Back to Journals » Cancer Management and Research » Volume 10
Comparison of gene expression in liver regeneration and hepatocellular carcinoma formation
Authors Yin L ![]() , Wang Y, Guo X, Xu C, Yu G
, Wang Y, Guo X, Xu C, Yu G ![]()
Received 3 May 2018
Accepted for publication 26 July 2018
Published 15 November 2018 Volume 2018:10 Pages 5691—5708
DOI https://doi.org/10.2147/CMAR.S172945
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Antonella D'Anneo
Li Yin,1–3 Yahao Wang,1,2 Xueqiang Guo,1,2 Cunshuan Xu,1,2 Guoying Yu1,2
1College of Life Science, Henan Normal University, Xinxiang, Henan 453007, China; 2State Key Laboratory Cultivation Base for Cell Differentiation Regulation and Henan Engineering Laboratory for Bioengineering and Drug Development, Henan Normal University, Xinxiang, Henan 453007, China; 3Laboratory of Tropical Biomedicine and Biotechnology, School of Tropical Medicine and Laboratory Medicine, Hainan Medical University, Haikou 571199, China
Background: Liver -cell proliferation occurs in hepatocellular carcinoma (HCC) and liver regeneration (LR). The development and progression of HCC and LR have many similar molecular pathways with very different results. In simple terms, LR is a controllable process of organ recovery and function reconstruction, whereas liver cancer is uncontrollable. Do they share common key pathways and genes?
Methods: In this study, the dynamic transcriptome profile at ten time points (0, 2, 6, 12, 24, 30, 36, 72, 120, and 168 hours) during LR in rats after two-thirds hepatectomy and eight stages (normal, cirrhosis without HCC, cirrhosis, low-grade dysplastic, high-grade dysplastic, and very early, early advanced, and very advanced HCC) representing a stepwise carcinogenic process from preneoplastic lesions to end-stage HCC were analyzed in detail. A variety of bioinformatic methods, including MaSigPro, weighted gene-coexpression network analysis, and spatial analysis of functional enrichment, were used to analyze, elucidate, and compare similarities and differences between LR and HCC formation.
Results: Key biological processes and genes were identified. From the comparison, we found that cell proliferation and angiogenesis were the most significantly dysregulated processes shared by LR and HCC. The pattern of cell-proliferation-related gene expression in progression stage during LR is similar to the transition process from dysplasia to early-stage HCC. LR and HCC showed different expression patterns as a whole. Some key genes, including FYN, XPO1, FOXM1, EZH2, and NRF1, were identified as playing critical roles in both LR and HCC.
Conclusion: These findings could contribute to revealing the molecular mechanism of development and regulation mechanism of normal and abnormal proliferation, which could provide new ideas and treatment methods for regenerative medicine, oncological drug development, and oncological treatment.
Keywords: liver regeneration, hepatocellular carcinoma, angiogenesis, MaSigPro, microarray, FYN
Background
The liver plays a vital role in the maintenance of body homeostasis and is essential for the coordination of normal metabolism of carbohydrates, lipids, proteins, and vitamins as well as for biochemical defense against toxic chemicals. The liver is a magic organ, due to its powerful regenerative ability. Two-thirds partial hepatectomy (PH) is one of the most effective surgical models in rodents for the study of liver regeneration (LR). After resection of two of the six lobes, quiescent hepatocytes, which stay in the G0 phase, can rapidly reenter the G1 phase under the influence of different cytokines and growth factors, which play roles of activating downstream kinases and transcription factors (TFs). After one or two rounds of hepatocyte proliferation, the remnant liver lobes undergo compensatory hyperplasia via cell proliferation, thereby restoring the liver to its original presurgical size within 7 days, the process which is called LR.1,2
Hepatocellular carcinoma (HCC) is the fifth most common cancer in men and the seventh in women across the world and the second leading cause of cancer-related deaths worldwide, accounting for around 11% of all cancer deaths.3 Major risk factors for HCC development include chronic viral hepatitis, metabolic disease, and autoimmune hepatitis. Because of the increase in hepatitis C virus infections, the incidence of liver cancer is gradually increasing. HCVs can cause acute and chronic infections that can lead to liver cirrhosis and HCC.4 Cirrhosis is the most important risk factor for developing HCC, and the majority of virus-associated HCC cases develop from liver cirrhosis, therefore, the clarification of the mechanism of liver cancer caused by hepatitis B virus infection will contribute to the new understanding of liver cancer formation. The incidence of HCC in individuals with hepatitis C virus cirrhosis is about 3%–5% per year.5,6
Currently, potential curative options for HCC patients include radiofrequency ablation, liver transplantation, and tumor resection. However, many factors, including tumor staging, donor deficiencies, graft rejection, and opportunistic infections, affect these adaptations. The pathological stage of liver cancer is very important for the development of treatment plans and patient prospects.7,8 For early HCC without cirrhosis, liver resection or liver transplantation can be used, but recurrence occurs frequently. In the end stage of HCC, patient survival is usually <3 months. It is necessary to understand the mechanism of progression from cirrhosis to HCC.9–12
Organ regeneration has been observed in many animals and the human liver is able to regenerate after moderate injury. Liver transplantation is a typical application of regenerative capacity of the liver. Since 50%–70% of the normal liver is excised and exchanged with the recipient’s diseased liver, donor and recipient liver will not regenerate if the liver does not have the regenerative capacity. The powerful regenerative capacity of the liver has been used to cure patients with liver removal due to liver disease, such as liver cancer, and provide a safe and effective transplantable tissue source in the case of liver donation.13–15 The development and progression of HCC and LR have many similar molecular pathways with very different results. In simple terms, LR is a controllable process of organ recovery and function reconstruction, whereas liver cancer is uncontrollable. In this study, gene-expression patterns in stepwise HCC formation were compared with LR, which elucidated the similarity and differences between them. Several key pathways and genes involved in this pathway were identified.
Methods
Microarray-data normalization
The mRNA-expression profile of LR came from what we have done before, the microarray data have been deposited in the Gene Expression Omnibus under accession number GSE55434, and details about the experiment have been described in a study published previously. The platform is GPL1355, and all samples are hybridized on the Affymetrix Rat Genome 230 2.0 Array. The gene-expression data set GSE6764 provided by Wurmbach et al (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE6764) was downloaded from the Gene Expression Omnibus database. A total of 75 tissue samples were divided into eight consecutive pathological stages from preneoplastic lesions to HCC, including normal, cirrhosis (cirrhosis with and without HCC), dysplastic (low- and high-grade dysplastic), very early HCC, early HCC, advanced HCC, and very advanced HCC. All tissue samples are hybridized on the human U133 plus 2.0 array (Affymetrix). These are all raw .cel files and were converted into expression data using the robust multiarray average (RMA) algorithm using the Bioconductor Affy package.
Identification of differentially expressed genes during LR
MaSigPro software was used to identify differentially expressed genes (DEGs) across different time points in rat LR and stepwise carcinogenic process in HCC. MaSigPro can be used to perform analyses of single and multiseries time-course data. A two-regression-step approach was defined by MaSigPro to find genes with significantly changed expression profiles between experimental groups by dummy variables. The first step is to identify DEGs by constructing a global regression model with all the defined variables. The second is to find statistically significant, differentially expressed profiles using a variable selection method to identify the differences between groups. Different pathological stages were considered at different time points. Quaternary regression models (df=4 in LR and df=3 in HCC) were defined across the samples that had enough power to compute the reduced number of time points. The threshold of a false discovery rate of 0.05 (Q=0.01) was set to sift the significantly differentially transcribed genes. We defined a stepwise regression method (“two ways backward”, a=0.05), with an R2-cutoff value fit of 0.7 in LR and 0.5 in HCC. The hierarchical approach based on correlation distances was used to cluster the significantly differentially transcribed genes across the samples.
Construction of weighted gene-coexpression networks and identification of modules associated with external traits in LR and HCC
DEGs obtained from MaSigPro were subjected to further analysis using weighted gene-coexpression network analysis (WGCNA) to test the clustering results from MaSigPro.16 From thousands of genes, interesting gene modules were identified with WGCNA, and then intramodular connectivity and gene significance based on the correlation of a gene-expression profile with a sample trait were used to identify hub genes in LR and HCC for further validation. WGCNA is a freely accessible R package for the construction of WGCNs.
Gene-enrichment analysis
The ClusterProfiler package was employed to perform the enrichment analysis of the biological process (BP) Gene Ontology (GO) term (GO:BP) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) for all DEGs. For each module with a cutoff value of P=0.05, the q-value cutoff is 0.2, the pAdjustMethod is BH, and the minimum and maximum number of genes annotated by the ontology term for testing are 10 and 500, respectively. Functional similarity among GO terms was computed using the SimRel method, which is based on Resnik’s and Lin’s similarity measures using the REVIGO and visualized with R language.17
Comparison and merging of results from MaSigPro and WGCNA
The algorithm’s mathematical essence of WGCNA is the exponentiation of correlation coefficients between paired genes, which makes the strong stronger and the weak weaker. In theory, the DEGs, grouped into one cluster by MaSigPro based on their similar expression profiles, will be inclined to occur in the same module in WGCNA. As such, the results were redesigned systematically, and the new clustered DEGs were defined for the next analysis.
Comparative analyses of all new modules in LR and HCC
Owing to the dynamic progression of LR and HCC, ToppCluster was used to perform multimodule gene functional enrichment analysis, which shows shared and list-specific functional features in the results.18 The BioMart package was used to perform homologous matching from rats to humans.19,20 Unmatched genes by BioMart are confirmed one by one manually. A hypergeometric test was applied in the analysis. GO:BP, human phenotype (HP), mouse phenotype (MP), pathways, transcription factor binding site, and microRNA were selected for annotations with a P-value cutoff of 0.05 and multiple-testing correction, ie, module-specific phenotype associations, BP, pathways, genes whose mRNAs were targets of microRNAs, and genes whose promoters contained known TFs. Results were visualized in Cytoscape.
Identification and key modules in LR and HCC
Genes belonging to the same biological process or pathway tend to share similar expression patterns of genetic interactions, and relationships among them can be defined in biological networks. Spatial analysis of functional enrichment (SAFE) is a systematic and quantitative method mapping local enrichment for functions and phenotypes and has uncovered a new biological mechanism in budding-yeast Saccharomyces cerevisiae.21–23 We constructed a protein–protein interaction (PPI) global network for LR with similar interaction patterns grouped together: nodes in this network represented genes including all known LR-related genes (from NCBI) and all DEGs, and edges represented interactions among genes from the STRING (search tool for the retrieval of interacting genes/proteins) database. The network was displayed by applying the edge-weighted spring-embedded layout in Cytoscape, and all nodes were positioned based on their occurrence. The same method was used for the construction of the HCC network. Dysplasia-, infiltration-, and metastasis-related genes are the known genes for HCC. Genes in the same modules with similar expression profiles from MaSigPro and WGCNA were overlaid onto the network. Key modules were identified according to the position, and they overlap with all known LR- and HCC-related genes.
