Back to Journals » Cancer Management and Research » Volume 10
Comparison of clinical features and outcomes in patients with extraskeletal vs skeletal Ewing sarcoma: an SEER database analysis of 3,178 cases
Authors Jiang S, Wang G, Chen J, Dong Y
Received 3 July 2018
Accepted for publication 12 October 2018
Published 23 November 2018 Volume 2018:10 Pages 6227—6236
DOI https://doi.org/10.2147/CMAR.S178979
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Chien-Feng Li
Sujing Jiang,1 Guannan Wang,1 Jieyu Chen,2 Ying Dong1
1Department of Medical Oncology, The Second Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, Zhejiang, China; 2Department of Medical Radiology, The Second Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, Zhejiang, China
Background: The clinicopathological characteristics, outcomes and prognostic factors of primary extraskeletal Ewing sarcoma (EES) remained insufficiently explored. We aimed to examine these aspects and compared the same with skeletal Ewing sarcoma (SES).
Patients and methods: We identified Ewing sarcoma, peripheral primitive neuroectodermal tumors or Askin tumor patients who were registered in the Surveillance, Epidemiology, and End Results database from 1973 to 2014. Clinicopathological features were assessed by using Fisher’s exact tests. Cancer-specific survival (CSS) and overall survival (OS) were estimated by using the Kaplan–Meier method and the Cox proportional hazards model. Prognostic factors were identified by multivariate Cox regression analysis.
Results: The age of patients with EES was diagnosed to be higher and they were more likely to be female (46.1% vs 36.2%; P<0.001), have tumor <10 cm (49.8% vs 35.4%; P<0.001), have regional node involvement (5.4% vs 1.0%; P<0.001) and receive surgery (69.1% vs 53.8%; P<0.001) compared to patients with skeletal tumors. Metastatic status did not differ by origin. Kaplan–Meier analysis showed that the origin had significant difference in CSS and OS among patients aged 0–19 years and with metastatic stage at presentation, but not in patients aged 20–39, ≥40 years and with no-metastatic stage. A Cox multivariable model controlling for differences between groups confirmed inferior survival for patients with EES. Age, tumor size, tumor stage and surgery were the most important factors significantly influencing both CSS and OS in the EES and SES patients. Race, year of diagnosis and tumor site were associated with CSS and OS among patients with SES, but failed in EES.
Conclusion: The clinicopathological characteristics, outcomes and prognostic factors differed among patients with EES compared to patients with SES. Extraskeletal origin was an unfavorable prognostic factor.
Keywords: Ewing sarcoma, extraskeletal, skeletal, PNET, outcomes
Introduction
The Ewing sarcoma family of tumors (ESFT) is a family of morphologically similar malignancies that include Ewing tumors, peripheral primitive neuroectodermal tumors (PNET) and Askin tumors, representing the second most frequent bone malignancies in children.1 These tumors can arise from bone or extraskeletal. The extraskeletal Ewing sarcoma (EES) accounts for ~6% to 47% of all Ewing tumors.2 The principles of management of EES have been extrapolated from the treatment of skeletal Ewing sarcoma (SES). It is unclear if the approach of the primary tumor differs based upon tissue of origin. There are few clinical studies available to describe the clinical features, therapeutic approaches and prognostic factors of EES and SES. Moreover, the majority of the studies include patients with small sample size, heterogeneous chemotherapy protocols, conflicting results of outcome and lack of prognostication.3,4 The aim of this study is to evaluate clinicopathological characteristics, outcomes and prognostic factors of EES by using the data from the Surveillance, Epidemiology, and End Results (SEER) database. This may develop a better understanding of the relationship between EES and SES, which is important for the development of future clinical trials and therapeutic protocols.
Patients and methods
Patient population
We gathered patient information from the SEER program between 1973 and 2014. The SEER program is used in the regions that cover 28% of the US population. The SEER system routinely collected data on patient demographics, primary tumor site, tumor morphology and histology, stage at diagnosis, limited treatment data and follow-up for vital status and survival. These data accessed from the SEER database were freely available. Patients with histologically confirmed Ewing sarcoma (EWS, 9260/3), PNET (9364/3) or Askin tumor (9365/3) according to the International Classification of Disease for Oncology, third revision were eligible for the study. Clinicopathological characteristics, outcomes and prognostic factors of EES and SES were compared. Parameters included race, ethnicity, age, sex, year of diagnosis, tumor size, primary tumor site, stage, regional lymph node involvement, surgery data, survival status, cancer-specific survival (CSS) and overall survival (OS). CSS was calculated from the date of diagnosis until the date of disease progression, relapse, death or last follow-up visit. OS was calculated from the date of diagnosis to the date of death or last follow-up. Exclusion criteria include patients with other cancers during lifespan; unknown primary tumor site; lack of race or ethnicity; survival <1 month or unknown; tumor arose within the central nervous system (although primary central nervous system tumors have the same name as peripheral PNET, they are not thought to be related biologically).5
Statistical methods
The Fisher’s exact test was used to identify difference between categorical variables. CSS and OS were estimated by the Kaplan–Meier method, together with 95% CI. Cumulative survival rates were compared using the log-rank test, with P<0.05 considered to be significant. The analysis was also stratified by age and metastatic classification. Cox proportional hazard models were carried out to assess the effect of extraskeletal vs skeletal origin on CSS and OS while controlling for known prognostic factors. The missing data from the analysis included 1,315/3,178 (41.38%) patients with a missing tumor size variable and 2,290/3,178 (72.06%) with a missing regional node involvement variable. A second model was constructed that eliminated tumor size and regional node involvement, which yielded similar results. Back stepwise multivariate Cox regression analysis was done to identify the independent prognostic factors of EES and SES. Statistical analysis was performed using SPSS version 20.0. All P-values were two sided, and P<0.05 was considered statistically significant.
