Back to Journals » Journal of Pain Research » Volume 8
Comparison of single-dose and multiple-dose pharmacokinetics between two formulations of hydrocodone bitartrate/acetaminophen: immediate-release versus biphasic immediate- release/extended release
Authors Devarakonda K, Kostenbader K, Giuliani MJ, Young J
Received 19 December 2014
Accepted for publication 20 April 2015
Published 9 September 2015 Volume 2015:8 Pages 607—618
DOI https://doi.org/10.2147/JPR.S79578
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Richard Robinson
Krishna Devarakonda,1 Kenneth Kostenbader,2 Michael J Giuliani,3 Jim L Young4
1Department of Clinical Pharmacology, Mallinckrodt Pharmaceuticals, Hazelwood, MO, USA; 2Independent Pharmaceuticals Professional, Mallinckrodt Pharmaceuticals, Hazelwood, MO, USA; 3Research and Development, 4Clinical Affairs and Program Management, Mallinckrodt Pharmaceuticals, Hazelwood, MO, USA
Objective: This study aimed to compare the single-dose and steady-state pharmacokinetics (PK) of biphasic immediate-release (IR)/extended-release (ER) hydrocodone bitartrate (HB)/acetaminophen (APAP) and IR HB/APAP.
Setting: The study was conducted in a contract research center.
Participants: The study included healthy adults.
Interventions: In a three-way crossover study, Study 1, participants received the following treatments: (A1) a single dose of IR/ER HB/APAP 7.5/325 mg one tablet, followed by one tablet every 12 hours (q12h); (B1) a single dose of IR/ER HB/APAP 7.5/325 mg two tablets, followed by two tablets q12h; (C1) a single dose of IR HB/APAP 7.5/325 mg two tablets (one tablet at hours 0 and 6), followed by one tablet q6h. In a two-way crossover study, Study 2, participants received the following treatments: (A2) an initial dose of IR/ER HB/APAP 7.5/325 mg three tablets, followed by two tablets q12h; (B2) three doses of IR HB/APAP 7.5/325 mg one tablet q4h, followed by one tablet q6h.
Main outcome measures: PK values were compared, and adverse events were assessed.
Results: Single-dose and steady-state area under the concentration–time curves for hydrocodone and APAP were similar for IR/ER and IR HB/APAP; the steady-state peak plasma concentrations (Cmax) at steady state were also similar, but single-dose Cmax for hydrocodone was lower for IR/ER HB/APAP. For most PK parameters, 90% confidence intervals for geometric least squares mean ratios were not meaningfully different (80%–125%). Steady state was achieved in 2-3 days for IR/ER HB/APAP and in 2 days for IR HB/APAP. Median time to Cmax was longer for IR/ER HB/APAP versus IR HB/APAP (P<0.05). Adverse events were similar across treatments.
Conclusion: PK outcomes and tolerability were similar for IR/ER HB/APAP and IR HB/APAP.
Keywords: acute pain, extended release, fixed-dose combination, hydrocodone, pharmacokinetics, opioid analgesic
Introduction
Coadministration of acetaminophen (APAP) with hydrocodone bitartrate (HB) has long been known to provide additive analgesic effects while reducing the risk of dose-related adverse events (AEs) associated with each component as monotherapy.1,2 Acute pain severe enough to require opioid therapy is typically treated with an immediate-release (IR) formulation, such as IR HB/APAP, to ensure rapid onset of relief. However, extended-release (ER) opioids provide less-frequent dosing, fewer troughs and peaks of plasma opioid concentration, and potentially less sleep disruption.3
Biphasic IR/ER HB/APAP 7.5/325 mg tablets (MNK-155; Mallinckrodt Pharmaceuticals, Hazelwood, MO, USA) are being developed for management of moderate to severe acute pain for which nonopioid analgesics are inadequate. IR/ER HB/APAP has a biphasic delivery mechanism that consists of 1) an IR layer delivering 25% of the HB and 50% of the APAP for rapid pain relief and 2) an ER layer delivering the remainder of the HB/APAP over a 12-hour dosing period. The APAP dose was selected in accordance with a request from the US Food and Drug Administration that drug manufacturers limit APAP to 325 mg per dosage unit in order to limit the risk of liver injury,4 which has been observed in patients receiving more than 4 g total dose per day.5 A similarly formulated IR/ER oxycodone/APAP tablet has been approved for management of moderate to severe acute pain for which alternative treatment options are inadequate.6
To ensure safety, it is essential to characterize the extent and rate of exposure to hydrocodone and APAP during treatment with IR/ER HB/APAP. This manuscript presents the combined data from two Phase 1 clinical trials to describe the single-dose and steady-state pharmacokinetics (PK) of the hydrocodone and APAP components of IR/ER HB/APAP and IR HB/APAP. In addition, the safety of IR/ER HB/APAP tablets administered for 4.5 days was assessed in healthy adult participants.
Methods
Study design
Study 1 was a single-center, open-label, randomized, three-period crossover study that evaluated the single- and multiple-dose PK of IR/ER HB/APAP. Participants underwent screening to determine eligibility and were randomly assigned to receive single- and multiple-dose treatment in sequences of A1, B1, and C1, with a minimum 7-day washout interval separating each period. During each of the three treatment periods, participants were confined to the study facility for approximately 8 days.