Identification of hub-bottleneck genes
In a scale-free network like the gene-coexpression one, highly connected genes are called hub genes (high-degree genes) and are important for the robustness of the network. In WGCNA, the identification of hub genes is based mainly on intramodular connectivity without any statistical criteria.24 Except for degree, betweenness is one of the most important topological properties of a network and represents the number of shortest paths going through a certain node. Nodes with high betweenness are called bottlenecks and control the information flow in a network. Two types of hubs were defined by Han et al: date hubs (hub bottlenecks) and party hubs (hub nonbottlenecks).25 They proposed that hub bottlenecks played a more important role in the organization of biological modules in the whole interaction network.26 Hub-bottleneck genes are identified by calculating the degree and betweenness of every gene in a network using the CytoHubba plug-in. The top 5% of genes ranked by degree score are defined as the hub genes and the top 5% of the betweenness genes as the bottlenecks. Genes that appear in both hub and betweenness lists are the hub-bottleneck genes.
Results
DEGs in LR and HCC
A total of 30 tissue samples from GSE54534 and 75 from (.cel format) GSE6764 were downloaded from the NCBI. The raw files were converted into expression data using the RMA algorithm based on R language, including background correction, normalization, and summary. Quality control was performed using the SimpleAffy package. QC-plot and box-plot results before and after normalization are shown in Figures S1 and S2. DEGs were identified using MaSigPro according to the details described in the Methods section for LR and HCC.
Based on the expression profiles across ten regenerative time points from hepatic resection to liver-structure restoration, the 1,362 DEGs identified using MaSigPro were grouped into nine clusters of 104, 225, 154, 102, 211, 130, 171, 217, and 48 features, as shown in Figure 1A. The nine clusters were further grouped by patterns into “up-down-up” (clusters 1 and 3), “down-up” (clusters 2 and 4), “down-up-down” (clusters 5 and 6), and up-down (clusters 7–9), corresponding to changes during LR. The most significant turning points during LR were at 12, 36, and 72 hours after liver resection.
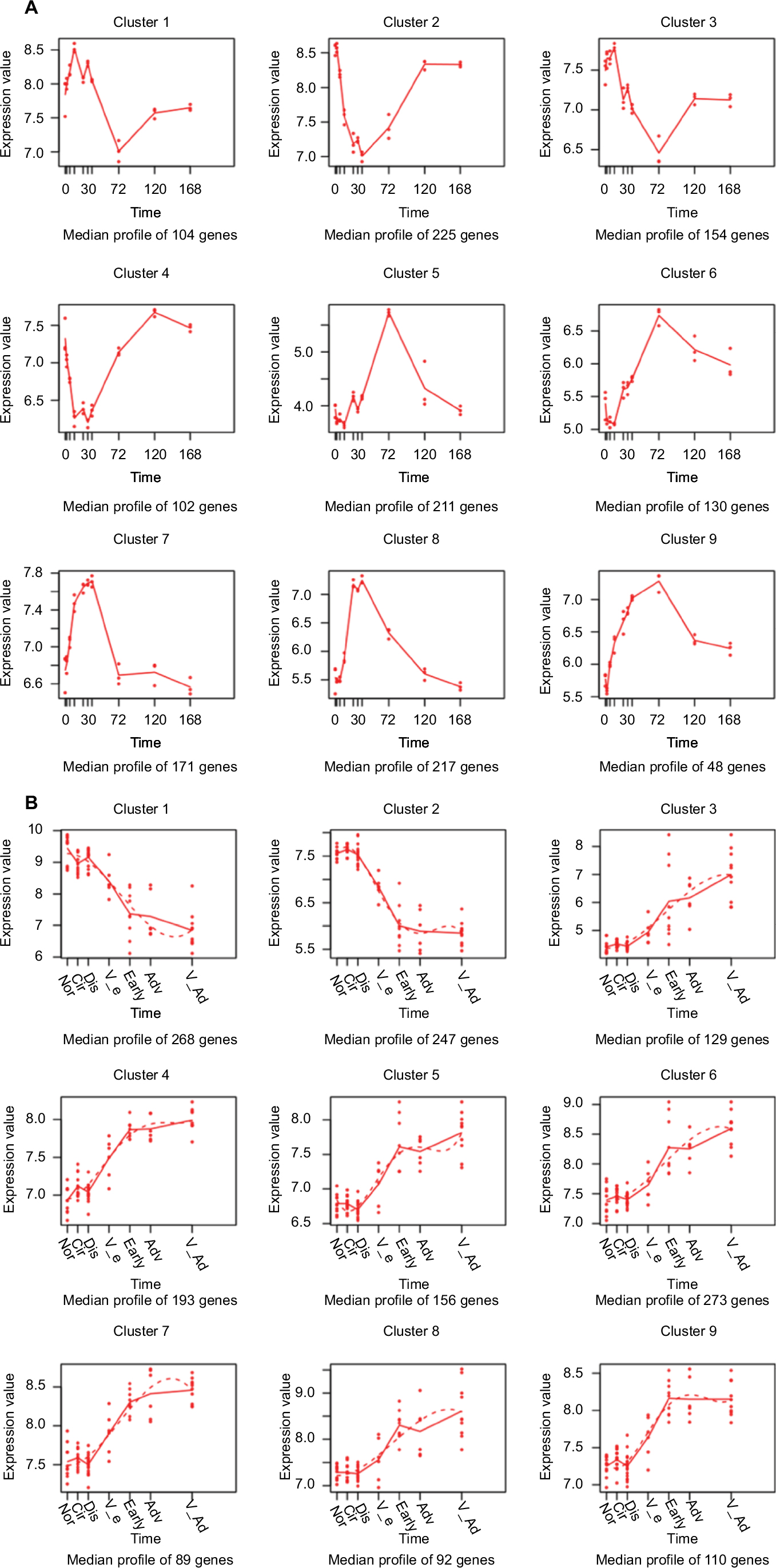 | Figure 1 Differentially expressed gene-expression patterns. Note: (A) Liver regeneration (B) hepatocellular carcinoma. |
Based on the expression profiles across six consecutive pathological stages from preneoplastic lesions to end-stage HCC – normal, cirrhosis, dysplastic, very early HCC, early HCC, advanced HCC, and very advanced HCC – the 1,557 DEGs identified using MaSigPro were grouped into nine clusters of 268, 247, 129, 193, 156, 273, 89, 92, and 110 features, as shown in Figure 1B. The nine clusters were further grouped by patterns into down (clusters 1 and 2) and up (clusters 3–9), corresponding to changes during the dynamic progression of HCC. The most significant turning phase occurred at the dysplastic and early stages, representing tumorigenesis and tumor aggression.
Functional enrichment analysis for DEGs in each cluster obtained from MaSigPro
To explore the relationship between the enrichment results from all the DEGs and genes within each module, ClusterProfiler was used to perform GO:BP and KEGG enrichment analyses for all DEGs and genes in each cluster. A total of 101 GO terms were identified to be significantly enriched for all DEGs (Table S1). The final GO enrichment result was visualized using R language after calculating semantic similarity of GO terms (Figure 2A). Enriched GO terms in all DEGs were associated with carboxylic acid catabolism, which included the metabolic processes of amino acids, fatty acids, and small molecules, RNA localization, which included RNA localization and transport and protein transport, DNA replication, which included DNA replication and repair, nuclear division, which included nuclear and organelle fission, telomere maintenance, and extracellular matrix (ECM) organization, aging, which included aging, liver development, and vasculogenesis, and response to hypoxia, which included response to oxygen levels and nutrients. All DEGs were grouped into nine clusters according to their expression profiles, and GO enrichment terms were grouped into several modules. As we know, the nine clusters were related to the dynamic process of LR with different meanings. This indicated that the clusters did not occur randomly if their enrichment results were consistent with the results of all DEGs. As such, we extracted all genes in every cluster and performed GO-term enrichment analysis using ClusterProfiler, as shown in Figure 3. Six of the nine clusters stood for special meanings, and clusters 4, 6, and 9 had relatively fewer genes. It was obvious that each module occurred sequentially in LR. KEGG enrichment terms for all DEGs in LR included mainly the cell cycle, DNA replication and repair, RNA transport, spliceosomes, amino-acid metabolism, and ribosome biogenesis (Figure S3). The enrichment result for every module was consistent with all DEGs, which indicated an orderly process occurring during LR (Figure S4).
