Back to Journals » Journal of Asthma and Allergy » Volume 14
Comparison of Gut Microbiota Between Infants with Atopic Dermatitis and Healthy Controls in Guangzhou, China
Authors Yu L, Deng YH, Huang YH ![]() , Ke HJ, Guo Y
, Ke HJ, Guo Y ![]() , Wu JL
, Wu JL ![]()
Received 12 February 2021
Accepted for publication 9 April 2021
Published 10 May 2021 Volume 2021:14 Pages 493—500
DOI https://doi.org/10.2147/JAA.S304685
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Luis Garcia-Marcos
Li Yu, Yu-Hong Deng, Yuan-Hui Huang, Hai-Jin Ke, Yong Guo, Jie-Ling Wu
Department of Children’s Health Care, Guangdong Women and Children Hospital, Guangzhou Medical University, Guangzhou, Guangdong, People’s Republic of China
Correspondence: Jie-Ling Wu; Yong Guo
Department of Children’s Health Care, Guangdong Women and Children Hospital, Guangzhou Medical University, Guangzhou, 511400, Guangdong, People’s Republic of China
Tel +86 20 39151521
Email [email protected]; [email protected]
Purpose: Evidence on the role of the gut microbiota in atopic dermatitis is inconsistent as human intestinal microbiota is influenced by geography. This cross-sectional study therefore aimed to compare differences in the gut microbiota of infants with atopic dermatitis and healthy infants in Guangzhou, China, by analyzing their stool.
Patients and Methods: The composition of the intestinal microbiota was analyzed from the stool samples of 20 infants with atopic dermatitis (AD group) and 25 healthy infants (non-AD group) (1– 6 months old), using full-length 16S rRNA gene sequencing. The Wilcoxon test was used to analyze the relative abundance of bacteria by phylum, family, genus, and species between groups; microbial community richness and diversity were compared between the two groups.
Results: There were no significant differences in the microbial community richness and diversity between the two groups. At the phylum level, 11 bacterial phyla were found; most sequences belonged to one of the three dominant bacterial phyla – Firmicutes, Proteobacteria, and Bacteroidetes. The top 10 microbes at the phylum, family, and genus levels showed no significant changes in their composition within the gut microbiota between the AD and non-AD groups. A decrease in the ratio of the Streptococcus genus was found in atopic dermatitis group when compared with healthy controls (p=0.048).
Conclusion: A decrease in the abundance of Streptococcus was found in children with AD. The role of Streptococcus in the development of AD needs to be confirmed in a large cohort study.
Keywords: allergy, infant, gut microbiome, Streptococcus
Introduction
Atopic dermatitis (AD) is characterized by recurrent, eczematous lesions and intense itching, with a prevalence of 10% in adults and 20% in children in developed countries,1 and the highest disease burden among skin diseases.2 The pathogenesis of AD is affected by barrier dysfunction and immune dysregulation; microbial dysbiosis, which affects the development of AD, has therefore gained more attention.3 However, reports regarding the influence of changes in intestinal microbiota on the development and progression of AD are inconsistent. Abrahamsson et al reported that low intestinal microbial diversity early in life was associated with subsequent AD.4 Wang et al found that in infants with eczema, Deinococcaceae and Bacteroidaceae were significantly enriched, while the co-abundance network of Clostridiaceae, Veillonellaceae, and Lactobacillaceae were reduced.5
Intestinal microbiota is affected by various factors, including age and geographical location;6 due to different traditions, lifestyles, and genetic backgrounds, there are differences in the human intestinal microbiomes from different geographical locations.7 Few studies have focused on the characteristics of the intestinal microbiota of infants with AD in Guangzhou, China; therefore, we conducted this case-control study to compare the abundance and characteristics of the intestinal microbiota between infants with and without AD.
Patients and Methods
This case-control study and its protocols were approved by the Medical Research Ethics Committee of the Guangdong Women and Children Hospital (No. 201,701,068), and carried out in accordance with the principles of the Helsinki Declaration. Written informed consent was obtained from the parents of each participant.
Healthy children who underwent routine physical examination and children with AD, aged between 1 and 6 months, were recruited through convenience sampling. The diagnosis of AD included pruritus, typical morphology and distribution, chronic or chronically relapsing course, and atopic personal or family history according to the Hanifin and Rajka criteria.8 All healthy control infants had no personal or family history of allergic disease. Exclusion criteria for research subjects were: intestinal diseases such as diarrhea and gastroenteritis; congenital genetic diseases; endocrine and metabolic diseases; blood system diseases; heart, liver, and kidney diseases or dysfunction; and having taken pro- or antibiotics within 1 month before the start of the study.
