Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Comparative study on solid self-nanoemulsifying drug delivery and solid dispersion system for enhanced solubility and bioavailability of ezetimibe
Authors Rashid R, Kim D, Yousaf AM, Mustapha O ![]() , Din FU, Park JH
, Din FU, Park JH ![]() , Yong CS, Oh Y, Youn YS
, Yong CS, Oh Y, Youn YS ![]() , Kim JO, Choi H
, Kim JO, Choi H
Received 26 June 2015
Accepted for publication 1 September 2015
Published 30 September 2015 Volume 2015:10(1) Pages 6147—6159
DOI https://doi.org/10.2147/IJN.S91216
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Carlos Rinaldi
Rehmana Rashid,1 Dong Wuk Kim,1 Abid Mehmood Yousaf,1 Omer Mustapha,1 Fakhar ud Din,1 Jong Hyuck Park,1 Chul Soon Yong,2 Yu-Kyoung Oh,3 Yu Seok Youn,4 Jong Oh Kim,2 Han-Gon Choi1
1College of Pharmacy and Institute of Pharmaceutical Science and Technology, Hanyang University, Ansan, South Korea; 2College of Pharmacy, Yeungnam University, Gyeongsan, South Korea; 3College of Pharmacy, Seoul National University, Seoul, South Korea; 4School of Pharmacy, Sungkyunkwan University, Suwon, South Korea
Background: The objective of this study was to compare the physicochemical characteristics, solubility, dissolution, and oral bioavailability of an ezetimibe-loaded solid self-nanoemulsifying drug delivery system (SNEDDS), surface modified solid dispersion (SMSD), and solvent evaporated solid dispersion (SESD) to identify the best drug delivery system with the highest oral bioavailability.
Methods: For the liquid SNEDDS formulation, Capryol 90, Cremophor EL, and Tween 80 were selected as the oil, surfactant, and cosurfactant, respectively. The nanoemulsion-forming region was sketched using a pseudoternary phase diagram on the basis of reduced emulsion size. The optimized liquid SNEDDS was converted to solid SNEDDS by spray drying with silicon dioxide. Furthermore, SMSDs were prepared using the spray drying technique with various amounts of hydroxypropylcellulose and Tween 80, optimized on the basis of their drug solubility. The SESD formulation was prepared with the same composition of optimized SMSD. The aqueous solubility, dissolution, physicochemical properties, and pharmacokinetics of all of the formulations were investigated and compared with the drug powder.
Results: The drug existed in the crystalline form in SMSD, but was changed into an amorphous form in SNEDDS and SESD, giving particle sizes of approximately 24, 6, and 11 µm, respectively. All of these formulations significantly improved the aqueous solubility and dissolution in the order of solid SNEDDS ≥ SESD > SMSD, and showed a total higher plasma concentration than did the drug powder. Moreover, SESD gave a higher area under the drug concentration time curve from zero to infinity than did SNEDDS and SMSD, even if they were not significantly different, suggesting more improved oral bioavailability.
Conclusion: Among the various formulations tested in this study, the SESD system would be strongly recommended as a drug delivery system for the oral administration of ezetimibe with poor water solubility.
Keywords: ezetimibe, solid self-nanoemulsifying drug delivery system, solid dispersion, solubility, bioavailability
Introduction
The main risk factor for coronary heart disease is having elevated low-density lipoproteins. High cholesterol or hyperlipidemia refers to a high degree of cholesterol in the blood, which raises the risk of heart disease, stroke, and atherosclerosis, plus other serious conditions. The discovery of the novel cholesterol-lowering agent ezetimibe represents a different position from other developments. The first new therapy for the treatment of hypercholesterolemia since the discovery of the statins, ezetimibe represents an important discovery.1 Ezetimibe, [1-(4-fluorophenyl)-3(R)-[3-(4-fluorophenyl)-3(S)-hydroxypropyl]-4(S)-(4 hydroxyphenyl)-2-azetidinone], which selectively obstructs the absorption of bile and dietary cholesterol, as well as related phytosterols, from the intestine without affecting the concentration of fat-soluble vitamins, triglycerides, or bile acids, is the first member of a new class of lipid-lowering agents.2,3 The US Food and Drug Administration has accepted ezetimibe as a new medication.4 Ezetimibe is a class II molecule as per the Biopharmaceutics Classification System due to its poor water solubility and high permeability. It has a fast first-pass metabolism and P-glycoprotein efflux. Moreover, due to its hydrophobic character, this drug has an extremely irregular and very low dissolution profile in the gastrointestinal (GI) fluids,5 resulting in highly unpredictable bioavailability.6,7
The bioavailability of orally administered drugs can be improved by using lipid-based formulations.8 Despite a few restrictions, such as sensitivity to humidity and temperature, a high cost of production, and incompatibility with soft gelatin capsules,9 the self-nanoemulsifying drug delivery system (SNEDDS) is a well-known strategy for the delivery of hydrophobic drugs.10 A solidification system is used to prepare solid SNEDDS by incorporating the liquid excipients into powders; is a favorable drug delivery system for compounds with poor water solubility as it combines the benefits of liquid SNEDDS (improved solubility and bioavailability) with those of solid dosage forms (high stability with various dosage form options).11 Solid SNEDDS produced oil-in-water nanoemulsions upon slight agitation in aqueous media and GI fluids.12,13
The solid dispersion system is a renowned method for improving the solubility and bioavailability of the drugs with poor water solubility.14 The solubility and dissolution of a hydrophobic drug can be increased by incorporating it into hydrophilic polymers. Such a system improves the solubility, either by a surface modified method where the drug is dispersed in the aqueous polymer solution15 or by the solvent evaporation method where the drug and polymer are dissolved in a common solvent, followed by removal of the solvent.16 The drug dispersed in polymeric carriers has a small particle size and a large surface area, resulting in increased dissolution rates.17 Several methods, such as the kneading, melting, and solvent wetting methods, have been previously used for the preparation of solid dispersions.18 However, the use of these methods has certain drawbacks, such as chemical decomposition, environmental problems, and stability problems. Solid dispersion by the surface modified technique and the solvent evaporation method seem to be the most suitable methods for enhancing the solubility and bioavailability of drugs with poor water solubility.18
In this study, the physicochemical characteristics, solubility, and bioavailability of an ezetimibe-loaded solid SNEDDS, a surface modified solid dispersion (SMSD), and a solvent evaporated solid dispersion (SESD) were compared in order to select the best drug delivery system with the highest oral bioavailability. The physicochemical characteristics were assessed using differential scanning calorimetry (DSC), scanning electron microscopy (SEM), Fourier-transform infrared spectroscopy (FTIR), and powder x-ray diffraction (PXRD). Additionally, their aqueous solubility, dissolution, and pharmacokinetics in rats were investigated compared with the drug powder.