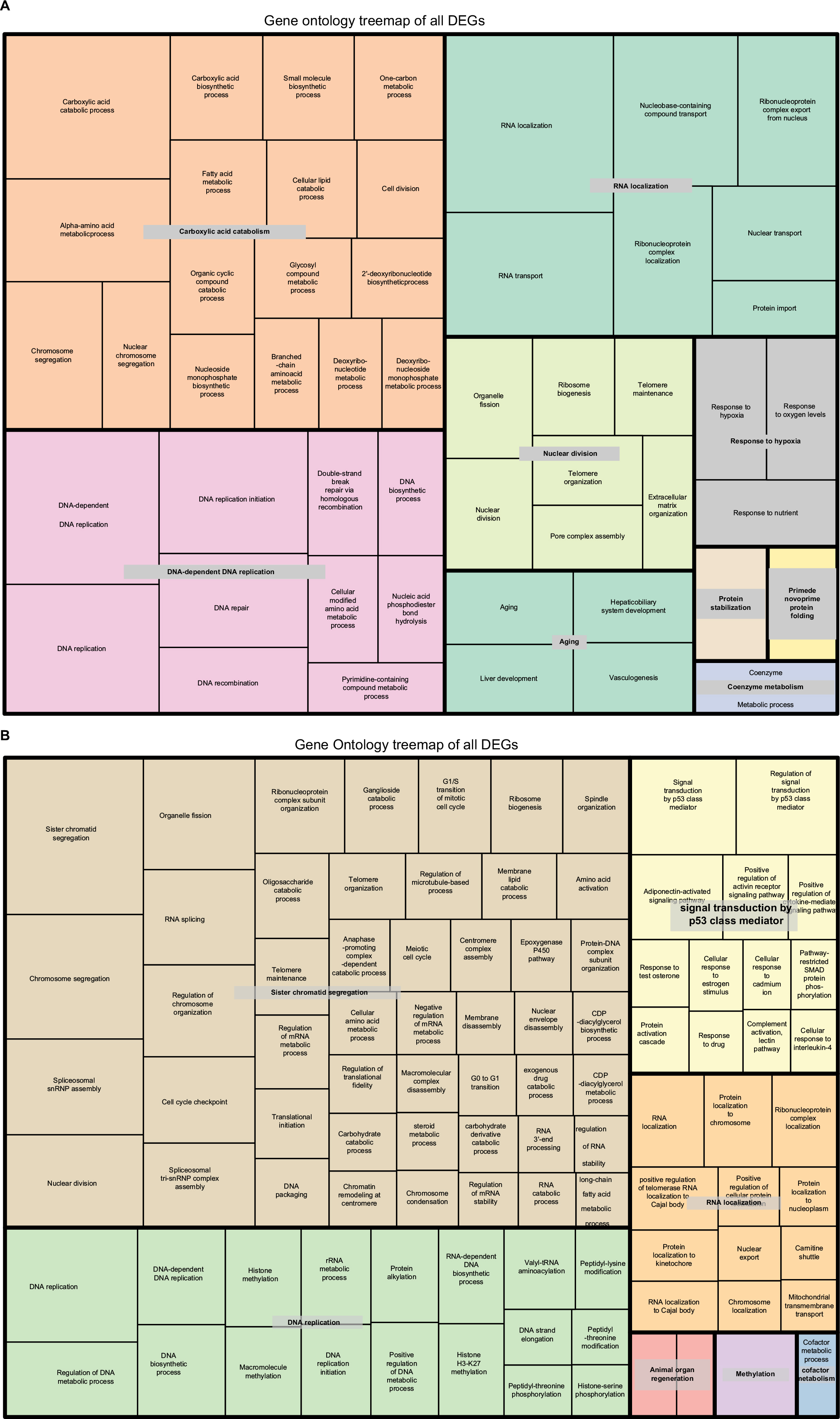 | Figure 2 Gene functional enrichment analysis (GO:BP) of all DEGs during LR and HCC formation. Note: (A) LR (B) HCC. Abbreviations: BP, biological process; DEGs, differentially expressed genes; GO, Gene Ontology; HCC, hepatocellular carcinoma; LR, liver regeneration. |
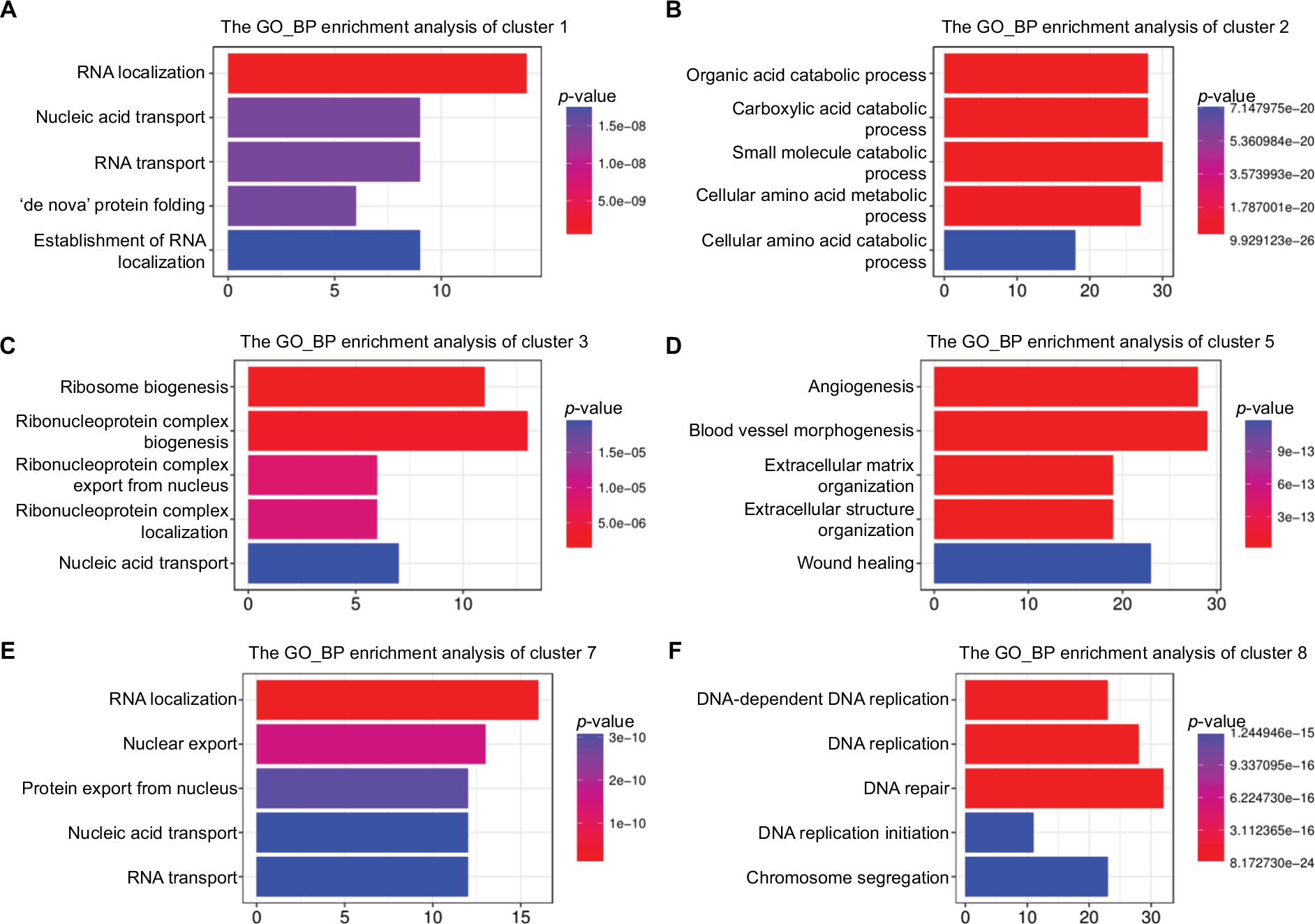 | Figure 3 Functional enrichment analysis (GO:BP) of DEGs from six clusters in LR. Abbreviations: BP, biological process; DEGs, differentially expressed genes; GO, Gene Ontology; LR, liver regeneration. |
The same workflow described in this section was applied to the enrichment analysis of all DEGs and genes in each module in HCC. A total of 250 GO terms were identified to be significantly enriched for all DEGs (Table S1). The final GO enrichment result was visualized using R language after calculating GO-term semantic similarity (Figure 2B). Enriched GO terms in all DEGs were associated with cell-cycle-related processes, which included chromosome segregation, cell-cycle checkpoint, nuclear and organelle fission, and ribosome biogenesis, DNA replication, which included histone (H3-K27) methylation, DNA-replication initiation and elongation, and protein alkylation, RNA localization, which included RNA localization to Cajal bodies, protein localization to chromosomes, kinetochores, and nucleoplasm, and carnitine shuttle, p53-signaling pathway, animal-organ regeneration, and methylation. We extracted all genes in the nine clusters and performed GO-term enrichment analysis using ClusterProfiler, as shown in Figure 4. Six of the nine clusters had special meaning. It was obvious that each module occurred sequentially in HCC. The KEGG enrichment terms for all DEGs in LR included mainly the cell cycle, DNA replication, RNA transport, spliceosomes, p53-signaling pathway, and oocyte meiosis (Figure S5). Enrichment results for every module were consistent with all DEGs, which indicated an orderly process occurring during the development of HCC (Figure S6).
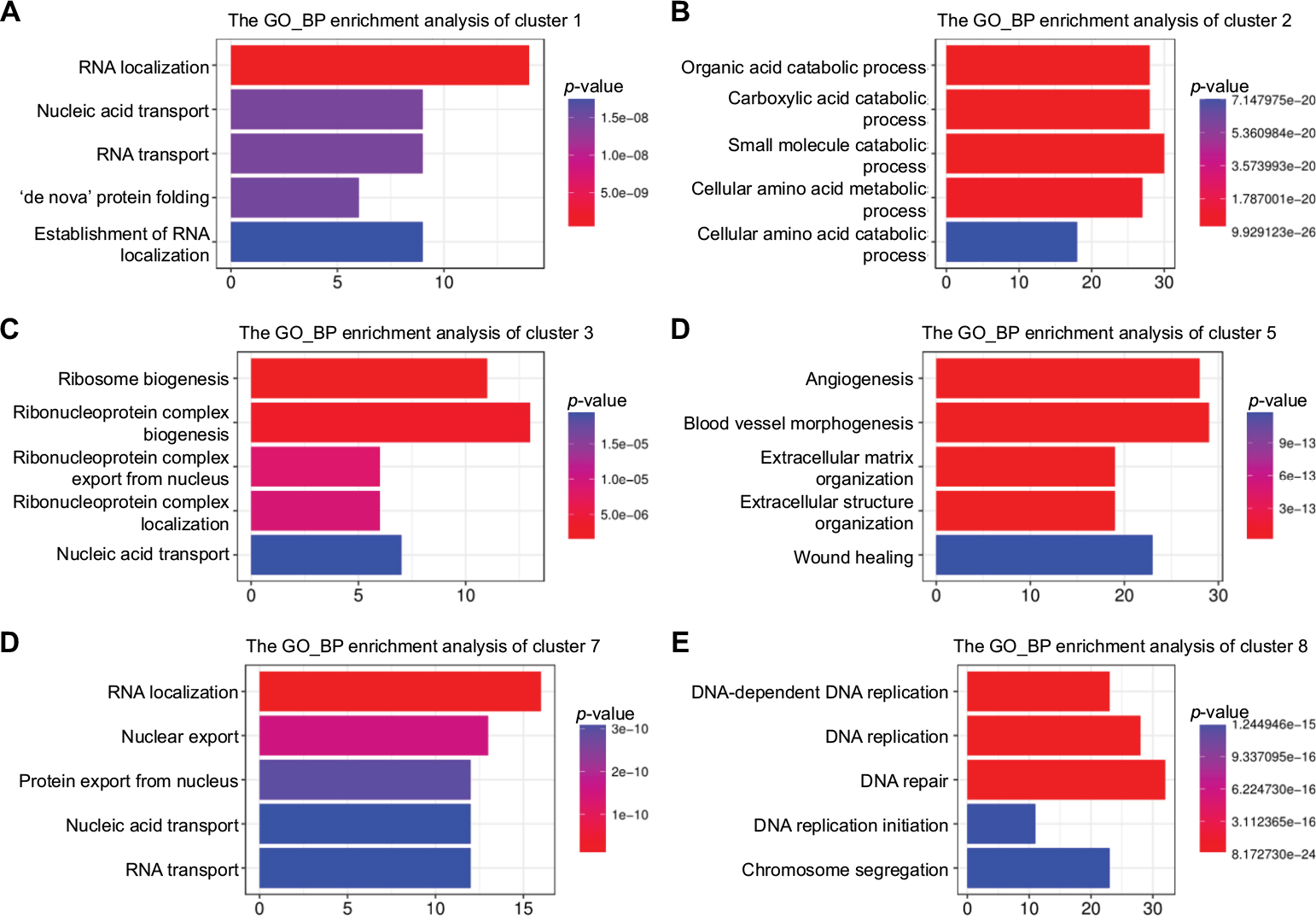 | Figure 4 Functional enrichment analysis (GO:BP) of DEGs from six clusters in HCC. Abbreviations: BP, biological process; DEGs, differentially expressed genes; GO, Gene Ontology; HCC, hepatocellular carcinoma. |
Construction of WGCNA and identification of modules associated with external traits in LR and HCC
The coexpression network was constructed from DEGs obtained from MaSigPro in LR and HCC. In LR, seven modules were identified, including a gray module. We chose soft-threshold power 18 to define the adjacency matrix based on the criterion of approximate scale-free topology, with a minimum module size 30, module detection sensitivity DeepSplit 2, and a cutoff height for merging of modules 0.2, which meant that modules whose eigengenes were correlated >0.8 were merged. Modules associated with different time points during LR were identified according to module–trait relationships (Figure 5A). The cyan module was positively correlated with PH at 72 hours. The green and green/yellow modules were also similar. In order to explore the biological meaning of every module, functional enrichment analysis was performed for each module in LR (Figures S7 and S8). DEGs from MaSigPro were recalculated according to amplified correlations between paired genes. It was obvious that enrichment results were consistent with results from MaSigPro. Overlapping analysis of genes in the nine clusters from MaSigPro and seven modules from WGCNA was performed (Figure S9). Results showed that each module overlapped with a specific cluster. For example, the overlapping rate of genes between the cyan module and cluster 5 reached 68%. Because of the nature of unsigned networks, there were possibilities where one module overlapped with several clusters.