Study 2 was a single-center, open-label, randomized, two-period crossover study that evaluated the steady-state PK of IR/ER HB/APAP. This study included a screening visit and two treatment periods. However, unlike Study 1, the two confinement periods during the treatment periods were approximately 6 days each, and the washout periods between treatments were 14 days before period 1 and 6 days before period 2. Participants received a 7-day poststudy telephone call.
This research was carried out in accordance with the International Conference on Harmonization Good Clinical Practice (GCP) guidelines, ethical principles that have their origin in the Declaration of Helsinki, and US clinical research regulations and guidelines. Prior to initiation of these two single-center studies (both conducted at Pharmaceutical Product Development, LLC’s Phase 1 Clinic, Austin, TX, USA), the protocol for each study was reviewed and approved by the IntegReview Ethical Review Board (Austin, TX, USA).
Participants
In both studies, eligible participants included normal healthy men and nonlactating/nonpregnant women from 18 years to 55 years of age with a body mass index between 19 kg/m2 and 30 kg/m2 and a body weight of ≥50 kg for women and ≥59 kg for men.
Key exclusion criteria included use of any prescription medication, over-the-counter drugs, vitamins, minerals, or other supplements within 14 days of study check-in; use of or treatment for drugs or alcohol, narcotic addiction or positive urine test for drugs of abuse, or use of nicotine-containing products; or a history of any condition that could interfere with the absorption, distribution, metabolism, or excretion of the study drug. Individuals were also excluded if they had a history of gastric bypass surgery or gastric band implantation because of the possibility that such procedures might interfere with the gastroretentive technology responsible for the ER properties of IR/ER HB/APAP.
Study treatments
In Study 1, a single-dose analysis was performed on study Day 1 and a multiple-dose analysis was performed over study days 3−7 (Figure 1A). For this reason, study Day 3 was the first day of multiple-dose treatment and study hour 72 was taken as time =0 with respect to the calculation of multiple-dose PK parameters such as attainment of steady state. Participants were randomly assigned to receive each of the following three treatments under fasted conditions.
- (A1) IR/ER HB/APAP 7.5/325 mg one tablet at hour 0, then fasted 4 hours posttreatment. On Day 3 (hour 72), dosing every 12 hours (q12h) began following ≥1-hour fast; last dose was administered at hour 144.
- (B1) IR/ER HB/APAP 7.5/325 mg two tablets (15/650 mg total dose) at hour 0, then fasted 4 hours posttreatment. On Day 3 (hour 72), dosing q12h began following ≥1-hour fast; last dose was administered at hour 144.
- (C1) IR HB/APAP 7.5/325 mg (Norco®; Watson Laboratories, Inc., Corona, CA, USA) one tablet at hours 0 and 6 on Day 1, then fasted for 4 hours postdosing. On Day 3 (hour 72), dosing every 6 hours (q6h) began following ≥1-hour fast; last dose was administered at hour 144.
 | Figure 1 Timelines of (A) Study 1 and (B) Study 2. |
In Study 2, a multiple-dose analysis was performed over study days 1−7 (Figure 1B). For this reason, study Day 1 was the first day of the multiple-dose treatment and study hour 0 was time =0 with respect to the calculation of multiple-dose PK parameters such as attainment of steady state. Participants were randomly assigned to receive each of the following treatments under fasting conditions.
- (A2) Initial dose of IR/ER HB/APAP 7.5/325 mg three tablets (22.5/975 mg total dose), followed 12 hours later by two tablets taken q12h (eight doses) for 4.5 days.
- (B2) IR HB/APAP 7.5/325 mg one tablet taken q4h for three doses, followed at 12 hours after the last dose by one tablet taken q6h (16 doses) for 4.5 days.
Treatment A2 corresponds to the planned commercial dosing regimen that incorporates this three-tablet initial dose. Treatment B2 differs from the IR regimen in Study 1, for which patients received their initial tablets at 6-hour intervals rather than 4-hour intervals.
Plasma sampling and assessments
In Study 1, blood samples for PK assessment were taken as follows:
- Day 1: Samples were taken at 15 minutes, 30 minutes, 45 minutes, 1 hour, 2 hours, 3 hours, 4 hours, 6 hours, 8 hours, 9 hours, 10 hours, 12 hours, 16 hours, and 18 hours postdose; an additional sample was collected 30 minutes after the dose given at 6 hours (treatment C1).
- Day 2: Samples were taken at 24 hours and 36 hours.
- Days 3–6: Samples were taken before the morning dose (48 hours, 72 hours, 96 hours, and 120 hours).
- Day 7: Samples were taken predose; at 15 minutes, 30 minutes, and 45 minutes after the dose given at 144 hours; and at hours 145, 146, 147, 148, 150, 152, 154, and 156, with additional sample collected 30 minutes after the dose given at 150 hours (treatment C1).
At study exit (hour 48 for single-dose period, Day 8 for multiple-dose period) or early termination of study participation, participants underwent end-of-study assessments, including physical examinations and AE assessment.
In Study 2, blood samples for PK assessments were taken at the following times:
- Day 1: Samples were taken before dosing; at 15 minutes, 30 minutes, 45 minutes; and at 1 hour, 2 hours, 3 hours, 4 hours, 6 hours, 8 hours, 10 hours, and 12 hours after the initial dose. For treatment B2, additional samples were collected after the second dose at 4.5 hours and after the third dose at 8.5 hours.
