac)/DIABLO9 indicates that it inhibits apoptosis via the intrinsic pathway by interfering with the post-mitochondrial events.10 It is well established that endogenous survivin is highly over-expressed in most cancer types including colon,11 liver,12 skin,13 prostate,14 breast,15 ovarian,16 lung,17 pancreas,18 and neuronal19 cancers. Thus, survivin has proven to be the most important target for cancer cells (Figure 1), and various studies have targeted survivin in order to combat cancer.20–22
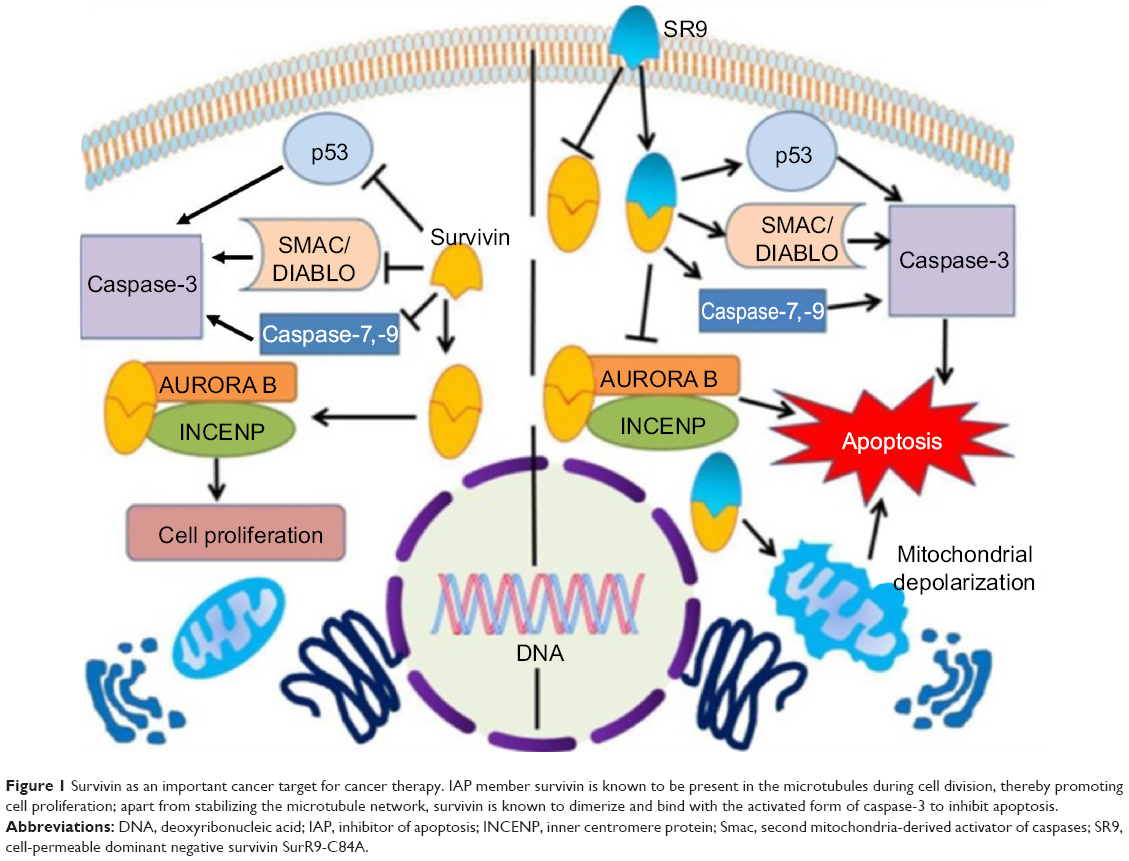 | Figure 1 Survivin as an important cancer target for cancer therapy. IAP member survivin is known to be present in the microtubules during cell division, thereby promoting cell proliferation; apart from stabilizing the microtubule network, survivin is known to dimerize and bind with the activated form of caspase-3 to inhibit apoptosis. |
Previously, we injected tumors from mouse EL-4 thymic lymphoma with plasmids encoding dNSurC84A and the T-cell costimulator B7-1 and observed that the gene therapy by dNSurC84A plasmids suppressed survivin expression, rendering large tumors susceptible to B7-1-mediated immunotherapy.23 By incorporating a Cysteine to Alanine mutation at the 84th position a mutant-type dominant negative protein was formed (SurR9-C84A, hereafter referred to as SR9).24 The apoptotic effects of SR9 have been evaluated in prostate cancer cells where it sensitized the cells to tumor necrosis factor alpha (TNF-α) and induced apoptosis.25 Recently, we have shown that SR9 displays dual actions: it is cytotoxic to cancerous cells and helps in proliferation of normal non-cancerous cells. This is because tumor cells have a high endogenous survivin pool, and treatment with SR9 leads to forceful survivin expression.26
Chitosan obtained from chitin is a naturally occurring muco-polysaccharide present in the exoskeleton of crustaceans.27 It is biodegradable, thus ensuring its safe administration and degradation once applied and absorbed by cells.28 Ionotropic crosslinking or interpolymer linking of chitosan with sodium tripolyphosphate (STPP) was used more recently for formation of chitosan nanoparticles (CHNP).29 The mechanism of interaction of chitosan with human cells has been studied elaborately, and it was proposed that chitosan enters the target cells by a combination of bioadhesion and a transient widening of tight junctions.30 In Caco-2 cells, it was observed that chitosan molecules were able to penetrate through the tight junctions between the epithelial cells to facilitate paracellular transport of large hydrophilic drugs.31
In this article, we have evaluated the anti-cancer efficacy of dominant negative survivin for the first time in nano-free as well nano-encapsulated form using CHNP in two-dimensional (monolayer), and three-dimensional (3D) (tumor spheroid) models, along with a mouse xenograft colon cancer model.
Materials and methods
Cell lines and cell culture conditions
The Caco-2, SW480, FHs 74 Int, and MCF 10A cell lines were obtained from the American type culture collection (ATCC) and were grown in tissue culture flasks using Dulbecco’s Modified Eagle’s Medium or minimum essential medium containing high glucose, L-glutamine, and sodium pyruvate (Thermo Fisher Scientific, Waltham, MA, USA), supplemented with 20% heat-inactivated fetal bovine serum (FBS) (Interpath, Heidelberg West, VIC, Australia Penicillin/streptomycin was added at a final concentration of 0.1 mg/mL to prevent growth of microorganisms, and the culture was maintained at 37°C in 5% CO2 to ensure a saturated humid atmosphere. Culture media were changed every 2–3 days.
Confocal microscopy
Cells were seeded in 8-well culture slides, and once confluent, the cells were treated with respective treatments for the fixed time interval and fixed using 4% paraformaldehyde (PF) for 20 minutes at 37°C. Cells were permeabilized using 0.01% Triton-X100 for 2 minutes on ice. Cells were then blocked with 3% bovine serum albumin for 30 minutes. Following blocking, the cells were washed and incubated with primary antibody (1:100) for 1 hour at 37°C. After washing with 1× phosphate-buffered saline (PBS) three times, the cells were incubated with fluorescein isothiocyanate/tetramethylrhodamine isothiocyanate conjugated secondary antibody (1:100) for 1 hour at 37°C in the dark. The cells were washed and mounting media with 4′,6-diamidino-2-phenylindole was added to the slide. The slide was analyzed with a Leica TCS SP5 laser immunoconfocal microscope.
Flow cytometric analysis
Cells were seeded in 6-well plates, and once confluent, they were treated with respective treatments for a fixed time interval, trypsinized, and fixed using 4% PF for 20 minutes at 37°C. Cells were permeabilized using 0.01% Triton-X100 for 5 minutes. Non-permeabilized cells were maintained as control. Cells were further blocked with 3% BSA for 30 minutes. The cells were washed and incubated with primary antibody (1:100) for 1 hour at 37°C. After washing with 1× PBS thrice, the cells were incubated with secondary fluorescein isothiocyanate/tetramethylrhodamine isothiocyanate conjugated antibody (1:100) for 1 hour in the dark. The cells were then washed and acquired using BD FACSCanto™ II (BD Biosciences, San Jose, CA, USA).
Western blotting
Approximately 106 cells were seeded in 6-well plates and treated with desired treatments for the specific time interval. The lysates were collected and run on a 10%–12.5% gel. The proteins were then transferred from the gel onto the polyvinylidene difluoride membrane using Bio-Rad (Bio-Rad Laboratories Inc., Hercules, CA, USA) Trans-Blot® Turbo™ Transfer System. The membrane was blocked using 2% skimmed milk for 1 hour following washes with Tris-buffer saline with Tween 20 and Tris-buffer saline (three times each). After washing, the membrane was incubated with primary antibody for 1 hour at 37°C and washed again, after which it was incubated with horseradish peroxidase conjugated secondary antibody (1:40,000). The washing steps were repeated, and the membrane was developed using horseradish peroxidase substrates (GE Healthcare, Silverwater, NSW, Australia). The membrane was visualized using a Bio-Rad ChemiDoc™ XRS camera.
Preparation and characterization of SR9-loaded CHNP
Varying concentrations of chitosan was dissolved in an acetic aqueous solution (pH 5). A small amount of glacial acetic acid was added to bring the pH down to 4.5 to dissolve the chitosan. The entrapment method was used for loading the protein, and SR9 was added drop-wise in a known concentration to the chitosan suspension under constant magnetic stirring at 4°C. For ionotropic gelation of chitosan, varying concentrations of STPP were added to the chitosan–protein suspension. The solution was stirred for 24 hours at various stirring speeds at 4°C. After 24 hours, the nanoparticle suspension was centrifuged at 2,000 rpm for 30 minutes at 4°C. The pellet obtained was frozen at −80°C overnight and further subjected to lyophilization (FreeZone 2.5 Liter Benchtop Freeze Dryer; Labconco, Kansas City, MO, USA) for a period of 24 hours. The nanoparticle powder obtained after lyophilization was stored at 4°C.
Scanning electron microscopy (Supra 55vp; Zeiss, Oberkochen, Germany) was used to observe the surface morphology, size, and shape of the nanoparticles. Fourier transform infra-red spectroscopy and X-ray diffraction were performed to study the chemical nature and complex structure of both void and CHNP–SR9. Protein release with and stability of nanoparticles with change at different pH values was studied by suspending the nanoparticles at pH 2, 3, 4, 5, 6, 7, and 8 solutions. The protein release was calculated by Bradford Coomassie method at 0, 1, 2, 4, 6, 12, 24, 48, 72, and 96 hours.
Using the following equations, the percentage loading capacity and the percentage association efficiency for CHNP–SR9 was calculated:
|
|
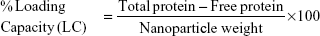
|
|
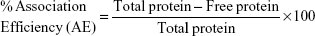
Nanoparticle uptake studies
Cells were seeded in 6-well plates and treated with rhodamine-labeled CHNP–SR9 and void CHNP at a concentration of 5 mg/mL and 10 mg/mL for 1 hour. After treatment, cells were washed; lysates were obtained and checked for optical diffraction (OD) at 530 nm. The nanoparticle uptake was calculated (in milligrams) using a standard graph plotted using OD of various concentrations of rhodamine-labeled CHNP–SR9 and void CHNP.
To understand the mechanism of protein release and transport of nanoparticles in a complete system, transepithelial electrical resistance (TEER) assay was performed. The TEER protocol was followed from our previous study.32 The ex vivo loop assay was performed by injecting rhodamine-labeled nanoparticles in rat intestinal loops (detailed methodology is described in the “Supplementary materials” section)
Gene expression analysis
RNA was isolated from cells treated with 50 μg/mL of SR9, CHNP–SR9 and equal weight of the void CHNP after 6 hours of treatment, using TRIzol® reagent (Thermo Fisher Scientific, Waltham, MA, USA). The isolated RNA was immediately converted to complementary DNA (cDNA). The synthesized cDNA was stored at −80°C and was further used for quantitative real time polymerase chain reaction (qRT-PCR) (detailed methodology can be found in the “Supplementary materials” section).
Cytotoxicity studies
Lactate dehydrogenase assay was performed for cytotoxicity analysis as per the manufacturer’s instructions (Invitrogen). The OD was measured at 492 nm, and percentage cytotoxicity was calculated.
Approximately 104 Caco-2 and 104 SW480 cells were plated in 6-well plates. Once confluent, the cells were treated with 50 and 100 μg/mL SR9, 50 and 100 μg/mL CHNP–SR9, and 50 μg/mL void CHNP for a period of 24 hours. The cells were then washed, trypsinized, and counted using a hemocytometer. Two hundred cells per treatment were plated out in fresh 6-well plates and left until 7 days to grow in normal growth media. The media was removed, and the cells were fixed using 4% PF for 20 minutes. The cells were then washed using PBS, and stained with 0.1% crystal violet stain for 3–5 minutes. The colony-forming units were washed again with PBS and counted, using a colony counter.
Apoptosis studies
The mitochondrial membrane potential assay kit (Sigma-Aldrich Co., St Louis, MO, USA) was used to calculate the mitochondrial potential of SR9 and CHNP–SR9-treated Caco-2 and SW480 cells. The apoptotic array kit was used as per the manufacturer’s instructions (R&D Systems, Inc., Minneapolis, MN, USA), and the array was developed using gel-dock (Bio-Rad).
