Back to Journals » Drug Design, Development and Therapy » Volume 10
Comparisons of the pharmacokinetics and tolerability of fixed-dose combinations of amlodipine besylate/losartan and amlodipine camsylate/losartan in healthy subjects: a randomized, open-label, single-dose, two-period, two-sequence crossover study
Authors Choi Y, Lee S ![]() , Cho S, Kang W, Nam K, Jang I
, Cho S, Kang W, Nam K, Jang I ![]() , Yu K
, Yu K ![]()
Received 30 May 2016
Accepted for publication 27 July 2016
Published 20 September 2016 Volume 2016:10 Pages 3021—3028
DOI https://doi.org/10.2147/DDDT.S113891
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
YoonJung Choi,1 SeungHwan Lee,2 Sang-Min Cho,3 Won-Ho Kang,3 Kyu-Yeol Nam,4 In-Jin Jang,1 Kyung-Sang Yu1
1Department of Clinical Pharmacology and Therapeutics, Seoul National University College of Medicine, 2Clinical Trials Center, Seoul National University Hospital, 3Research Institute, 4Global R&D, Korea United Pharm Inc., Seoul, Republic of Korea
Background: A fixed-dose combination (FDC) of amlodipine and losartan has been used to reduce blood pressure in patients whose hypertension is not sufficiently controlled with either drug alone. The aim of this study was to evaluate the pharmacokinetic (PK) characteristics and tolerability of an FDC of 6.94 mg amlodipine besylate (5 mg as amlodipine)/50 mg losartan potassium compared to an FDC of 5 mg amlodipine camsylate/50 mg losartan potassium in healthy subjects.
Subjects and methods: A randomized, open-label, single-dose, two-period, two-sequence crossover study was conducted on 46 healthy male subjects. Blood concentrations were measured by liquid chromatography–tandem mass spectrometry. Blood samples were collected up to 144 hours post dose for each period. PK parameters were calculated in each treatment group using a noncompartmental method. The 90% confidence intervals (CIs) of the geometric mean ratios of the two treatments for the maximum plasma concentration (Cmax) and the area under the concentration curve from time zero to the last quantifiable time point (AUC0–t) were estimated. Tolerability assessments were performed for all subjects who received the drug at least once.
Results: The PK profiles of the two treatments were similar. For amlodipine, the geometric mean ratios (90% CIs) of amlodipine besylate to amlodipine camsylate for the Cmax and AUC0–t were 0.98 (0.94-1.01) and 0.97 (0.93-1.01), respectively. The corresponding values for losartan were 0.91 (0.81-1.02) and 1.05 (0.98-1.12), respectively. The incidence of adverse events was not significantly different between the two treatments, and both were well tolerated.
Conclusion: An FDC of 6.94 mg amlodipine besylate (5 mg as amlodipine)/50 mg losartan potassium produced similar results to an FDC of 5 mg amlodipine camsylate/50 mg losartan potassium treatment with respect to the PK parameters of amlodipine and losartan based on Cmax and AUC0–t values. The amlodipine besylate/losartan potassium combination was well tolerated by healthy male subjects.
Keywords: comparative pharmacokinetics, amlodipine, losartan, drug development
Introduction
High blood pressure (BP) is a risk factor for cardiovascular disease. The main objective of hypertension treatment is to normalize BP to prevent complications such as stroke and renal failure.1 In patients with hypertension, the first-line treatments consist of angiotensin-converting-enzyme inhibitors, angiotensin II receptor blockers (ARBs), calcium-channel blockers (CCBs), diuretics, and beta-blockers.2 Accordingly, the hypertension guidelines from the National Institute for Health and Care Excellence recommend ARBs or CCBs as first-line drugs for hypertension treatment.3 If BP control is not achieved by monotherapy, combination therapy with antihypertensive drugs is a useful and appropriate treatment option that can be more effective at lowering BP than high-dose monotherapy in hypertensive patients.4–6 Moreover, using a fixed-dose combination (FDC) of drugs reduces the burden of taking multiple drugs and decreases the financial burden for patients, which leads to improved medication compliance.7,8 Currently, several FDCs of antihypertensive agents are available on the pharmaceutical market (Co-Diovan®, Exforge®, and Tekturna HCT®), and new FDCs are under development.9
Amlodipine is a CCB that is prescribed for the treatment of high BP.10 Based on its mechanism of action, amlodipine inhibits the movement of calcium ions into cardiac and vascular smooth muscles.11 Because it acts directly on vascular smooth muscle, it reduces arterial BP and peripheral vascular resistance.12 Losartan, similar to amlodipine, is an ARB that is also used to treat high BP.13 The major metabolic pathway for losartan involves cytochrome P450 (CYP) 2C9 and 3A4, which are converted into active metabolites that also show antihypertensive activity similar to the parent compound.14 By competitively blocking the binding of angiotensin II, losartan relaxes vascular smooth muscle and dilates blood vessels, thereby reducing vascular resistance and BP.15,16