Stool samples were freshly collected and stored at −80°C until extraction. Total genome DNA was isolated from fecal samples using the cetyltrimethyl ammonium bromide/sodium dodecyl sulphate method. DNA samples were amplified by polymerase chain reaction (PCR) using primers 5′-AGAGTTTGATCCTGGCTCAG-3′ and 5′- GNTACCTTGTTACGACTT-3ʹ specific for the full-length of the 16S ribosomal RNA (rRNA) gene. Each PCR was performed in TransStart® FastPfu DNA Polymerase (TransGen Biotech). Same volume of 1X loading buffer was mixed with the PCR products which were then detected by 2% agarose gel operate electrophoresis. QIAquick@ Gel Extraction Kit (QIAGEN) was used to purify the mixture of PCR products. We generated sequencing libraries using SMRTbell TM Template Prep Kit (PacBio) and assessed the library quality by using the Qubit @ 2.0 Fluorometer (Thermo Scientific) and FEMTO Pulse system. At last, the full-length16S rRNA sequencing using a PacBio Sequel platform was conducted after library construction, as previously described.9 Processing of the raw data included filtering for a minimum of three passes, deleting the circular consensus sequence, removing sequences outside the expected amplicon size (min length: 1340 bp, max length: 1640 bp), and removing chimeric sequences. Clean reads were finally obtained. UPARSE software v7.0.1001 (Robert Edgar, http://drive5.com/uparse/) was used to assign sequences with a similarity of ≥97% to the same operational taxonomic units (OTUs).10 Based on the Mothur algorithm, SSUrRNA datasets from the Silva database (https://www.arb-silva.de/) were used to annotate taxonomic information, obtain taxonomic information, and count each sample at each classification level, ie, kingdom, phylum, class, order, family, genus, and species community composition.11,12 The raw sequences reported in this article have been deposited in the NCBI BioProject: PRJNA718759.
To identify community richness and diversity, the Chao1 and Shannon indices were calculated, whereas to evaluate differences in species complexity, the weighted UniFrac algorithm was used for beta diversity.13 We then assessed the similarity of the bacterial community compositions among samples using a Venn diagram. Continuous variables were presented as means and standard deviations, while categorical variables were presented as absolute values and relative frequencies. The Chi-squared test was applied to evaluate the characteristics of infants with and without AD. Alpha and beta diversity index differences between groups were analyzed using the t-test or Wilcoxon test. The relative bacterial abundance with respect to phylum, family, genus, and species between groups was analyzed using the Wilcoxon test; p-values <0.05 were considered statistically significant. All indices were calculated using QIIME (v1.9.1), and displayed with R software (v2.15.3, http://www.R-project.org).
Results
We recruited 45 children aged between 1 and 6 months – 20 children with AD and 25 healthy children; their characteristics are presented in Table 1. Stool samples were collected for 16S rRNA sequencing, and a total of 393,391 optimized reads were obtained. There were 131 unique OTUs for infants without AD, 123 unique OTUs for infants with AD, and 101 OTUs shared by both groups (Figure 1). The species accumulation boxplot suggests that the boxplot tends toward a more gradual position, indicating that the sample size was sufficient for analysis of fecal microbiota (Figure 2).