Materials and methods
Materials
Ezetimibe was obtained from MSN Laboratories Ltd (Medak, Telangana, India). Cotton oil, corn oil, peanut oil, sesame oil, soybean oil, capryol glyceryl monocaprylate, mineral oil, linseed oil, olive oil, Tween 20 (polysorbate 20), Tween 80 (polysorbate 80), Span 20 (sorbitan monooleate 20), and Span 80 (sorbitan monooleate 20) were obtained from Daejung Chem. Co. (Siheung, Gyeonggi, South Korea). Capryol 90 (propanediol monocaprylate), Labrafil M 2125 CS (linoleoyl polyoxyl-6 glycerides), and Labrasol (caprylocaproyl polyoxyl-8 glycerides) were obtained from Gattefossé (Saint-Priest Cedex, France). Hydroxypropylmethylcellulose, polyvinyl alcohol, sodium carboxymethylcellulose, and hydroxypropylcellulose (HPC) were provided by Shin-Etsu Co. (Tokyo, Japan). Dextran was obtained from Sigma-Aldrich Co. (St Louis, MO, USA). Gelatin, sodium alginate, carbopol, and the Cremophor series (EL, A6, A25, RH40, and PH60) were obtained from BASF (Ludwigshafen, Germany). Silicon dioxide (Aerosil® 200) was obtained from Degussa (Frankfurt, Germany). All other chemicals and solvents were of reagent grade and were used without additional purification.
Solubility
An excess amount of ezetimibe (nearly 20 mg) was placed in a 2 mL microtube (Axygen MCT-200) with 1 mL of the oils, 1% (w/v) surfactant, and 0.1% (w/v) polymer in aqueous solutions. These surfactant and polymer aqueous solutions were used in the solubility test because they are easily prepared and treated due to their appropriate viscosities.10,15,17,18 After vortexing, the mixtures were kept for 5 days at 25°C in a shaking water bath to facilitate solubilization. Centrifugation of the samples was performed at 10,000× g for 10 minutes (Hanil Science Industrial Co, Incheon, South Korea) before filtering through a syringe filter (0.45 μm, number 6789-1304; Whatman Co., Shrewsbury, MA, USA). The supernatant was diluted with acetonitrile. The diluted solution was analyzed for the amount of ezetimibe using a high-performance liquid chromatography (HPLC) system (Agilent 1220 Infinity; Agilent Technologies, Santa Clara, CA, USA) consisting of a Capcell Pak C18 column (Shiseido, 4.6 mm ID ×250 mm, 5 μm; Tokyo, Japan). The mobile phase, a mixture of triple distilled water and acetonitrile (30:70, v/v), was used at a flow rate of 1 mL/min, and 20 μL eluent was monitored at 232 nm.19 The interday and intraday variances were within the acceptable limits (R2=0.999).
Construction of the ternary phase diagram
The presence of self-nanoemulsifying oil formulation fields that could self-emulsify after dilution and slight agitation is recognized using a ternary phase diagram. On the basis of the solubility study, Capryol 90, Cremophor EL, and Tween 80 were selected as the oil, surfactant, and cosurfactant, respectively. Different concentrations of Capryol 90, Cremophor EL, and Tween 80 were used to develop a number of self-emulsifying systems. Each formulation (approximately 0.2 mL) was added to 500 mL of distilled water in a beaker at 37°C and was gently blended with a magnetic bar. After the investigation of the tendency to emulsify immediately and the propagation of emulsion droplets, we could easily differentiate between the nanoemulsions, which were clear, and the simple emulsions, which had a white appearance. The phase diagram was created by identifying the self-nanoemulsifying region. All studies were performed three times. The self-nanoemulsifying performance was visually checked after immense dilution using distilled water.15
Preparation of solid SNEDDS formulations
The liquid SNEDDS was prepared by dissolving 5 g of ezetimibe in 100 mL of the mixture of Capryol 90, Cremophor EL, and Tween 80 at a weight ratio of 10:35:55. A clear solution was obtained by vortexing the final mixture. The final drug content was analyzed in the liquid SNEDDS. The self-emulsification and particle size studies were assessed after the inspection of formulations for signs of turbidity or phase separation. Liquid SNEDDS (3 mL) was added with constant stirring to 300 mL of an ethanol solution of well-suspended silicon dioxide (1.5 g). This suspension was continuously stirred at room temperature for 15 minutes to obtain a homogeneous dispersion. Employing a mini spray dryer (Büchi 190 nozzle-type; Büchi Co., Flawil, Switzerland), the following conditions were used to spray dry the suspension with continuous stirring: outlet temperature, 55°C; inlet temperature, 85°C; aspiration, 100%; feeding rate of the suspension, 5 mL/min.