In this context, amlodipine and losartan, which have different but synergistic mechanisms of action for controlling hypertension, are commonly combined to treat patients with hypertension.17 Both of these agents exert a protective effect on the heart and blood vessels, which are target organs in treating hypertension.18 Moreover, these drugs have complementary actions on electrolytes in the body: amlodipine tends to cause retention of potassium and losartan tends to suppress the loss of potassium. Therefore, the coadministration of amlodipine and losartan can act as a complementary therapy for hypertension.19
Although the drug–drug interactions between amlodipine and losartan have not been reported, amlodipine and losartan are routinely coadministered. In addition, several studies of FDCs of amlodipine and losartan have been reported.17,20,21 An FDC of amlodipine camsylate (5 mg) and losartan (50 mg) was approved for the treatment of hypertension in the domestic South Korean market. This FDC was used to treat high BP in patients whose hypertension was not sufficiently controlled with either drug alone.22 Recently, a new FDC formulation of amlodipine and losartan that incorporates a besylate salt rather than a camsylate salt was developed by Korea United Pharm Inc., Ltd. (Seoul, South Korea). Amlodipine has been incorporated into various salt forms to provide improved physicochemical properties because its base is not soluble in water.23 The besylate salt is the most common form and is known to be highly suitable for the preparation of amlodipine in pharmaceutical formulations.24 Furthermore, a clinical study has shown that the efficacy and tolerability of amlodipine besylate is not different from that of amlodipine camyslate.25
This study evaluated the pharmacokinetic (PK) profile and tolerability of a single oral administration of a newly developed FDC formulation containing amlodipine besylate (6.94 mg, 5 mg as amlodipine) and losartan potassium (50 mg; test drug) and compared these values to those obtained from a single oral dose of a commercially available FDC of amlodipine camsylate (5 mg) and losartan potassium (50 mg; reference drug) in healthy male subjects.
Subjects and methods
This study was performed after approval was obtained from the institutional review board of Seoul National University Hospital (Seoul, Korea) and the Korea Ministry of Food and Drug Safety (ClinicalTrials.gov registry number: NCT02166398) and was conducted in accordance with the ethical guidelines of the Declaration of Helsinki and Korean Good Clinical Practice. All subjects provided informed consent prior to undergoing a screening test for study participation.
Study subjects
The present study included healthy male subjects between 20 years and 45 years of age with a body mass index between 19.0 kg/m2 and 27.0 kg/m2. The health status of each subject was determined based on medical history, a physical examination, electrocardiogram (ECG) monitoring, vital signs, and laboratory tests, which were examined within 3 weeks of the first product administration. The subjects with a history of hypersensitivity to amlodipine or losartan were excluded in this study. Additionally, the subjects with a sitting systolic blood pressure (SBP) <100 mmHg or >150 mmHg or a sitting diastolic blood pressure (DBP) <60 mmHg or >100 mmHg in the screening test were excluded.
The sample size of this study was obtained based on a previous bioequivalence study of losartan potassium. The previous study reported that the highest value of the coefficient of variation for intrasubject variability was 35.44% for the Cmax of losartan. Based upon this estimate of variability, a total sample size of 46 subjects, accounting for a 10% dropout rate, was estimated to achieve the bioequivalence criteria based on a power of 80% and a 5% significance level, assuming no difference between the two treatments.
Study design
This study was designed as a randomized, open-label, single-dose, two-period, two-sequence, crossover study. The enrolled subjects were administered a single oral dose of either the test or reference drug during each study period with a 14-day washout period between treatments.
All eligible subjects were hospitalized at the clinical trials center, Seoul National University Hospital, 1 day before drug dosing. After overnight fasting, the subjects randomly received a single oral dose of the test or reference drug during each period with 240 mL of water. The washout period followed the first treatment period, after which the subjects received the alternate treatment. Water was not permitted for 2 hours before and 2 hours after dosing during each period. Standard meals were provided at 4 hours post treatment.
Blood samples for the PK evaluations of amlodipine and losartan were collected pre dose and at 0.25 hours (losartan only), 0.5 hours (losartan only), 0.75 hours (losartan only), 1 hour, 1.25 hours (losartan only), 1.5 hours (losartan only), 2 hours, 3 hours (losartan only), 4 hours, 6 hours, 8 hours, 10 hours, 12 hours, 24 hours (amlodipine only), 48 hours (amlodipine only), 72 hours (amlodipine only), 96 hours (amlodipine only), and 144 hours (amlodipine only) post dose during each period. The blood samples were collected in heparin tubes and centrifuged at 3,000 rpm for 10 minutes at 4°C. Plasma supernatants were transferred to plastic aliquot tubes and immediately stored at −70°C until analysis.