- Days 2–4: Samples were taken before the morning dose on Day 2 (hour 24), Day 3 (hour 48), and Day 4 (hour 72).
- Day 5: Samples were taken before dosing (hour 96) and at 96:15 hours, 96:30 hours, 96:45 hours, 97 hours, 98 hours, 99 hours, 100 hours, 102 hours, 104 hours, 106 hours, 108 hours, and 112 hours. For treatment B, additional samples were collected at 102:25 hours, 102:5 hours, and 102:75 hours.
- Days 6–7: Samples were taken on Day 6 at hours 120 and 132 and on Day 7 at hour 144.
AEs were monitored throughout both studies and rated based on severity and potential relationship to study drug. Patients were also monitored for changes in clinical laboratory test values (hematology, clinical chemistry, and urinalysis), vital signs, and pulse oximetry. Physical examinations were performed and 12-lead electrocardiograms were taken at each study visit. In both studies, any patient who experienced the AE of vomiting was discontinued from the study.
Statistical analysis
Analysis of variance was used to compare treatments A1, B1, and C1 (Study 1) or treatments A2 and B2 (Study 2). Geometric least squares (LS) mean, percentage ratio of the geometric LS mean, and 90% confidence interval (CI) for the ratio of the geometric LS mean (treatment comparisons, A1/B1; A1/C1; B1/C1 [Study 1] and A2/B2 [Study 2]) were calculated. A 90% CI of the geometric LS mean ratio that was fully within 80% to 125% indicated no meaningful difference between treatments.
In both studies, steady-state attainment was confirmed using the Helmert transformation method, which compares the geometric mean concentration on a given study day to the geometric mean concentration pooled over all the remaining study days. Comparison continues until the comparison is no longer statistically significant (α=0.05), at which time steady state has been attained. In Study 1, Wilcoxon signed-rank test was used to analyze the difference between treatments for median time to peak concentration (tmax) and steady-state tmax (tssmax). In Study 2, Wilcoxon signed-rank test was used to analyze the median difference between treatments for tssmax.
Results
Study populations