One hundred μL of 0.1% agarose solution was added in each well of 96-well plates. Once the agar was solidified, 10³ Caco-2 cells/well were added to the plate and incubated for 7 days at 37°C with 5% CO2. After 7 days, cells had formed nearly uniform spheroids in all the wells; the spheroids were treated with two different concentrations of SR9 and CHNP-SR9 (50 μg/ml, 100 μg/ml) and 100 μg/ml of void CHNP for 24 hours. The surface area of tumor spheroids was measured using ImageJ software and plotted as a graph.
Cancer xenograft animal model of human colon cancer and in vivo studies
Caco-2 cells were used to establish the human colon cancer xenograft model by subcutaneously injecting 2×105 cells into the left flank of 5–6-week-old female nude mice (Mus musculus; BALB/c nu/nu). All the animal work was done according to institutions guidelines. The mice were fed with AIN93G diet supplemented with void CHNP or CHNP–SR9 (1.2% of SR9 w/w). Regular checks were conducted for any sign of physiological or physical distress thrice a week. Vernier calipers were used to measure the tumor size every week. At the end of the experimental period, the mice were euthanized. Biodistribution studies of targeted and non-targeted nanocarriers were conducted by injecting mice with fluorescence-tagged void CHNP and CHNP–SR9 and obtaining the fluorescent signals from the various parts of the mice such as the ovary, muscles, bones, eyes, blood, liver, spleen, kidney, lung, heart, brain, stomach, small intestine, large intestine, spinal cord, leg muscles, arm muscles, mesenteric lymph nodes, tumor site, and tissues around the tumor, using confocal microscopy.
After 7 days of treatment, the tumors were excised, sectioned and stained with annexin V (apoptosis marker), tunnel stain (apoptosis marker), and propidium iodide (necrosis marker). The respective indices were measured using a 40× objective from three tumor sections from the regions of tumor that consisted primarily of healthy proliferating cells, mainly found at the margins of the tumor. The necrotic index was measured by counting the cells present in the center of each tumor. For each tumor specimen, five to ten images were analyzed and the average cells positive for annexin V, tunnel stain, and propidium iodide were calculated. Only the cancer stem cells which were double positive for both CD44 and CD133 (cancer stem cell markers) were taken into account for calculating the number of cells that showed uptake of the nanocarriers.
Statistical analysis
All the results provided in the present study are expressed as the mean values ± standard error of at least three experiments, unless otherwise stated. The Student’s -test was used for evaluating the level of significance for statistical analysis of the numerical data. A value of <0.05 denotes statistical significance, <0.005 denotes a high level of statistical significance, and <0.0005 denotes a very high level of statistical significance.
Results
Dominant negative protein inhibits survivin expression
Purified form of SR9 was obtained using affinity chromatography, as shown in sodium dodecyl sulfate polyacrylamide gel electrophoresis and Western blotting (Figure S1). The confocal images revealed that the Texas Red®-SR9 was internalized in Caco-2 cells within 30 minutes of treatment (Figure 2A). The images obtained from Caco-2, HepG2, and MDA-MB231 cell lines showed a high level of colocalization (yellow color) and interaction between the two proteins (wild-type survivin and SR9) (Figure 2B). Flow cytometry analysis revealed that the maximum uptake of SR9 was observed in mimosine-fixed cells in the G1/S phase (Figure 2C). Confocal microscopy also confirmed that maximum co-localization of SR9 with β-tubulin was observed in mimosine (the G1/S phase)-fixed cells (Figure 2D). Western blotting results showed that even though 50 μg/mL of SR9 failed to reduce the survivin expression in 1 hour, 100 μg/mL of SR9 led to a substantial decrease in the expression of wild-type (WT) survivin in Caco-2 cells along with an increase in Bax, caspase-9, and caspase-3 (Figure 2E). Bcl-2 was found to be downregulated with both 50 and 100 μg/mL of SR9.
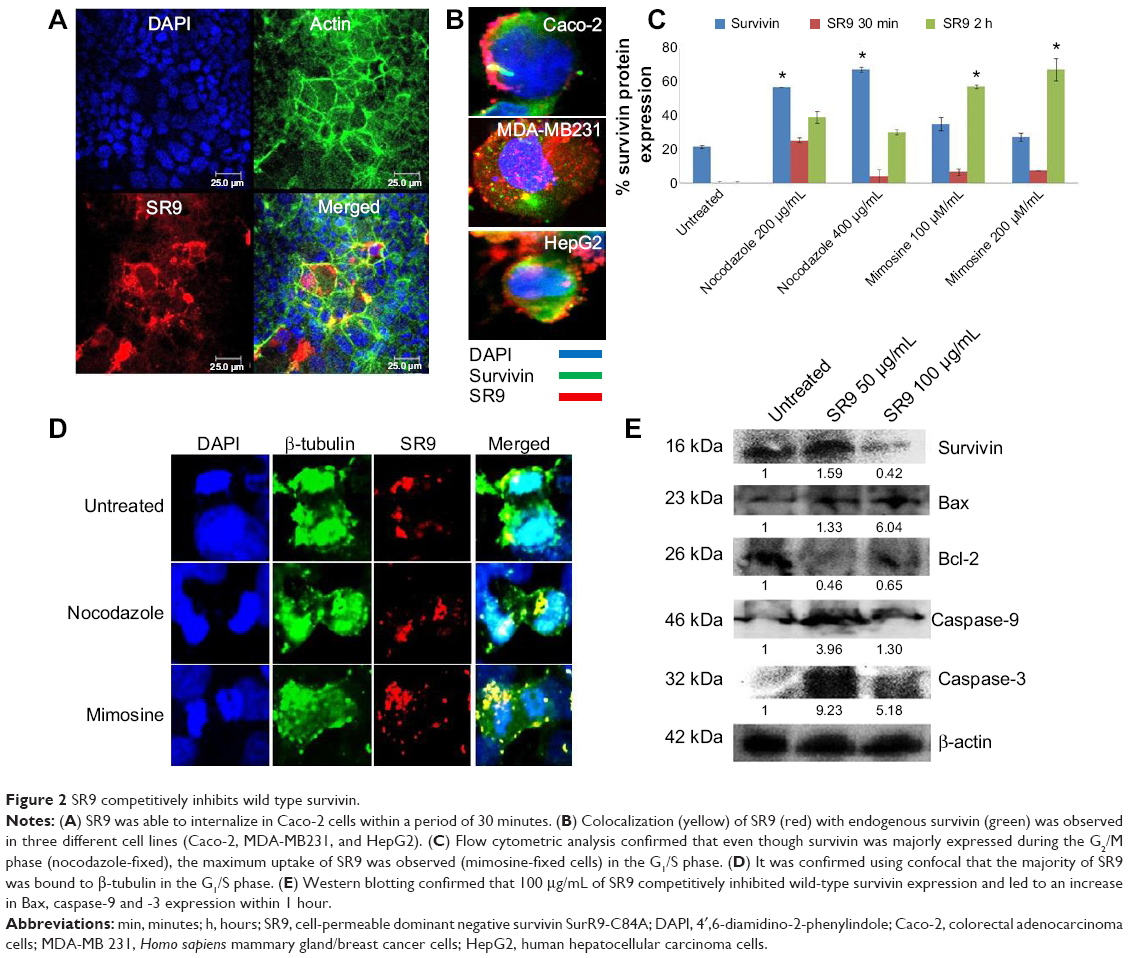 | Figure 2 SR9 competitively inhibits wild type survivin. |
Preparation and characterization of SR9-loaded CHNP
The SR9 was encapsulated in low-molecular-weight CHNP using the ionotropic gelation procedure. The scanning electron microscopy images confirmed uniformity in shape and size of the synthesized CHNP (Figure 3A). Western blotting confirmed that SR9 was degraded in the presence of 1% FBS within 2 hours, whereas nano-encapsulated SR9 (CHNP–SR9) was stable in 1% FBS for over a 24-hour period (Figure 3B). It was observed from the graph that the maximum protein release from the CHNP–SR9 was in between the 4–12 hour interval at pH 4 (Figure 3C). The percentage loading capacity for CHNP–SR9 was calculated to be 15.36%, whereas the percentage association efficiency was found to be 92.192%. It was also observed that the Fourier transform infra-red spectroscopy spectra of void CHNP were almost similar to that of chitosan powder, whereas there were significant differences in the spectra of CHNP–SR9 nanoparticles as expected, due to binding of the protein (Figure 3D). X-ray diffraction analysis showed the characteristic peaks of chitosan powder at 10° (2θ) and at 20° (2θ). Decreases in the peak intensities was observed in the case of void and CHNP-SR9 nanoparticles, which was due to the cross-linking of CHNP–SR9 with STPP and encapsulation of protein (Figure 3E). The differential scanning colorimetry was also used to characterize the nanoparticles (Figure S2).
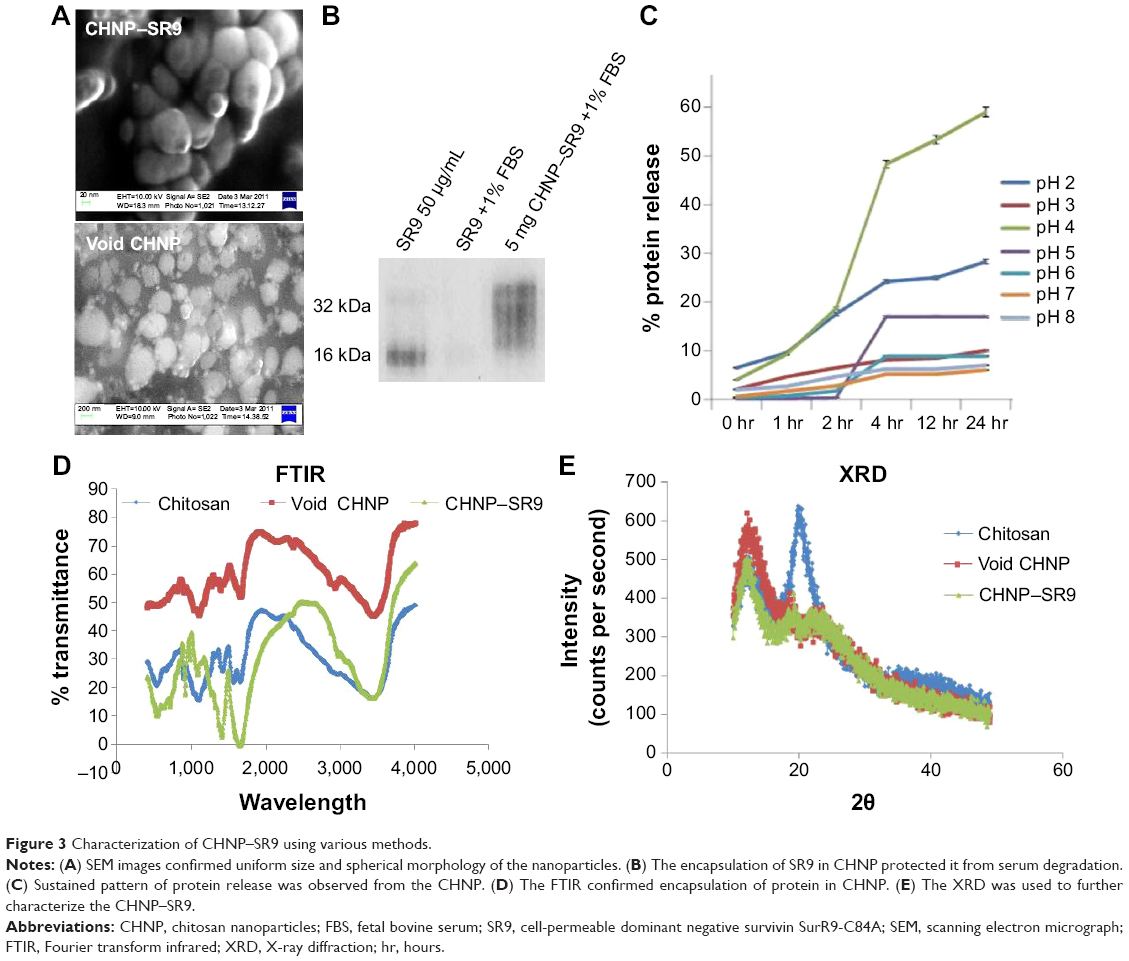 | Figure 3 Characterization of CHNP–SR9 using various methods. |
Nanoformulated-SR9 internalized within 2 hours using mucin-1 (Muc-1) receptors
The rhodamine-labeled SR9-loaded CHNP (red color) were best internalized in Caco-2 cells (blue color) in 2 hours (Figure 4A). A high expression of Muc-1 was seen in the case of both Caco-2 and SW480 (Figure S3), and a clear interaction between the Muc-1 (green color) and CHNP–SR9 (red color) was observed in the confocal images in both the cell lines (Figure 4B). It was observed that both Caco-2 (0.5 mg/mL) and SW480 cells showed (0.74 mg/mL) significantly (≤0.05; 2.63-fold and 3.89-fold, respectively) higher uptake of CHNP–SR9 when compared to FHs-74 Int cells (0.19 mg/mL) (Figures 4C and S4). The TEER values of CHNP–SR9, on the other hand, showed a significant time-dependent decrease when compared to the untreated cells and the void CHNP treated cells (Figure 4D). It was observed that the maximum absorption of CHNP–SR9 took place in the jejunum at 24 hours (Figure 4E). It was clear that the CHNP–SR9 did not cause any damage to the intestinal tissues and was efficiently absorbed within 2 hours (Figures S5 and 4F).