Preparation of SMSD and SESD
A mini spray dryer (Büchi 190 nozzle-type; Büchi Co.) was used to prepare two kinds of ezetimibe-loaded solid dispersions, SMSD and SESD. The SMSD was prepared as follows: Different quantities of Tween 80 and HPC were dissolved in distilled water. Then, 3 g of ezetimibe that had been presieved through a 60-mesh screen was dispersed in this solution (Table 1). A pneumatic nozzle (0.7 mm) with a peristaltic pump was used to deliver and spray dry the resulting dispersion, which was constantly stirred, under the given conditions: inlet temperature, 115°C; outlet temperature, 75°C–85°C; aspiration, 100%; suspension feeding rate of 5 mL/min.
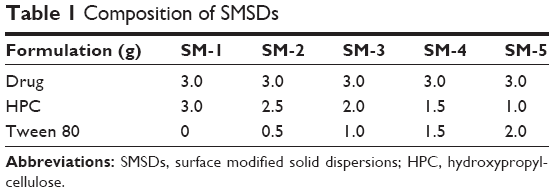 | Table 1 Composition of SMSDs |
The SESD was also prepared using the spray drying technique. A clear solution containing ezetimibe (3 g), HPC (1.5 g), and Tween 80 (1.5 g), entirely dissolved in 300 mL of ethanol and distilled water (1:1, v/v), was spray dried under the following conditions: inlet temperature, 100°C; outlet temperature, 65°C–55°C; aspiration, 100%; solution feeding rate of 5 mL/min.
Solid state characterization
The droplet size of the emulsion in liquid and solid SNEDDS was determined using a Zetasizer Nano ZS (Malvern Instruments, Malvern, UK) dynamic light scattering particle size analyzer to measure the droplet size of the emulsion at a scattering angle of 90° at 25°C with a wavelength of 635 nm. All studies were performed thrice, and the values of the z-average diameters were used. The results were analyzed using Automeasure software (Malvern Instruments). The particle size was determined using a laser diffraction Helos particle size analyzer (H1918; Sympatec GmbH, System-Partikel-Technik, Clausthal-Zellerfeld Saxony, Germany) to describe the particle size and distribution of the solid SNEDDS, SESD, and SMSD formulations.
A morphological study was performed using an S-4800 SEM (Hitachi, Tokyo, Japan) to observe the outer microscopic structures of the three formulations. A brass stub with double-sided adhesive tape was used to fix the samples and was made electrically conductive by coating with platinum (6 nm/min) in a vacuum (0.8 Pa) using an EmiTech Sputter Coater (Emitech Ltd, Ashford, Kent, UK) (K575 K) for 3 minutes at 25 mA.
The crystalline state of ezetimibe in the solid SNEDDS and solid dispersion formulations was described using DSC (DSC Q200; TA Instruments, New Castle, DE, USA). Approximately 5 mg of every sample, sealed in an aluminum pan, was exposed to heating at a rate of 10°C/min in the range of 10°C–220°C under a nitrogen flow of 25 mL/min. Their PXRD shapes were recorded using a Rigaku X-ray diffractometer (D/MAX-2500 PC, Rigaku Corporation, Tokyo, Japan) with a copper tube anode with 2θ angles over the interval of 10°–40°. The operational data were as follows: generator current 40 mA; generator tension (voltage) 40 kV; scanning speed 5°/min. FTIR was used to obtain the infrared (IR) spectra. The IR spectra of the drug powder and ezetimibe-loaded formulations were recorded on a Nicolet-6700 spectrophotometer (Thermo Scientific, Pittsburgh, PA, USA). The analysis reports were acquired using OMNIC software (Thermo Scientific). Each sample, properly loaded onto the sample disc, was scanned in the range of 400–4,000 cm−1.
Drug loading
The drug loading in the solid SNEDDS, SESD, and SMSD formulations was assessed using the following equation:
Drug loading (%) = Wd/WS ×100 |
where Wd is the weight of drug in milligrams present in the formulations, and WS is the weight of formulations in milligrams.
Drug release
Drug release studies were performed by placing ezetimibe-loaded SNEDDS, SESD, and SMSD (equal to 10 mg of ezetimibe) in the dissolution apparatus (Vision G2 Classic 6; Hanson Technology, Chatsworth, CA, USA) with 900 mL of pH 1.2, 4.0, and 6.8 and distilled water as the dissolution media. The dissolution test was accomplished at 37°C±0.5°C. The paddle speed was adjusted to 100× g. Each sample (1 mL) was collected at scheduled time intervals, filtered through a Whatman nylon filter (0.45 μm), and assayed for the content of ezetimibe using the HPLC method described above.
Pharmacokinetics
Male Sprague–Dawley rats (6–8 weeks old, 300±20 g), purchased from Nara Biotech (Seoul, South Korea), were allowed free access to the usual standard laboratory food and tap water. Throughout the investigation, the animals were housed under controlled conditions of 23°C–24°C/50%–60% relative humidity. The procedures for the animal studies were implemented consistent with the National Institutes of Health Policy and Animal Welfare Act under the approval of the Institutional Animal Care and Use Committee at Hanyang University.
Twenty-four rats were randomly separated into four groups. The right femoral artery of the rats was catheterized using polyethylene tubing (PE-50, Clay Adams, Parsippany, NJ, USA) filled with 50 IU/mL of heparin in saline under anesthesia by ketamine in the supine position. Ezetimibe-loaded SESD, solid SNEDDS, SMSD formulations, and the drug powder (at a dose of 3 mg/kg) enclosed in small hard gelatin capsules (#9, Suheung Capsule Co., Seoul, Korea) were orally administered to the rats in each group. Then, 0.4 mL of blood was obtained from the right femoral artery in heparinized syringes at scheduled time intervals (0, 0.5, 1, 1.5, 2, 3, 4, 5, 6, 12, and 24 hours) and was centrifuged at 3,000× g for 10 minutes. Plasma samples were stored at −20°C until further analysis. Plasma samples (160 μL) were then mixed with 10 μL of itraconazole (100 μg/mL in methanol) as an internal standard and were vortexed. Acetonitrile (0.2 mL) was used to deproteinize the plasma, which was then vortexed for 4 minutes and centrifuged at 8,000× g for 10 minutes. The supernatant was collected, and the free amount of ezetimibe was analyzed using the HPLC method described above.