Determination of amlodipine and losartan plasma concentrations
Plasma sample preparation for amlodipine was performed by liquid–liquid extraction using methyl tertiary butyl ether. Amlodipine-d5 was used as an internal standard (IS) for the quantitation of amlodipine. The mobile phase was 10 mM ammonium formate in distilled water (DW) and methanol (60:40, v/v). Amlodipine and amlodipine-d5 were separated on a Unison UK-C18 column (3.0×50 mm, 3 μm; Imatakt, Japan) and detected via electrospray ionization in the multiple reaction monitoring (MRM) mode. The MRM was based on an m/z transition of 409.1→238.0 for amlodipine and an m/z transition of 413.1→238.0 for amlodipine-d5 (IS). The analytical method demonstrated linearity and allowed the quantification of amlodipine in human plasma within the range of 0.1–10 ng/mL. The intra- and interday precision values of amlodipine were all <7.62%, and the accuracies were found to be within the ranges of 96.8%–100.63% and 93.2%–99.9%, respectively.
The sample preparation for losartan involved a simple protein precipitation with acetonitrile. Losartan-d4 was used as an IS for the quantitation of losartan. The mobile phase consisted of 10 mM ammonium acetate in DW and acetonitrile (10:90, v/v). Losartan and losartan-d4 were separated on a Zorbax SB-Aq column (4.6×100 mm, 3.5 μm; Agilent Technologies, Santa Clara, CA, USA) and detected using positive electrospray ionization with MRM. The MRM was based on an m/z transition of 423.2→207.2 for losartan and an m/z transition of 427.2→211.2 for losartan-d4 (IS). The calibration curves for losartan were linear over the plasma concentration range of 5–1,000 ng/mL. The intra- and interday precision values of losartan were all <8.09%, and the accuracies ranged from 97.7% to 106.8% and from 97.6% to 105.6%, respectively.
Safety and tolerability evaluation
Tolerability evaluations were conducted from the screening period until the end of the study on all subjects who received at least one dose of the test or reference drug. Tolerability assessments included vital sign measurements (SBP, DBP, and pulse rate [PR]), physical examinations, ECG monitoring, clinical laboratory tests, and adverse events (AEs). All AEs were categorized per system organ class and treatment, and the incidences and causal relationships of all AEs were summarized. AE data, including number of events, number of subjects, severity, seriousness, and causality, were summarized by treatment group using descriptive statistics.
PK analysis
PK parameters were estimated by a noncompartmental method using Phoenix WinNonlin Version 6.3 (Pharsight Corporation, St Louis, MO, USA). The maximum observed plasma concentration (Cmax) and the time to the peak concentration (Tmax) were determined for each subject for each treatment. The area under the plasma concentration time curve from time 0 to the last measurable time point (AUC0–t) was calculated by the linear trapezoidal rule. The area under the concentration time curve from time 0 to infinity (AUC0–∞) was determined by the linear trapezoidal rule with extrapolation to infinity based on the last quantifiable concentration divided by the elimination rate constant. The plasma half-life (t1/2) was estimated from the slope of the terminal elimination phase.
Statistical analysis
Statistical analysis was performed using SAS software, Version 9.3 (SAS Institute Inc., Cary, NC, USA). Analysis of variance was used on log-transformed Cmax and AUC0–t values and on the geometric mean ratios of the test to reference drugs obtained when the Cmax and AUC0–t values were calculated. The two study drugs were considered bioequivalent if the 90% confidence interval (CI) of the mean difference for each PK variable of the test drug and the reference drug was within 80%–125%.
Results
Demographic characteristics
A total of 46 healthy subjects were enrolled in the study and randomized into each of the sequences. All the subjects had normal clinical and laboratory parameters and were eligible based on the inclusion/exclusion criteria of the study. The average age, height, body weight, and body mass index were 28.6±5.1 years (mean ± standard deviation), 174.8±6.1 cm, 69.1±7.0 kg, and 22.6±1.8 kg/m2, respectively. All the subjects were included in the tolerability/safety assessment, but only 45 subjects were included in the PK evaluation because one subject withdrew his consent after the first treatment.
Pharmacokinetics
The plasma concentration–time profiles of amlodipine and losartan were similar for both FDCs (Figure 1A and B). The Cmax values of amlodipine and losartan in both FDCs were reached at ~6 hours and 1 hour, respectively, after dosing. The terminal elimination t1/2 values for amlodipine and losartan in both FDCs were 43.6–46.4 hours and 1.8–1.9 hours, respectively. The individual differences in the Cmax and AUC0–t values of amlodipine and losartan showed no obvious trends between the two treatments (Figure 2A–D). The PK parameters of amlodipine and losartan for the test and reference treatments are presented in Table 1. Both drugs had similar PK properties for each component.