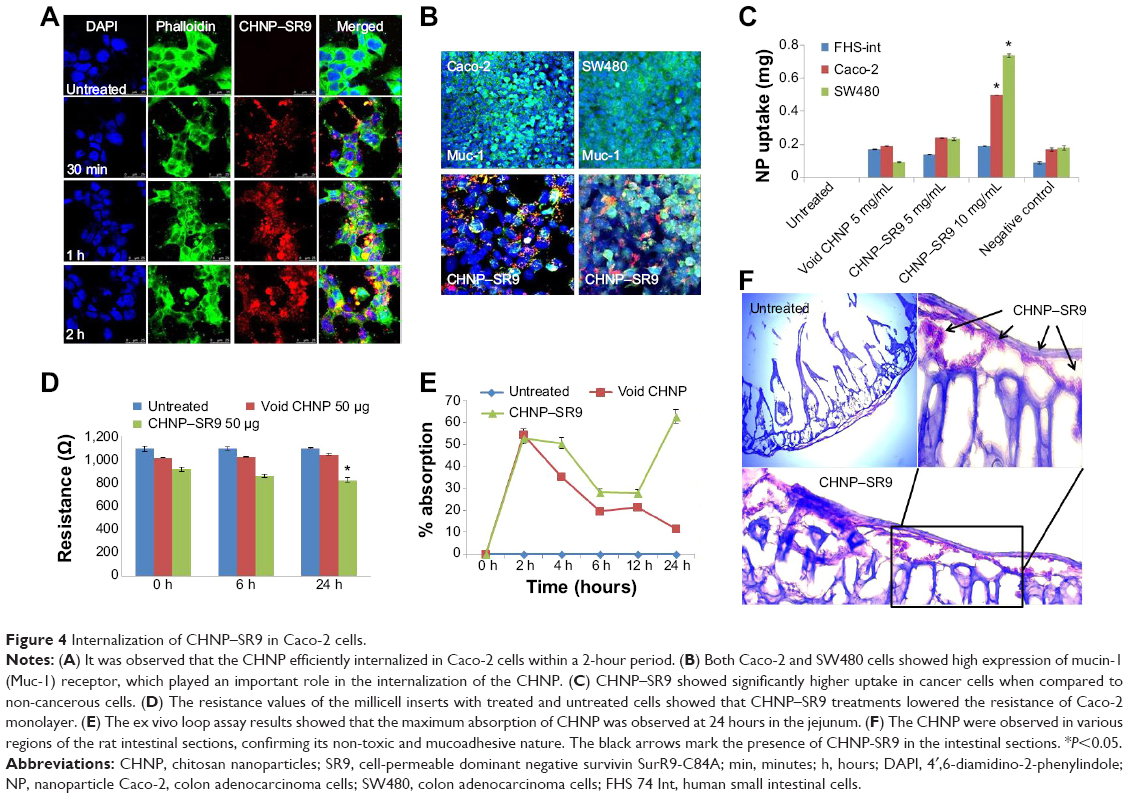 | Figure 4 Internalization of CHNP–SR9 in Caco-2 cells. |
Cytotoxicity studies using SR9 and CHNP–SR9
It was observed that the expression of pro-apoptotic molecules (Bad and Bax) was upregulated with SR9 and CHNP–SR9 treatments. FAS, TRAIL, caspases-3, -7, -8, and -9, and cytochrome-C were significantly upregulated by both SR9 and CHNP–SR9; however, pro-caspase 7 was only upregulated by CHNP–SR9 (Figure 5A and B; Figure S6). In the case of FHs 74 Int cells treated with SR9, a dose-dependent decrease in the protein expression of LC3-II (autophagosomal marker) was observed. In the case of Caco-2, the SR9 treatments significantly reduced the expression of LC3-II (Figure 5C). The level of lactose dehydrogenase released in primary cells FHs 74 Int and MCF-10A was nearly 2–3-folds less than in the Caco-2 and SW480 cells (Figure 5D). CyQUANT® (Thermo Fisher Scientific, Waltham, MA, USA). cell proliferation studies were also conducted in order to determine effects of SR9 on cancer and normal cell proliferation (Figure S7).
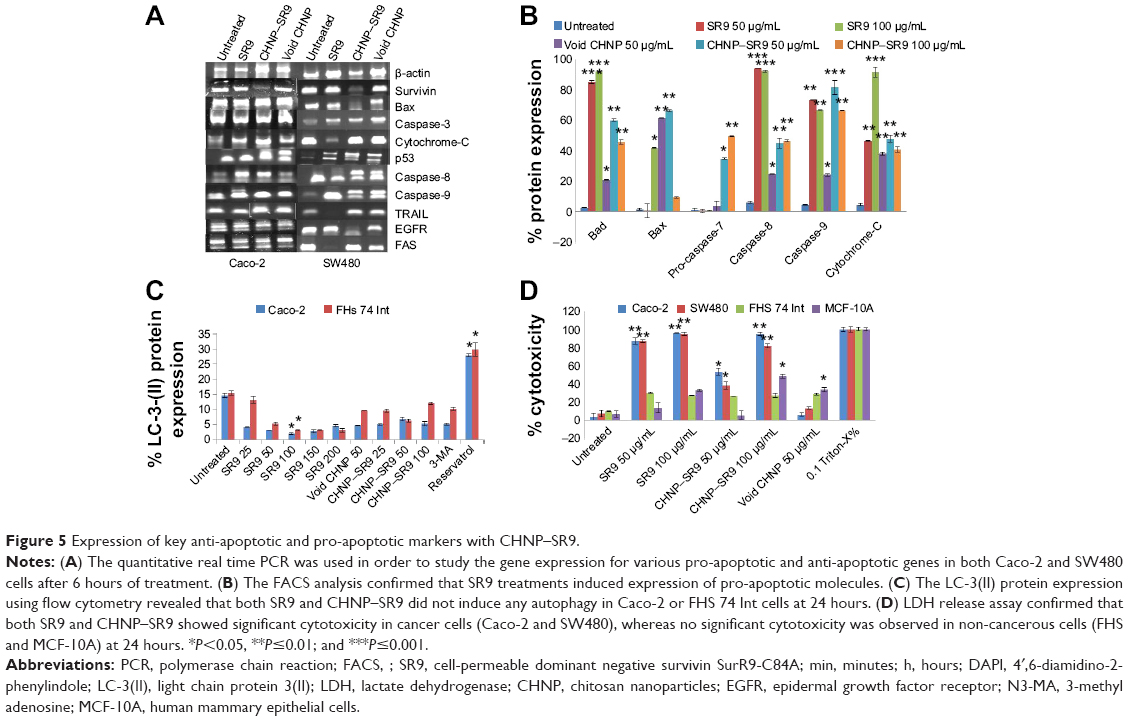 | Figure 5 Expression of key anti-apoptotic and pro-apoptotic markers with CHNP–SR9. |
CHNP–SR9 and SR9 induce apoptosis in colon cancer cells
More than 2-fold reduction in colony-forming ability was observed in both Caco-2 and SW480 cells with treatments of SR9 (50 μg/mL) (Figure S8). The CHNP–SR9, on the other hand, significantly reduced the clonogenic potential by 5-fold in Caco-2, and 3-fold in SW480 cells (Figure 6A). In healthy cells, the JC1 dye (mitochondrial membrane potential probe) accumulated and formed a red-colored complex in mitochondria, whereas in the apoptotic cells due to lowering of mitochondrial membrane potential, the JC1 was retained as a monomer-emitting green fluorescence (Figure S9), and was spread throughout the cells. It was evident that the cytochrome-C was released from mitochondria after treatment with SR9 and CHNP–SR9 nanoparticle treatments (Figure S10). Caspase-3 was upregulated (Figures S11 and S12) when the Caco-2 cells were treated with SR9, and an enhanced upregulation was observed with the higher-dose treatments. However, there was no caspase-3 upregulation observed with the treatments of void CHNP (Figure 6B). It was observed that Caco-2 cells were capable of forming a tumor spheroid (Figure S13) (three-dimensional model) on agar base (Figure 6C). On treatment with SR9, the tumor volume reduced significantly by 3-fold, thus confirming the anti-cancer property of SR9. CHNP–SR9 also showed a similar reduction in the tumor spheroid volume when compared to the void nanoparticles. The void CHNP also showed significant reduction in the tumor spheroid surface area, thus proving in vitro the anti-cancer activity of chitosan. The key molecules which showed significant upregulation in their expression due to treatments of SR9 were Bax, claspin, clusterin, phosphorylated p53 TNFRSF1A, cytochrome-C, caspase-3, Smac/Diablo, and TRAIL (Figure 6D). The molecules that showed increased expression post-treatment with CHNP–SR9 were Bax, claspin, cytochrome-C, caspase-3, Smac/Diablo, FADD, and FAS. The void nanoparticles did not cause any significant changes other than in the expression of caspase-3, TRAIL, and FAS. Other important molecules in the array (Figure S14A) were Heat shock proteins heme oxygenases (HO-1 and HO-2; Figure S14B), forms of p53 (Figure S14C), and anti-apoptotic molecules such as Bcl-2, Bcl-xL, HIF-1α, Livin, survivin, PON2, XIAP, and Catalase.
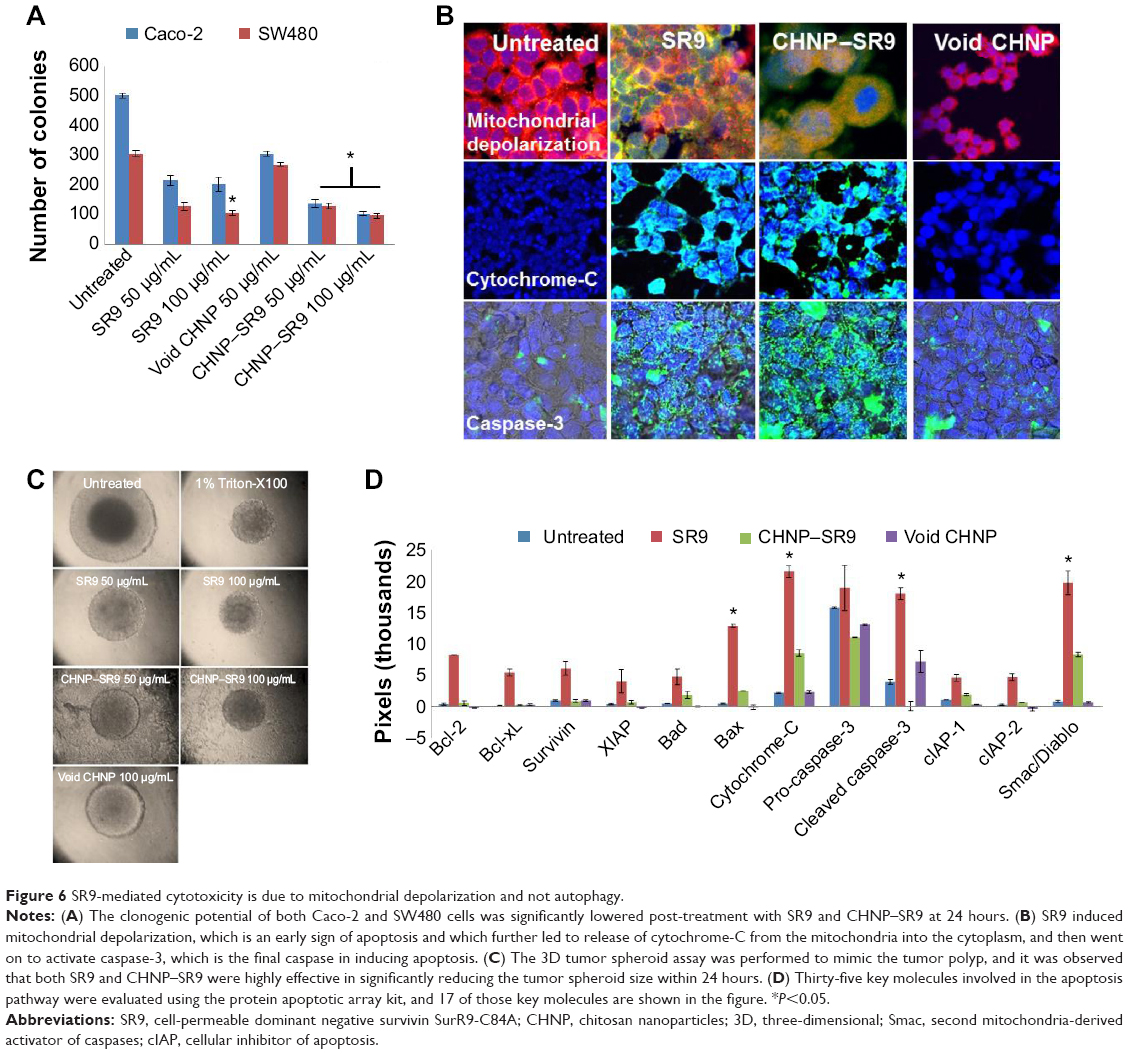 | Figure 6 SR9-mediated cytotoxicity is due to mitochondrial depolarization and not autophagy. |
In vivo anti-cancer efficacy of SR9
It was observed that the tumor regression was highly sustained in mice treated with CHNP–SR9 (4.26-fold reduction at the 70th day). The mice fed with the void nanocarrier-supplemented diet showed increased tumor volume with time (Figure 7A). The CHNP–SR9 showed significant (≤0.005) in vivo internalization in CD44- and CD133-positive cancer stem cells (4.5-fold) when compared to the void nanoparticles. The CHNP–SR9 also showed a significantly high apoptotic (6.25-fold, ≤0.005) and necrotic index (3.5-fold, ≤0.05) in the tumor cells when compared to void nanoparticles treatment (Figure 7B). The apoptotic index obtained by both annexin V and tunnel staining showed a similar pattern with treatments. The biodistribution studies confirmed that CHNP–SR9 was majorly present in the tumor when compared to other organs of the mice, and showed a comparatively better tumor-specific internalization when compared to void nanoparticles (Figure 7C), showing significantly (≤0.005) higher uptake in tumor when compared to other organs of the mice.