Results
Patients’ characteristics
A total of 3,663 patients with EWS, PNET or Askin tumor diagnosed between 1973 and 2014 were registered in the SEER program. We excluded 340 patients with non-primary EWS, PNET or Askin tumor, 18 patients with unknown primary tumor site, 57 patients with tumor within the central nervous system, 12 patients with unknown race or ethnicity and 58 patients with survival less than 1 month or unknown. The remaining 3,178 patients were included in our analysis; of whom, 981 (30.87%) had EES whereas 2,197 (69.13%) had SES. The demographic and clinical characteristics of the two cohorts are shown in Table 1. The majority of the patients were White non-Hispanic in both EES and SES. The age of patients with EES was diagnosed to be higher compared with patients with SES. The most common age range at initial diagnosis was 20–39 years for EES (35.8% vs 25.8%; P<0.001), whereas ≥40 years (22.1% vs 6.5%; P<0.001) for SES. Patients with EES were more likely to be female (46.1% vs 36.2%; P<0.001), have tumor <10 cm (49.8% vs 35.4%; P<0.001), have regional node involvement (5.4% vs 1.0%; P<0.001) and receive surgery (69.1% vs 53.8%; P<0.001) compared to SES patients. EES was also more likely to have a histological classification of PNET/Askin compared with SES (42.2% vs 3.6%; P<0.001). There were no differences in tumor stage between EES and SES.
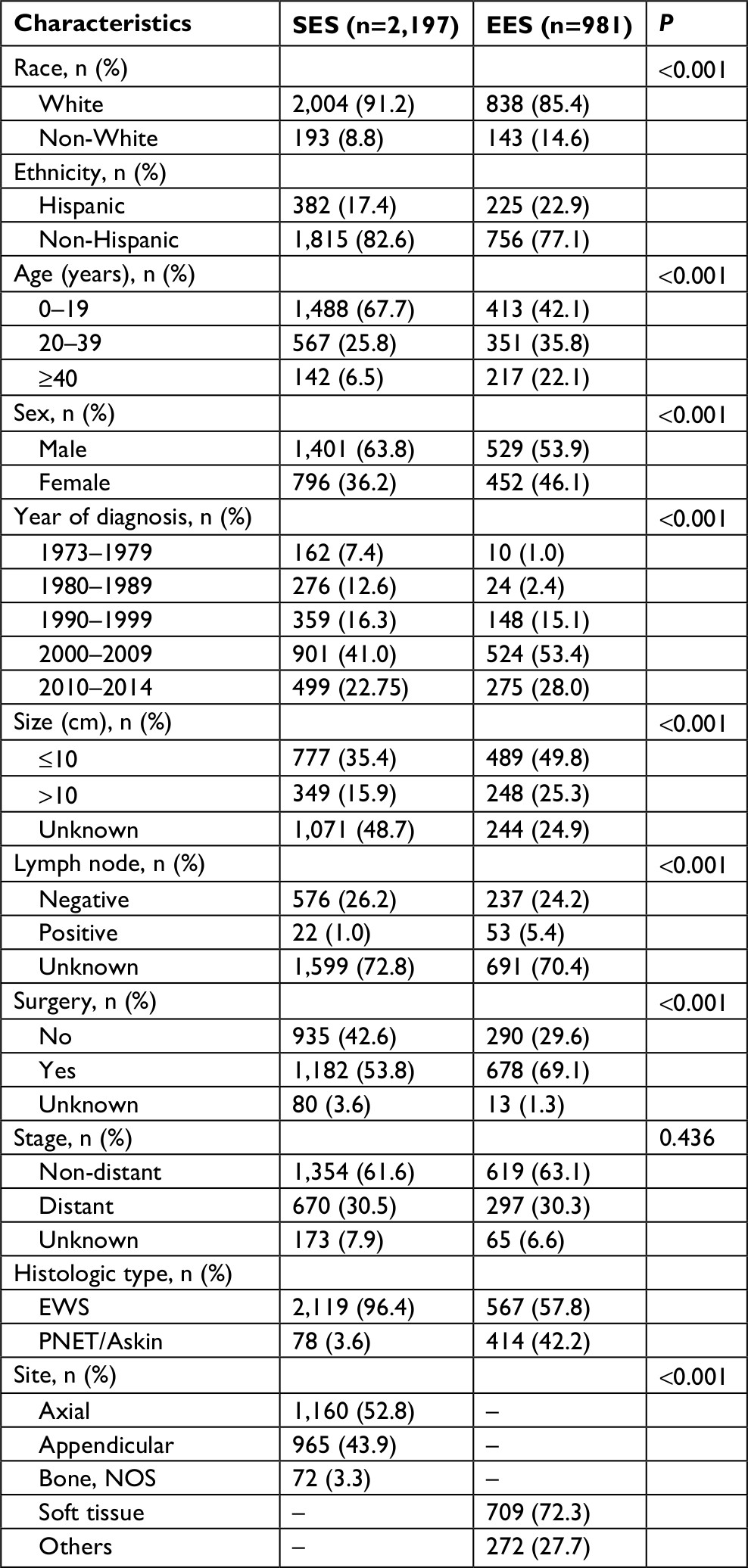 | Table 1 Comparison of demographic and clinical characteristics of patients with EES and SES Notes: “–” indicates not applicable. Abbreviations: EES, extraskeletal Ewing sarcoma; EWS, Ewing sarcoma; NOS, not otherwise specified; PNET, peripheral primitive neuroectodermal tumors; SES, skeletal Ewing sarcoma. |
Clinical outcomes differ between EES and SES
CSS and OS estimates are shown in Figure 1. In all patients, Kaplan–Meier prediction of CSS and OS was not affected by origin. We then analyzed the effects of origin on CSS and OS in each subgroup of ages and metastatic stage. Among patients aged 0–19 years at presentation, the unadjusted CCS and OS for patients with EES were superior compared with those with SES, whereas the prognosis factors for those aged 20–39 and ≥40 years were not affected by origin (Figure 1). The unadjusted 5-year CSS for patients with metastatic stage EES was 26.18% (95% CI, 20.70%–31.67%) compared to 32.47% (95% CI, 28.55%–36.39%) for patients with metastatic stage SES. The unadjusted 5-year OS for patients with metastatic stage EES was 24.70% (95% CI, 19.41%–29.99%) compared to 31.40% (95% CI, 27.48%–35.32%) for patients with metastatic stage SES (Figure 2). To further evaluate these survival differences, we constructed a Cox proportional hazards model in the full analytic that also controlled for other known prognostic factors. After controlling for race, ethnicity, age, sex, year of diagnosis, tumor size, primary tumor site, stage, regional lymph node involvement and surgery data, patients with EES had significantly higher HRs for both CSS and OS compared to SES (HR: 1.157; 95% CI, 1.018–1.315; P=0.025 for CSS; HR: 1.160; 95% CI, 1.026–1.312; P=0.018 for OS; respectively) (Table 2). However, tumor size was not available for approximately half of the analyzed population in SES and ~70% of the patients had incomplete data for regional node involvement in both EES and SES. A second model was constructed that eliminated tumor size and regional node involvement. This model included only ~56% of the patients due to missing data. In this sensitivity model, EES still had significantly higher HRs for both CSS and OS compared to SES (HR: 1.171; 95% CI, 1.032–1.329; P=0.014 for CSS; HR: 1.161; 95% CI, 1.028–1.311; P=0.016 for OS; respectively) (Table S1).