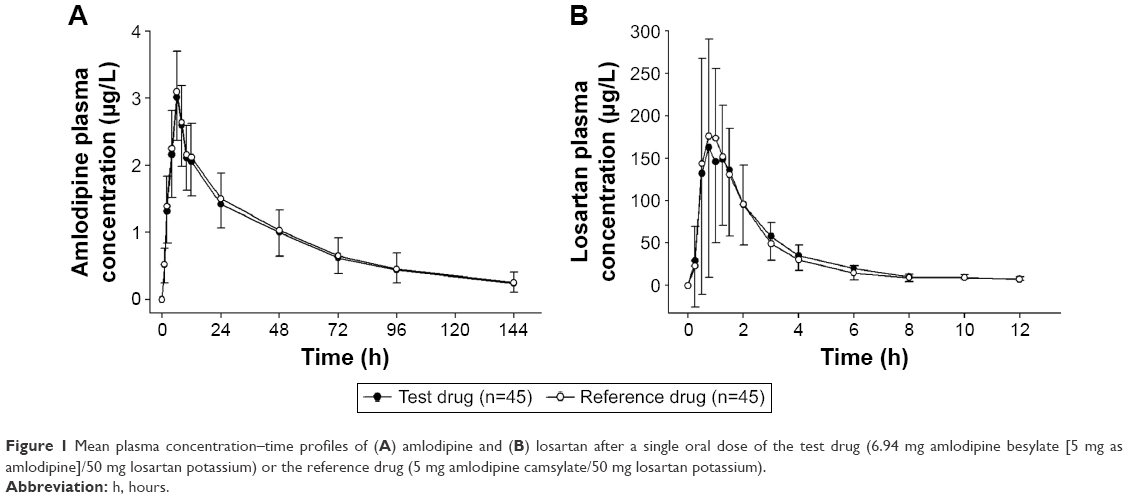 | Figure 1 Mean plasma concentration–time profiles of (A) amlodipine and (B) losartan after a single oral dose of the test drug (6.94 mg amlodipine besylate [5 mg as amlodipine]/50 mg losartan potassium) or the reference drug (5 mg amlodipine camsylate/50 mg losartan potassium). |
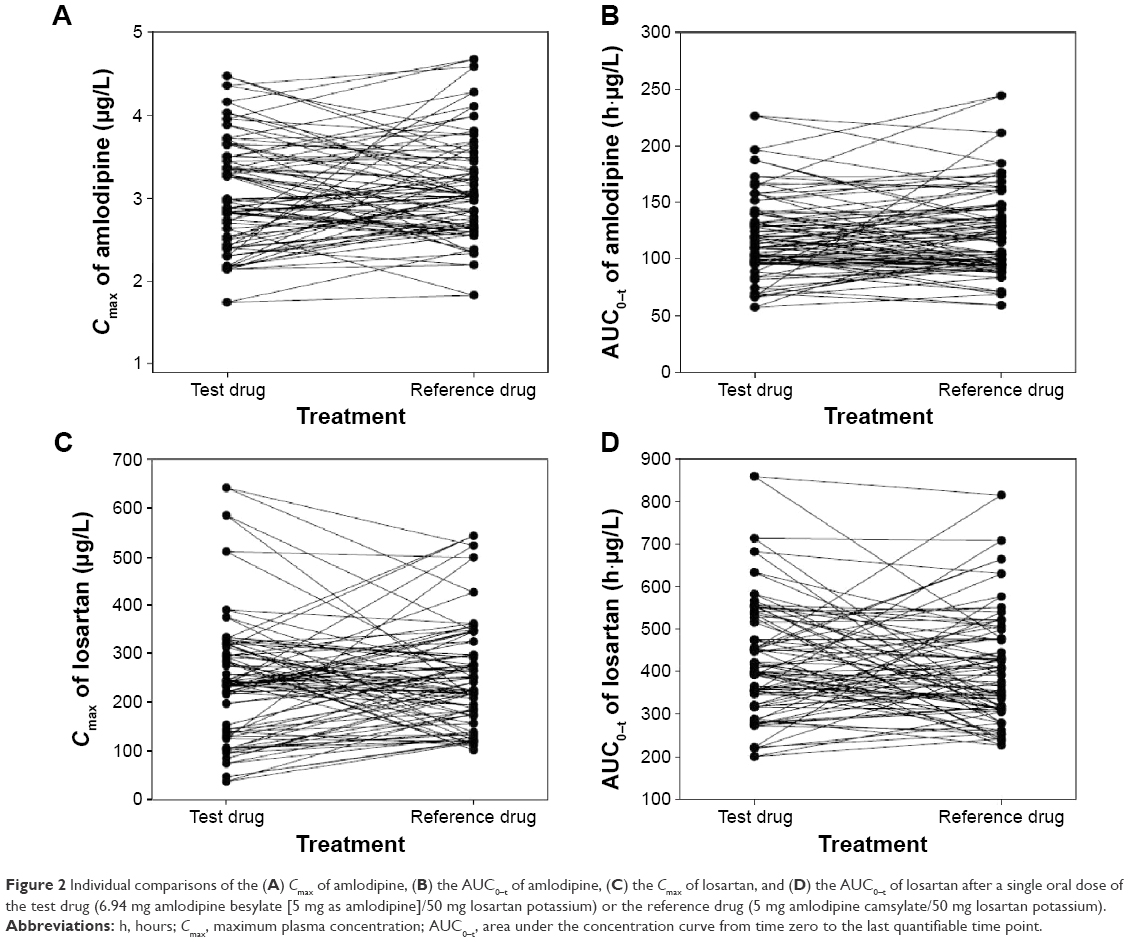 | Figure 2 Individual comparisons of the (A) Cmax of amlodipine, (B) the AUC0–t of amlodipine, (C) the Cmax of losartan, and (D) the AUC0–t of losartan after a single oral dose of the test drug (6.94 mg amlodipine besylate [5 mg as amlodipine]/50 mg losartan potassium) or the reference drug (5 mg amlodipine camsylate/50 mg losartan potassium). |
 | Table 1 Summary of the PK results of amlodipine and losartan for the test drug (6.94 mg amlodipine besylate [5 mg as amlodipine]/50 mg losartan potassium) and the reference drug (5 mg amlodipine camsylate/50 mg losartan potassium) |
The 90% CIs for the ratio of the geometric mean of the test to reference drug for amlodipine were 0.98 (0.94–1.01) for Cmax and 0.97 (0.93–1.01) for AUC0–t. The corresponding values for losartan were 0.91 (0.81–1.02) for Cmax and 1.05 (0.98–1.12) for AUC0–t. Therefore, the 90% CIs for the Cmax and AUC0–t of amlodipine and losartan were within the commonly accepted bioequivalence criteria of 0.8–1.25.
Tolerability assessment