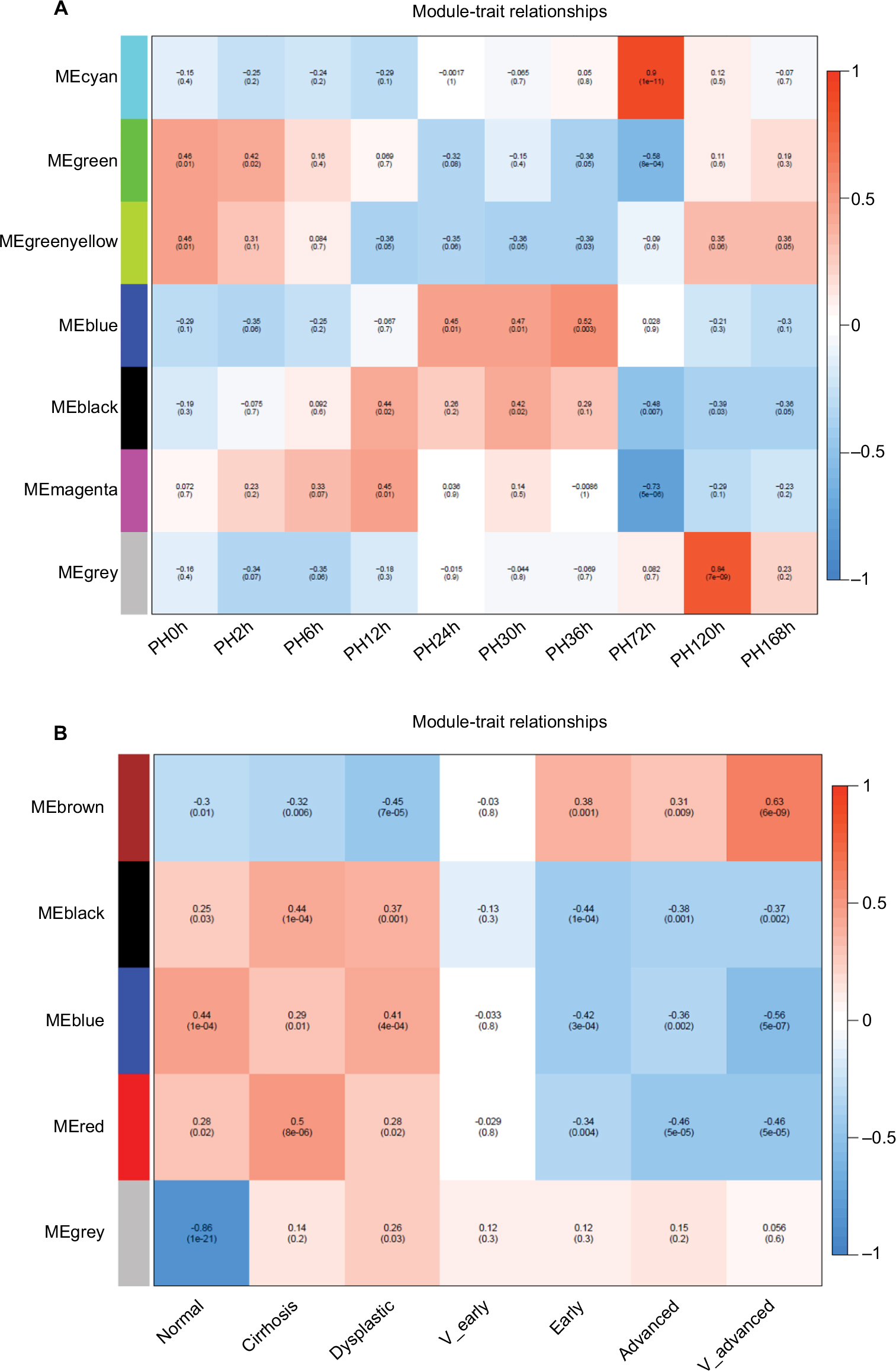 | Figure 5 Relationships of consensus-module eigengenes and different time points/stages. Notes: Relationships of consensus-module eigengenes and ten time points in (A) LR and (B) HCC. Each row in the table corresponds to a consensus module, and each column to a time point/stage. The module name is shown at the left of each cell. Numbers in the table report the correlations of the corresponding module eigengenes and time points/stage, with P-values in parentheses below the correlations. The table is color-coded by correlation according to the color legend. Intensity and direction of correlations are indicated at the right of the heat map (red, positive; green, negative). Abbreviations: HCC, hepatocellular carcinoma; LR, liver regeneration; ME, module eigengene; PH, partial hepatectomy. |
In the same way, the DEGs from HCC were assessed by WGCNA (Figure 5B). A soft-threshold power of 12 was selected to define the adjacency matrix based on the criterion of approximate scale-free topology with other parameters same as the LR. Functional enrichment analysis results were consistent with MaSigPro results (Figures S10 and S11). Overlap analysis of genes in the nine clusters from MaSigPro and seven modules from WGCNA was also performed (Figure S12). The brown module had the most genes overlap, with several clusters.
WGCNA and MaSigPro results were in good agreement in this analysis, so genes that appeared simultaneously in modules and clusters played an important role in biological function. Based on this information, genes were regrouped into new modules in LR and WGCNA, with some genes filtered. Finally, 1,004 and 1,275 probes were obtained in LR and HCC, respectively. Six modules – blue, magenta, green, green/yellow, black, and cyan – were generated in LR. Seven modules – blue_c12, blue_c4, brown_c12, brown_c56, brown_c7, black, and red – were obtained in HCC. Enrichment analysis for genes in the new modules in LR and HCC was performed and showed consistency in modules before and after merging (Figures S13 and S14). All probes in the new module were matched to symbols for the next analysis using ClusterProfiler. The probe with the smallest P-value was selected if several probes matched the same symbol. Finally, 1,004 probes matched 849 unique genes in LR and 1,275 probes to 1,047 unique genes in HCC.
Comparative analyses of all new modules in LR and HCC
The abstracted network of LR is shown in Figure 6. Notably, the magenta, black, and blue modules shared general ribonucleoprotein-complex export and localization and RNA transport and localization biological pathways, but differed with respect to phenotypes, which included embryonic lethality prior to organogenesis, increased carcinoma incidence, and small gonads; GO:BP, which included DNA geometric and conformation change, DNA replication and repair, G1/S and G2/M transition, and the p53-signaling pathway; TFs, including E2F1, E2F4, and KTGGYRSGAA_unknown, and Has-miR24 for the blue module; and GO:BP, which included translation initiation, ribosomal large-subunit biogenesis, ER to Golgi vesicle-mediated transport and posttranscription regulation of gene expression, and NRF1 TF for the magenta module; and GO:BP, which included RNA splicing, termination of transcription and telomere maintenance via telomerase, and ELK1 TF. Genes in these three modules shared similar expression profiles at the second cell cycle. The green and yellow/green modules shared the general abnormality of amino-acid and carboxylic acid metabolism, fatty acid and organic acid metabolism, and oxidation–reduction processes, but differed with respect to MP, which included decreased cholesterol and abnormal lipid levels, GO:BP, which included lipid metabolism and fatty acid oxidation in the green/yellow module, HP, which included abnormality of acid–base homeostasis, acidosis, and brain atrophy (very small), and GO:BP, which included cerebral hypoplasia and purine metabolism. Genes in the cyan module were enriched in MP, which included abnormal angiogenesis, abnormal ECM morphology, and decreased angiogenesis and respiratory distress, HP, which included atrophic scars and gastrointestinal angiodysplasia, GO:BP, which included angiogenesis, cell migration and mobility, and endothelial cell proliferation and cell localization, and TF, which included PU1, ELK1, and Has-miR29a–c.