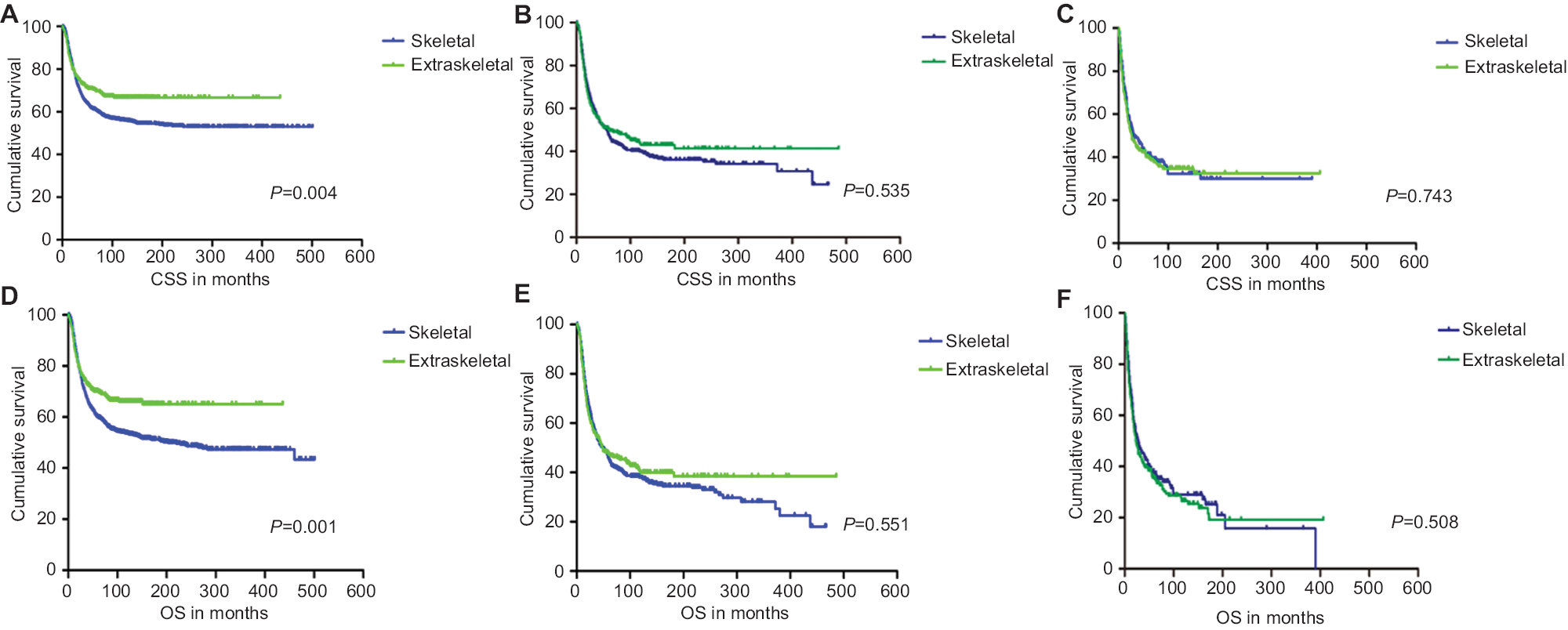 | Figure 1 Kaplan–Meier curves of CSS and OS for patients aged 0–19 years (A and D), 20–39 years (B and E) and ≥40 years (C and F) with Ewing sarcoma. Abbreviations: CSS, cancer-specific survival; OS, overall survival. |
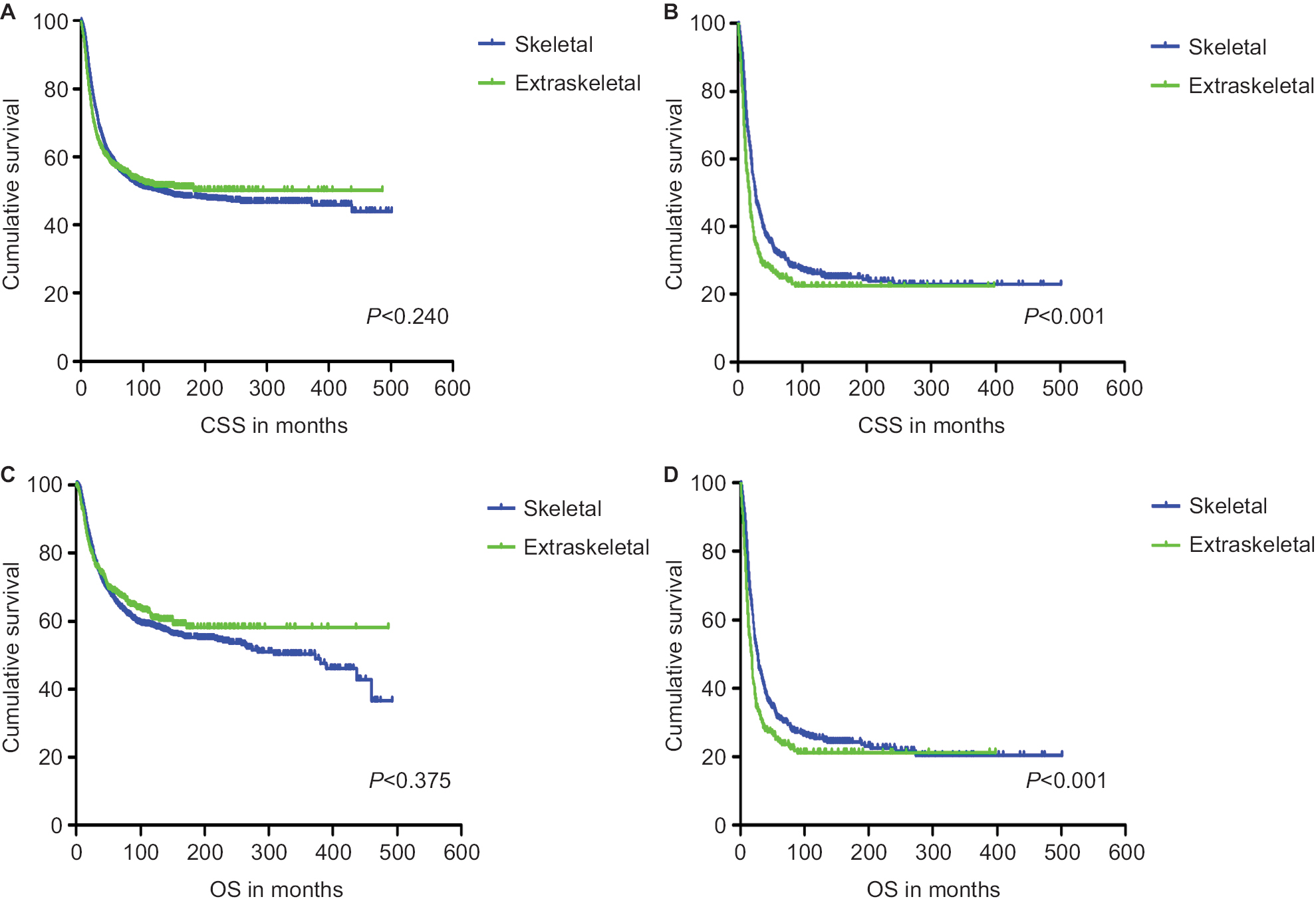 | Figure 2 Kaplan–Meier curves of CSS and OS for patients with non-metastatic stage (A and C) and metastatic stage (B and D) Ewing sarcoma. Abbreviations: CSS, cancer-specific survival; OS, overall survival. |
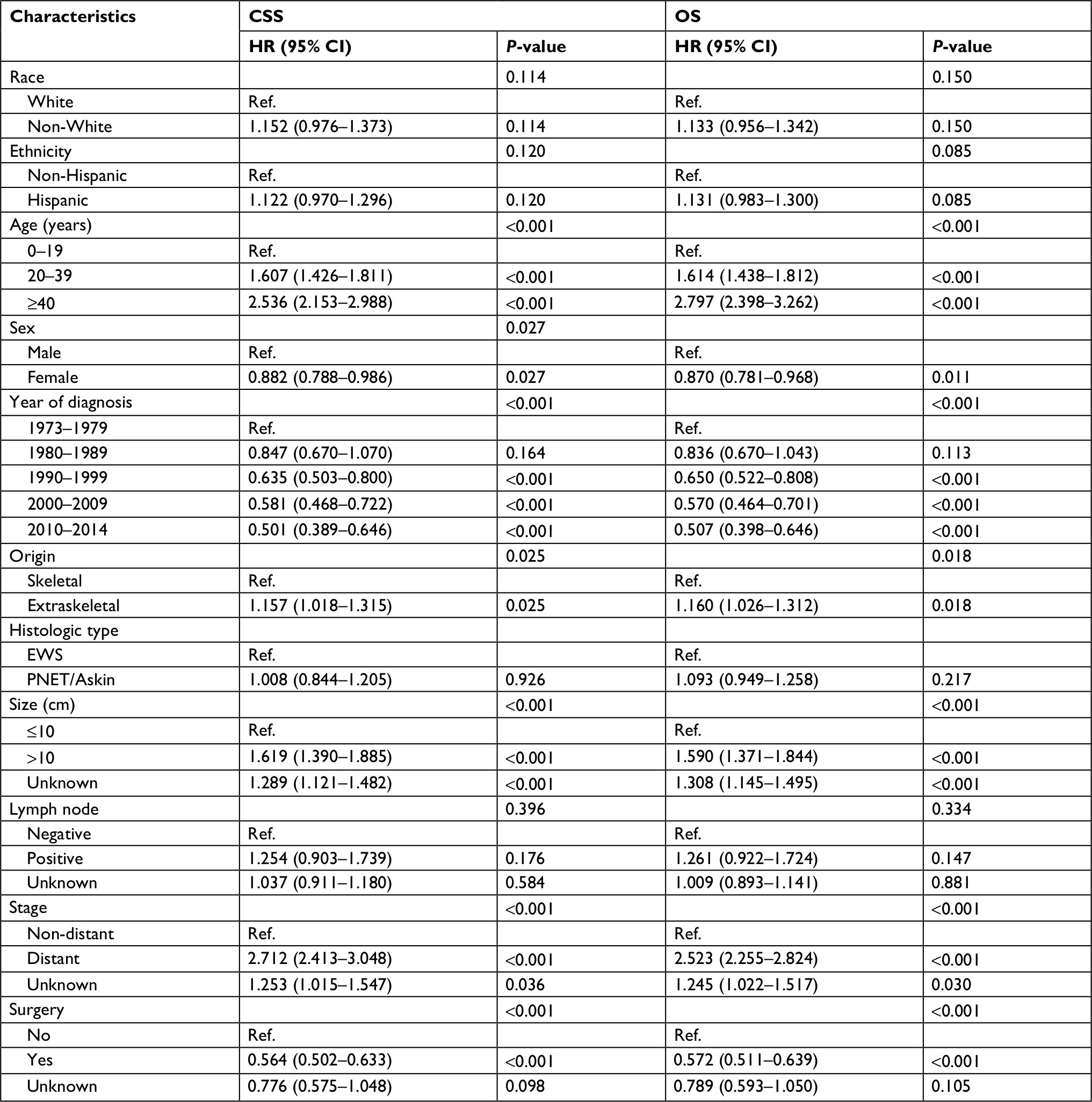 | Table 2 Cox proportional hazards analysis of CSS and OS for patients with extraskeletal and skeletal Ewing sarcoma Abbreviations: CSS, cancer-specific survival; EWS, Ewing sarcoma; OS, overall survival; PNET, peripheral primitive neuroectodermal tumors; Ref., reference. |
Prognostic factors differ between EES and SES
Multivariate Cox regression analysis was used to evaluate associations of race, ethnicity, age, sex, year of diagnosis, tumor size, primary tumor site, stage, regional lymph node involvement and surgery data. In multivariate analysis, age, size, stage and surgery were independent risk factors for CSS and OS in both EES and SES (Table 3). Patients with non-White, earlier years of diagnoses and axial location were associated with a significantly worse CSS and OS in SES (Table 4).