No clinically significant changes were observed regarding vital signs, physical examinations, ECG findings, clinical laboratory results, or AEs. During the study, a total of 46 AEs were reported in 22 subjects. The most frequently noted AEs were headache (eleven cases) and dizziness (seven cases). The percentage difference between the two treatments for the occurrence of AEs was insignificant (Table 2). No serious AEs occurred during the course of the study; all events were mild to moderate and resolved spontaneously, except for one case of blurred vision in one subject. In this case, the symptom occurred after the final visit, and a relationship between it and the study drugs was considered unlikely.
 | Table 2 AEs reported following a single oral dose of the test drug (6.94 mg amlodipine besylate [5 mg as amlodipine]/50 mg losartan potassium) or the reference drug (5 mg amlodipine camsylate/50 mg losartan potassium) |
As shown in Figure 3A–C, similar reductions in the 24-hour SBP, DBP, and mean PR were observed for the test drug and reference drug. The mean SBP and DBP were lower than the baseline values at 6–8 hours after dosing (Figure 3A–C). The mean PR showed a tendency to increase 6 hours after dosing. The lowest mean values (standard deviation) for SBP were 102.2 mmHg (9.4 mmHg) and 100.1 mmHg (8.8 mmHg), which were measured at 8 hours after administration of the test and reference drugs, respectively. The lowest mean values (standard deviation) for DBP were 57.0 mmHg (6.2 mmHg), measured 6 hours after the administration of the test drug, and 57.2 mmHg (6.2 mmHg), measured 8 hours after the administration of the reference drug. No clinically significant symptoms related to BP reduction were observed in our study.