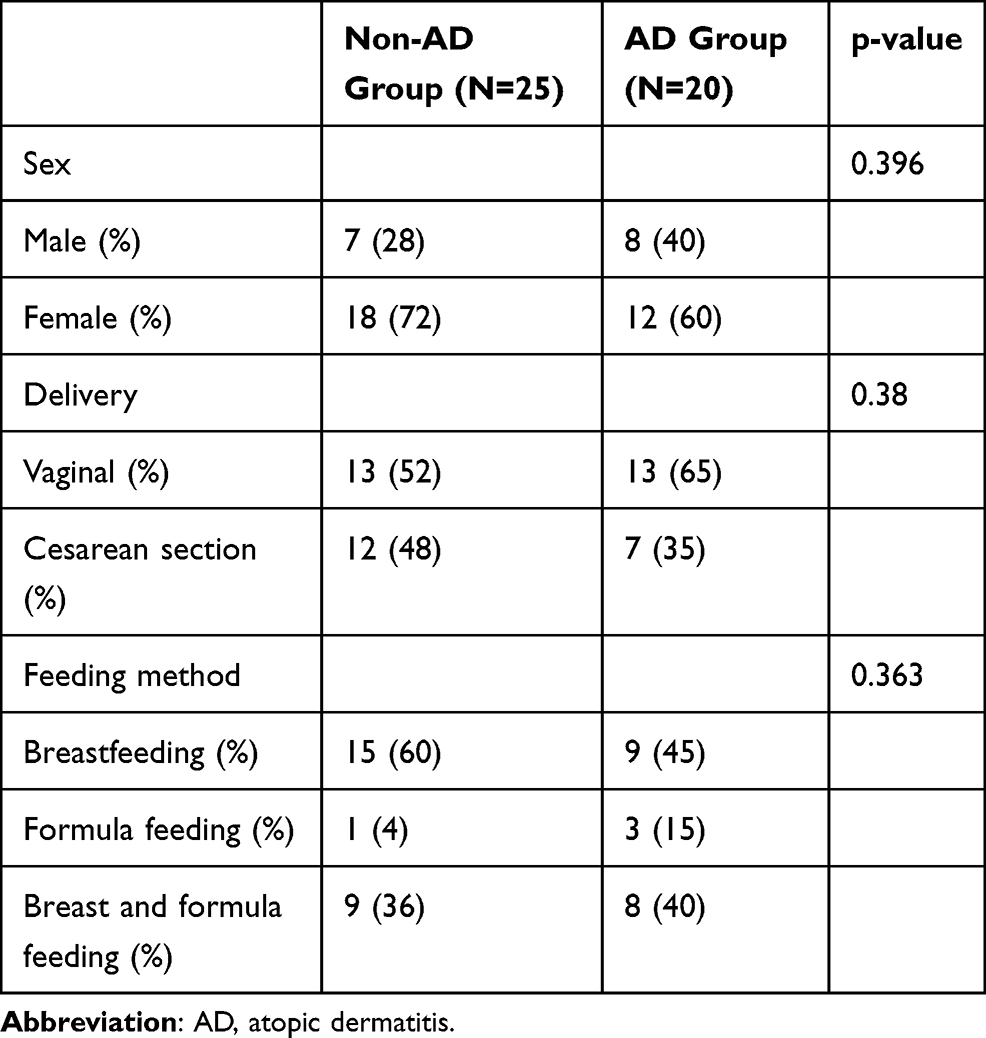 |
Table 1 Characteristics of Infants with and without Atopic Dermatitis |
 |
Figure 1 Venn diagram of the observed operational taxonomic units in the non-AD (NG) and AD (AG) groups. |
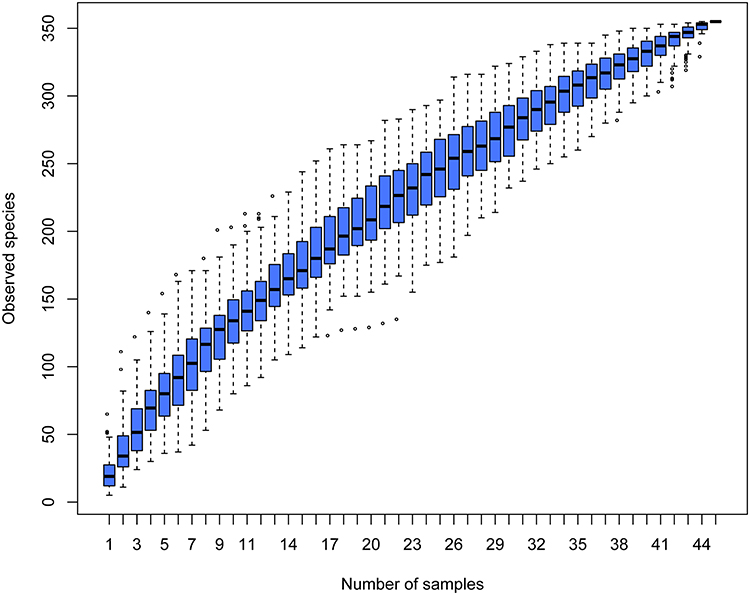 |
Figure 2 Species accumulation boxplot. |
The top 10 microbes at the phylum (Figure 3A), family (Figure 3B), genus (Figure 3C) and species (Figure 3D) levels showed no significant changes in composition within the gut microbiota between the non-AD and AD groups (Table 1S). At the phylum level, most sequences belonged to one of the three dominant bacterial phyla, ie, Firmicutes, Proteobacteria, and Bacteroidetes. These three bacterial phyla accounted for 97.44% and 97.34% of the total sequence in infants with and without AD, respectively. Other bacterial phyla, such as Verrucomicrobia, Actinobacteria, Fusobacteria, Nitrospirae, Rokubacteria, and Planctomycetes accounted for a much lower proportion than the dominant bacterial phyla; however, there were no differences in the proportions of Firmicutes, Proteobacteria, and Bacteroidetes between the two groups (p>0.05).