Noncompartmental analysis (WinNonlin; professional edition, version 2.1; Pharsight Co., Mountain View, CA, USA) was used to assess the area under the drug concentration time curve from zero to infinity (AUC), the time taken to reach the maximum plasma concentration (Tmax) and the maximum plasma concentration of the drug (Cmax), the elimination constant (Kel), and the half-life (t1/2). Levels of statistical significance (P<0.05) were determined using an analysis of variance test among the three means for unpaired data. All data are denoted as the mean ± standard deviation or as the median (ranges) for Tmax.
Results
A mixture of oil, surfactant/cosurfactant, and drug with monophasic liquid state at room temperature is considered a SNEDDS; these formulations are widely used to enhance the bioavailability of drugs with poor water solubility.20 These mixtures should form a fine emulsion when added to an aqueous phase with mild agitation.21 Therefore, to select the SNEDDS composition for ezetimibe, its solubility in various carriers was studied; the results are presented in Figure 1. Among the oils tested, Capryol 90 gave the maximum drug solubility (Figure 1A). Capryol 90, a medium chain fatty acid, has been widely used in pharmaceutical applications, especially the SNEDDS formulation due to its good solubilizing capacity.22 Moreover, Cremophor EL and Tween 80 showed the highest solubility as compared with the other surfactants (Figure 1B). Cremophor EL, a widely used emulsifying and solubilizing agent, could lead to an improvement in drug loading and the formation of an extemporaneous nanoemulsion together with Tween 80.22 Thus, in the SNEDDS preparation, Cremophor EL, Tween 80, and Capryol 90 were used as the surfactant, cosurfactant, and oil.
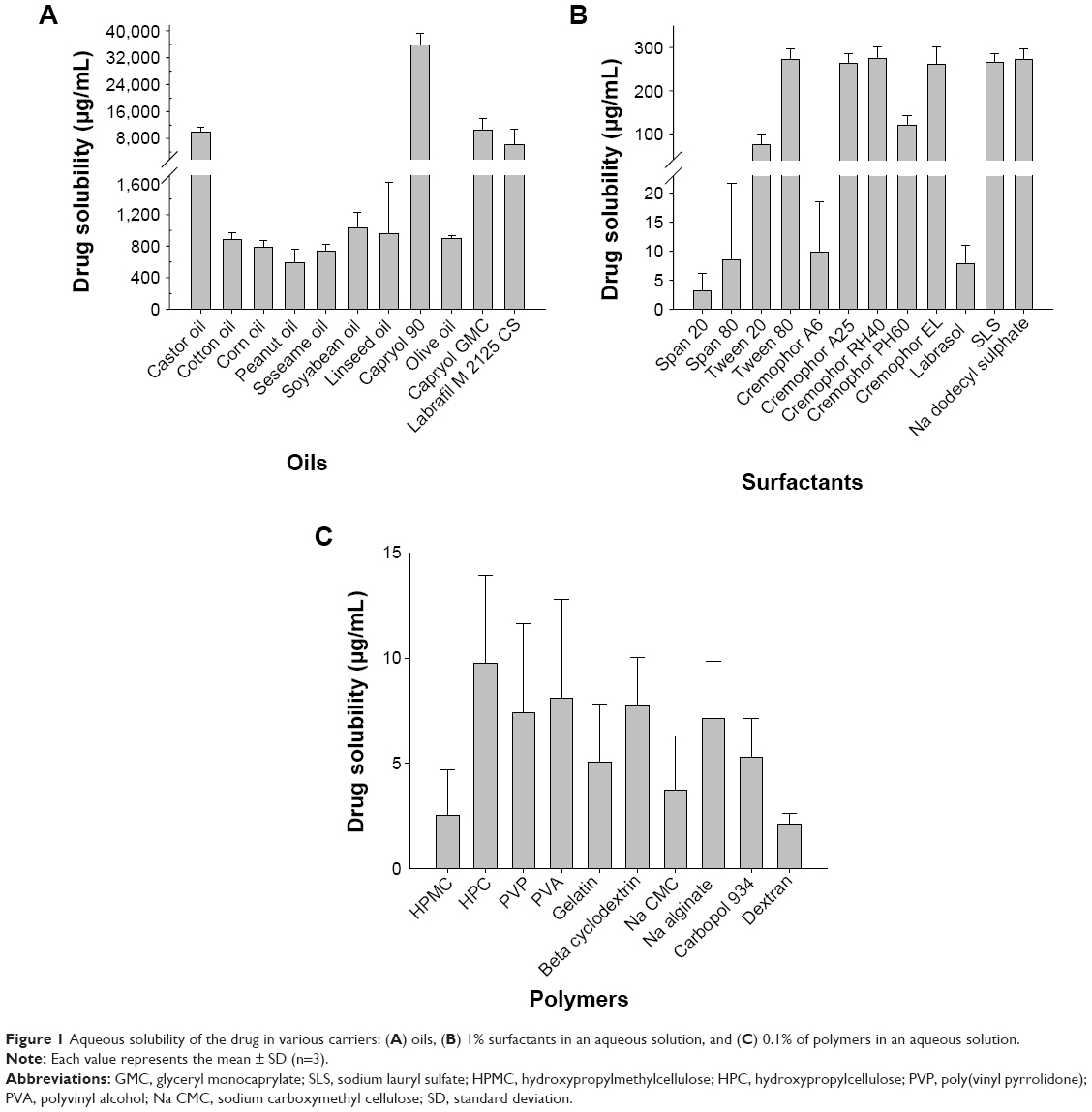 | Figure 1 Aqueous solubility of the drug in various carriers: (A) oils, (B) 1% surfactants in an aqueous solution, and (C) 0.1% of polymers in an aqueous solution. |
The determination of self-emulsification can be performed visually by observing the emulsification and droplet formation of the prepared SNEDDS formulations.23 The size of the droplets in the SNEDDS emulsion plays a significant role, as it determines the bioavailability of the formulated drug.24,25 A number of SNEDDSs were prepared and their self-emulsification properties were checked visually. To categorize the optimized concentrations of oil, surfactant, and cosurfactant in the SNEDDS formulation, a pseudoternary phase diagram was assembled in the absence of ezetimibe. The phase diagram of the system having Cremophor EL as the surfactant, Capryol 90 as the oil, and Tween 80 as the cosurfactant is shown in Figure 2. The pseudoternary phase diagram with these components had a reasonable self-nanoemulsification region.