Demographics and baseline characteristics for participants in the two studies are listed in Table 1. The populations of both studies were generally equally divided between men and women. Almost all participants were white or black, with a mean age of 31.8 years (Study 1) and 36.7 years (Study 2). As required by the study protocol, no pediatric or geriatric participants were included and none were substantially overweight or underweight.
 | Table 1 Baseline demographic and clinical characteristics |
In Study 1, 48 participants enrolled, four discontinued due to AEs (vomiting, n=3; anxiety, n=1), and 44 (91.7%) completed all three treatment phases. In Study 2, 26 participants enrolled, seven discontinued due to AEs (vomiting), and 19 (73.1%) completed all treatment periods. There were no notable differences in study completion rates based on age, race, weight, height, or body mass index. Men were slightly more likely to complete treatment compared with women. In the two studies combined, eleven participants discontinued due to AEs. Ten of these participants (nine female) discontinued after vomiting, as required by the study protocols. The remaining participant discontinued owing to moderate anxiety.
Study 1: single-dose and steady-state PK
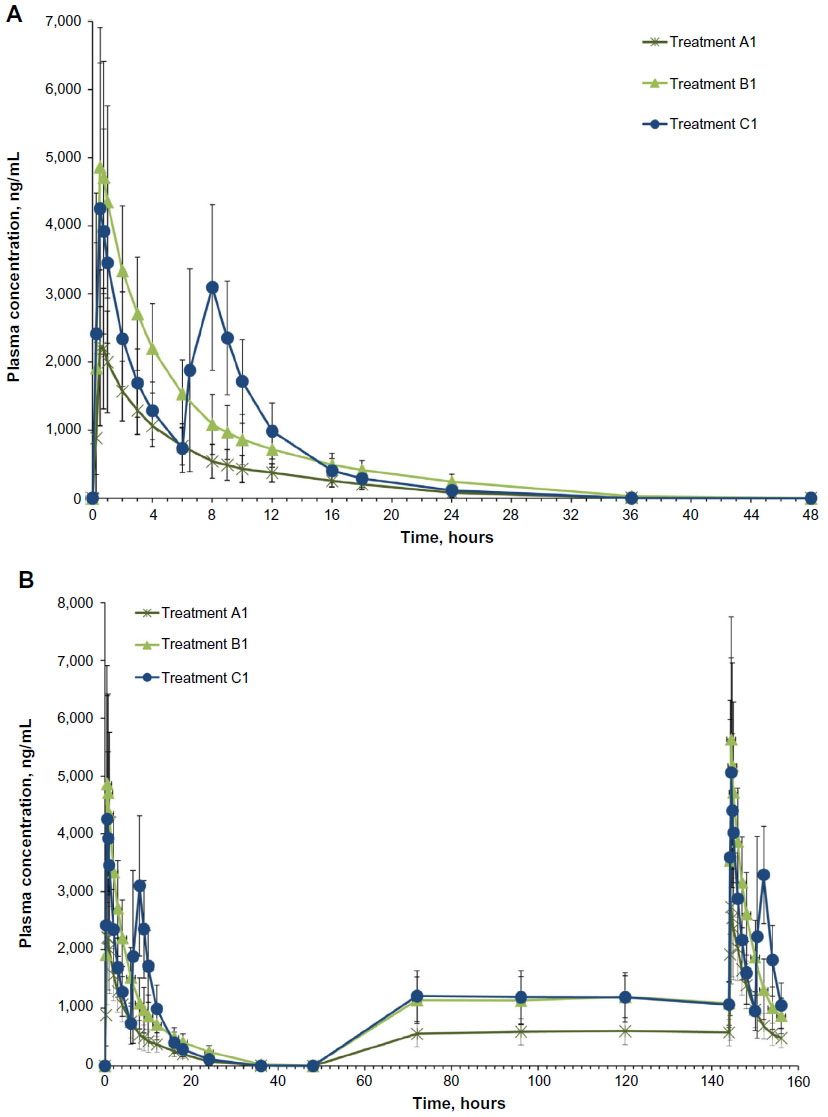
Hydrocodone: single dose
After a single dose, plasma hydrocodone concentrations increased rapidly and remained detectable for 48 hours with all three treatments (Figure 2A). Single-dose hydrocodone tmax was 3.1 hours for treatment A1, 3.4 hours for treatment B1, and 7.2 hours for treatment C1 (Table 2). Counterintuitively, the single-dose tmax value was longer for IR HB/APAP (C1) versus IR/ER HB/APAP (A1, B1) because Study 1 defined a “single dose” of IR HB/APAP as two tablets separated by 6 hours (ie, dosed at hours 0 and 6). Thus, for single-dose IR HB/APAP, Cmax refers to the higher of the two peaks observed with this dosing protocol, which occurred after the IR HB/APAP tablet administered at hour 6. In contrast, in Study 2 on Day 1, Cmax for IR HB/APAP followed the tablet administered at hour 0 and tmax was reported as 0.75 hours in that study.
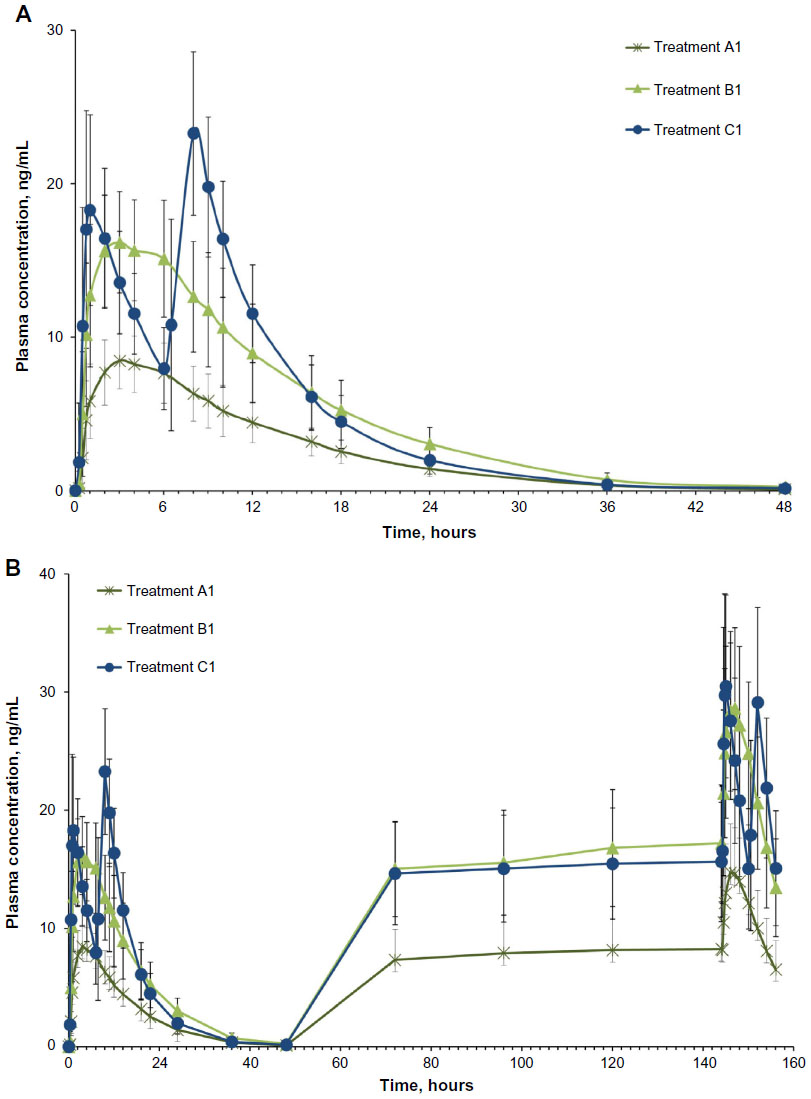 | Figure 2 Mean plasma concentrations of hydrocodone in (A) single-dose and (B) steady-state completers. |
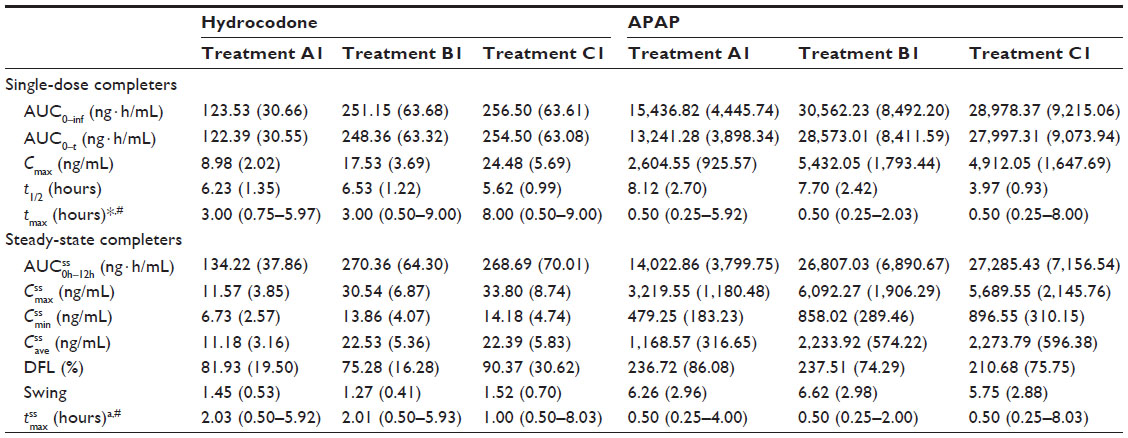 | Table 2 Mean (SD) plasma pharmacokinetic parameters by treatment (Study 1) |
 . #Median (range).