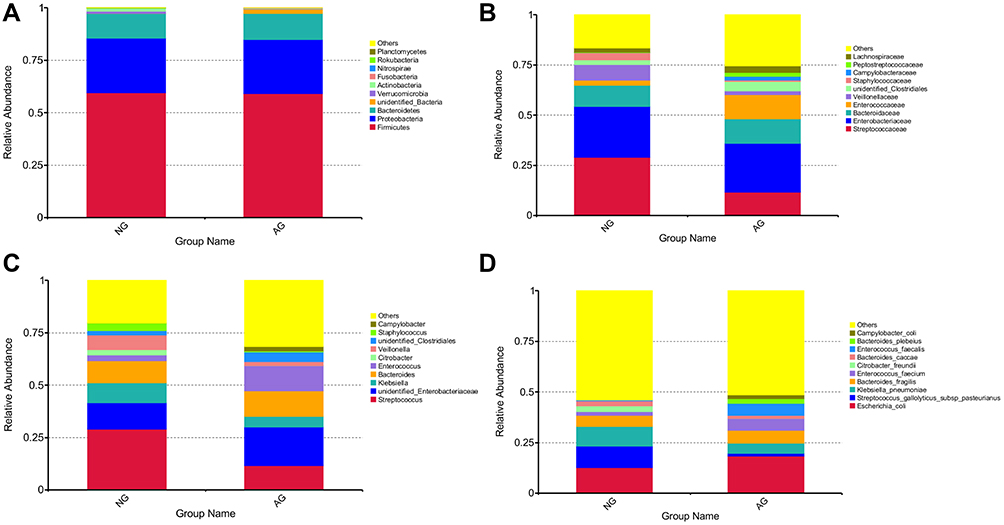 |
Figure 3 The top 10 relative abundances of bacteria at phylum (A), family (B) genus (C) and species (D) levels in fecal samples from the non-AD (NG) and AD (AG) groups. |
A total of 41 bacterial families were found in infants both with and without AD. The top 10 shared families included Streptococcaceae, Enterobacteriaceae, Bacteroidaceae, Enterococcaceae, Veillonellaceae, unidentified_Clostridiales Staphylococcaceae, Campylobacteraceae, Peptostreptococcaceae, and Lachnospiraceae; these accounted for 74.51% and 83.48% of the total sequence in infants with and without AD, respectively. Additionally, children with and without AD were found to have nine and six unique bacterial families, respectively.
In total, 51 bacterial genera were shared between the two groups; 19 unique bacterial genera were identified in infants with AD, and 21 unique bacterial genera were identified in infants without AD. The top 10 shared bacterial genera included Streptococcus, unidentified_Enterobacteriaceae, Klebsiella, Bacteroides, Enterococcus, Citrobacter, Veillonella, unidentified_Clostridiales, Staphylococcus, and Campylobacter, accounting for 68.34% and 79.67% of the total DNA sequence in infants with and without AD, respectively. Excluding Staphylococcus, Citrobacter, and Campylobacter, the shared bacterial genera accounted for greater than 1% of the total DNA sequence in the two groups. Of the 41 bacterial families and 51 bacterial genera in the gut microbiota, the ratio of Streptococcaceae family and the Streptococcus genus between the two groups were significantly different (Figure 4); we found significant decreases in the ratio of Streptococcus in the AD group (p=0.048).