 | Figure 2 Pseudoternary phase diagram using Capryol 90 as an oil, Cremophor EL as a surfactant, and Tween 80 as a cosurfactant. |
As shown in Figure 3A, the z-average diameters of the nanoemulsions decreased with an increased quantity of surfactant at 30%, and then at 45% v/v with that of oil, but a further increase in the surfactant ratio had little effect on droplet size. Furthermore, the z-average diameter was further decreased with the addition of Tween 80 as a cosurfactant (Figure 3B). It was observed that the formulation comprised of 10% Tween 80, 35% Cremophor EL, and 55% Capryol 90 gave the smallest z-average diameter among the formulations tested in this study. In addition, Capryol 90 was sufficient for dissolving the drug in this formulation. There were no significant differences in the self-nanoemulsifying performance when compared with the formulation containing 5% ezetimibe. Thus, this formulation was selected as the liquid SNEDDS for further studies. Afterward, liquid SNEDDS in the ethanolic solution containing silicon dioxide was spray dried to convert it into an ezetimibe-loaded solid SNEDDS. Additionally, the liquid SNEDDS without the drug showed a small droplet size of around 86.95±3.12 nm (polydispersity index [PDI], 0.28±0.04). The z-average diameter of the solid SNEDDS (177.36±20.15 nm; PDI, 0.29±0.06) was higher than that of liquid SNEDDS loaded with the drug (109.56±2.93 nm; PDI, 0.22±0.04). Both the z-average diameters of the liquid and solid SNEDDS were less than 200 nm.
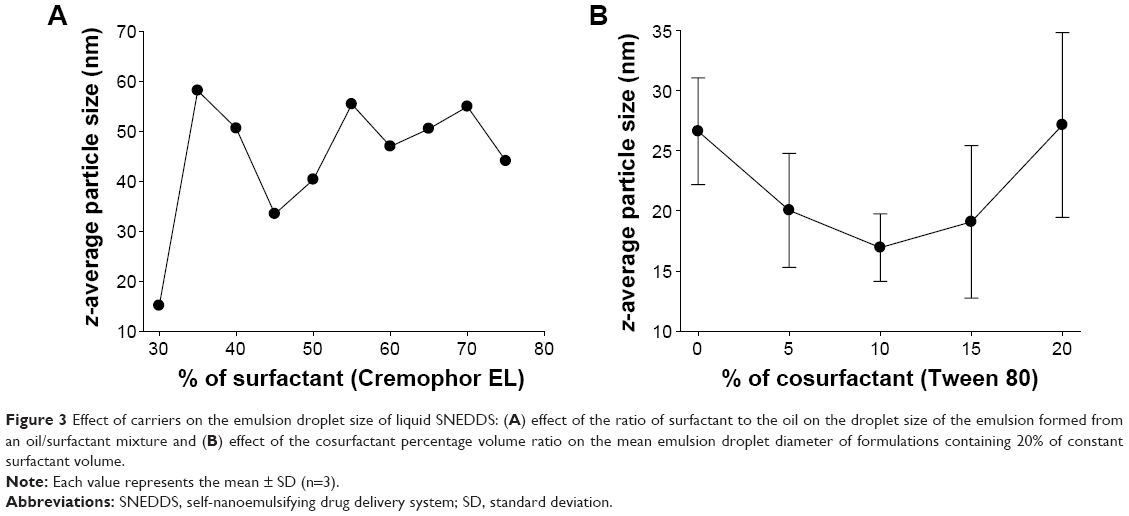 | Figure 3 Effect of carriers on the emulsion droplet size of liquid SNEDDS: (A) effect of the ratio of surfactant to the oil on the droplet size of the emulsion formed from an oil/surfactant mixture and (B) effect of the cosurfactant percentage volume ratio on the mean emulsion droplet diameter of formulations containing 20% of constant surfactant volume. |
In order to choose the appropriate hydrophilic polymer and surfactant as carriers for ezetimibe-loaded SMSD and SESD, distilled water containing 0.1% polymers or 1% surfactants was used to assess the solubility of the drug (Figure 1). Among the carriers assessed in this test, Tween 80 (Figure 1B) and HPC (Figure 1C) showed the highest solubility. Tween 80, a nonionic surfactant, is considered safe to take orally because it has an oral acceptance of up to 25 mg/kg of human body weight.26,27 Thus, this polymer and surfactant were selected for further study due to their better drug solubility.28
Generally, large ratios of the polymer and drug are used in the conventional solid dispersion technique.8 However, optimized amounts of the surfactant and polymer mixture are used in the SMSD technique in order to avoid a relatively high polymer/drug ratio, which does not increase drug solubility and dissolution.29 As shown in Table 1, various SMSD formulations were prepared with various ratios of the drug with HPC, Tween 80, and distilled water through the surface modified method. The aqueous solubility of ezetimibe in different SMSDs is presented in Figure 4. All of the formulations significantly improved drug solubility as compared with the drug powder. Because the ratio of HPC to Tween 80 was decreased, the drug solubility was increased. Among these formulations, the formulations SM-5 and SM-4 gave the highest and second highest drug solubility, respectively. However, SM-5 was very sticky due to the presence of a high amount of surfactant (Tween 80). Thus, SM-4, composed of the drug, HPC, and Tween 80 at a weight ratio of 3/1.5/1.5, was chosen as an optimized SMSD formulation due to its enhanced drug solubility and nonsticky property. In addition, the optimized SESD formulation was prepared using the same composition of SMSD with a combination of ethanol and distilled water (1:1, v/v) in place of water for dissolving the drug and carriers.