. #Median (range). , steady-state area under the concentration–time curve from time 0 to 12 hours postdose; AUC0–inf, AUC from time 0 extrapolated to infinity; AUC0–t, AUC from time 0 to time t;
, steady-state area under the concentration–time curve from time 0 to 12 hours postdose; AUC0–inf, AUC from time 0 extrapolated to infinity; AUC0–t, AUC from time 0 to time t; 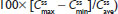 ; IR/ER HB/APAP, immediate-release/extended-release hydrocodone bitartrate/acetaminophen; IR HB/APAP, immediate-release hydrocodone bitartrate/acetaminophen; SD, standard deviation.
; IR/ER HB/APAP, immediate-release/extended-release hydrocodone bitartrate/acetaminophen; IR HB/APAP, immediate-release hydrocodone bitartrate/acetaminophen; SD, standard deviation.Because the single dose administered in treatment A1 of HB/APAP (total dose of 7.5/325 mg) was half that of treatments B1 and C1 (total dose of 15/650 mg), dose-normalized values were used for comparison of treatments. The 90% CIs of dose-normalized geometric LS mean ratios for AUC0–t and AUC0–inf were within the prespecified 80%–125% range for all treatment comparisons, indicating that dose-normalized exposure to hydrocodone was equivalent (Table 3). The dose-normalized geometric LS mean ratio of Cmax was 26% lower for treatment A1 versus C1 (73.53 ng/mL/mg) and 28% lower for treatment B1 versus C1 (71.86 ng/mL/mg) compared with the ratio for treatment A1 versus B1 (102.33 ng/mL/mg), with a 90% CI that was below the lower threshold for treatment equivalence (Table 3).
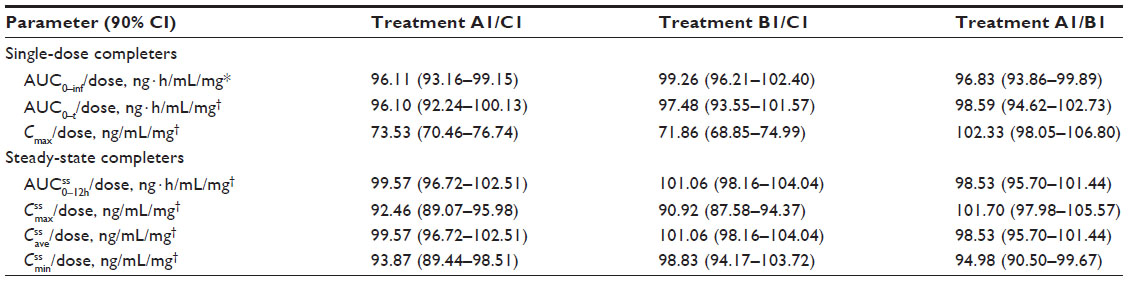 | Table 3 Dose-normalized hydrocodone geometric LS mean ratios and 90% CIs (Study 1) |
Hydrocodone: steady state
Hydrocodone steady state was attained on the third day of the multiple-dose treatment (study Day 5) with treatments A1 and C1 and on the fourth day of the multiple-dose treatment (study Day 6) with treatment B1 (Figure 2B). Time to steady-state median 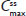 (
(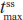 ) was 2 hours for treatments A1 and B1 and 1 hour for treatment C1 (Table 2). Steady-state, dose-normalized hydrocodone exposure was similar between all treatments, as the 90% CIs of mean ratios for geometric LS mean ratios for the AUCss value and were within the prespecified 80%–125% range (Table 3).
) was 2 hours for treatments A1 and B1 and 1 hour for treatment C1 (Table 2). Steady-state, dose-normalized hydrocodone exposure was similar between all treatments, as the 90% CIs of mean ratios for geometric LS mean ratios for the AUCss value and were within the prespecified 80%–125% range (Table 3).