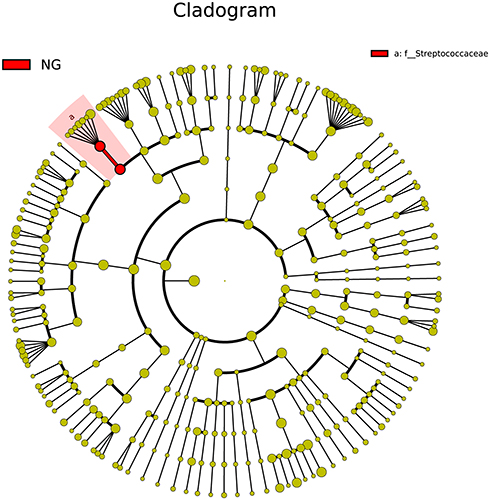 |
Figure 4 Cladogram of biomarkers for the AD group. The red circle represented biomarkers. Concentric rings from outside to inside were genus, family, order, class, and phylum. |
A total of 52 bacterial species were found in infants with and without AD. In addition, children with and without AD each had 26 unique bacterial species. Among the shared bacterial species, the most abundant in the AD group were Escherichia coli, Bacteroides fragilis, and Enterococcus faecalis; the most abundant bacterial species in the non-AD group were Escherichia coli, Streptococcus gallolyticus subsp pasteurianus, and Klebsiella pneumoniae. No significant differences were found between the two groups at the species level (p>0.05).
The Shannon and Chao1 indices were used to evaluate microbial community diversity and richness. According to the OTUs distribution, differences in the Chao1 index were insignificant, with averages of 28.5±22.9 and 25.0±15.4 in children without and with AD, respectively (p=0.5497). The mean Shannon index values of the infants without and with AD were 1.63±0.95 and 1.63±0.85, respectively (p=0.9867). The Chao1 and Shannon indices indicated that there was no significant difference in microbial diversity between infants with and without AD (Figure 5), while the analysis of differences in beta diversity indicated that there was no significant difference between the two groups (Figure 6).
 |
Figure 5 Distribution of the Chao1 index (A) and Shannon index (B) in the non-AD (NG) and AD (AG) group infants. |
 |
Figure 6 Gut microbial β diversity in the non-AD (NG) and AD group (AG) infants. |
Discussion
Although the pathogenesis of AD is both complex and multifactorial, recent evidence has implicated the gut microbiota. Similar to previous observations, the findings of this study demonstrated that at the phylum level, the intestinal microbiota mainly comprises Firmicutes, Proteobacteria, and Bacteroidetes.14,15
Chen et al analyzed the stools of children with food allergies and revealed that compared with healthy infants, the abundance of Firmicutes was higher in infants with food allergies, whereas the abundance of Bacteroidetes was lower.16 Abrahamsson et al reported that children with IgE-associated AD had a lower diversity of Bacteroidetes in the first month of age, and a lower diversity of Proteobacteria at 1 year of age.4 Inconsistent with these reports, our study did not reveal dramatic changes in the microbial composition of infants with AD in contrast to healthy infants at the phylum level.
Geographical location is a key factor that influences intestinal microbiota. For example, compared with children from central Thailand, children from northeastern Thailand have a greater abundance of Bacteroides fragilis, Clostridium leptum, Prevotella, Clostridium coccoides, Eubacterium rectale, and lactobacilli.17 Lin et al explored the gut microbiota of 394 healthy subjects from seven different cities in China; they found that the influence of ethnicity on the diversity and composition of the gut microbiota was less than that of geographical location.18 Fontana et al studied the intestinal microbiota of healthy volunteers of the same ethnicity from Apulia, Lazio, and Lombardy (three different regions of Italy), and found that both alpha and beta diversity did not change.19 This notion was consistent in our study, in which we showed that the gut microbial community richness in infants with and without AD remained similar; recruiting both infants with AD and healthy controls within the same geographic area may have contributed most to these results.
Colonization of the intestinal flora is a dynamic and complex process that continues to develop in the first few years of life.20 A previous study suggested that low intestinal microbial diversity in infants at one month of age is related to subsequent atopic eczema; however, we did not observe a change in the diversity of gut microbes in the AD group.4 Lee MJ compared the intestinal microbial composition of stool samples from healthy infants and infants with AD at the age of 6 months; no significant differences in OTUs, PCoA plot, and Shannon diversity indices were found between the two groups.21
Zhang et al assessed the relationship between the gut microbiome profile of healthy infants and infants with AD aged <3 years. They found that the decreased abundance of Bifidobacterium was not significant in children under 6 months, while the decline in microbial diversity was most obvious in children aged 2–3 years.22 A longitudinal study by Galazzo et al suggested that fecal microbiota diversity did not differ significantly between children 5–13 weeks of age, but increased at 21–31 weeks;23 the different ages of the research subjects are believed to have caused these inconsistent results.