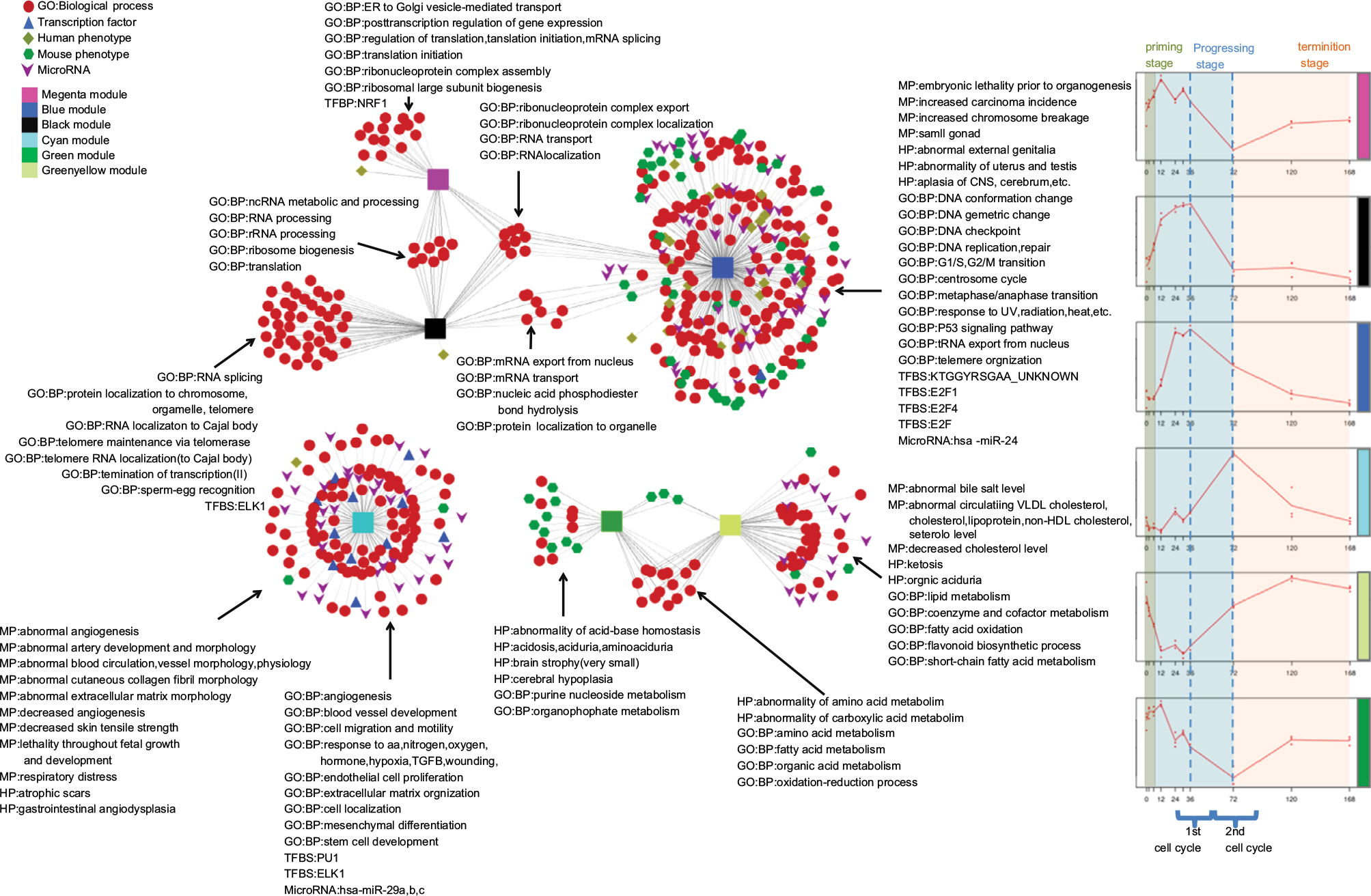 | Figure 6 An abstracted network showing enriched MP, HP, GO:BP, microRNAs, and TFs associated with five modules identified during LR in rat. Abbreviations: BP, biological process; CNS, central nervous system; ER, endoplasmic reticulum; GO, Gene Ontology; HDL, high-density lipoprotein; HP, human phenotype; LR, liver regeneration; MP, mouse phenotype; UV, ultraviolet; VLDL, very-low-density lipoprotein. |
The abstracted network of HCC is shown in Figure 7. Notably, the blue_c1 and brown_c12 modules shared general GO:BP, which included lipid metabolic and oxidation–reduction processes, but differed with respect to phenotypes, which included liver fibrosis and abnormal circulating amino-acid level, GO:BP, which included organic-acid metabolism in the blue_c12 module, phenotypes, which included abnormal homeostasis and hepatobiliary system physiology, and GO:BP, which included fatty acid oxidation, complement action, and inflammatory response in the brown_c12 module. Genes in the cyan module were enriched in the respiratory system phenotype, GO:BP, which included angiogenesis, cell motility and migration, cell localization, and response to growth factor, and pathways, which included ECM proteoglycans and ensembles of genes encoding the extracellular matrix. Genes in the brown_c56 module were enriched in MP, which included embryonic lethality and abnormal tumor incidence, HP, which included myelodysplasia and aplasia cerebrum, GO:BP, which included cell-cycle arrest, DNA replication, and ribosome biogenesis, pathways, which included TP53 activity and AURKA activation by TPX2, TF, which included USF, NFY, MAX, E2F, E2F1, E2F4, YY1, NRF1, and KTGGYRSGAA_unknown, and microRNAs, which included miR24 and miR108a. The genes in brown_c7, black, and blue_c4 enriched few pathways.
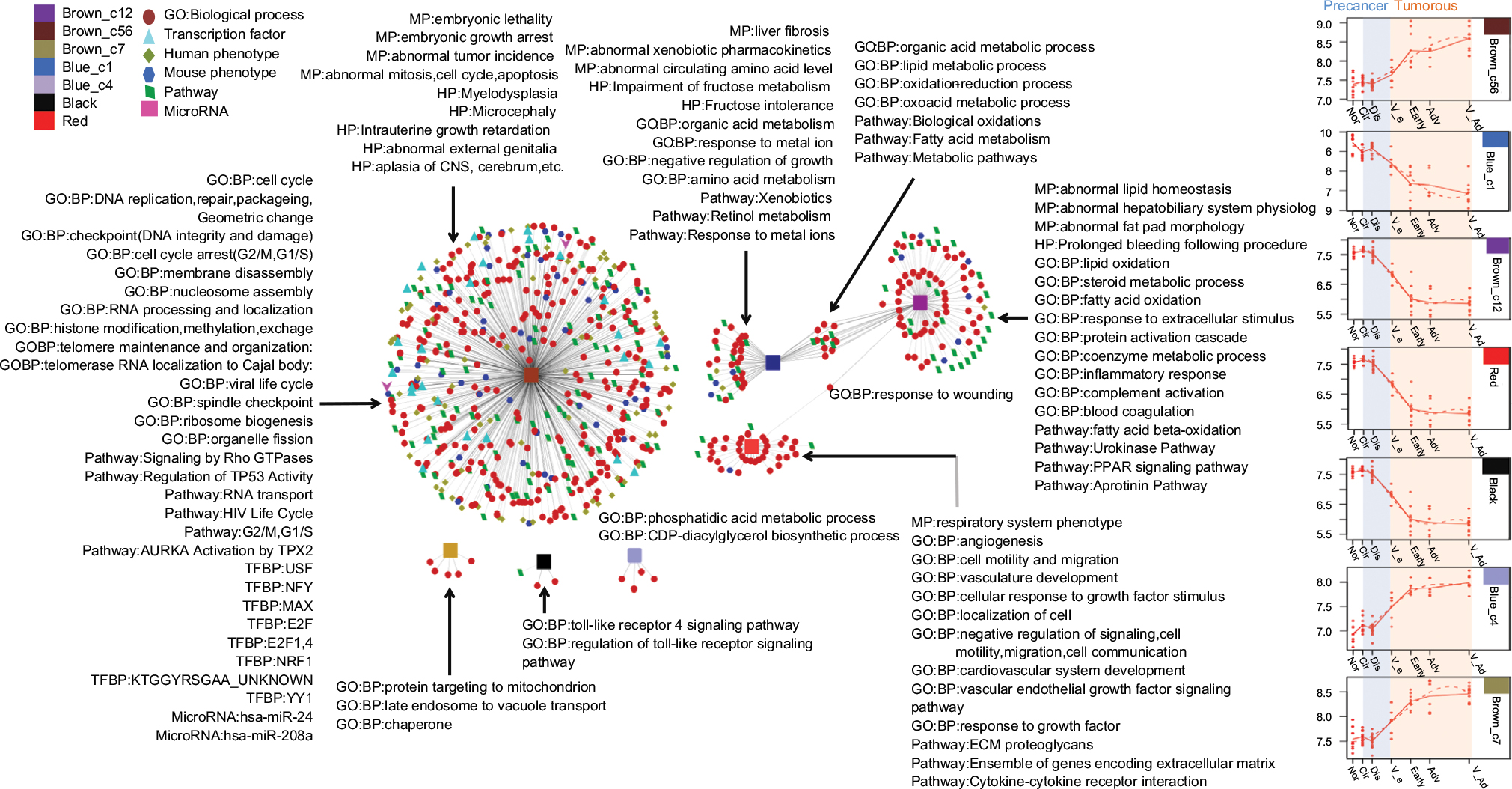 | Figure 7 Abstracted network showing enriched MP, HP, pathways, GO:BP, microRNAs, and TFs associated with seven modules identified during HCC progression. Abbreviations: BP, biological process; CNS, central nervous system; ECM, extracellular matrix; GO, Gene Ontology; HCC, hepatocellular carcinoma; HP, human phenotype; MP, mouse phenotype. |
Identification of key modules and key genes in LR and HCC
The PPI network for LR, containing 990 nodes and 7,755 edges, was constructed by connecting the 219 known LR-related genes downloaded from the NCBI and the 849 DEGs during LR. The network was visualized by applying the spring-embedded layout algorithm in Cytoscape, as shown in Figure 8A. Next, genes sharing similar expression profiles in each module were overlapped onto the network with different colors corresponding to different modules, as shown in Figure 8B. Four functional domains in the network were identified. With opposite expression profiles, the cyan and magenta modules were mutually exclusive in the network based on their interaction, and the same was applied to the blue and green modules. Known LR-related genes (the yellow dot pointed to by arrows) were overlapped onto the network, as shown in Figure 8C and D. Note that all known genes related to LR were overlapped mostly onto the cyan module, which represented angiogenesis. The degree and betweenness of every gene in the network were calculated using the CytoHubba plug-in. Hub-bottleneck genes were overlapped onto the network, as shown in Figure 9A. Notably, hub-bottleneck genes overlapped with the cyan module most. FYN was the only hub-bottleneck gene that did not belong to known LR-related genes in the cyan module. FYN is a member of the protein tyrosine-kinase oncogene family, which is involved in angiogenesis, cell growth, cell motility, and tumor invasion.27–31 However, FYN has not been reported to play a role in LR.