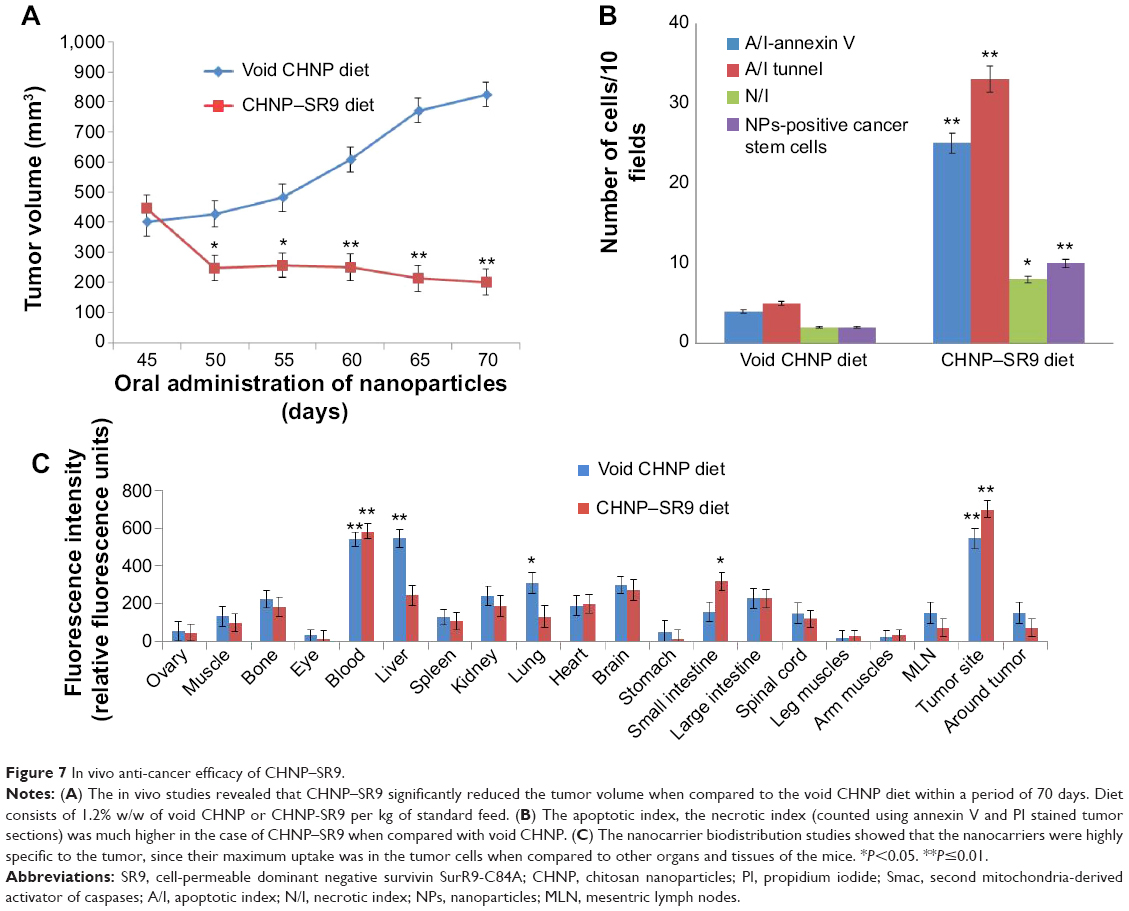 | Figure 7 In vivo anti-cancer efficacy of CHNP–SR9. |
Discussion
Studies have shown that it is possible to prevent the dimerization of survivin by inducing a point mutation in the BIR domain at the 84th position (Cysteine to Alanine) that can produce a dominant negative form of survivin (SR9) that interferes with the anti-apoptotic function of WT survivin.23 Although studies have been conducted with the C84A form of dominant negative survivin in both gene therapy23 and protein therapy,25 no interaction between the two forms of protein has been demonstrated. This is the first report that has showed interaction of dominant negative survivin protein SR9 with WT survivin in colon, liver, and breast cancer cells, and was further confirmed by Western blotting. It has been proposed that this protein competitively inhibits the WT survivin by dimerizing with WT survivin, inhibiting the ability of WT survivin to interact with the activated form of caspases. It has been reported that a potential dimerization of survivin-C84A with WT survivin promoted accelerated degradation of the complex, thus reducing survivin levels below a crucial anti-apoptotic threshold.33 The half-life of survivin was estimated in 293 cells by cycloheximide, and it was reported that half the quantity of protein was degraded within 30 minutes.33
Therefore, due to poor serum stability and low half-life of SR9, it was essential to ensure its protection and increase its bioavailability. Owing to its mucoadhesive nature, chitosan was the apparent choice for targeting colorectal cancer. Apart from being non-toxic and biocompatible, chitosan is also biodegradable (in the intestine) due to the presence of pancreatic enzymes and the enzymes secreted by healthy bacteria in the gut.34 Chitosan reversibly opens the tight junctions in epithelial cells by interacting with the protein kinase C system, and internalizes via the paracellular transport.35 Several standard techniques have been established to synthesize chitosan/STPP particles, including by Bhumkar and Pokharkar,36 Wu et al,37 and Bowman and Leong.35
A study using ten patients suffering with colon cancer showed that all patients showed high expression of Muc-1 receptors.39 Our results confirmed that both SW480 and Caco-2 expressed high amounts of Muc-1 receptors, and a clear colocalization of SR9-loaded CHNP was observed with the Muc-1 receptors, proving the mucoadhesive nature of the particles. The confluent Caco-2 monolayer acts as the most suitable model for measuring intestinal permeability, because its polarity, tight junctions, and transport systems are the same as that of the human small intestine.40 Thus, the TEER assay was performed to mimic the intestinal epithelial barrier, in order to understand the transport of nanoparticles across it with respect to time. Another approach to study the drug and particle transport across the small intestine is the rat intestinal loop model.41 Therefore, the ex vivo loop assay using rat intestinal segments was performed to mimic the absorption of the nanoparticles by the intestinal cells.
A previous study has shown that YM155 (inhibitor of survivin) was capable of inducing both apoptosis as well as autophagy by conversion of cytosolic light chain protein (LC3-I) to LC3-II, which is autophagosome-associated.42 It has been reported that survivin plays an important role in control of autophagy.43 However, we observed that both SR9 and CHNP–SR9 failed to induce autophagy in Caco-2, whereas a dose-dependent decrease in expression of LC3-II was observed in FHs 74 Int cells, which confirmed that SR9 prevented autophagy in healthy intestinal cells. Using qRT-PCR, flow cytometry, and apoptotic array studies, the over-expression of FAS, TRAIL and caspase-8 were observed with treatments of CHNP–SR9. It is known that the stimulation of death receptors of TNF family, such as FAS and TRAIL, results in activation of caspase-8, which further propagates apoptosis by activating caspase-3, and this pathway is termed the extrinsic apoptotic pathway.44 Interestingly, the apoptotic array of expression analysis for 35 key proteins in the apoptotic pathway showed that along with DIABLO, Bax, cleaved caspase-3, and cytochrome-C were the essential proteins that were significantly upregulated with treatments of SR9 and CHNP–SR9. Therefore, our results confirmed that SR9 and CHNP–SR9 worked through both extrinsic and intrinsic apoptotic pathways to induce apoptosis in colon cancer cells (Figure 8).
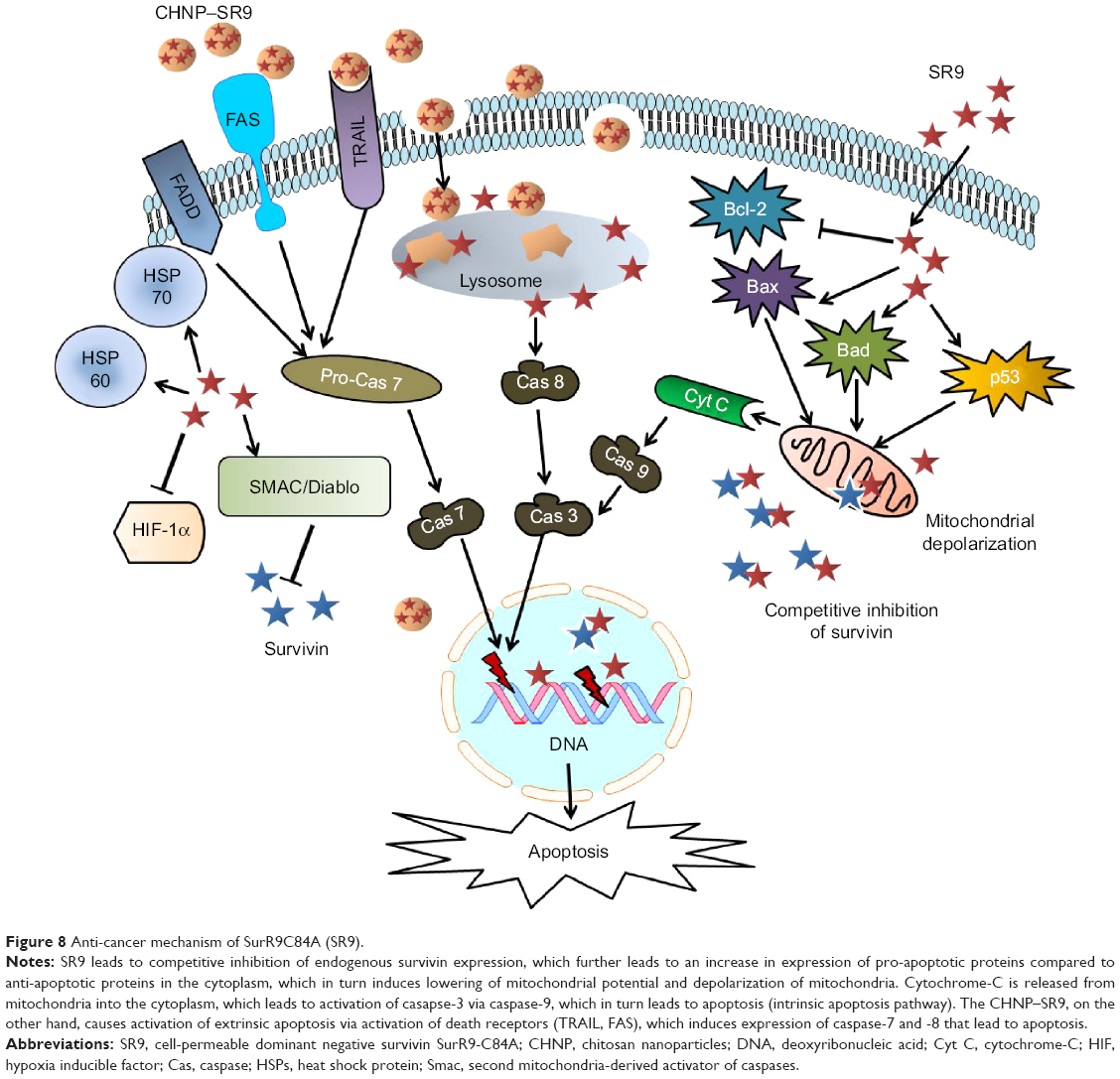 | Figure 8 Anti-cancer mechanism of SurR9C84A (SR9). |
The current in vivo studies also proved that the apoptotic index in CHNP–SR9-treated cells was comparatively higher when compared to void nanocarriers. The CHNP–SR9 was majorly taken up by the cancer stem cells, which overexpress survivin. Therefore, SR9 on its own is a cancer-specific approach and can be proven to specifically target cancer cells and cancer stem cells as they overexpress survivin.