APAP: single dose
With each treatment, plasma concentrations of APAP were detectable through 24 hours postdose but declined to <1% by 36 hours (Figure 3A). Single-dose Cmax of APAP was achieved in 0.50 hour for all treatments (Table 2). In contrast to the hydrocodone PK results, APAP Cmax followed administration of the IR HB/APAP tablet at hour 0. The single-dose 90% CIs of the ratios of dose-normalized geometric LS mean ratios for AUC0–t, AUC0–inf, and Cmax for all treatments were within the prespecified 80%–125% range (Table 4).
 | Figure 3 Mean plasma concentrations of acetaminophen in (A) single-dose and (B) steady-state completers. |
 | Table 4 Dose-normalized acetaminophen geometric LS mean ratios and 90% CIs (Study 1) |
APAP: steady state
Steady state for APAP was attained on the first day of multiple-dose treatment (study Day 4) with treatment B1 and on the second day of multiple-dose treatment (study Day 5) with treatment A1, whereas it was not attained after 4 days (end of study, Day 7) with treatment C1 (Figure 3B). The of APAP was 0.50 hours for all treatments (Table 2). The 90% CIs of the dose-normalized geometric LS mean ratios for 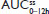 , ,
, , 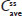 , and
, and 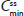 were each within the prespecified 80%–125% range (Table 4).
were each within the prespecified 80%–125% range (Table 4).
Study 2: steady-state PK
Hydrocodone
Steady state was achieved by Day 3 for treatment A2 and Day 2 for treatment B2. was achieved rapidly for both treatments (Figure 4A). Steady-state for hydrocodone was significantly longer for IR/ER HB/APAP compared with IR HB/APAP (2.00 hours versus 1.00 hour; P<0.05, Table 5). and were similar for both treatments. At steady state, the ratios of the geometric LS means of nearly all parameters were entirely within the 80%–125% CI intervals, implying no treatment difference; however, the 90% CI for swing (ie, 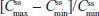 ) extended slightly above the upper threshold (125%) of the bioequivalence range (Table 6). The differences between exposures to hydrocodone with IR/ER HB/APAP versus IR HB/APAP were not considered clinically meaningful.
) extended slightly above the upper threshold (125%) of the bioequivalence range (Table 6). The differences between exposures to hydrocodone with IR/ER HB/APAP versus IR HB/APAP were not considered clinically meaningful.
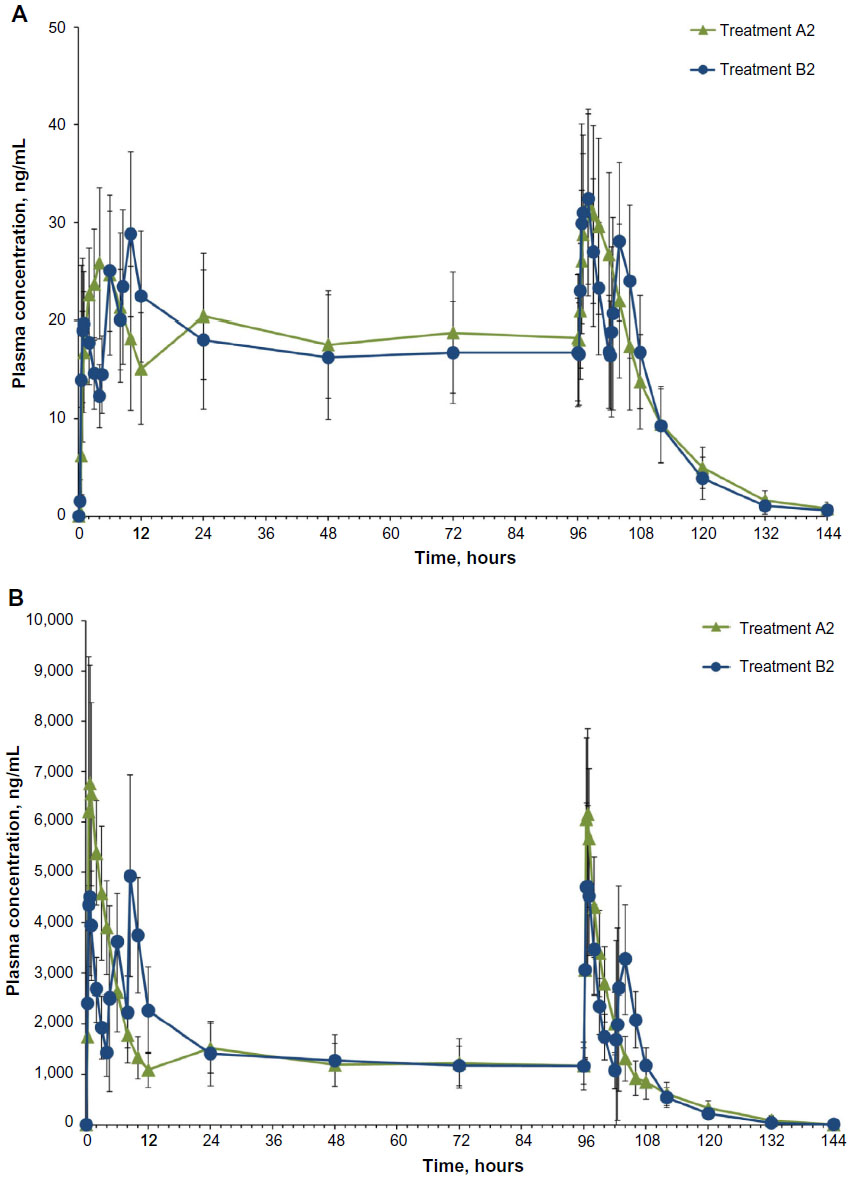 | Figure 4 Mean plasma concentrations of (A) hydrocodone and (B) acetaminophen by treatment in completers. |
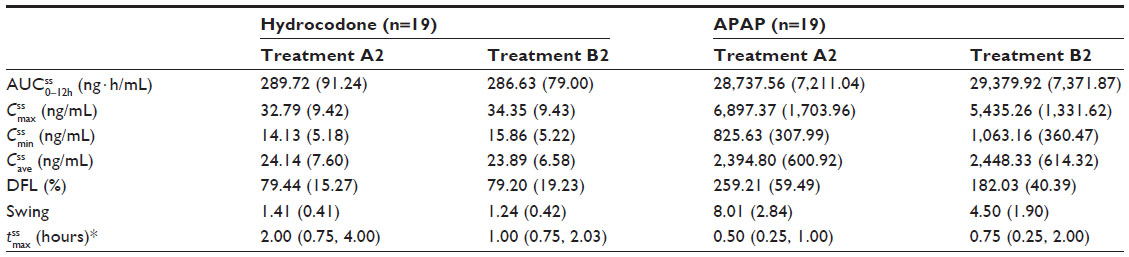 | Table 5 Mean (SD) plasma pharmacokinetic parameters by treatment (Study 2) |
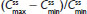 .