Interestingly, the abundance of Streptococcus was significantly decreased in the AD group, compared with the healthy controls in our study. Similarly, Chen et al reported significant decreases in the number of streptococci in the food sensitization group.16 Park et al studied the intestinal microbiota of infants with AD and found that high levels of Akkermansia and low levels of Streptococcus were evident in transient AD cases.24 Although there were no significant differences in the microbial community richness and diversity between the AD and non-AD groups, the abundance of Streptococcus was underrepresented in children with AD. The reduction Streptococcus may be a sign of dysbiosis associated with impending AD onset and might contribute to the prevention of AD development in infancy. Streptococci are widely distributed in the oral cavity, nasal cavity, and intestinal tract. How changes in streptococci abundance affect the occurrence of AD is not clear; however, previous studies have suggested that streptococci might be involved in the intestinal Th17 response.25
The present study has several major limitations; first, this was a case-control study that did not analyze stool samples before the onset of AD, making it difficult to prove a time connection. Second, the samples were selected by convenience in our study, which can lead to bias. Third, our research subjects were infants aged 1–6 months; as intestinal microbiota varies greatly among individuals, the intestinal microbiota of this age group is still in the process of dynamic development. Fourth, many factors that affect the intestinal microbiota, including maternal prebiotic and antibiotic exposure, number of siblings, and animal exposure; since this information was not collected, the description of potential confounding factors was insufficient. Finally, our analysis of the intestinal microbiota was limited to species-level analysis of the bacterial communities; fungi were not identified. Nevertheless, studies have maintained the importance of gut fungal communities during the early stages of life with respect to allergic diseases.26,27
Conclusion
In summary, our study suggests that diversity may not be as important as specific microorganisms in the pathogenesis of AD. The genus and family differences between infants with AD and healthy controls observed in our study may provide insight regarding the link between the gut microbiome and atopic dermatitis. Still, the role of Streptococcus in the development of AD needs to be confirmed in a large cohort study.
Abbreviations
AD, atopic dermatitis; OTU, operational taxonomic unit; 16SrRNA, 16S ribosomal RNA.
Funding
This work was supported in part by the Medical Scientific Research Foundation of Guangdong Province, China (grant number: A2019482).
Disclosure
The authors declare that they have no competing interests.
References
1. Langan SM, Irvine AD, Weidinger S. Atopic dermatitis. Lancet. 2020;396(10247):345–360. doi:10.1016/S0140-6736(20)31286-1
2. Laughter MR, Maymone MB, Mashayekhi S, et al. The global burden of atopic dermatitis: lessons from the Global Burden of Disease Study 1990–2017. Br J Dermatol. 2021;184(2):304–309. doi:10.1111/bjd.19580
3. Patrick GJ, Archer NK, Miller LS. Which way do we go? Complex interactions in atopic dermatitis pathogenesis. J Invest Dermatol. 2020;14.
4. Abrahamsson TR, Jakobsson HE, Andersson AF, Björkstén B, Engstrand L, Jenmalm MC. Low diversity of the gut microbiota in infants with atopic eczema. J Allergy Clin Immunol. 2012;129(2):
5. Wang H, Li Y, Feng X, et al. Dysfunctional gut microbiota and relative co-abundance network in infantile eczema. Gut Pathog. 2016;8(1):36. doi:10.1186/s13099-016-0118-0
6. Rehman A, Rausch P, Wang J, et al. Geographical patterns of the standing and active human gut microbiome in health and IBD. Gut. 2016;65(2):238–248. doi:10.1136/gutjnl-2014-308341
7. Magne F, O’Ryan ML, Vidal R, Farfan M. The human gut microbiome of Latin America populations: a landscape to be discovered. Curr Opin Infect Dis. 2016;29(5):528–537. doi:10.1097/QCO.0000000000000300