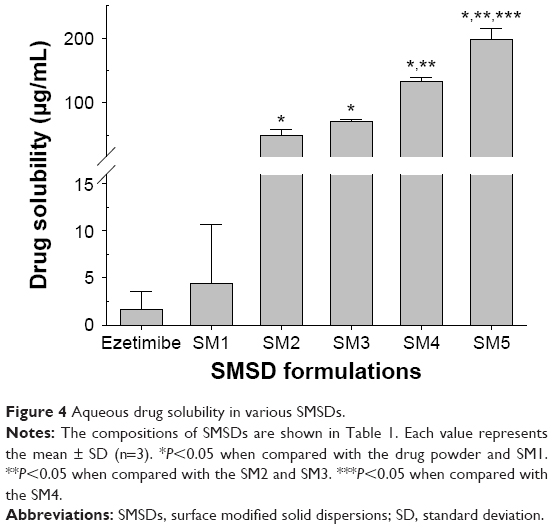 | Figure 4 Aqueous drug solubility in various SMSDs. |
The solid state characteristics of solid SNEDDS, SMSD, and SESD were compared. The SEM images of the drug powder, solid SNEDDS, SMSD, and SESD are shown in Figure 5. The drug powder (Figure 5A) appeared as rectangular crystals in different sizes. Solid SNEDDS (Figure 5B) appeared as relatively smooth and round particles of silicon dioxide.15 The SMSD had a relatively coarse and fractured surface (Figure 5C). On the other hand, SESD had a relatively smooth and round surface (Figure 5D).
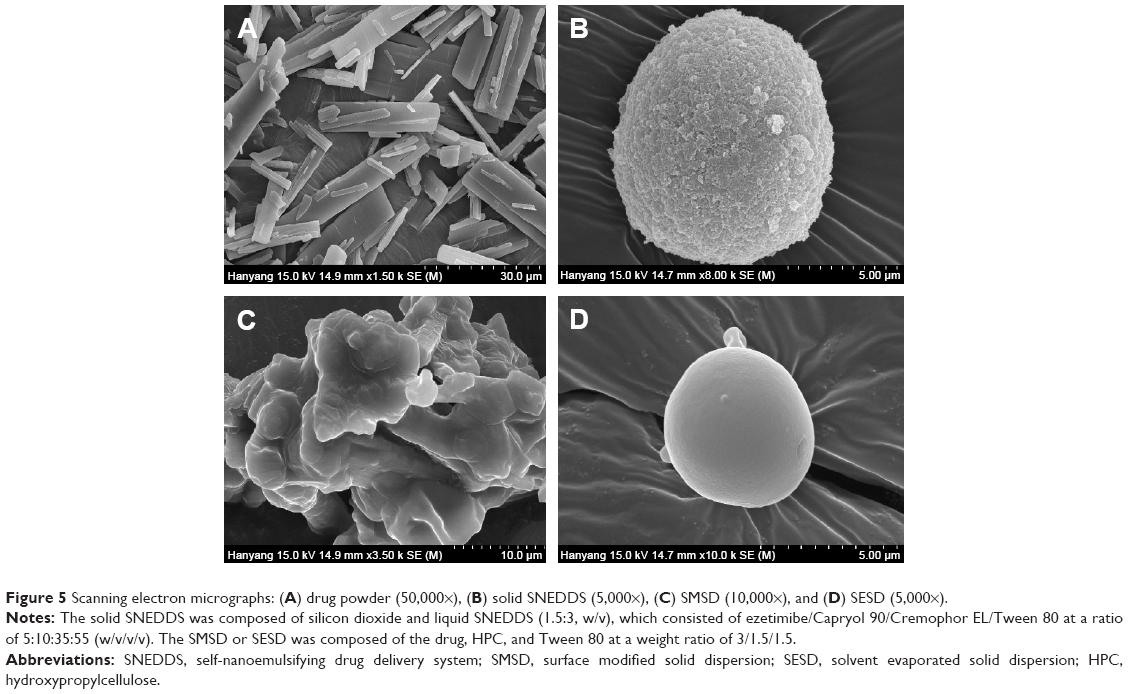 | Figure 5 Scanning electron micrographs: (A) drug powder (50,000×), (B) solid SNEDDS (5,000×), (C) SMSD (10,000×), and (D) SESD (5,000×). |
The thermal performance of the drug powder, physical mixture, carriers, and three formulations are presented in Figure 6A. The drug powder gave a sharp endothermic peak at approximately 160°C in the DSC thermograph, indicating its exact melting point due to its crystalline nature (Figure 6Aa).30 Silicon dioxide (Figure 6Ab) showed no peak in the tested region. A small endothermic peak matching the drug was also detected in the physical mixture of solid SNEDDS (Figure 6Ac). No drug peak was observed for solid SNEDDS (Figure 6Ad), suggesting that the amorphous drug was present in a molecularly dissolved state.23 Furthermore, HPC did not show any peak in the tested region (Figure 6Ae). The physical mixture of the solid dispersion showed a small endothermic peak (Figure 6Af). SMSD showed a small endothermic peak around the drug melting point (Figure 6Ag), whereas SESD produced no peak (Figure 6Ah). Thus, our results suggest that the drug existed in a crystalline and amorphous form in SMSD and SESD, respectively.31,32
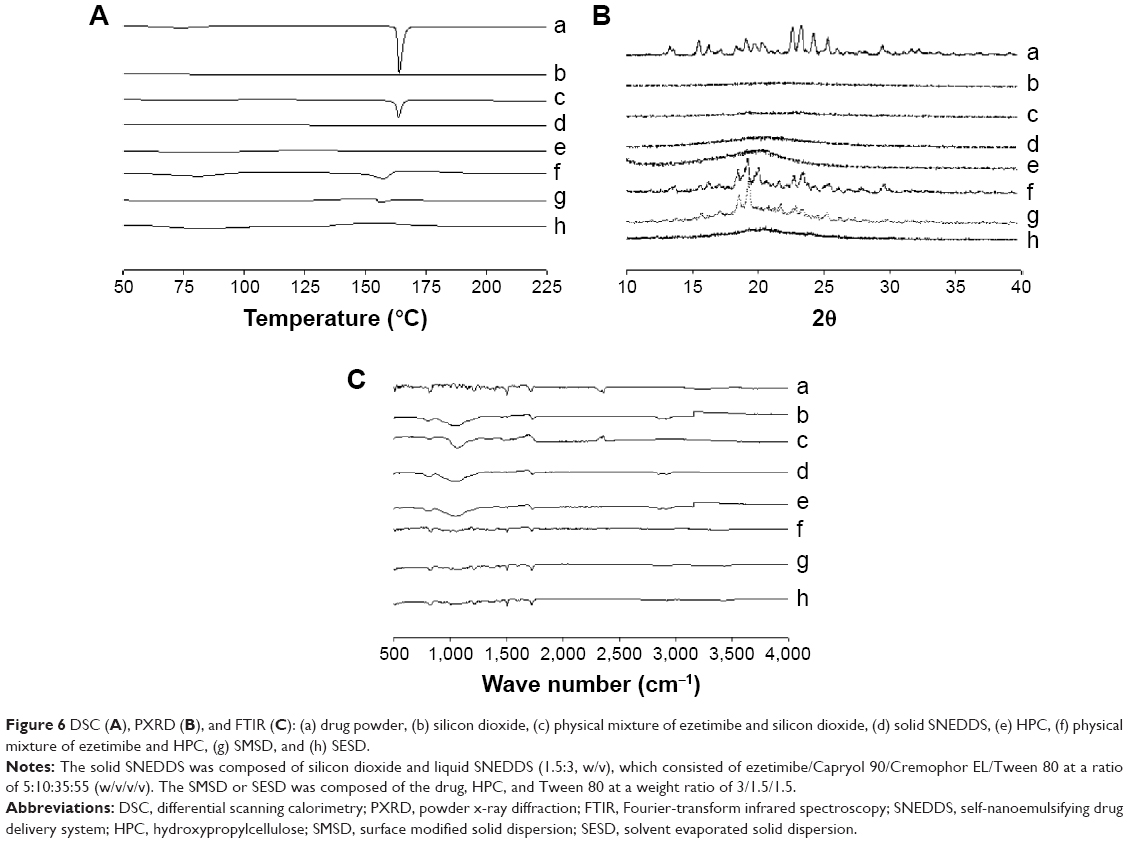 | Figure 6 DSC (A), PXRD (B), and FTIR (C): (a) drug powder, (b) silicon dioxide, (c) physical mixture of ezetimibe and silicon dioxide, (d) solid SNEDDS, (e) HPC, (f) physical mixture of ezetimibe and HPC, (g) SMSD, and (h) SESD. |