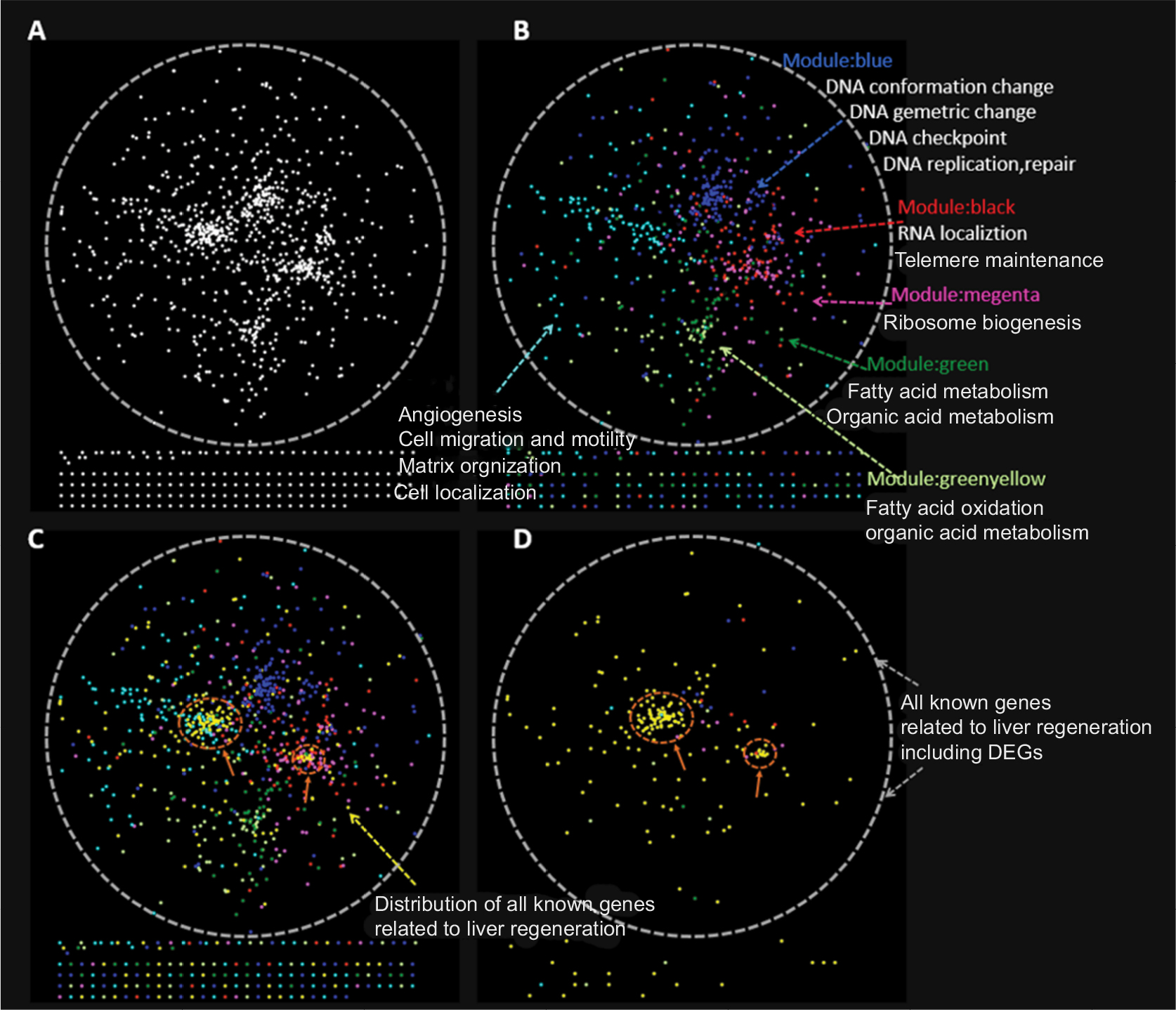 | Figure 8 Distribution of known LR-related genes and DEGs in the spatial protein–protein interaction (PPI) network, generated in Cytoscape. Notes: (A) The PPI network was constructed by connecting all the known LR-related genes and DEGs in our study. (B) All DEGs from six modules were colored differently. The cyan module was opposite the to magenta module l. The cyan module is located in the middle of the blue and pink modules. (C) All known LR-related genes were mapped to the PPI network, shown in yellow. These genes overlapped with the cyan model most. (D) Distribution of all known LR-related genes, including DEGs, in our study. Abbreviations: DEGs, differentially expressed genes; LR, liver regeneration. |
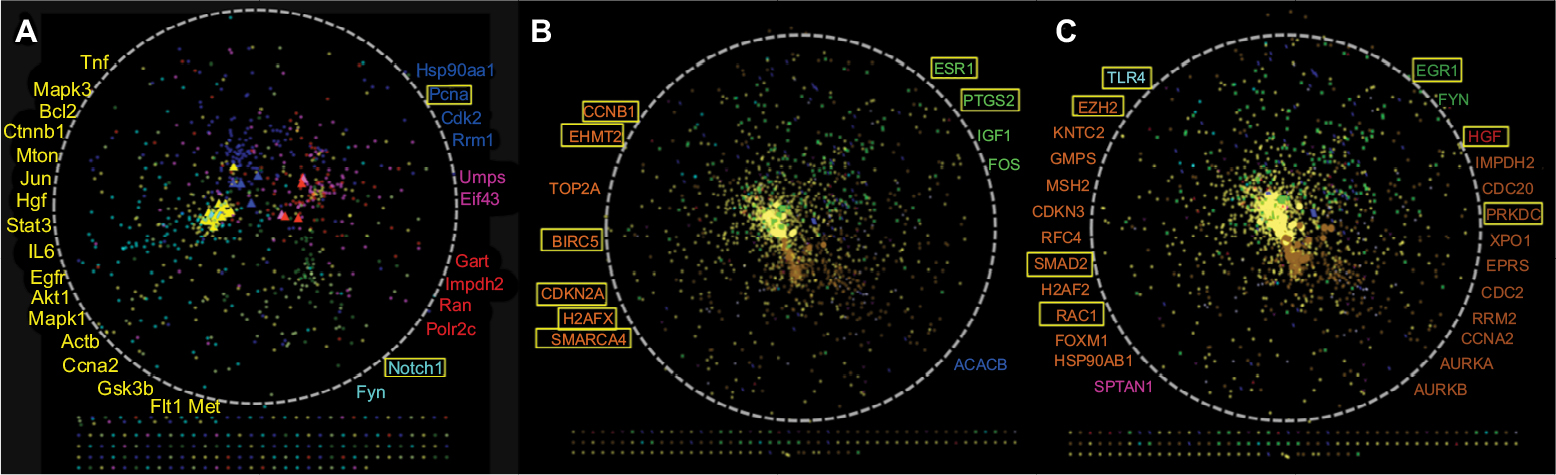 | Figure 9 Distribution of hub-bottleneck genes in the spatial protein–protein interaction (PPI) network. Notes: (A) Distribution of top 5% hub-bottleneck genes in the spatial PPI network in LR. Those in yellow are the known LR-related genes. The rest represent the DEGs in every model, and the color of every gene is consistent with Figure 8. (B) Distribution of top 5% hub bottlenecks belonging to DEGs in the spatial PPI network in HCC. (C) Distribution of top 5%–10% hub bottlenecks belonging to DEGs in the spatial PPI network in HCC. Those in yellow are the known HCC-related genes. The rest represent the DEGs in every model, and the color of every gene is consistent with Figure 8. Yellow rectangles represent the known HCC-related DEGs, and the color of every gene is consistent with Figure 10. Abbreviations: DEGs, differentially expressed genes; HCC, hepatocellular carcinoma; LR, liver regeneration. |
The same method was used for PPI-network construction, which contained 1,963 nodes and 45,499 edges for liver cancer, as shown in Figure 10A. Next, genes belonging to the same module were overlapped onto the network (Figure 10B). Notably, the brown_c56 module formed a central and independent area that represented cell-cycle-related processes. The distribution of genes in the red and brown_c12 modules was also very concentrated. In fact, the genes in red and brown_c12 were from cluster 2, identified by MaSigPro software. They were further divided into two different categories according to the power law by WGCNA and given different physiological meanings. It can be inferred that angiogenesis and fatty acid oxidation may share common regulators or co-occurrence mechanisms, due to their similar expression profiles and close PPI. To explore the spatial distribution of known genes associated with the stepwise carcinogenic process from preneoplastic lesions to end-stage HCC, including cirrhosis, dysplasia, infiltration, and metastasis in the network, the known genes were overlapped onto the network, as shown in Figure 10C–F. It can be concluded that angiogenesis, fatty acid metabolism, and cell proliferation play vital roles during the progression of HCC and that angiogenesis and fatty acid metabolism may be the initial inducing factors. Studies have shown the relationship among fatty acid synthesis, cell proliferation, angiogenesis, and autophagy.32–36 The degree and betweenness of every gene in the network was calculated using the CytoHubba plug-in. The top-ranked 100 and 200 hub-bottleneck genes were overlapped onto the network, as shown in Figure 9B and C. Hub-bottleneck genes belonging to known liver-disease-related genes were not listed in the figure. Notably, the hub-bottleneck genes overlapped mostly with the red, brown_c12, and brown_c56 modules.
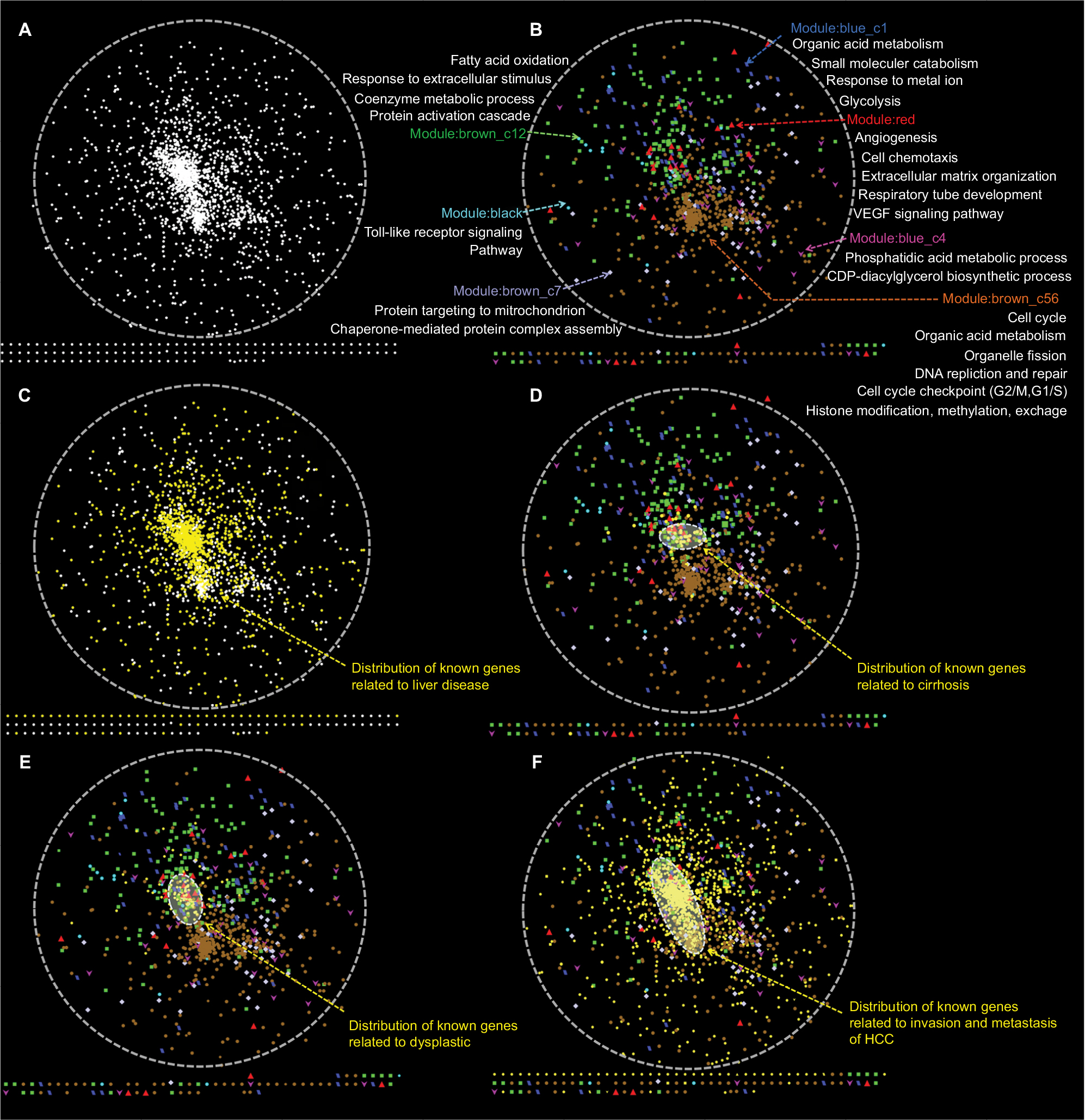 | Figure 10 Distribution of known liver disease-related genes and differentially expressed genes (DEGs) in the spatial protein–protein interaction (PPI) network, generated in Cytoscape. Notes: (A) A PPI network was constructed by connecting all the known liver disease-related genes and DEGs in our study. (B) All DEGs from seven modules were colored differently. The brown_c56 module was in opposition to brown_c12. (C) All known liver-disease-related genes were mapped to the PPI network, shown in yellow. (D) Distribution of all known cirrhosis-related genes, including DEGs, in our study. (E) Distribution of all known dysplasia-related genes, including DEGs, in our study. (F) Distribution of all known invasion and metastasis-related genes, including DEGs, in our study. |
Discussion
With a series of bioinformatic methods and tools, possible key pathways, genes, enriched TFs, and microRNAs that may regulate DEGs were analyzed during the dynamic progression of LR and HCC formation. The fundamental difference between LR and HCC is that the former will eventually return to its 0-h level after PH, whereas the latter will never come back (Figures 6 and 7).