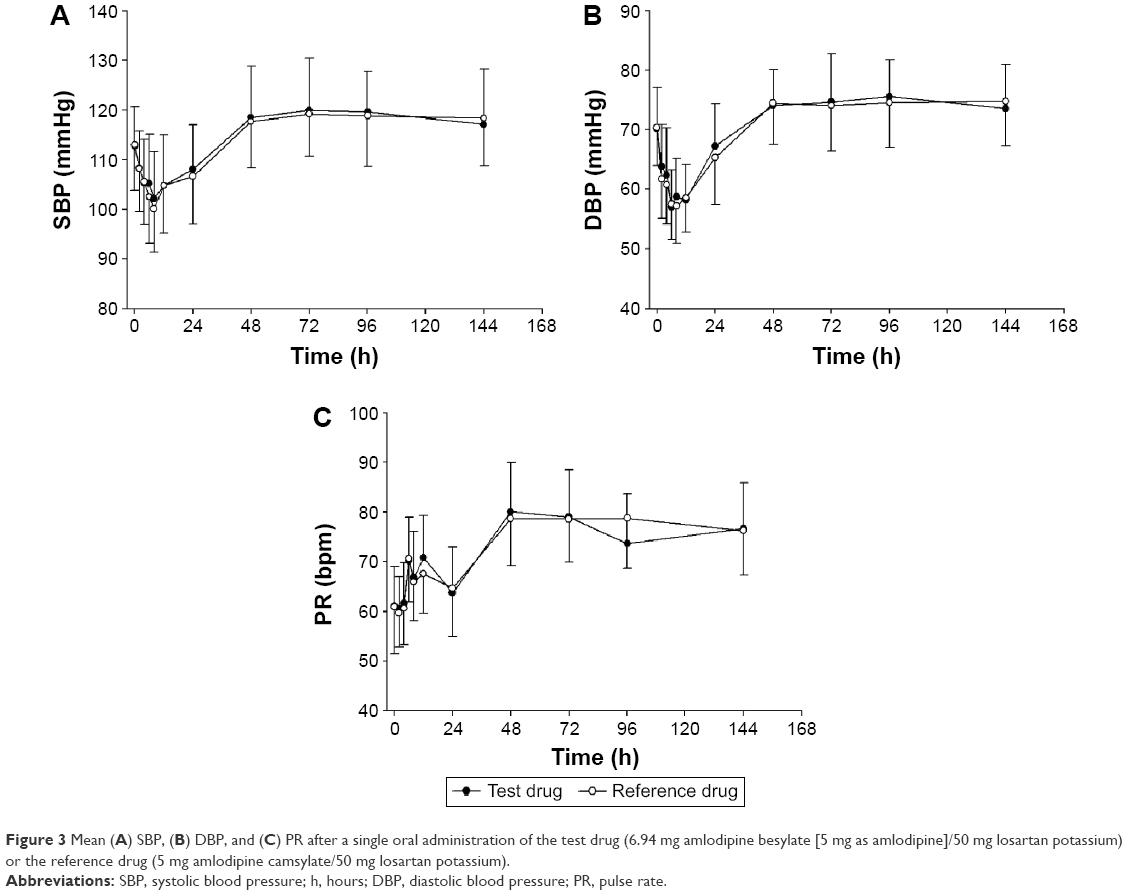 | Figure 3 Mean (A) SBP, (B) DBP, and (C) PR after a single oral administration of the test drug (6.94 mg amlodipine besylate [5 mg as amlodipine]/50 mg losartan potassium) or the reference drug (5 mg amlodipine camsylate/50 mg losartan potassium). |
Discussion
In this study, the concentration–time profiles for the two study drugs were well characterized, and the sampling design included a sufficient duration to characterize the elimination phases of both study drugs. For amlodipine, the AUC0–t/AUC0–∞ ratio was greater than 80% in 40 of 45 and 43 of 45 subjects for the test and reference drugs, respectively. For losartan, all 45 subjects had an AUC0–t/AUC0–∞ ratio >80%. These findings indicate that the sampling point was adequately reflected in the determination of the Cmax and AUC0–t for amlodipine and losartan, allowing full assessment of the elimination of these drugs. Moreover, an adequate washout period of 14 days, which was based on the long t1/2 of amlodipine (45 hours), was used to ensure complete elimination of the drug from the blood after the first treatment period. The concentrations of amlodipine and losartan in the predose samples for Period 2 were not detectable, and no carryover effect from the previous period was observed.
In most cases, bioequivalence assessment is performed by focusing only on the measurement of parent drug concentrations.26 Accordingly, this study was conducted based on the PK properties of the parent compound. The bioequivalence acceptance criteria that are applied to comparative bioavailability studies and the bioequivalence requirements of the US Food and Drug Administration and European Medicines Agency (EMA) guidelines recommend that the evaluation of bioequivalence should be based upon measured plasma concentrations of the parent compound.27,28 Additionally, it is recommended that both the parent compound and its major active metabolite should be measured in case the metabolite contributes to the clinical safety and/or efficacy of the drug. For bioequivalence studies of any FDC containing losartan, the active metabolite largely contributes to the antihypertensive effect. Therefore, this metabolite should be included in the assessment of bioequivalence.29 However, the role of metabolites in bioequivalence assessments is a controversial issue.30 According to the Efficacy Working Party therapeutic subgroup on PKs of the EMA,31 the bioequivalence for losartan should be proven based on parent data. As indicated in the results of this study, the primary end points for the parent compound, losartan, met the bioequivalence criteria. Future studies examining the PK parameters of losartan metabolites in patients could show comparable therapeutic outcomes for the two drugs.