The PXRD profiles shown in Figure 6B were investigated for further verification of the internal physical state of the drug in these formulations. The drug (Figure 6Ba) showed a typical crystalline pattern, but carriers such as HPC (Figure 6Be) and silicon dioxide (Figure 6Bb) showed no peaks. The physical mixture of solid SNEDDS (Figure 6Bc) and the physical mixture of the solid dispersions (Figure 6Bf) showed the same drug peak pattern. However, no clear peaks were observed, representing crystals of ezetimibe in solid SNEDDS (Figure 6Bd) and SESD (Figure 6Bh), which further confirmed the amorphous nature of the drug in these formulations.5,30 However, there was a small crystalline peak observed in SMSD (Figure 6Bg).
FTIR was performed to check for any possible drug/carrier chemical interaction.31 The FTIR spectra of ezetimibe, carriers, and the formulations are shown in Figure 6C. The IR spectrum of ezetimibe (Figure 6Ca) was characterized by principal absorption peaks at 3,264.41, 2,913.81, 2,855.57, 1,879.87, 1,718.04, 1,602.50, 1,509.34, 1,445.82, 1,432.02, 1,354.03, 1,221.51, and 830.98 cm−1.32 Silicon dioxide (Figure 6Cb) and HPC (Figure 6Ce) showed their specific patterns. The physical mixture of solid SNEDDS (Figure 6Cc) and the physical mixture of the solid dispersions (Figure 6Cf) showed major peaks of ezetimibe. The FTIR spectra of solid SNEDDS (Figure 6Cd), SMSD (Figure 6Cg), and SESD (Figure 6Ch) showed that there was no shift in the drug peaks in these formulations, indicating that the drug and carriers had no chemical interactions in all formulations.32–34
Among the prepared formulations, SESD showed more uniform behavior and a smaller particle size as compared with SMSD and solid SNEDDS (Figure 7). The ezetimibe powder did not show a uniform particle size distribution as compared with the formulations. The solid SNEDDS, SESD, and SMSD formulations had smaller particle sizes compared with the drug powder (approximately 11, 6, and 24 vs 33 μm, respectively). Additionally, the drug loadings in the solid SNEDDS, SESD, and SMSD formulations were 4.2%±0.85%, 47.3%±5.19%, and 52.9%±2.18%, respectively.