Conclusion
Targeting survivin in cancer cells reduces the chances of non-specific cytotoxic effects of SR9, as survivin is widely expressed in most cancers and is scarcely present in healthy cells. Dominant negative protein isolated in our laboratory is a perfect example of a tumor-specific approach, as it protects the healthy cells and causes apoptosis in cancer cells. Nanoformulation of proteins that are susceptible to degradation is an efficient method to provide stability and increase their bioavailability. We have successfully shown that nano-formulated survivin mutant protein not only targets cancer cells but also has the potential to target cancer stem cells. However, further in vivo studies and pre-clinical studies need to be conducted to validate the therapeutic potential of SR9.
Acknowledgments
The authors would like to thank the Australia–India Strategic Research Fund (AISRF) and the National Health and Medical Research Council (NHMRC) for financial support.
Disclosure
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript, apart from those disclosed in the “Acknowledgments” section. No writing assistance was utilized in the production of this manuscript. The authors report no other conflicts of interest in this work.
References
Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2006. CA Cancer J. Clinic. 2006;56:106–130. | ||
Kanwar JR, Roy K, Kanwar RK. Colon cancer genetics, nanotherapy and individualized chemotherapy. In Zhou S, Sneed KB, Whelan G, Pathak YV, editors. Clinical and translational pharmacogenomics. Boca Raton, Florida: CRC Press; 2014. | ||
Alteri DC. Survivin, cancer networks and pathway-directed drug discovery. Nat Rev Cancer. 2008;8(1):61–70. | ||
Kanwar JR, Kamalapuram SK, Kanwar RK. Survivin signaling in clinical oncology: a multifaceted dragon. Med Res Rev. 2013;33(4):765–789. | ||
Konno R, Yamakawa H, Utsunomiya H, Ito K, Sato S, Yajima A. Expression of survivin and Bcl-2 in the normal human endometrium. Mol Hum Reprod. 2000;6(6):529–534. | ||
Li F, Ambrosini G, Chu EY, et al. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature. 1998;396(6711):580–584. | ||
Altieri DC, Marchisio C. Surviving apoptosis: an interloper between cell death and cell proliferation. Lab Invest. 1999;79(11):1327–1333. | ||
Verdecia MA, Huang H, Dutil E, Kaiser DA, Hunter T, Noel JP. Structure of the human anti-apoptotic protein survivin reveals a dimeric arrangement. Nat Struct Biol. 2000;7(7):602–608. | ||
Song Z, Yao X, Wu M. Direct interaction between smac/DIABLO is essential for anti-apoptotic activity of survivin during taxol-induced apoptosis. J Biol Chem. 2003;278(25):23130–23140. | ||
Tamm I, Wang Y, Sausville E, et al. IAP family protein survivin inhibits caspase activity and apoptosis induced by Fas (CD95), bax, caspases, and anti-cancer drugs. Cancer Res. 1998;58(23):5315–5320. | ||
Kim PJ, Plesica J, Clevers H, Feaaron ER, Altieri DC. Survivin and molecular pathogenesis of colorectal cancer. Lancet. 2003;362(9379):205–209. | ||
Fields AC, Cotsonis G, Sexton D, Santoianni R, Cohen C. Survivin expression in hepatocellular carcinoma: correlation with proliferation, prognostic parameters, and outcome. Mod Pathol. 2004;17(11):1378–1385. | ||
Grossman D, Kim PJ, Schechner JS, Altieri DC. Inhibition of melanoma tumor growth in vivo by survivin targeting. Proc Natl Acad Sci USA. 2001;98(2):635–640. | ||
Zhang M, Latham DE, Delaney MA, Chakravarti A. Survivin mediates resistance to antiandrogen therapy in prostate cancer. Oncogene. 2005;24(15):2474–2482. | ||
Vegran F, Boidot R, Bonnetain F, Cadouot M, Chevrier S, Lizaed-Nacol S. Apoptosis gene signature of survivin and its splice variant expression in breast carcinoma. Endocr Relat Cancer. 2011;18(6):783–792. | ||
Liguang Z, Peishu L, Honglua M, et al. Survivin expression in ovarian cancer. Exp Oncol. 2007;29(2):121–125. | ||
Olie RA, Simões-Wüst PS, Baumann B, et al. A novel antisense oligonucleotide targeting survivin expression induces apoptosis and sensitizes lung cancer cells to chemotherapy. Cancer Res. 2000;60(11):2805–2809. | ||
Lee MA, Park GS, Lee HJ, et al. Survivin expression and its clinical significance in pancreatic cancer. BMC cancer. 2005;5:127. | ||
Shankar SL, Mani S, O’Guin KN, Kandimalla ER, Agrawal S, Shafit-Zagardo BS. Survivin inhibition induces human neuronal tumor cell death through caspase-independent and -dependent pathways. J Neurochem. 2001;79(2):426–436. | ||
Kanwar JR, Kamalapuram SK, Kanwar RK. Targeting survivin in cancer: the cell-signalling perspective. Drug Discov Today. 2011;16(11–12):485–494. | ||
Kanwar RK, Cheung CH, Chang JY, Kanwar JR. Recent advances in anti-survivin treatments for cancer. Curr Med Chem. 2010;17(15):1509–1515. | ||
Kanwar JR, Kamalapuram SK, Kanwar RK. Targeting survivin in cancer: patent review. Expert Opin Ther Pat. 2010;20(12):1723–1737. | ||
Kanwar JR, Shen WP, Kanwar RK, Berg RW, Krissansen GW. Effects of survivin antagonists on growth of established tumors and B7-1 immunogene therapy. J Natl Cancer Inst. 2001;93(20):1541–1552. | ||
Baratchi S, Kanwar RK, Cheung CH, Kanwar JR. Proliferative and protective effects of SurR9-c84A on differentiated neural cells. J Neuroimmunol. 2010;227(1–2):120–132. | ||
Cheung CHA, Sun X, Kanwar JR, Bai JZ, Cheng L, Krissansen GW. A cell-permeable dominant-negative survivin protein induces apoptosis and sensitizes prostate cancer cells to TNF-α therapy. Cancer Cell Int. 2010;10(36):1–11. | ||
Sriramoju B, Kanwar RK, Kanwar JR. Nanoformulated cell-penetrating survivin mutant and its dual actions. Int J Nanomedicine. 2014;9:3279–3298. | ||
Ravi Kumar MN. A review of chitin and chitosan applications. React Funct Polymers. 2000;46(1):1–27. | ||
Pangburn SH, Trescony PV, Heller J. Lysozyme degradation of partially deacetylated chitin, its films and hydrogels. Biomaterials. 1982;3(2):105–108. | ||
Mi FL, Shyu SS, Wong TB, Jang SF, Lee ST, Lu KT. Chitosan–polyelectrolyte complexation for the preparation of gel beads and controlled release of anticancer drug. II. Effect of pH-dependant ionic crosslinking or interpolymer complex using tripolyphosphate or polyphosphate as reagent. J Appl Poly Sci. 1999;74(5):1093–1107. | ||
Artursson P, Lindmark T, Davis SS, Illum L. Effect of chitosan on the permeability of monolayers of intestinal epithelial cells (Caco-2). Pharm Res. 1994;11(9):1358–1361. | ||
Schipper NG, Vârum KM, Stenberg P, Ocklind G, Lennernäs H, Artursson P. Chitosan as absorption enhancers of poorly absorbable drugs. 3: Influence of mucus as absorption enhancement. Eur J Pharm Sci. 1999;8(4):335–343. | ||
Kanwar JR, Kanwar RK. Gut health immunomodulatory and anti-inflammatory functions of gut enzyme digested high protein micro-nutrient dietary supplement-Enprocal. BMC Immunol. 2009;10(7):1–19. | ||
Zhao J, Tenev T, Martins LM, Downward J, Lemoine NR. The ubiquitin-proteasome pathway regulates survivin degradation in a cell cycle-dependent manner. J Cell Sci. 2000;113(Pt 23):4363–4371. | ||
Bowman K, Leong KW. Chitosan nanoparticles for oral drug and gene delivery. Int J Nanomedicine. 2006;1(2):117–128. | ||
Bhumkar DR, Joshi HM, Sastry M, Pokharkar VB. Chitosan reduced gold nanoparticles as novel carriers for transmucosal delivery of insulin. Pharm Res. 2007;24(8):1415–1426. | ||
Bhumkar DR, Pokharkar VB. Studies on effect of pH on cross-linking of chitosan with sodium tripolyphosphate: a technical note. AAPS Pharm Sci Tech. 2006;7(2):138–143. | ||
Wu Y, Yang W, Wang C, Hu J, Fu S. Chitosan nanoparticles as a novel delivery system for ammonium glycyrrhizinate. Int J Pharmaceutics. 2005;295(1):235–245. | ||
Ji J, Hao S, Wu D, Huang R, Xu Y. Preparation, characterization and in vitro release of chitosan nanoparticles loaded with gentamicin and salicylic acid. Carbohydrate Polym. 2011;85:803–808. | ||
Kotera Y, Fontenot JD, Pecher G, Metzgar RS, Finn OJ. Humoral immunity against a tandem repeat epitope of human mucin MUC-1 in sera from breast, pancreatic, and colon cancer patients. Cancer Res. 1994;54(11):2856–2860. | ||
Van Breemen RB, Li Y. Caco-2 cell permeability assays to measure drug absorption. Expert Opin Drug Metab Toxicol. 2005;1(2):175–185. | ||
Lueßen HL, Rentel CO, Kotzé AF, et al. Mucoadhesive polymers in peroral peptide drug delivery. IV. Polycarbophil and chitosan are potent enhancers of peptide transport across intestinal mucosae in vitro. J Contr Rel. 1997;45(1):15–23. | ||
Wang Q, Chen Z, Diao X, Huang S. Induction of autophagy-dependent apoptosis by the survivin suppressant YM155 in prostate cancer cells. Cancer Lett. 2011;302(1):29–36. | ||
Roca H, Varsos Z, Pienta KJ. CCL2 protects prostate cancer PC3 cells from autophagic death via phosphatidylinositol 3-kinase/AKT-dependent survivin up-regulation. J Biol Chem. 2008;283(36):25057–25073. | ||
Walczak H, Krammer PH. The CD95 (APO-1/Fas) and the TRAIL (APO-2L) apoptosis systems. Exp Cell Res. 2000;256(1):58–66. |
Supplementary materials
Trans-epithelial endodermal resistance and nanoparticle transport
Caco-2 cells were seeded in trans-well plates and were allowed to grow and differentiate for a period of 21 days. The media was changed every third day in the plates. Once the cell monolayer was found to be intact in all spaces, the cells were treated with rhodamine-labeled chitosan nanoparticles/cell-permeable dominant negative survivin SurR9-C84A (CHNP–SR9) and void CHNP (an untreated well was maintained as a control). The resistance of each trans-well was measured using Millicell® cell culture inserts (Merck Millipore, Billerica, MA, USA) without disturbing the cell monolayer. Equal amount of media was collected from the base of the trans-well at regular time intervals of 0 minutes, 30 minutes, and 1, 4, 6, 24, 48, 72, and 96 hours, and replaced with fresh media. The optical density (OD) of the collected sample was measured and a graph was plotted between OD and time. The OD of rhodamine-labeled nanoparticles was measured and used as a control.
The trans-well plate was then washed twice with phosphate-buffered saline and fixed with 4% paraformaldehyde. After washing again with phosphate-buffered saline, the trans-well was dehydrated in 70%, 90%, and 100% ethanol for 2 minutes each. The membrane was then cut from the trans-well, air dried on a scanning electron microscopy sample stub, and was coated with gold and viewed under scanning electron microscope.
Gene expression analysis
A ready-to-use TRIzol® reagent (Thermo Fisher Scientific, Waltham, MA, USA) was used to isolate the total ribonucleic acid (RNA) from both Caco-2 and SW480 cells plated in 6-well plates, treated with 50 μg/mL of SR9, CHNP–SR9, and equal weight of the void CHNP. Since the majority of the treated cells failed to survive after 12 and 24 hours, the RNA extraction had to be done after 6 hours of treatment. The concentration of RNA was determined by taking absorbance readings at 260 nm, using a Corona SH-1000 lab absorbance microplate reader and further multiplying the absorbance values by 40 μg/mL. The isolated RNA had to be immediately subjected to complementary deoxyribonucleic acid (cDNA) synthesis. The cDNA thus synthesized was stored at −80°C and was further used for quantitative real time polymerase chain reaction (qRT-PCR). The purpose of qRT-PCR is to detect the amplified DNA during a real-time reaction, thus enabling its detection and quantification simultaneously. Depending on the annealing temperatures of each primer used, a suitable cycle was created in the qRT-PCR software, iQ5 (Bio-Rad Laboratories Inc., Hercules, CA, USA). The qRT-PCR reaction mixture (7.5 μL SYBR® green premix (Bio-Rad), 0.4 μL forward primer, 0.4 μL reverse primer, 1 μL cDNA templet per sample, and RNase-free water to make the volume of reaction mixture up to 15 μL) was prepared in standard Bio-Rad PCR plates and further incubated in iQ5 optical system software for quantitative expression analysis. The cycles consisted of 95°C for 5 minutes followed by 60 repeats of 95°C for 30 seconds, then incubated at respective primer annealing temperatures for 45 seconds and at 72°C for 45 seconds. The iQ5 can detect and quantify the amplified DNA in each sample during real time followed by an extension of 10 minutes at 72°C. The 2-^^ct (fold change gene expression) values were calculated by subtracting treatment values from untreated samples and comparing them with control values, and the graph was plotted with these values. The amplified DNA of each sample was run on 1% agarose gel, and the values from the iQ5 were further analyzed for gene expression. The experiment was done thrice to obtain consistency.