During LR, within 2 h after two-thirds hepatectomy, the residual liver grows relatively slowly, but its growth rate is extremely obvious from 36 to 72 h and then relatively slow. As shown in Figure 6, cell-proliferation-related activities increased significantly between 36 and 72 h, whereas the genes associated with angiogenesis and ECM remodeling and fatty acid oxidation increased significantly during this time period, suggesting that cell proliferation had been completed by 72 h, and angiogenesis and fatty acid metabolism played an active role in the process of liver restoration. At the termination stage of LR, active protein synthesis implies recovery of the physiological function of the liver. Based on this analysis, we propose that liver cells may have some kind of memory signal: once the liver is resected, the signal is activated or induced, and the cell numbers reach original levels in a short period through cell proliferation. In our study, LR was accompanied by acid–base balance disorders. Was the change in the extracellular environment leading to changes in biological processes in the cells or vice versa? In addition, LR was accompanied by brain atrophy and sexual dysplasia. Was this a redistribution of energy after PH or an evolutionary sacrifice to survive? These questions need to be explored further. Some key genes, which included FYN, GART, IMPDH2, RAN, POLR2C, ACACB, CAV1, VIM, and NRP1, some microRNAs, which included miR24 and miR29a–c, and some TFs, including PU1, ELK1, E2F1/4, NRF1, and KTGGYRSGAA_unknown, may play important roles in LR.
During the stepwise carcinogenic process from preneoplastic lesions to end-stage HCC, the most significant turning point is from dysplastic nodules to early HCC. As shown in Figure 7, genes associated with cell-proliferation-related activities increased significantly at very early and early HCC points, whereas genes involved in fatty acid metabolism and angiogenesis were the opposite. All of these three biological processes played vital roles in HCC formation through spatial network analysis. Some key genes, which included FYN, GYS2,CENPF, FYN, CCNB2, KIF4A, H2AFV, BUB1, H2AFZ, DCN, TOP2A, RRM2, HSP90AB1, and XPO1, some microRNAs, which included miR24 and miR208a, and some TFs, which included USF, MAX, E2F1/4, NRF1, YY1, and KTGGYRSGAA_unknown, may play a critical role during HCC formation.
Comparative analysis of LR and HCC formation was performed. LR and the formation of HCC share three common biological processes – cell proliferation, angiogenesis, and fatty acid metabolism – of which angiogenesis may play the most important role based on spatial network analysis. Some key genes, such as FYN, XPO1, FOXM1, and EZH2, were identified and need further study to determine their mechanism in the pathogenesis of LR and HCC. As the most significant and most fundamental change in LR and HCC formation, what exactly determines the initiation and termination of the cell cycle? Why do almost all DEGs return to 0-h levels after PH, but continue to upregulate or downregulate in HCC? These questions need further study.
Angiogenesis is the formation of new microvessels from preexisting vessels and involves tumor growth and metastasis, organ development, regeneration, and wound healing when the distance between cells and vessels increases as the cell numbers increase.37–39 Angiogenesis is a dynamic process that is hypoxia-stimulated and growth-factor-dependent. Several reports have shown that angiogenesis plays a key role in LR after PH and confirmed that LR is an angiogenesis-related physiological process.40,41 It is a striking fact that many of the growth factors upregulated in LR are known to be proangiogenic.42,43 When antiangiogenic agents are administered, hepatocyte proliferation decreases after PH, leading to a delay in LR. In our study, angiogenesis played a vital role in both LR and HCC. Is it possible that angiogenesis is the key driver for normal or abnormal proliferation of cells?
Regardless of LR or the formation of liver cancer, initially small cell clusters form as a result of certain stimuli (eg, resection, virus) that initiate the cell cycle. As the number of cells increases, the distance between cells, especially internal cells and blood vessels, increases, resulting in the formation of a hypoxic environment inside the cells. Cells, including tumor cells, require blood supply to provide oxygen and nutrients while excreting metabolic waste. Hypoxia induces the production of certain factors, eventually leading to the formation of new blood vessels from the preexisting vascular network, which could provide energy and substances for cells. As we know, there is a huge difference between physiological and cancer-associated angiogenesis. For regeneration, regenerated hepatocytes are reconstructed structurally and functionally. Finally, cell proliferation was completed, cells reentered the G0 phase, and all returned to the 0-hour level after hepatectomy. In the advanced stage of HCC, cancer cells spread to nearby normal tissue as the establishment of neovascularization. We discovered some key genes involved in this process by a variety of bioinformatic methods. As one of the most promising developments in cancer treatment, more angiogenesis inhibitors have been evaluated in clinical trials.44–47 In our study, angiogenesis is at the core of LR and HCC formation. The tumor is highly vascularized with abnormal structure and function. Is there a different regulation mechanism in LR and tumor formation? In addition, almost all growth-factor cytokines involved in angiogenesis play an important role in LR, which is not just a coincidence. A further and thorough analysis of angiogenic mechanisms may provide new and effective approaches to augment LR and counter liver diseases, especially HCC.
As the most frequently occurring gene, the role of FYN in LR and HCC is crucial. The proto-oncogene FYN is a member of the Src family of kinases and is a non-receptor tyrosine kinase (nRTK). Most nRTKs are localized in the cytoplasm without a transmembrane structure, which is the fundamental difference between RTKs and the transmembrane domain.48,49 RTKs receive extracellular signals through membrane-bound proteins, including EGFR and VEGFR. Fyn is anchored at the cytoplasmic membrane, through the myristoylation of the second glycine at its N-terminal and palmitoylated myristic acid of the third cysteine. FYN plays an important role in cell growth and proliferation, cell-cycle entry, skeleton remodeling, and cell migration through Ras–Mek–ERK, FAK, and PI3K signaling pathways, is often highly expressed in many tumors, and is related to the initiation of and progression to some cancers.27,28,31 TSP1 is a natural antiangiogenic substance in vivo. It can inhibit angiogenesis by destroying newly formed microvascular endothelial cells and inducing apoptosis of endothelial cells. However, its role requires the transmembrane receptor CD36.31,50 CD36 itself lacks intracellular kinase and has no phosphorylation activity. It works in conjunction with nRTKs, such as Fyn. In other words, TSP1 inhibits angiogenesis by depending on CD36 and Fyn.
In our study, the expression of Fyn in LR was generally in a inverted v-shaped distribution (peak at 72 hours after PH; Figure 11A). In HCC formation, the expression of Fyn in cirrhotic samples increased significantly, then decreased until early-stage HCC, followed by a small upregulation in end-stage HCC (Figure 11B), which was inconsistent with the high expressions in some tumors. Presumably, this was due to the following factors. First, the expression of Fyn is related to the pathological process of the tumor. Second, there are many downstream factors of Fyn, which depend mainly on the upstream factors that act on it. In newly forming vascular endothelial cells, TSP1 inhibits angiogenesis through Fyn, thereby promoting apoptosis and inhibiting proliferation. Although angiogenesis must be present during regeneration and tumor formation, vascular structure and function are abnormal in tumors. We speculate that Fyn may regulate angiogenesis in two ways in two processes or stages. Third, Fyn has three types of alternative splicing: FynT, FynB, and FynC. Among these, FynT contains exon 7B, which is expressed mainly in blood-related cells, and FynB contains exon 7A, which is expressed widely, especially in eucalyptus. FynC does not contain exons, and its corresponding protein and physiological effects have not yet been found.49 Therefore, Fyn may has different spiceosomes and plays different roles in different time and space.
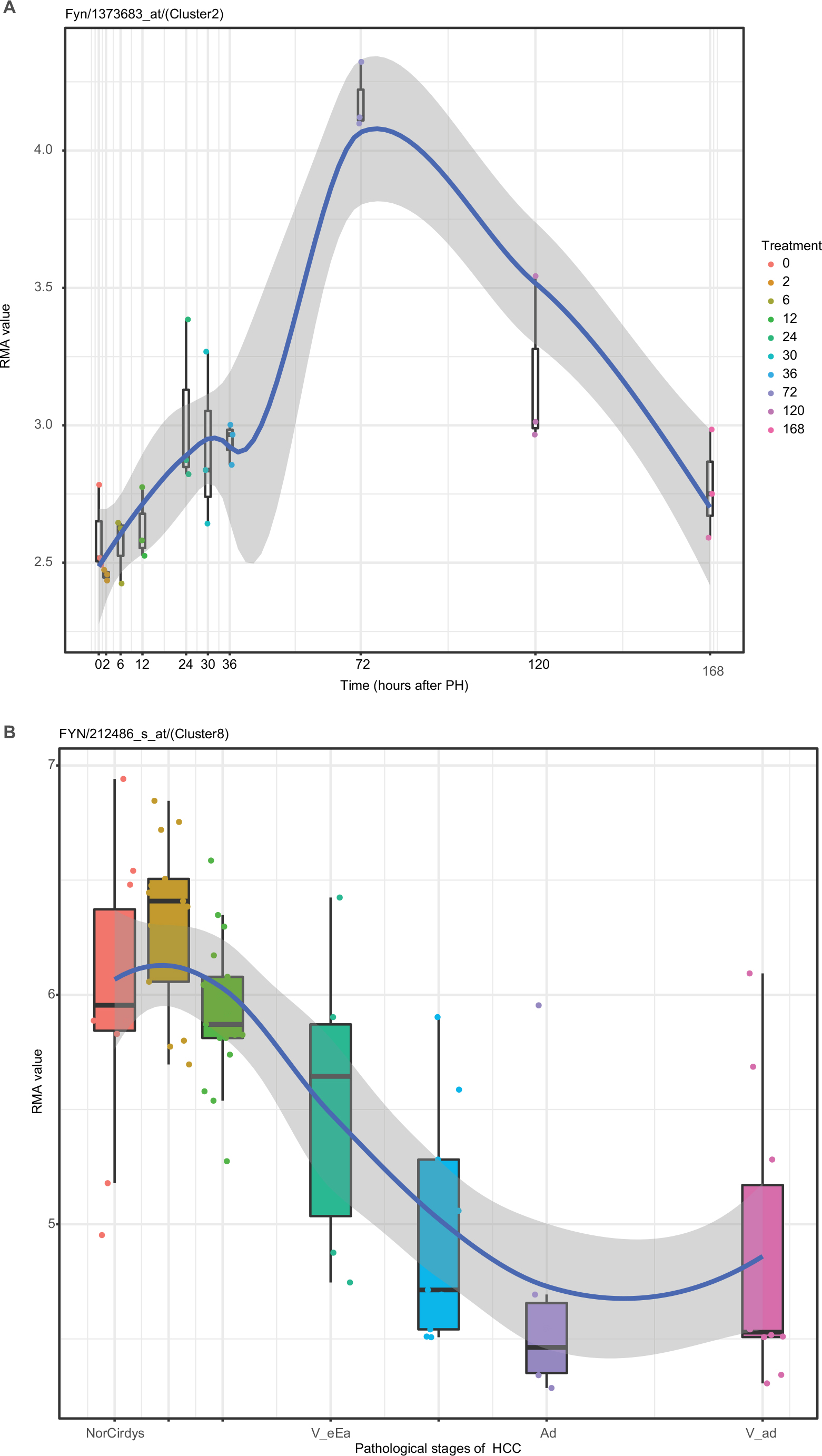 | Figure 11 Dynamic changes in Fyn expression: (A) LR. (B) HCC. Abbreviations: HCC, hepatocellular carcinoma; LR, liver regeneration; PH, partial hepatectomy. |
It is of great theoretical and practical significance to explore the key physiological processes and key genes that determine LR and liver cancer formation. For example, the elucidation of certain molecular mechanisms in LR can be used to promote the survival of patients with acute liver failure, and even artificial livers can be used to make up for donor deficiencies and transplant rejection in liver transplantation.