.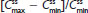 ); IR/ER HB/APAP, immediate-release/extended-release hydrocodone bitartrate/acetaminophen; IR HB/APAP, immediate-release hydrocodone bitartrate/acetaminophen; SD, standard deviation.
); IR/ER HB/APAP, immediate-release/extended-release hydrocodone bitartrate/acetaminophen; IR HB/APAP, immediate-release hydrocodone bitartrate/acetaminophen; SD, standard deviation.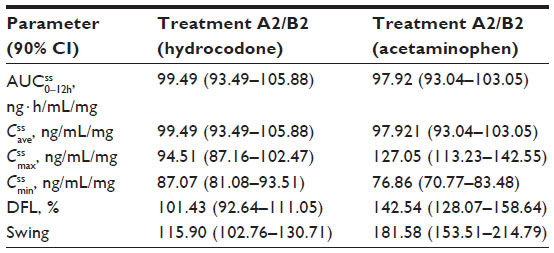 | Table 6 Geometric LS mean ratios and 90% CIs for hydrocodone and acetaminophen (Study 2) |
Acetaminophen
Plasma concentrations for APAP are shown in Figure 4B. As with hydrocodone, steady state for APAP was achieved by Day 3 for treatment A2 and Day 2 for treatment B2. At steady state, no significant difference in median was observed between treatments (Table 5). The geometric LS mean ratios for and for APAP were entirely within the 80%–125% CI intervals, implying no treatment difference in overall exposure (Table 6). Treatment A2 showed slightly higher and lower for APAP compared with treatment B2. Degree of fluctuation and swing were greater for treatment A2 compared with treatment B2.
Safety and tolerability
Study 1
Treatment-emergent AEs (TEAEs) were reported by 33 of 48 (68.8%) participants, and AEs were assessed to be treatment related in 27 of 48 (56.3%) participants. Four participants discontinued early due to AEs. Three discontinued due to vomiting: one each after receiving the first dose of treatment A1, B1, or C1. A fourth participant discontinued because of moderate anxiety during treatment B1.
The most prevalent TEAEs among all participants were nausea (29.2%), headache (22.9%), dizziness (18.8%), fatigue (12.5%), and pruritus (12.5%; Table 7). There were no serious or severe TEAEs. No clinically significant trends or changes in individual participants with respect to laboratory analyses, vital signs, electrocardiograms, or physical examination were reported.
 | Table 7 Treatment-emergent adverse events |
Study 2
The overall frequency of TEAEs and TEAEs reported by ≥5% of participants in either treatment group is summarized in Table 7. In seven participants (two for treatment A2 and five for treatment B2), vomiting led to early termination of study participation, as required by the study protocol. All of these participants who terminated early vomited during treatment period 1; of these, two individuals were withdrawn after receiving IR/ER HB/APAP (after one and three doses) and five individuals after receiving between 2 and 19 doses of IR HB/APAP.
TEAEs occurring in ≥5% of participants during treatment A2 included nausea, vomiting, dizziness, and headache, and during treatment B2, TEAEs included nausea, vomiting, and headaches. Vomiting was less frequent during treatment A2.
There were no severe or serious TEAEs reported. No abnormalities in mean clinical laboratory tests were noted by investigators. A single participant developed urogenital trichomoniasis of mild intensity that was not deemed related to study drug. Overall mean vital signs from baseline were similar for all treatments and at all points assessed. A single participant had a pulse oximetry reading of 82% while receiving treatment A2. Approximately 2 hours later, the participant’s reading was 100%; the event was not reported as a TEAE. No changes in physical examination findings were reported, and any electrocardiogram abnormalities observed during the study were apparent at screening rather than being treatment emergent.