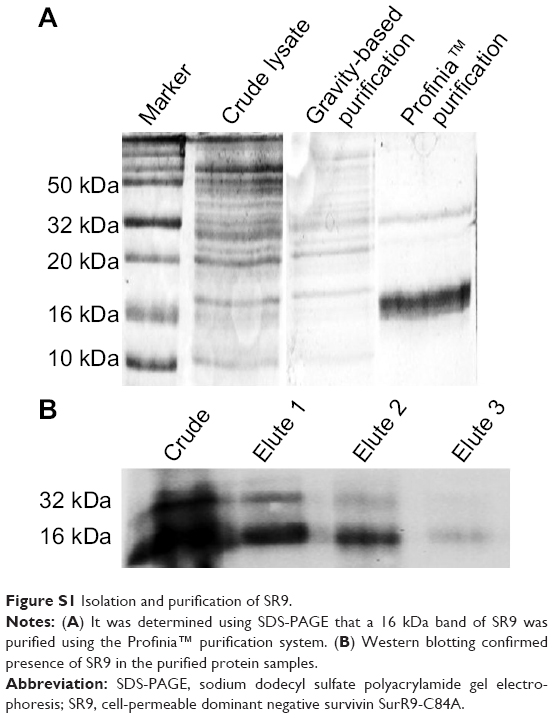 | Figure S1 Isolation and purification of SR9. |
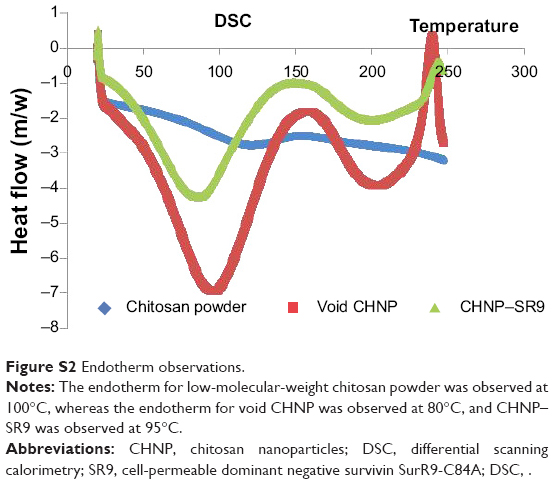 | Figure S2 Endotherm observations. |
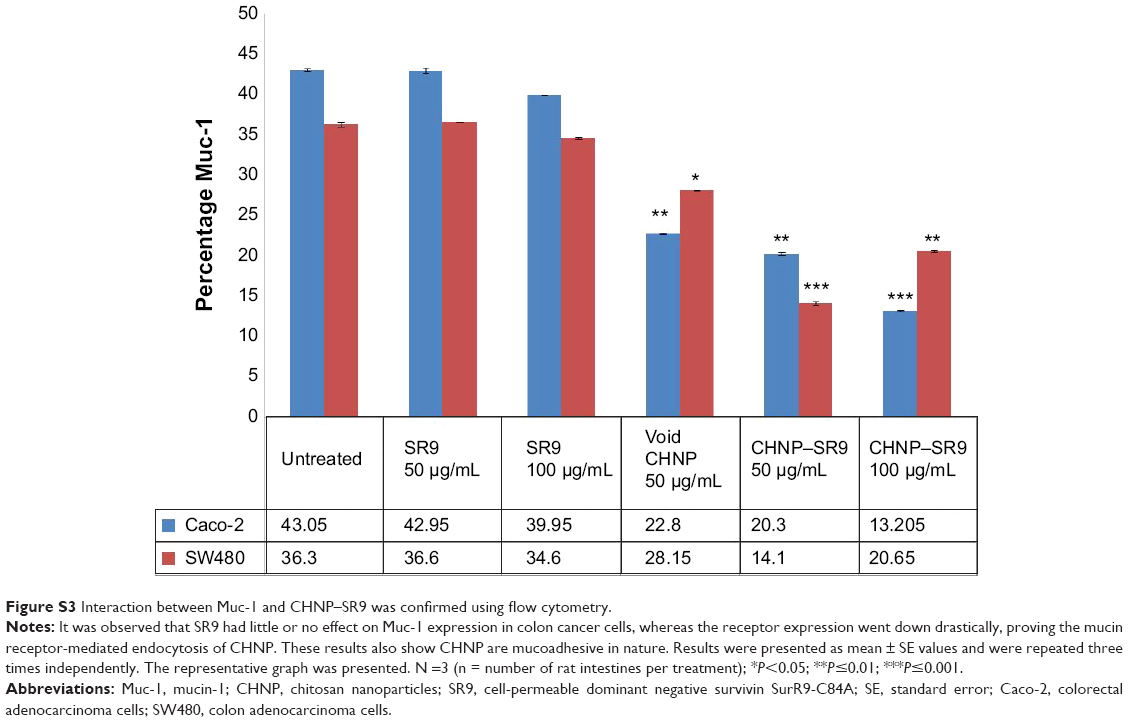 | Figure S3 Interaction between Muc-1 and CHNP–SR9 was confirmed using flow cytometry. |
 | Figure S4 Scanning electron microscopy (SEM) confirmed disruption in monolayer of CHNP–SR9-treated cells. The SEM images of the membranes confirmed a significant damage to the Caco-2 monolayer in CHNP–SR9-treated cells, whereas no damage was observed in untreated or void CHNP-treated cells. This illustrates that SR9 was highly cytotoxic to the cancerous cells, whereas the void CHNP showed no significant cytotoxicity on the Caco-2 cells. |
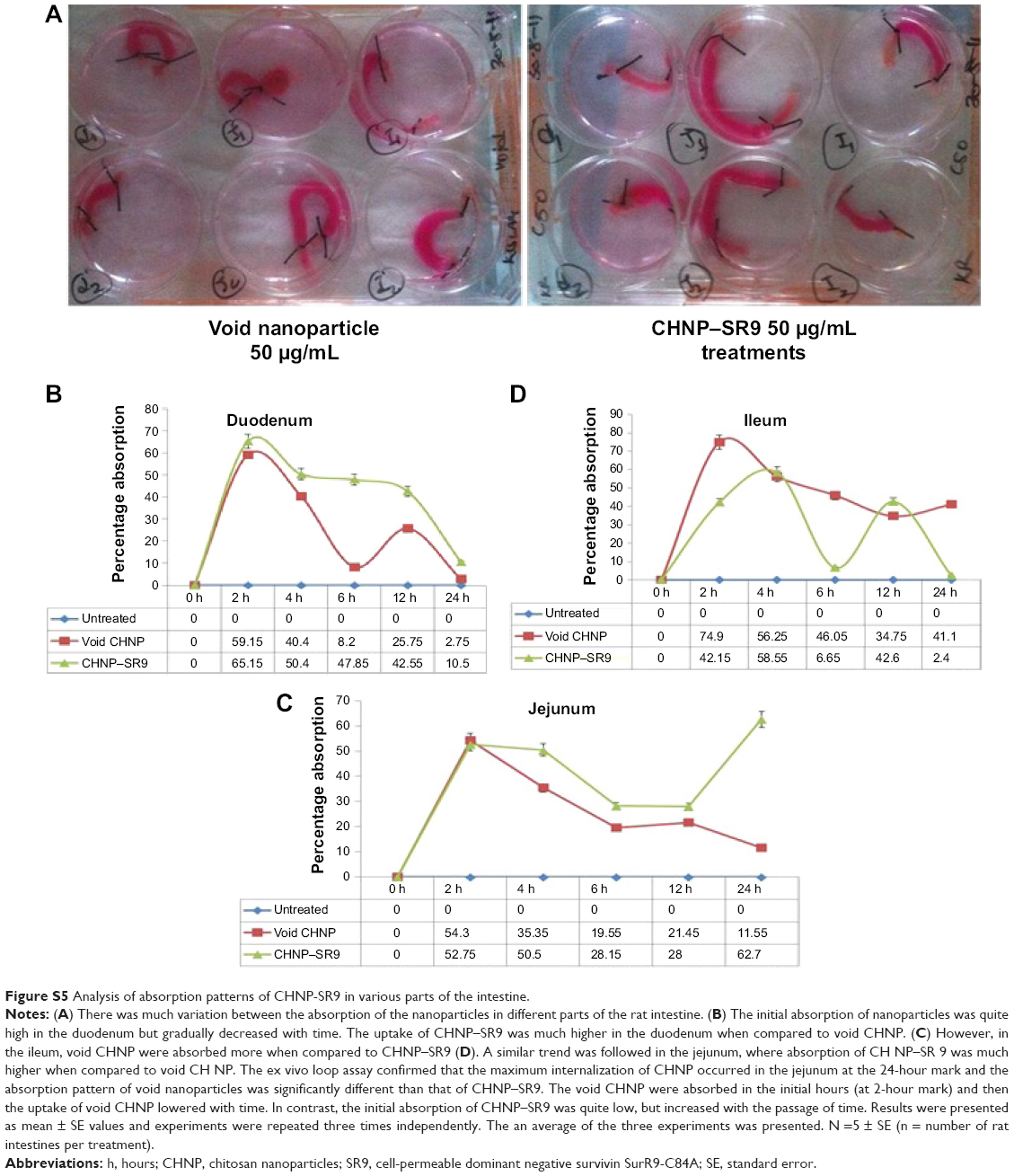 | Figure S5 Analysis of absorption patterns of CHNP-SR9 in various parts of the intestine. |
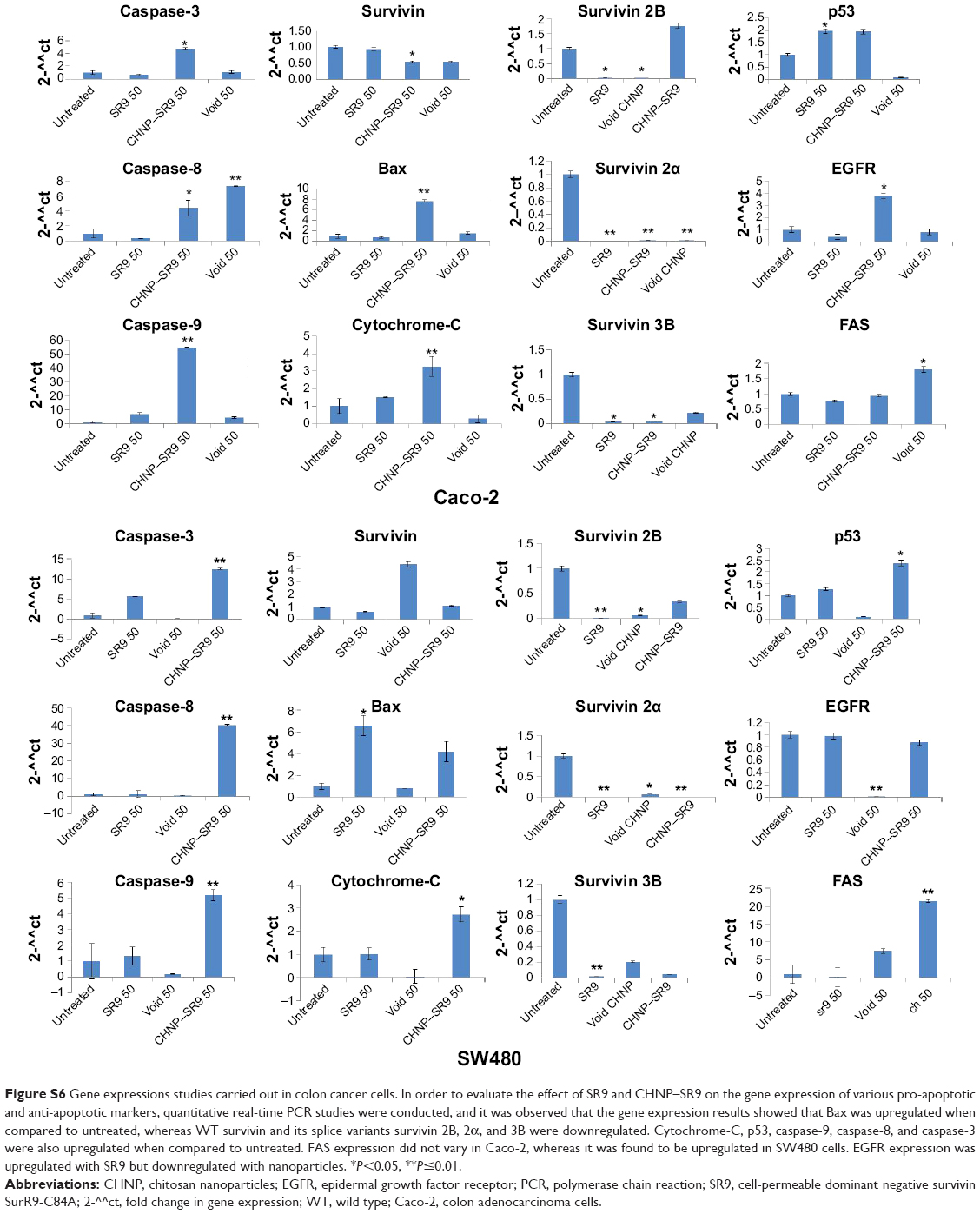 | Figure S6 Gene expressions studies carried out in colon cancer cells. In order to evaluate the effect of SR9 and CHNP–SR9 on the gene expression of various