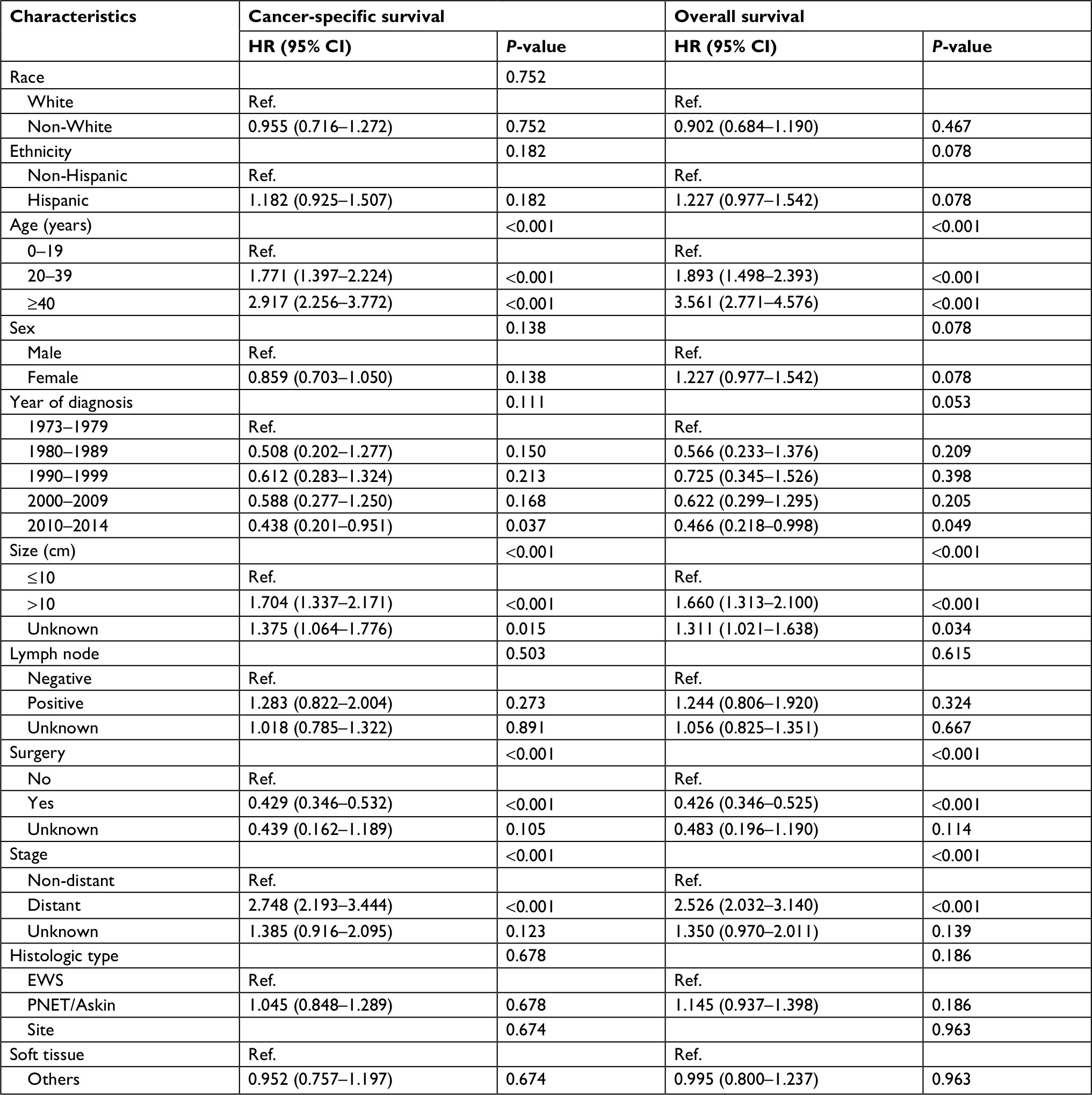 | Table 3 Multivariate Cox regression analysis for patients with extraskeletal Ewing sarcoma Abbreviations: EWS, Ewing sarcoma; PNET, peripheral primitive neuroectodermal tumors; Ref., reference. |
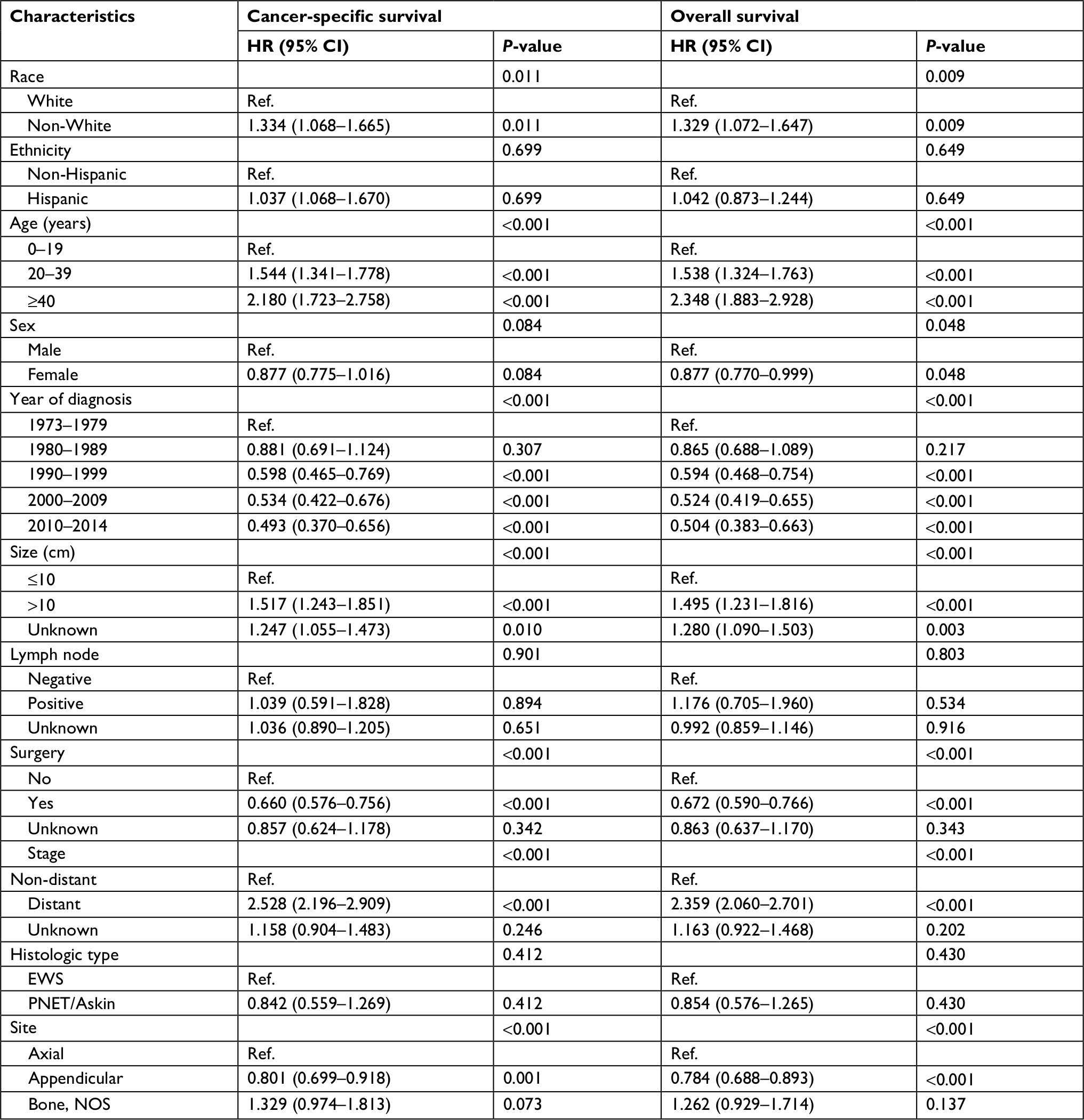 | Table 4 Multivariate Cox regression analysis for patients with skeletal Ewing sarcoma Abbreviations: EWS, Ewing sarcoma; PNET, peripheral primitive neuroectodermal tumors; Ref., reference. |
Discussion
EES is an aggressive type of tumor with a high incidence of local recurrence and distant metastasis. The clinical features, treatment and prognostic factors of EES were diverse and limited to small reports from few institutions. In the present study, we observed several notable differences in disease presentation between EES and SES. The differences in clinical characteristics and prognosis between EES and SES may be due to the differences in microenvironment and angiogenesis between the extraskeletal and skeletal origin.6,7
EES is relatively less common in children and adolescents. This finding was consistent with previous studies.8,9 Data comparing tumor size between EES and SES were conflicting. Some studies have shown that extraskeletal tumors were smaller at diagnosis, whereas others have shown no difference.10–12 We observed that extraskeletal patients were more likely to have smaller tumors compared to patients with SES. Patients with EES had a higher proportion of surgical resection as primary local therapy in comparison with the patients with SES. This is similar to the observation of Biswas et al.13 This difference in management may represent alternative decision-making by tumor site. Previous studies have shown that SES were more likely pelvic primary tumors.10,11 The resection and reconstruction of pelvic tumors were much more technically difficult than extremity lesions.14 Although complex and challenging, Puri et al15 indicated that the limb-sparing surgery in pelvic Ewing’s sarcoma was safe in oncology and had better functional and survival outcomes than amputation surgery. We also observed that patients with EES were more likely to have regional node involvement. Huh et al16 presented a wide spectrum of imaging features and metastatic patterns of EES in adult Asian patients, showing that lymph nodes were the most frequent site of metastases, followed by lungs, bones, solid organs, peritoneum and pleura. Völker et al17 also found that the incidence of EES with regional node involvement had consistently increased, particularly in recent years, likely reflecting that the development of imaging technology had improved detection. Applebaum et al18 indicated that regional node involvement may be an independent adverse prognostic factor in EWS and should be investigated carefully when diagnosed with EES. In contrast, Worch et al19 demonstrated that in patients with SES the tumors were more frequently transferred to the lung, followed by bone and bone marrow. Those findings suggested that the metastatic pattern may be different between EES and SES, although there was no difference in metastatic status in our study. It may reflect the underlying biological differences that we were unable to control for in this analysis, since the SEER database was limited in genetic and molecular information as mentioned above. EES was also more likely to have a histological classification of PNET/Askin compared with SES, as reported in the previous report.11 The increased proportion of extraskeletal tumors classified as PNET may reflect that the development of cytogenetic and molecular diagnostic techniques now allow for more reliable detection of genetic translocations characteristic of this disease.20