The test and reference drugs were both well tolerated. A total of 46 AEs considered probably or possibly related to treatment were observed. The most common AEs were headache and dizziness, which were associated with BP reduction due to the pharmacological effect of the antihypertensive agent. The AE profile was similar to that reported previously, in which the most common AEs were headache and dizziness.17 These AE profiles imply that the drug effects of amlodipine and losartan are significantly influenced by coadministration of the two drugs.
The BP-lowering effects of the test and reference drugs were identified throughout this study. In our study, reductions in SBP of −9.6 mmHg and −11.8 mmHg were observed at 8 hours for the test and reference drugs, respectively. Similarly, reductions in DBP of −11.1 mmHg and −10.7 mmHg were observed at 12 hours for the test and reference drugs, respectively. The test drug produced similar BP-lowering effects to the reference drug. Because the assessment of BP was not the primary objective of this study, sufficient evidence does not exist to support an equal effect of these two treatments. However, we found that the BP-lowering effect of the test drug was similar to that of the reference drug. It is generally known that BP fluctuates over a 24-hour period and tends to be high in the morning and low in the evening.32 Accordingly, several studies have reported that ambulatory BP monitoring can more accurately reflect diurnal variations in BP than spot measurements.33,34 Therefore, further Phase III clinical studies over longer periods of time and in a larger patient group are needed to assess the BP-lowering effects of the test drug.
Conclusion
This study demonstrated that an FDC of 6.94 mg amlodipine besylate (5 mg as amlodipine)/50 mg losartan potassium produced similar effects to an FDC of 5 mg amlodipine camsylate/50 mg losartan potassium with respect to the PK parameters of amlodipine and losartan based on the Cmax and AUC0–t values. These results meet the regulatory criteria for assuming bioequivalence. In addition, the 6.94 mg amlodipine besylate (5 mg as amlodipine)/50 mg losartan potassium combination was well tolerated and had a safety profile comparable to that of the 5 mg amlodipine camsylate/50 mg losartan potassium combination.
Acknowledgment
This study was sponsored by Korea United Pharm Inc., Seoul, Republic of Korea.
Disclosure
The authors report no conflicts of interest in this work.
References
Messerli FH, Williams B, Ritz E. Essential hypertension. Lancet. 2007;370(9587):591–603. | ||
James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA. 2014;311(5):507–520. | ||
National Institute of Health and Clinical Excellence. Hypertension: Clinical Management of Primary Hypertension in Adults. London: NICE; 2011. NICE Clinical Guideline 127. Available from: https://www.nice.org.uk/guidance/cg127/resources/hypertension-in-adults-diagnosis-and-management-35109454941637. Accessed January 25, 2016. | ||
Neutel JM. The role of combination therapy in the management of hypertension. Nephrol Dial Transplant. 2006;21(6):1469–1473. | ||
Chrysant SG. Using fixed-dose combination therapies to achieve blood pressure goals. Clin Drug Investig. 2008;28(11):713–734. | ||
Andreadis EA, Tsourous GI, Marakomichelakis GE, et al. High-dose monotherapy vs low-dose combination therapy of calcium channel blockers and angiotensin receptor blockers in mild to moderate hypertension. J Hum Hypertens. 2005;19(6):491–496. | ||
Erdine S. Compliance with the treatment of hypertension: the potential of combination therapy. J Clin Hypertens. 2010;12(1):40–46. | ||
Gupta AK, Arshad S, Poulter NR. Compliance, safety, and effectiveness of fixed-dose combinations of antihypertensive agents: a meta-analysis. Hypertension. 2010;55(2):399–407. | ||
Tony E, Neal H. A.9 FDCs and multiple dosage strengths. Pharmaceutical Lifecycle Management: Making the Most of Each and Every Brand. Hoboken, NJ: John Wilet & Sons, Inc.; 2012. | ||
Jeffers BW, Robbins J, Bhambri R, Wajsbrot D. A systematic review on the efficacy of amlodipine in the treatment of patients with hypertension with concomitant diabetes mellitus and/or renal dysfunction, when compared with other classes of antihypertensive medication. Am J Ther. 2015;22(5):322–341. | ||
US Food and Drug Administration, Drugs@FDA [webpage on the Internet]. Amlodipine Label. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2005/019787s038lbl.pdf. Accessed January 25, 2016. | ||
Stepien O, Zhang Y, Zhu D, Marche P. Dual mechanism of action of amlodipine in human vascular smooth muscle cells. J Hypertens. 2002;20(1):95–102. | ||