pro-apoptotic and anti-apoptotic markers, quantitative real-time PCR studies were conducted, and it was observed that the gene expression results showed that Bax was upregulated when compared to untreated, whereas WT survivin and its splice variants survivin 2B, 2α, and 3B were downregulated. Cytochrome-C, p53, caspase-9, caspase-8, and caspase-3 were also upregulated when compared to untreated. FAS expression did not vary in Caco-2, whereas it was found to be upregulated in SW480 cells. EGFR expression was upregulated with SR9 but downregulated with nanoparticles. *<0.05, **≤0.01. |
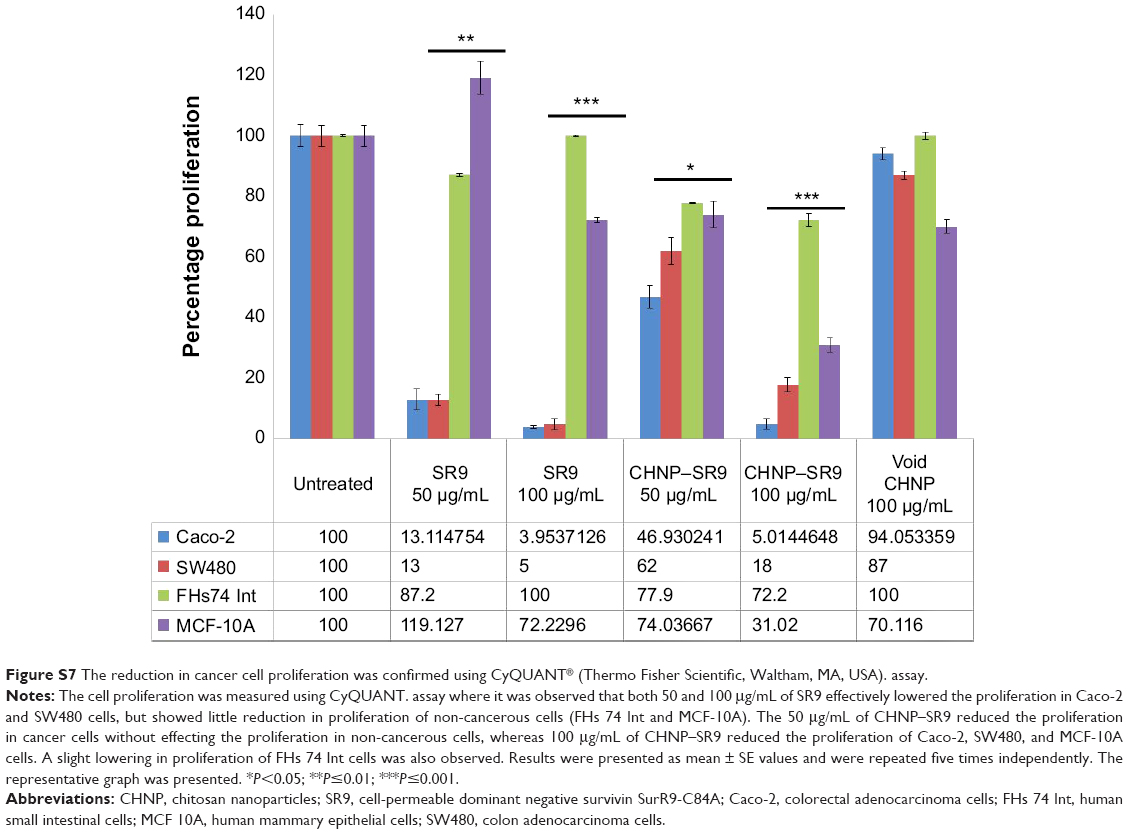 | Figure S7 The reduction in cancer cell proliferation was confirmed using CyQUANT® (Thermo Fisher Scientific, Waltham, MA, USA). assay. |
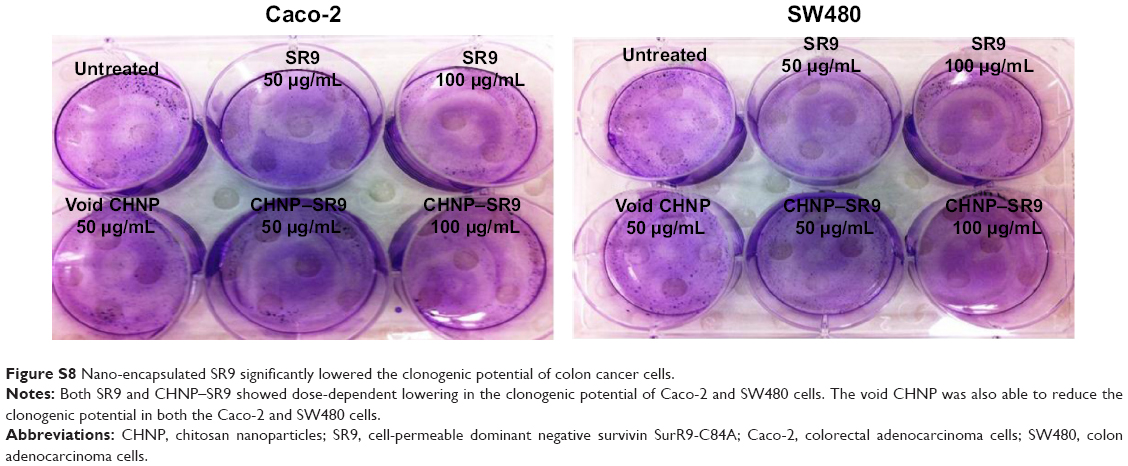 | Figure S8 Nano-encapsulated SR9 significantly lowered the clonogenic potential of colon cancer cells. |
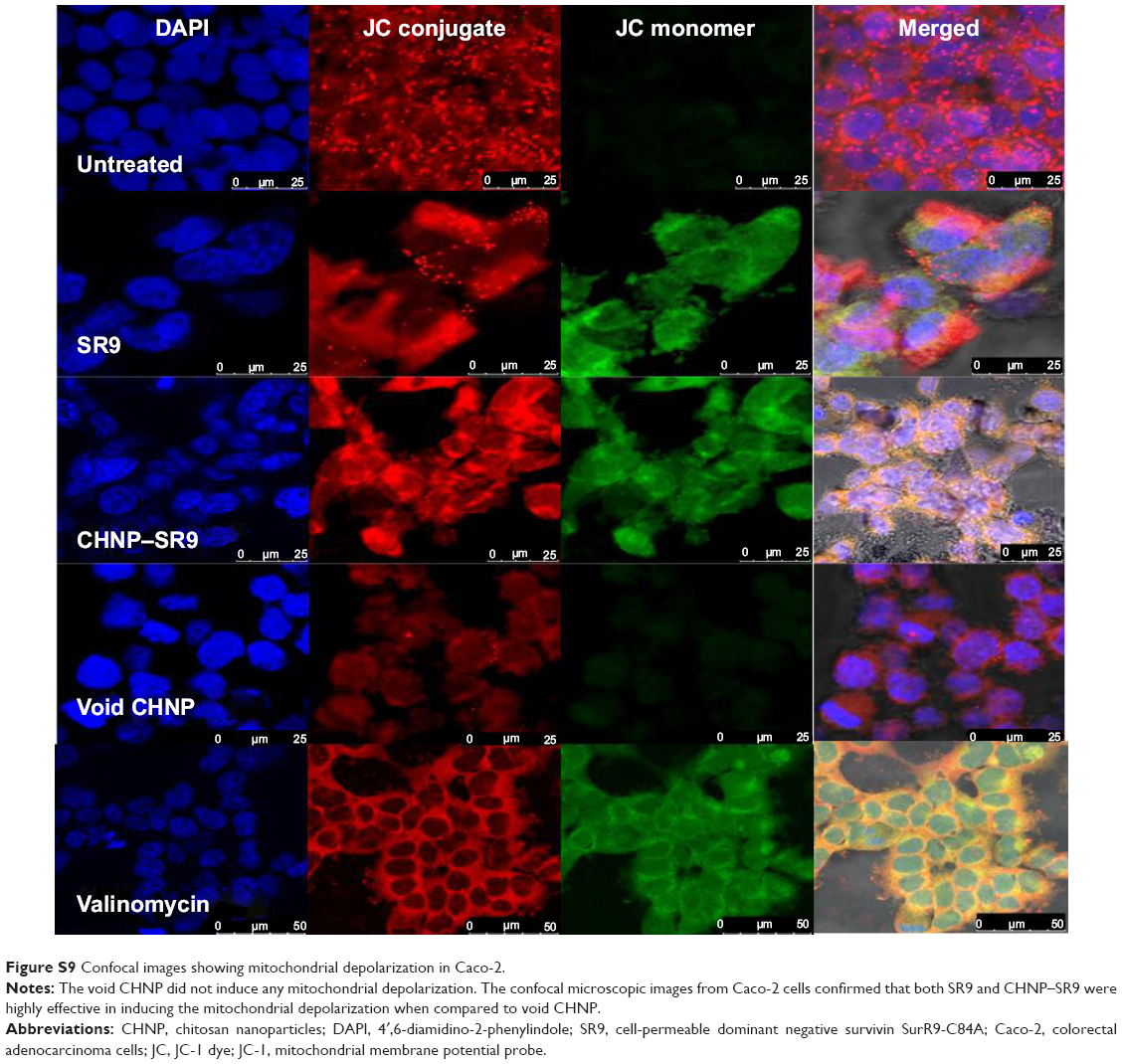 | Figure S9 Confocal images showing mitochondrial depolarization in Caco-2. |