Whether EES carried a worse prognosis compared with SES had been an issue of controversy in some studies.4,21,22 In the present study, origin was not a statistically significant prognostic factor for survival among all patients on univariate analysis. Subgroup univariate analysis evaluated that the CCS and OS were superior for patients aged 0–19 years in EES compared to those with SES, whereas the prognosis factors for patients aged 20–39 and ≥40 years were not affected by origin. Contrarily, the unadjusted 5-year CSS and OS for patients with metastatic stage SES were better than patients with metastatic stage EES. It had been confirmed that increasing age was an independent unfavorable prognostic factor in EWS.23,24 Few other studies also had demonstrated that there was no association between older age and worse prognosis when adult patients were treated by pediatric protocols.25,26 This may indicate the difference in treatment between youth and old patients with EWS. In the multivariate analysis, we found that patients with EES had an unfavorable prognosis compared with SES when combining all the patients. Due to the large numbers of missing values of tumor size and the regional node involvement, a second model was constructed that eliminated tumor size and regional node involvement, which yielded similar results. This was different from the observation in various published studies, which had suggested that the outcomes for EES and SES were similar.9,12,27 Applebaum et al found that patients with EES had an unfavorable prognosis prior to 2 years from initial diagnosis, but then the outcomes for EES are significantly better. This was also observed in the study by Biswas et al.13 These findings suggested that the outcome differences between patients with EES and SES were more complex. The etiology for this pattern was not clear. There may be other biological or therapeutic differences that contribute to mortality observed in EES. Unfortunately, no more biological and therapeutic data were available from the SEER database, and these possibilities cannot be further elucidated by this study.
Prognostic factors identified specifically in patients with EES were similar to those reported for SES.28,29 In this study, age, tumor size, tumor stage and surgery were the most important factors significantly influencing CSS and OS in the two cohorts. We also found that race and year of diagnosis were associated with CSS and OS among patients with SES, but failed in EES. Jawad et al30 found that woman constituted a survival benefit among the Caucasian, although the survival of EWS was not impacted by race. Over the past several decades, improvements in outcomes for patients with SES have been achieved through multidisciplinary approaches and multi-institutional trials.31–33 This may lead to the improved survival of SES. However, the principles of management of EES have been extrapolated from the treatment of skeletal tumors. It was still unclear if the approach of the primary tumor differs based upon tissue of origin.