8. Hanifin JM, Rajka G. Diagnostic features of atopic dermatitis. Acta Derm Venereol. 1980;Suppl 92:44–47.
9. Hu Y, Zhang L, Liu Q, Wang Y, Chen Q, Kong B. The potential correlation between bacterial diversity and the characteristic volatile flavour of traditional dry sausages from Northeast China. Food Microbiol. 2020;91:103505. doi:10.1016/j.fm.2020.103505
10. Li B, Zhang X, Guo F, Wu W, Zhang T. Characterization of tetracycline resistant bacterial community in saline activated sludge using batch stress incubation with high-throughput sequencing analysis. Water Res. 2013;47(13):4207–4216. doi:10.1016/j.watres.2013.04.021
11. Lundberg DS, Yourstone S, Mieczkowski P, Jones CD, Dangl JL. Practical innovations for high-throughput amplicon sequencing. Nat Methods. 2013;10(10):999–1002. doi:10.1038/nmeth.2634
12. Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71(12):8228–8235. doi:10.1128/AEM.71.12.8228-8235.2005
13. Mansour B, Monyók Á, Makra N, et al. Bladder cancer-related microbiota: examining differences in urine and tissue samples. Sci Rep. 2020;10(1):11042. doi:10.1038/s41598-020-67443-2
14. Donaldson GP, Lee SM, Mazmanian SK. Gut biogeography of the bacterial microbiota. Nat Rev Microbiol. 2016;14(1):20–32. doi:10.1038/nrmicro3552
15. Stojanov S, Berlec A, Štrukelj B. The influence of probiotics on the firmicutes/bacteroidetes ratio in the treatment of obesity and inflammatory bowel disease. Microorganisms. 2020;8(11):1715. doi:10.3390/microorganisms8111715
16. Chen CC, Chen KJ, Kong MS, Chang HJ, Huang JL. Alterations in the gut microbiotas of children with food sensitization in early life. Pediatr Allergy Immunol. 2016;27:254–262. doi:10.1111/pai.12522
17. La-ongkham O, Nakphaichit M, Leelavatcharamas V, Keawsompong S, Nitisinprasert S. Distinct gut microbiota of healthy children from two different geographic regions of Thailand. Arch Microbiol. 2015;197(4):561–573. doi:10.1007/s00203-015-1089-0
18. Lin D, Wang R, Luo J, et al. The core and distinction of the gut microbiota in Chinese populations across geography and ethnicity. Microorganisms. 2020;8(10):1579. doi:10.3390/microorganisms8101579
19. Fontana A, Panebianco C, Picchianti-Diamanti A, et al. Gut microbiota profiles differ among individuals depending on their region of origin: an Italian pilot study. Int J Environ Res Public Health. 2019;16:4065. doi:10.3390/ijerph16214065
20. Dzidic M, Boix-Amorós A, Selma-Royo M, Mira A, Collado MC, Microbiota G. Mucosal immunity in the neonate. Med Sci. 2018;6:56. doi:10.3390/medsci6030056
21. Lee MJ, Kang MJ, Lee SY, et al. Perturbations of gut microbiome genes in infants with atopic dermatitis according to feeding type. J Allergy Clin Immunol. 2018;141:1310–1319. doi:10.1016/j.jaci.2017.11.045
22. Zhang Y, Jin S, Wang J, et al. Variations in early gut microbiome are associated with childhood eczema. FEMS Microbiol Lett. 2019;366(9):fnz020. doi:10.1093/femsle/fnz020
23. Galazzo G, van Best N, Bervoets L, et al. Development of the microbiota and associations with birth mode, diet, and atopic disorders in a longitudinal analysis of stool samples, collected from infancy through early childhood. Gastroenterology. 2020;158(6):1584–1596. doi:10.1053/j.gastro.2020.01.024
24. Park YM, Lee SY, Kang MJ, et al. Imbalance of gut Streptococcus, Clostridium, and Akkermansia determines the natural course of atopic dermatitis in infant. Allergy Asthma Immunol Res. 2020;12:322–337. doi:10.4168/aair.2020.12.2.322
25. Engen SA, Schreurs O, Petersen F, Blix IJ, Baekkevold ES, Schenck K. The regulatory role of the oral commensal streptococcus mitis on human monocytes. Scand J Immunol. 2018;87(2):80–87. doi:10.1111/sji.12636
26. Fujimura KE, Sitarik AR, Havstad S, et al. Neonatal gut microbiota associates with childhood multisensitized atopy and T cell differentiation. Nat Med. 2016;22(10):1187–1191. doi:10.1038/nm.4176
27. Arrieta MC, Arévalo A, Stiemsma L, et al. Associations between infant fungal and bacterial dysbiosis and childhood atopic wheeze in a nonindustrialized setting. J Allergy Clin Immunol. 2018;142:424–434.e10. doi:10.1016/j.jaci.2017.08.041
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.