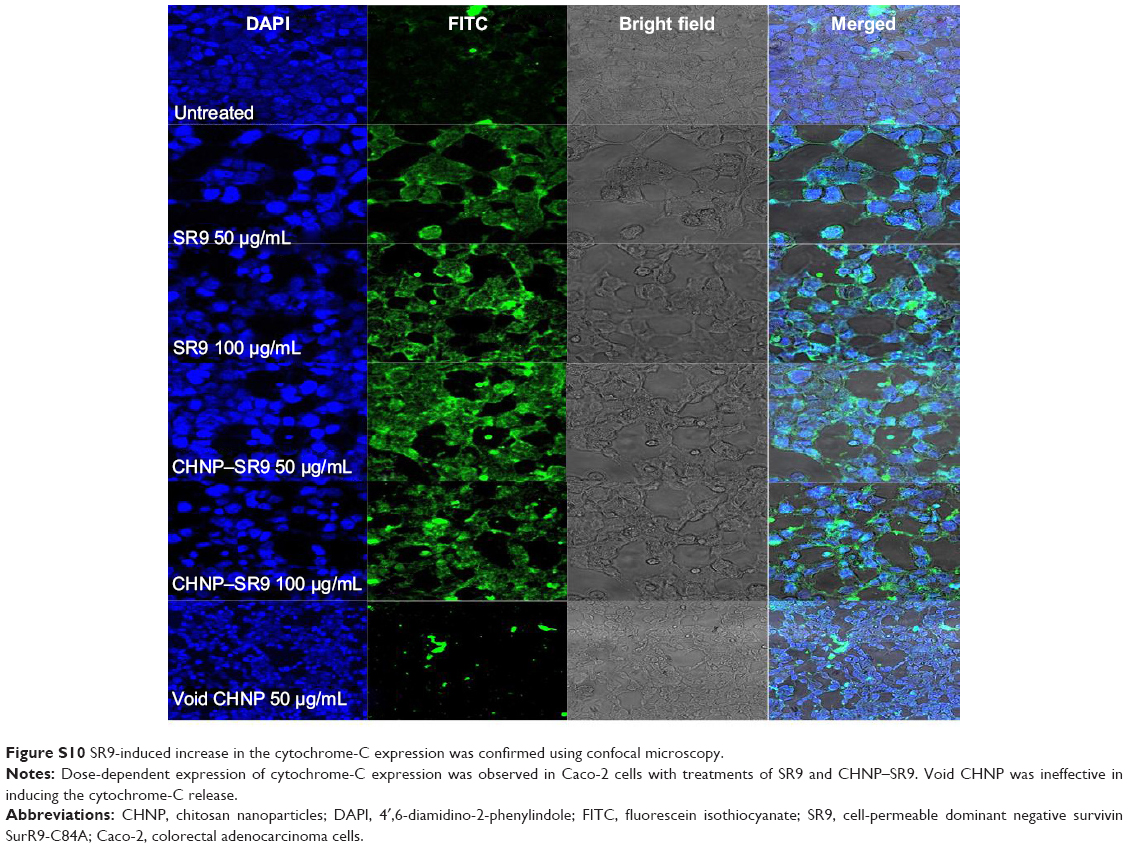 | Figure S10 SR9-induced increase in the cytochrome-C expression was confirmed using confocal microscopy. |
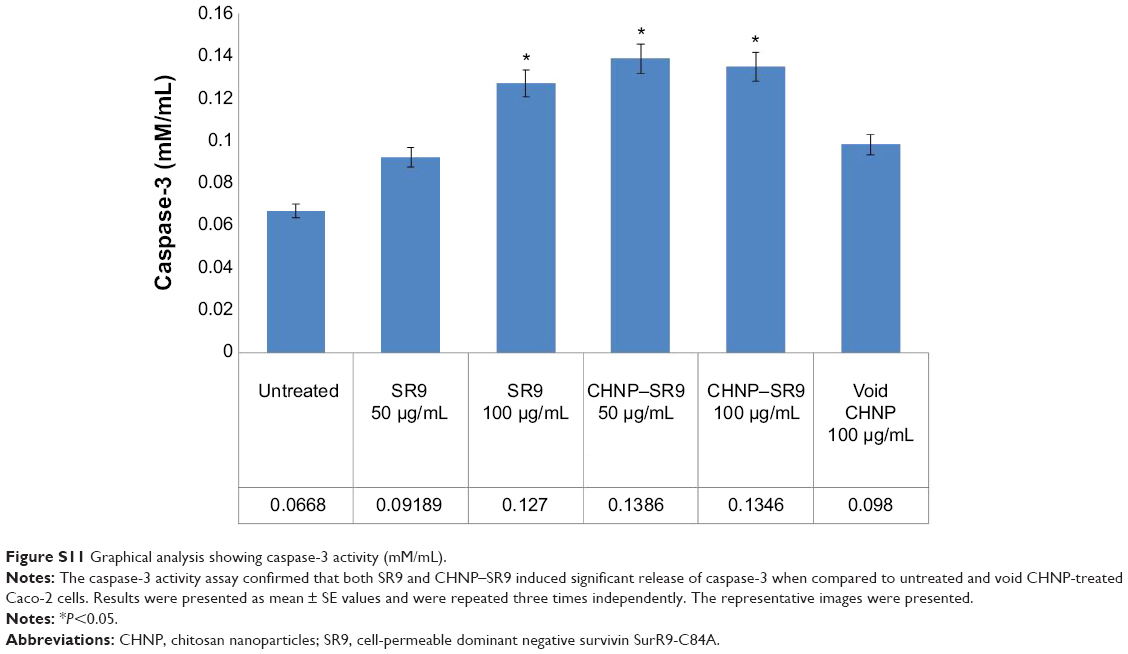 | Figure S11 Graphical analysis showing caspase-3 activity (mM/mL). |
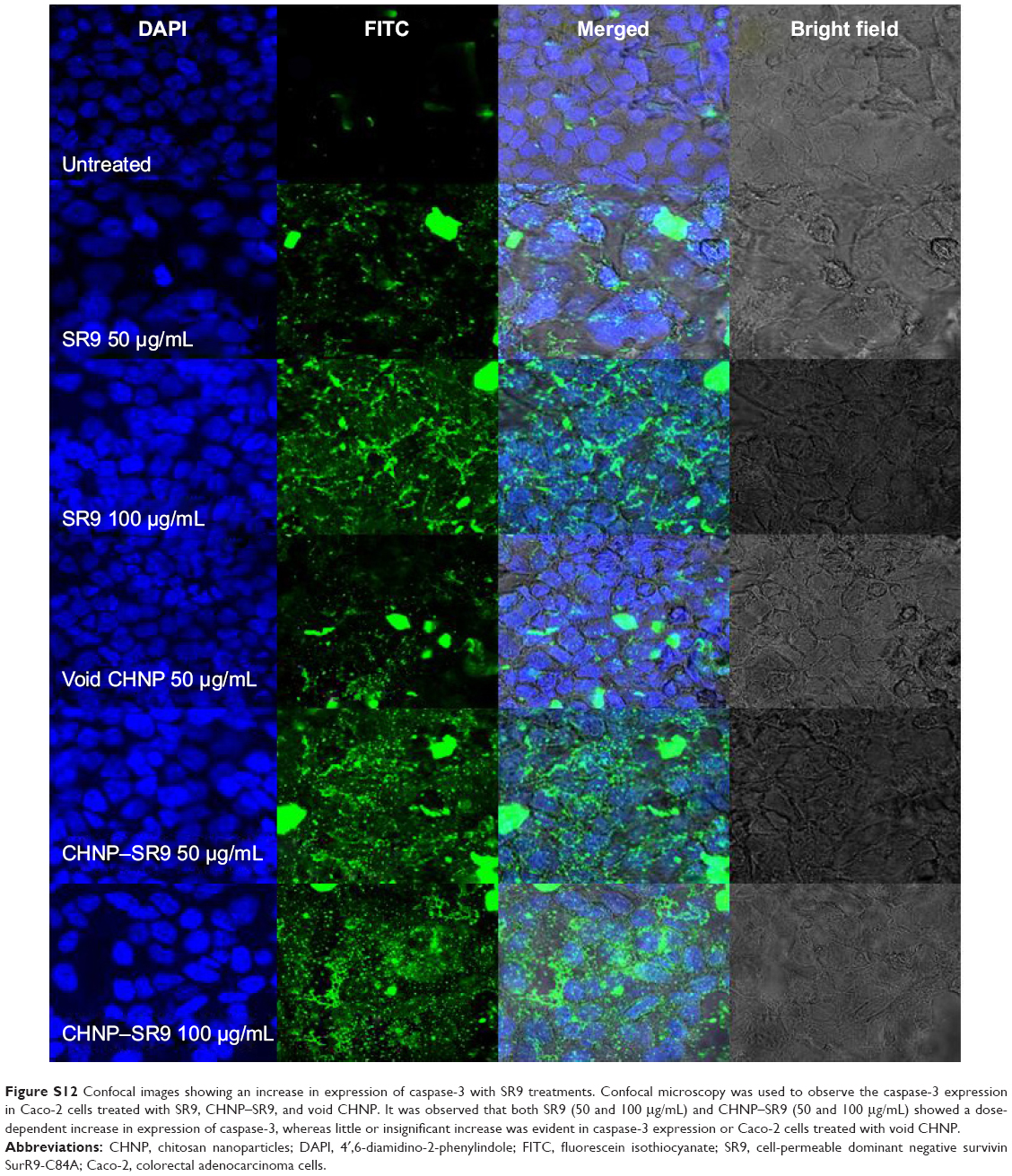 | Figure S12 Confocal images showing an increase in expression of caspase-3 with SR9 treatments. Confocal microscopy was used to observe the caspase-3 expression in Caco-2 cells treated with SR9, CHNP–SR9, and void CHNP. It was observed that both SR9 (50 and 100 μg/mL) and CHNP–SR9 (50 and 100 μg/mL) showed a dose-dependent increase in expression of caspase-3, whereas little or insignificant increase was evident in caspase-3 expression or Caco-2 cells treated with void CHNP. |
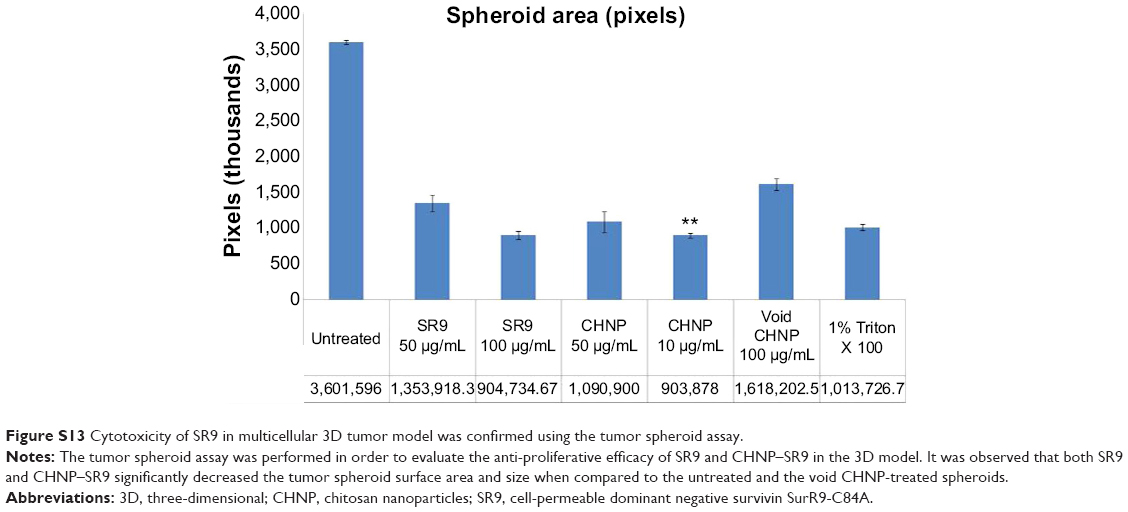 | Figure S13 Cytotoxicity of SR9 in multicellular 3D tumor model was confirmed using the tumor spheroid assay. |
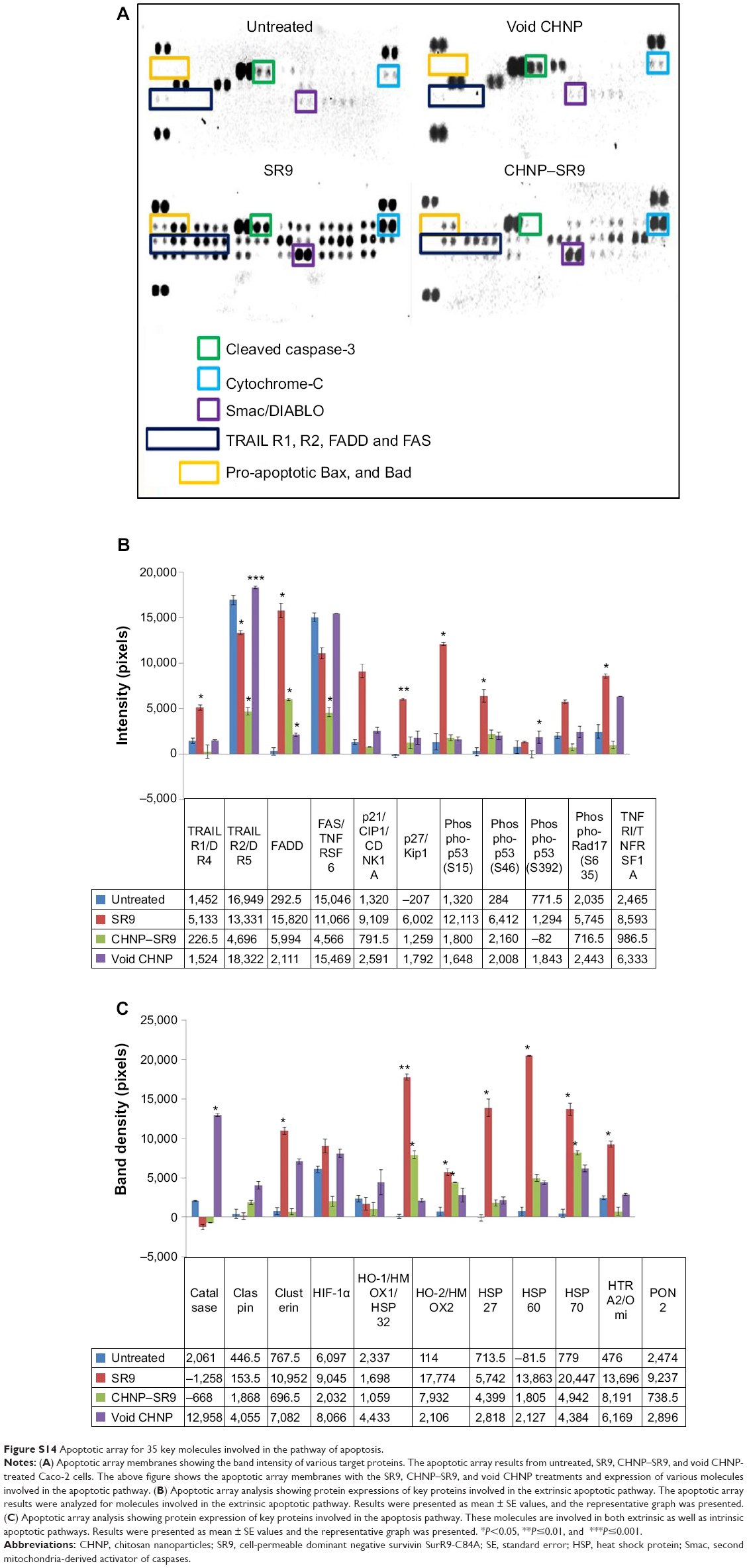 | Figure S14 Apoptotic array for 35 key molecules involved in the pathway of apoptosis. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.