Simpson KL, McClellan KJ. Losartan: a review of its use, with special focus on elderly patients. Drugs Aging. 2000;16(3):227–250. | ||
Sica DA, Gehr TW, Ghosh S. Clinical pharmacokinetics of losartan. Clin Pharmacokinet. 2005;44(8):797–814. | ||
Israili ZH. Clinical pharmacokinetics of angiotensin II (AT1) receptor blockers in hypertension. J Hum Hypertens. 2000;14(suppl 1):S73–S86. | ||
Watanabe T, Barker TA, Berk BC. Angiotensin II and the endothelium: diverse signals and effects. Hypertension. 2005;45(2):163–169. | ||
Kang SM, Youn JC, Chae SC, et al. Comparative efficacy and safety profile of amlodipine 5 mg/losartan 50 mg fixed-dose combination and amlodipine 10 mg monotherapy in hypertensive patients who respond poorly to amlodipine 5 mg monotherapy: an 8-week, multicenter, randomized, double-blind phase III noninferiority study. Clin Ther. 2011;33(12):1953–1963. | ||
Lawrence Gould A, Unniachan S, Wu D. Indirect treatment comparison between fixed-dose-combinations of amlodipine/losartan and amlodipine/valsartan in blood pressure control. Int J Clin Pract. 2014;68(2):163–172. | ||
Kim SW, Jun SS, Jo YG, Koo J-S, Sun SO inventor; Hanall Pharmaceutical Co., assignee. Combined preparation for the cardiovascular diseases based on chronotherapy theory. U.S. Patent 2010/0047341 A1. 25, 2010. | ||
Kohlmann O Jr, Oigman W, Mion D Jr, et al. The “LOTHAR” study: evaluation of efficacy and tolerability of the fixed combination of amlodipine and losartan in the treatment of essential hypertension. Arq Bras Cardiol. 2006;86(1):39–51. | ||
Park CG, Youn HJ, Chae SC, et al. Evaluation of the dose-response relationship of amlodipine and losartan combination in patients with essential hypertension: an 8-week, randomized, double-blind, factorial, phase II, multicenter study. Am J Cardiovasc Drugs. 2012;12(1):35–47. | ||
Kim SH, Ryu KH, Lee NH, et al. Efficacy of fixed-dose amlodipine and losartan combination compared with amlodipine monotherapy in stage 2 hypertension: a randomized, double blind, multicenter study. BMC Res Notes. 2011;4:461. | ||
Lee SY, Kim JR, Jung JA, Huh W, Bahng MY, Ko JW. Bioequivalence evaluation of two amlodipine salts, besylate and orotate, each in a fixed-dose combination with olmesartan in healthy subjects. Drug Des Devel Ther. 2015;9:2811–2817. | ||
Davison E, Wells JID, inventor; Pfizer Inc. Pharmaceutically, assignee. Salts of amlodipine. European patent EP 0244944 B1. Nov 7, 1989. | ||
Kim SH, Kim YD, Lim DS, et al. Results of a phase III, 8-week, multicenter, prospective, randomized, double-blind, parallel-group clinical trial to assess the effects of amlodipine camsylate versus amlodipine besylate in Korean adults with mild to moderate hypertension. Clin Ther. 2007;29(9):1924–1936. | ||
Karalis V, Macheras P. Examining the role of metabolites in bioequivalence assessment. J Pharm Pharm Sci. 2010;13(2):198–217. | ||
US Food and Drug Administration, Center for Drug Evaluation and Research. Guidance for Industry: Bioavailability and Bioequivalence Studies for Orally Administered Drug Products – General Considerations. Food and Drug Administration, Center for Drug Evaluation and Research; Rockville, 2003. | ||
European Medicines Agency (EMA), Committee for Medicinal Products for Human Use. Guideline on the Investigation of Bioequivalence. EMA, Committee for Medicinal Products for Human Use; London: 2010. | ||
US Food and Drug Administration [webpage on the Internet], Guidance on Losartan Potassium; 2008. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm088645.pdf. Accessed January 25, 2016. | ||
Midha KK, Rawson MJ, Hubbard JW. The role of metabolites in bioequivalence. Pharm Res. 2004;21(8):1331–1344. | ||
European Medicines Agency (EMA), Committee for Human Medicinal Products (CHMP) [webpage on the Internet]. Questions & Answers: Positions on Specific Questions Addressed to the Pharmacokinetics Working Party (PKWP). Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002963.pdf. Accessed January 25, 2016. | ||
Koroboki E, Manios E, Psaltopoulou T, et al. Circadian variation of blood pressure and heart rate in normotensives, white-coat, masked, treated and untreated hypertensives. Hellenic J Cardiol. 2012;53(6):432–438. | ||
Grossman E. Ambulatory blood pressure monitoring in the diagnosis and management of hypertension. Diabetes Care. 2013;36(suppl 2):S307–S311. | ||
Higashi Y, Nakagawa K, Kimura M, et al. Circadian variation of blood pressure and endothelial function in patients with essential hypertension: a comparison of dippers and non-dippers. J Am Coll Cardiol. 2002;40(11):2039–2043. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.