There were several limitations of this study. Our study was a retrospective study that employed the SEER database, which is limited to the available important variables, such as time to tumor recurrence, chemotherapy regimen utilized and molecular pathological characteristics, which may affect the prognosis of patients. However, this database is population based and practiced in the USA, which makes our findings broadly applicable. Most importantly, tumor size and the regional node involvement were not available for approximately half of the analyzed population, which may result in a limitation on the well-established prognostic factor in multivariate analysis.
Conclusion
We demonstrate that clinicopathological characteristics, outcomes and prognostic factors differ between EES and SES. The results of the present study show that EES was an aggressive disease with an inferior survival compared to SES. Future studies should be carried out to investigate the underlying biological differences between EES and SES to determine optimal treatment strategies to maximize outcomes in these tumors.
Abbreviations
EES, extraskeletal Ewing sarcoma; EWS, Ewing sarcoma; SES, skeletal Ewing sarcoma; PNET, peripheral primitive neuroectodermal tumors; SEER, Surveillance, Epidemiology, and End Results; CSS, cancer-specific survival; OS, overall survival; ESFT, Ewing sarcoma family of tumors.
Acknowledgments
All authors want to thank Weidong Han for statistical analysis guidance. In addition, Sujing Jiang particularly wants to thank QianHan Ye for her invaluable support over the years.
Author contributions
YD conceived the concept and designed the study; SJ and GW performed the experiments; JC analyzed the data and prepared the figures; and YD and SJ wrote the paper. All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Granowetter L, West DC. The Ewing’s sarcoma family of tumors: Ewing’s sarcoma and peripheral primitive neuroectodermal tumor of bone and soft tissue. Cancer Treat Res. 1997;92:253–308. | ||
Fizazi K, Dohollou N, Blay JY, et al. Ewing’s family of tumors in adults: multivariate analysis of survival and long-term results of multimodality therapy in 182 patients. J Clin Oncol. 1998;16(12):3736–3743. | ||
Martin RC, Brennan MF. Adult soft tissue Ewing sarcoma or primitive neuroectodermal tumors: predictors of survival? Arch Surg. 2003;138(3):281–285. | ||
Orr WS, Shannon Orr W, Denbo JW, et al. Analysis of prognostic factors in extraosseous Ewing sarcoma family of tumors: review of St. Jude Children’s Research Hospital experience. Ann Surg Oncol. 2012;19(12):3816–3822. | ||
Karski EE, Mcilvaine E, Segal MR, et al. Identification of discrete prognostic groups in Ewing sarcoma. Pediatr Blood Cancer. 2016;63(1):47–53. | ||
Ahmad R, Mayol BR, Davis M, Rougraff BT. Extraskeletal Ewing’s sarcoma. Cancer. 1999;85(3):725–731. | ||
Dehner LP. Primitive neuroectodermal tumor and Ewing’s sarcoma. Am J Surg Pathol. 1993;17(1):1–13. | ||
Lee JA, Kim DH, Lim JS, et al. Soft-tissue Ewing sarcoma in a low-incidence population: comparison to skeletal Ewing sarcoma for clinical characteristics and treatment outcome. Jpn J Clin Oncol. 2010;40(11):1060–1067. | ||
El Weshi A, Allam A, Ajarim D, et al. Extraskeletal Ewing’s sarcoma family of tumours in adults: analysis of 57 patients from a single institution. Clin Oncol. 2010;22(5):374–381. | ||
Applebaum MA, Worch J, Matthay KK, et al. Clinical features and outcomes in patients with extraskeletal Ewing sarcoma. Cancer. 2011;117(13):3027–3032. | ||
Cash T, Mcilvaine E, Krailo MD, et al. Comparison of clinical features and outcomes in patients with extraskeletal versus skeletal localized Ewing sarcoma: a report from the Children’s Oncology Group. Pediatr Blood Cancer. 2016;63(10):1771–1779. | ||
Castex MP, Rubie H, Stevens MC, et al. Extraosseous localized ewing tumors: improved outcome with anthracyclines – the French society of pediatric oncology and international society of pediatric oncology. J Clin Oncol. 2007;25(10):1176–1182. | ||
Biswas B, Shukla NK, Deo SV, et al. Evaluation of outcome and prognostic factors in extraosseous Ewing sarcoma. Pediatr Blood Cancer. 2014;61(11):1925–1931. | ||