Conclusion
In summary, a variety of bioinformatic methods, including MaSigPro, WGCNA, and SAFE, have been used to compare the similarities and differences between LR and HCC formation. Key biological processes and genes were identified. We found that angiogenesis plays a critical role in both LR and HCC. Some key genes, including FYN, XPO1, FOXM1, EZH2, and NRF1, were identified. These findings could contribute to revealing molecular mechanisms of development and regulation mechanisms of normal and abnormal proliferation, which could provide new ideas and treatment methods for regenerative medicine, oncological drug development, and oncological treatment.
Data sharing statement
Data sets generated and/or analyzed during the current study are available at https://www.ncbi.nlm.nih.gov/geo.
Acknowledgment
This research was funded by the Natural Science Foundation of China (31572270) and Natural Science Foundation of Henan (162300410181).
Disclosure
The authors report no conflicts of interest in this work.
References
Alison MR, Lin WR. Regenerating the liver: not so simple after all? F1000Res. 2016;5:1818. | ||
Forbes SJ, Newsome PN. Liver regeneration—mechanisms and models to clinical application. Nat Rev Gastroenterol Hepatol. 2016;13(8):473–485. | ||
Bosch FX, Ribes J, Díaz M, Cléries R. Primary liver cancer: worldwide incidence and trends. Gastroenterology. 2004;127(5 Suppl 1):S5–S16. | ||
Pons F, Varela M, Llovet JM. Staging systems in hepatocellular carcinoma. HPB. 2005;7(1):35–41. | ||
Jeong SW, Jang JY, Chung RT. Hepatitis C virus and hepatocarcinogenesis. Clin Mol Hepatol. 2012;18(4):347–356. | ||
Lin MV, King LY, Chung RT. Hepatitis C virus-associated cancer. Annu Rev Pathol. 2015;10:345–370. | ||
Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362(9399):1907–1917. | ||
Schafer DF, Sorrell MF. Hepatocellular carcinoma. Lancet. 1999;353(9160):1253–1257. | ||
Alves RC, Alves D, Guz B, et al. Advanced hepatocellular carcinoma. Review of targeted molecular drugs. Ann Hepatol. 2011;10(1):21–27. | ||
Chui AK, Rao AR, Mccaughan GW, et al. An active liver transplant programme for hepatocellular carcinoma in cirrhotic patients: is it justified? Clin Transplant. 1999;13(6):531–535. | ||
Intaraprasong P, Siramolpiwat S, Vilaichone RK. Advances in management of hepatocellular carcinoma. Asian Pac J Cancer Prev. 2016;17(8):3697–3703. | ||
Kim YS, Lim HK, Rhim H, Lee MW. Ablation of hepatocellular carcinoma. Best Pract Res Clin Gastroenterol. 2014;28(5):897–908. | ||
Bhoori S, Sposito C, Germini A, Coppa J, Mazzaferro V. The challenges of liver transplantation for hepatocellular carcinoma on cirrhosis. Transpl Int. 2010;23(7):712–722. | ||
Dutkowski P, Linecker M, Deoliveira ML, Müllhaupt B, Clavien PA. Challenges to liver transplantation and strategies to improve outcomes. Gastroenterology. 2015;148(2):307–323. | ||
Melloul E, Lesurtel M, Carr BI, Clavien PA. Developments in liver transplantation for hepatocellular carcinoma. Semin Oncol. 2012;39(4):510–521. | ||
Giulietti M, Occhipinti G, Principato G, Piva F. Weighted gene co-expression network analysis reveals key genes involved in pancreatic ductal adenocarcinoma development. Cell Oncol. 2016;39(4):379–388. | ||
Supek F, Bošnjak M, Škunca N, Šmuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE. 2011;6(7):e21800. | ||
Kaimal V, Bardes EE, Tabar SC, Jegga AG, Aronow BJ. ToppCluster: a multiple gene list feature analyzer for comparative enrichment clustering and network-based dissection of biological systems. Nucleic Acids Res. 2010;38(Web Server issue):W96–W102. | ||
Durinck S, Moreau Y, Kasprzyk A, et al. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics. 2005;21(16):3439–3440. | ||
Durinck S, Spellman PT, Birney E, Huber W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc. 2009;4(8):1184–1191. | ||
Baryshnikova A. Systematic functional annotation and visualization of biological networks. Cell Syst. 2016;2(6):412–421. | ||
Costanzo M, Vandersluis B, Koch EN, et al. A global genetic interaction network maps a wiring diagram of cellular function. Science. 2016;353(6306):aaf1420. | ||
Costanzo M, Baryshnikova A, Bellay J, et al. The genetic landscape of a cell. Science. 2010;327(5964):425–431. | ||
Bi D, Ning H, Liu S, Que X, Ding K. Gene expression patterns combined with network analysis identify hub genes associated with bladder cancer. Comput Biol Chem. 2015;56:71–83. | ||
Han JD, Bertin N, Hao T, et al. Evidence for dynamically organized modularity in the yeast protein–protein interaction network. Nature. 2004;430(6995):88–93. | ||
Yu H, Kim PM, Sprecher E, Trifonov V, Gerstein M. The importance of bottlenecks in protein networks: correlation with gene essentiality and expression dynamics. PLoS Comput Biol. 2007;3(4):e59. | ||
Jensen AR, David SY, Liao C, et al. Fyn is downstream of the HGF/MET signaling axis and affects cellular shape and tropism in PC3 cells. Clin Cancer Res. 2011;17(10):3112–3122. | ||
Posadas EM, Al-Ahmadie H, Robinson VL, et al. FYN is overexpressed in human prostate cancer. BJU Int. 2009;103(2):171–177. | ||
Eguchi R, Kubo S, Takeda H, et al. Deficiency of Fyn protein is prerequisite for apoptosis induced by Src family kinase inhibitors in human mesothelioma cells. Carcinogenesis. 2012;33(5):969–975. | ||
Saito YD, Jensen AR, Salgia R, Posadas EM. Fyn: a novel molecular target in cancer. Cancer. 2010;116(7):1629–1637. | ||
Yadav V, Denning MF. Fyn is induced by Ras/PI3K/Akt signaling and is required for enhanced invasion/migration. Mol Carcinog. 2011;50(5):346–352. | ||
Ku CY, Liu YH, Lin HY, Lu SC, Lin JY, Cy K, Sc L, Lin JY. Liver fatty acid-binding protein (L-FABP) promotes cellular angiogenesis and migration in hepatocellular carcinoma. Oncotarget. 2016;7(14):18229–18246. | ||
Menendez JA, Vellon L, Oza BP, Lupu R. Does endogenous fatty acid metabolism allow cancer cells to sense hypoxia and mediate hypoxic vasodilatation? Characterization of a novel molecular connection between fatty acid synthase (FAS) and hypoxia-inducible factor-1alpha (HIF-1alpha)-related expression of vascular endothelial growth factor (VEGF) in cancer cells overexpressing her-2/neu oncogene. J Cell Biochem. 2005;94(5):857–863. | ||
Rae C, Haberkorn U, Babich JW, Mairs RJ. Inhibition of fatty acid synthase sensitizes prostate cancer cells to radiotherapy. Radiat Res. 2015;184(5):482–493. | ||
Singh N, Manhas A, Kaur G, Jagavelu K, Hanif K. Inhibition of fatty acid synthase is protective in pulmonary hypertension. Br J Pharmacol. 2016;173(12):2030–2045. | ||
Singh N, Singh H, Jagavelu K, Wahajuddin M, Hanif K. Fatty acid synthase modulates proliferation, metabolic functions and angiogenesis in hypoxic pulmonary artery endothelial cells. Eur J Pharmacol. 2017;815:462–469. | ||
Baeriswyl V, Christofori G. The angiogenic switch in carcinogenesis. Semin Cancer Biol. 2009;19(5):329–337. | ||
Bishayee A, Darvesh AS. Angiogenesis in hepatocellular carcinoma: a potential target for chemoprevention and therapy. Curr Cancer Drug Targets. 2012;12(9):1095–1118. | ||
de Boüard S, Guillamo JS. Angiogenesis and anti-angiogenic strategies for glioblastoma. Bull Cancer. 2005;92(4):360–372. | ||
Uda Y, Hirano T, Son G, et al. Angiogenesis is crucial for liver regeneration after partial hepatectomy. Surgery. 2013;153(1):70–77. | ||
Drixler TA, Vogten MJ, Ritchie ED, et al. Liver regeneration is an angiogenesis-associated phenomenon. Ann Surg. 2002;236(6):703711–712702 [discussion]. | ||
Shimizu H, Mitsuhashi N, Ohtsuka M, et al. Vascular endothelial growth factor and angiopoietins regulate sinusoidal regeneration and remodeling after partial hepatectomy in rats. World J Gastroenterol. 2005;11(46):7254–7260. | ||
Bockhorn M, Goralski M, Prokofiev D, et al. VEGF is important for early liver regeneration after partial hepatectomy. J Surg Res. 2007;138(2):291–299. | ||
Fuso Nerini I, Cesca M, Bizzaro F, Giavazzi R. Combination therapy in cancer: effects of angiogenesis inhibitors on drug pharmacokinetics and pharmacodynamics. Chin J Cancer. 2016;35(1):61. | ||
Jain RK. Antiangiogenic therapy for cancer: current and emerging concepts. Oncology. 2005;19(4 Suppl 3):7–16. | ||
Niu G, Chen X. Vascular endothelial growth factor as an anti-angiogenic target for cancer therapy. Curr Drug Targets. 2010;11(8):1000–1017. | ||
Yano S, Takehara K, Tazawa H, et al. Cell-cycle-dependent drug-resistant quiescent cancer cells induce tumor angiogenesis after chemotherapy as visualized by real-time FUCCI imaging. Cell cycle. 2017; 16(5):406–414. | ||
Hubbard SR, Till JH. Protein tyrosine kinase structure and function. Annu Rev Biochem. 2000;69:373–398. | ||
Resh MD, Fyn RMD. Fyn, a Src family tyrosine kinase. Int J Biochem Cell Biol. 1998;30(11):1159–1162. | ||
Todorova D, Simoncini S, Lacroix R, Sabatier F, Dignat-George F. Extracellular vesicles in angiogenesis. Circ Res. 2017;120(10):1658–1673. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.