Discussion
In two clinical studies assessing single-dose and steady-state PK at equal doses, total exposure of hydrocodone and APAP from IR/ER HB/APAP was equivalent to IR HB/APAP as estimated by AUC0–inf and , as assessed by dose-normalized values for Study 1 and absolute values for Study 2. The single-dose analysis (Study 1) found that the dose-normalized geometric LS mean ratio of Cmax was 26% lower for treatment IR/ER HB/APAP (7.5/325 mg total dose) versus IR HB/APAP (15/650 mg total dose) and 28% lower for treatment IR/ER HB/APAP (15/650 mg total dose) versus IR HB/APAP (15/650 mg total dose), with a 90% CI that was below the lower threshold for treatment equivalence. However, the Study 1 steady-state values were equivalent for all treatments, normalized for dose. In steady-state Study 2, geometric LS mean ratios of and for hydrocodone and APAP were equivalent for IR/ER HB/APAP and IR HB/APAP administered at doses considered therapeutically comparable. Although swing for hydrocodone was slightly greater for IR/ER HB/APAP in Study 2, this difference was not considered clinically meaningful.
TEAEs observed during both studies were typical of those reported with low-dose opioid treatment. In Study 1, nausea was more frequent with IR HB/APAP than with IR/ER HB/APAP, and both nausea and vomiting were more frequent with IR HB/APAP in Study 2. In Study 2, the frequency of nausea and vomiting was higher than in Study 1 for IR/ER HB/APAP and for IR HB/APAP. Possibly, the increased frequency of these AEs was related to the difference in dosing regimens between the two studies, with Study 2 using a three-tablet initial dose of IR/ER HB/APAP and shortening of the initial dosing interval for IR HB/APAP. No serious or severe TEAEs were reported with any of the treatments.
The frequency of AEs observed in trials of healthy volunteers may not adequately represent AEs that may be observed in complicated patients in clinical practice, and AE rates reported in different trials cannot reliably compare the safety and tolerability of analgesic agents. However, in the absence of adequately powered, head-to-head clinical trials, noting the AE rates reported with different agents under similar conditions has some value. Generally, the tolerability results for IR/ER HB/APAP obtained in the current analyses appear to be similar to tolerability findings in studies of IR HB fixed-dose combination analgesics. In a study of patients with postoperative dental pain administered at a single dose of IR HB/APAP 7.5/500 mg, the frequency of nausea was 15.9% and that for vomiting was 7.9%.7 In patients with acute pain following a fracture who received a single dose of IR HB/APAP 5/325 mg followed by up to 12 additional doses over 3 days, the frequency of nausea was 35% and that for vomiting was 11%.8
A limitation of this study is that it was conducted in healthy volunteers and not in patients with medical conditions that might alter the PK of active treatments. The study also did not include pediatric or geriatric participants, individuals who were obese or underweight patients, or substantial numbers of individuals from races other than black and white. Moreover, the study populations were small, making it necessary to confirm findings in well-designed trials enrolling more participants.
In summary, administration of single and multiple doses of IR/ER HB/APAP was associated with similar levels of overall drug exposure compared with IR HB/APAP at similar doses.
Acknowledgments
Mallinckrodt Pharmaceuticals (Hazelwood, MO, USA) sponsored this research and funded editorial support provided by Jeffrey Coleman, MA, and Robert Axford-Gatley, MD, of C4 MedSolutions, LLC (Yardley, PA, USA), a CHC Group company.
Disclosure
All authors are employees of Mallinckrodt. Dr Kostenbader is a paid contract employee of Mallinckrodt. The authors report no other conflicts of interest in this work.
References
Beaver WT. Aspirin and acetaminophen as constituents of analgesic combinations. Arch Intern Med. 1981;141(3 Spec No):293–300. | |
Beaver WT, McMillan D. Methodological considerations in the evaluation of analgesic combinations: acetaminophen (paracetamol) and hydrocodone in postpartum pain. Br J Clin Pharmacol. 1980;10(Suppl 2):215S–223S. | |
McCarberg BH, Barkin RL. Long-acting opioids for chronic pain: pharmacotherapeutic opportunities to enhance compliance, quality of life, and analgesia. Am J Ther. 2001;8(3):181–186. | |
FDA drug safety communication: prescription acetaminophen products to be limited to 325 mg per dosage unit; boxed warning will highlight potential for severe liver failure; 2014. Available from: http://www.fda.gov/drugs/drugsafety/ucm239821.htm. Accessed March 21, 2014. | |
Watkins PB, Kaplowitz N, Slattery JT, et al. Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: a randomized controlled trial. JAMA. 2006;296(1):87–93. | |
Xartemis™ XR (oxycodone hydrochloride and acetaminophen extended- release tablets). Full Prescribing Information, Hazelwood, MO: Mallinckrodt Brand Pharmaceuticals, Inc.; 2014. | |
Litkowski LJ, Christensen SE, Adamson DN, Van Dyke T, Han SH, Newman KB. Analgesic efficacy and tolerability of oxycodone 5 mg/ibuprofen 400 mg compared with those of oxycodone 5 mg/acetaminophen 325 mg and hydrocodone 7.5 mg/acetaminophen 500 mg in patients with moderate to severe postoperative pain: a randomized, double-blind, placebo-controlled, single-dose, parallel-group study in a dental pain model. Clin Ther. 2005;27(4):418–429. | |
Marco CA, Plewa MC, Buderer N, Black C, Roberts A. Comparison of oxycodone and hydrocodone for the treatment of acute pain associated with fractures: a double-blind, randomized, controlled trial. Acad Emerg Med. 2005;12(4):282–288. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.