Indelicato DJ, Keole SR, Shahlaee AH, et al. Impact of local management on long-term outcomes in Ewing tumors of the pelvis and sacral bones: the University of Florida experience. Int J Radiat Oncol Biol Phys. 2008;72(1):41–48. | ||
Puri A, Pruthi M, Gulia A. Outcomes after limb sparing resection in primary malignant pelvic tumors. Eur J Surg Oncol. 2014;40(1):27–33. | ||
Huh J, Kim KW, Park SJ, et al. Imaging features of primary tumors and metastatic patterns of the extraskeletal Ewing sarcoma family of tumors in adults: a 17-year experience at a single institution. Korean J Radiol. 2015;16(4):783–790. | ||
Völker T, Denecke T, Steffen I, et al. Positron emission tomography for staging of pediatric sarcoma patients: results of a prospective multicenter trial. J Clin Oncol. 2007;25(34):5435–5441. | ||
Applebaum MA, Goldsby R, Neuhaus J, Dubois SG. Clinical features and outcomes in patients with Ewing sarcoma and regional lymph node involvement. Pediatr Blood Cancer. 2012;59(4):617–620. | ||
Worch J, Ranft A, Dubois SG, Paulussen M, Juergens H, Dirksen U. Age dependency of primary tumor sites and metastases in patients with Ewing sarcoma. Pediatr Blood Cancer. 2018;65(9):e27251. | ||
Bovée JV, Hogendoorn PC. Molecular pathology of sarcomas: concepts and clinical implications. Virchows Arch. 2010;456(2):193–199. | ||
Rud NP, Reiman HM, Pritchard DJ, Frassica FJ, Smithson WA. Extraosseous Ewing’s sarcoma. A study of 42 cases. Cancer. 1989;64(7):1548–1553. | ||
Baldini EH, Demetri GD, Fletcher CD, Foran J, Marcus KC, Singer S. Adults with Ewing’s sarcoma/primitive neuroectodermal tumor: adverse effect of older age and primary extraosseous disease on outcome. Ann Surg. 1999;230(1):79–86. | ||
Duchman KR, Gao Y, Miller BJ. Prognostic factors for survival in patients with Ewing’s sarcoma using the Surveillance, Epidemiology, and End Results (SEER) program database. Cancer Epidemiol. 2015;39(2):189–195. | ||
Karski EE, Matthay KK, Neuhaus JM, Goldsby RE, Dubois SG. Characteristics and outcomes of patients with Ewing sarcoma over 40 years of age at diagnosis. Cancer Epidemiol. 2013;37(1):29–33. | ||
Bacci G, Mercuri M, Longhi A, et al. Neoadjuvant chemotherapy for Ewing’s tumour of bone: recent experience at the Rizzoli Orthopaedic Institute. Eur J Cancer. 2002;38(17):2243–2251. | ||
Ahmed SK, Robinson SI, Okuno SH, Rose PS, Laack NN. Adult ewing sarcoma: survival and local control outcomes in 102 patients with localized disease. Sarcoma. 2013;2013:681425–7. | ||
van den Berg H, Heinen RC, van der Pal HJ, Merks JH. Extra-osseous Ewing sarcoma. Pediatr Hematol Oncol. 2009;26(4):175–185. | ||
Lee J, Hoang BH, Ziogas A, Zell JA. Analysis of prognostic factors in Ewing sarcoma using a population-based cancer registry. Cancer. 2010;116(8):1964–1973. | ||
Cotterill SJ, Ahrens S, Paulussen M, et al. Prognostic factors in Ewing’s tumor of bone: analysis of 975 patients from the European Intergroup Cooperative Ewing’s Sarcoma Study Group. J Clin Oncol. 2000;18(17):3108–3114. | ||
Jawad MU, Cheung MC, Min ES, Schneiderbauer MM, Koniaris LG, Scully SP. Ewing sarcoma demonstrates racial disparities in incidence-related and sex-related differences in outcome: an analysis of 1631 cases from the SEER database, 1973-2005. Cancer. 2009;115(15):3526–3536. | ||
Meyer WH, Kun L, Marina N, et al. Ifosfamide plus etoposide in newly diagnosed Ewing’s sarcoma of bone. J Clin Oncol. 1992;10(11):1737–1742. | ||
Paulussen M, Ahrens S, Dunst J, et al. Localized Ewing tumor of bone: final results of the cooperative Ewing’s Sarcoma Study CESS 86. J Clin Oncol. 2001;19(6):1818–1829. | ||
Grier HE, Krailo MD, Tarbell NJ, et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing’s sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med. 2003;348(8):694–701. |
Supplementary material
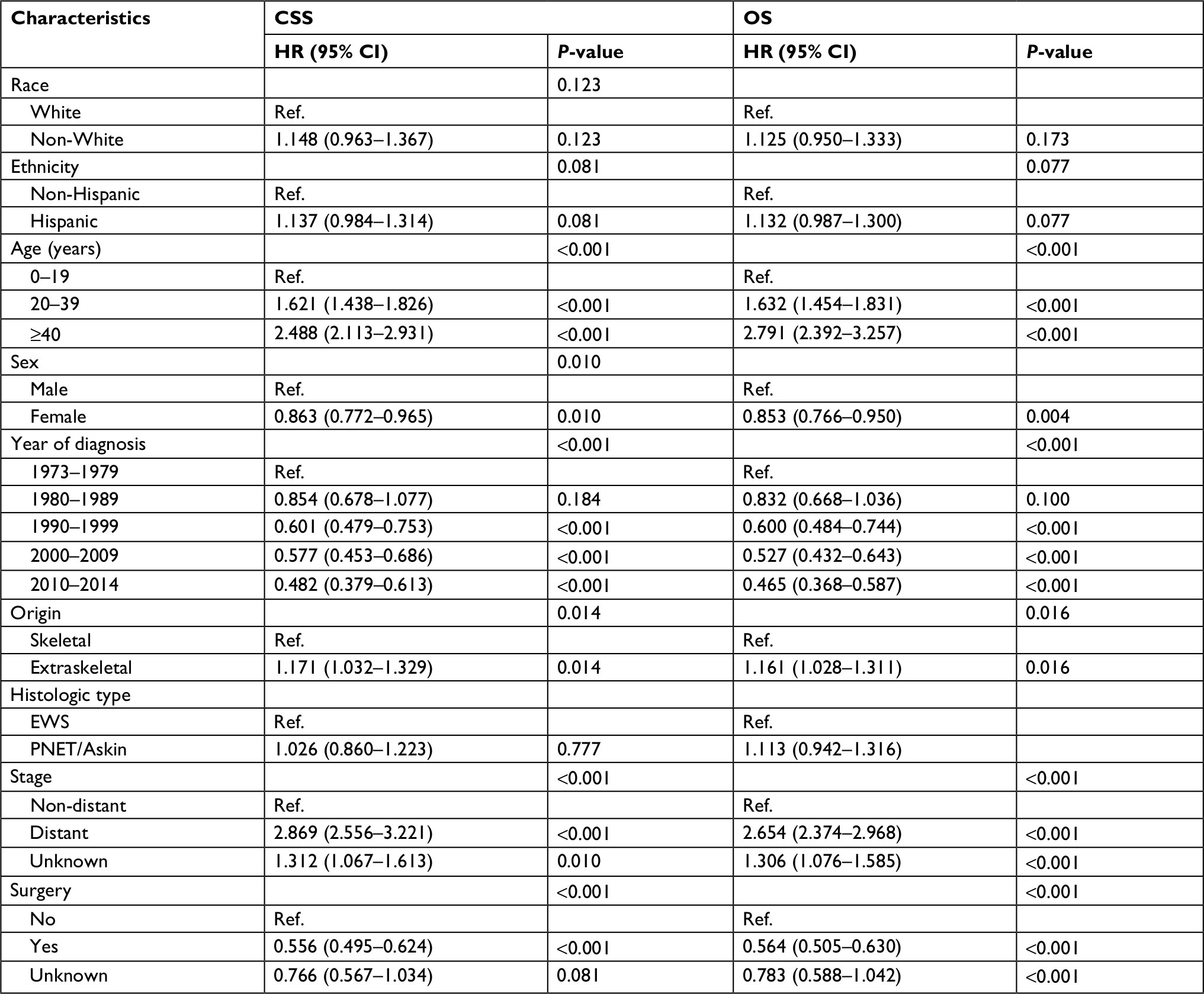 | Table S1 Cox proportional hazards analysisa of CSS and OS for patients with extraskeletal and skeletal Ewing sarcoma Notes: aModel adjusted for tumor size and regional node involvement. Abbreviations: CSS, cancer-specific survival; EWS, Ewing sarcoma; OS, overall survival; PNET, peripheral primitive neuroectodermal tumors; Ref., reference. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.