 | Figure 7 Cumulative undersize percentage of solid SNEDDS, SMSD, and SESD. |
The solid SNEDDS, SESD, and SMSD formulations significantly increased the drug solubility in water (337.54±8.19, 189.77±19.00, and 133.57±6.11 vs 1.69±1.91 μg/mL, respectively) (Figure 8A), indicating that solid SNEDDS had significantly higher aqueous solubility. However, at pH 1.2, pH 4.0, and pH 6.8, the three formulations showed more significantly increased drug solubility (3.5–3.9 μg/mL) (Figure 8B–D). In addition, the drug solubility for solid SNEDDS and SESD was significantly higher than that for SMSD, but no significant difference was found between solid SNEDDS and SESD (approximately 190–200, 190–200 vs 135–145 μg/mL, respectively).
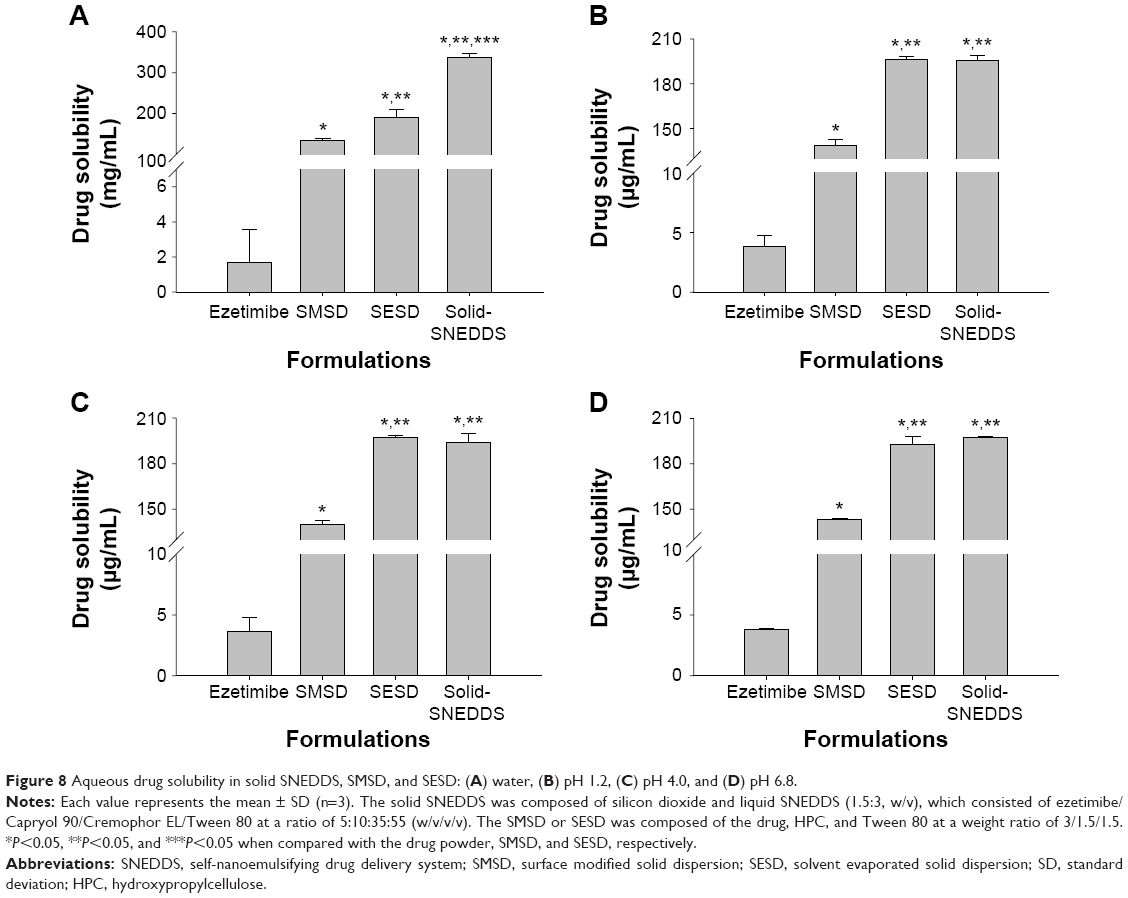 | Figure 8 Aqueous drug solubility in solid SNEDDS, SMSD, and SESD: (A) water, (B) pH 1.2, (C) pH 4.0, and (D) pH 6.8. |
The dissolution profiles of the drug powder and these formulations in water and solutions at pH 1.2, 4.0, and 6.8 are shown in Figure 9. In water and at pH 1.2, solid SNEDDS, SESD, and SMSD had a significantly increased dissolution rate as compared with the drug powder. In addition, there was improved dissolution of the drug at 30 minutes by approximately 1.8–3-fold (approximately 25% vs approximately 60%, 55%, and 45%, respectively) (Figure 9A and B). Similarly, at pH 4.0 and pH 6.8, these formulations significantly improved the dissolution of the drug in 30 minutes by approximately 1.8–3-fold (approximately 25% vs approximately 50%, 55%, and 43%, respectively) (Figure 9C and D).
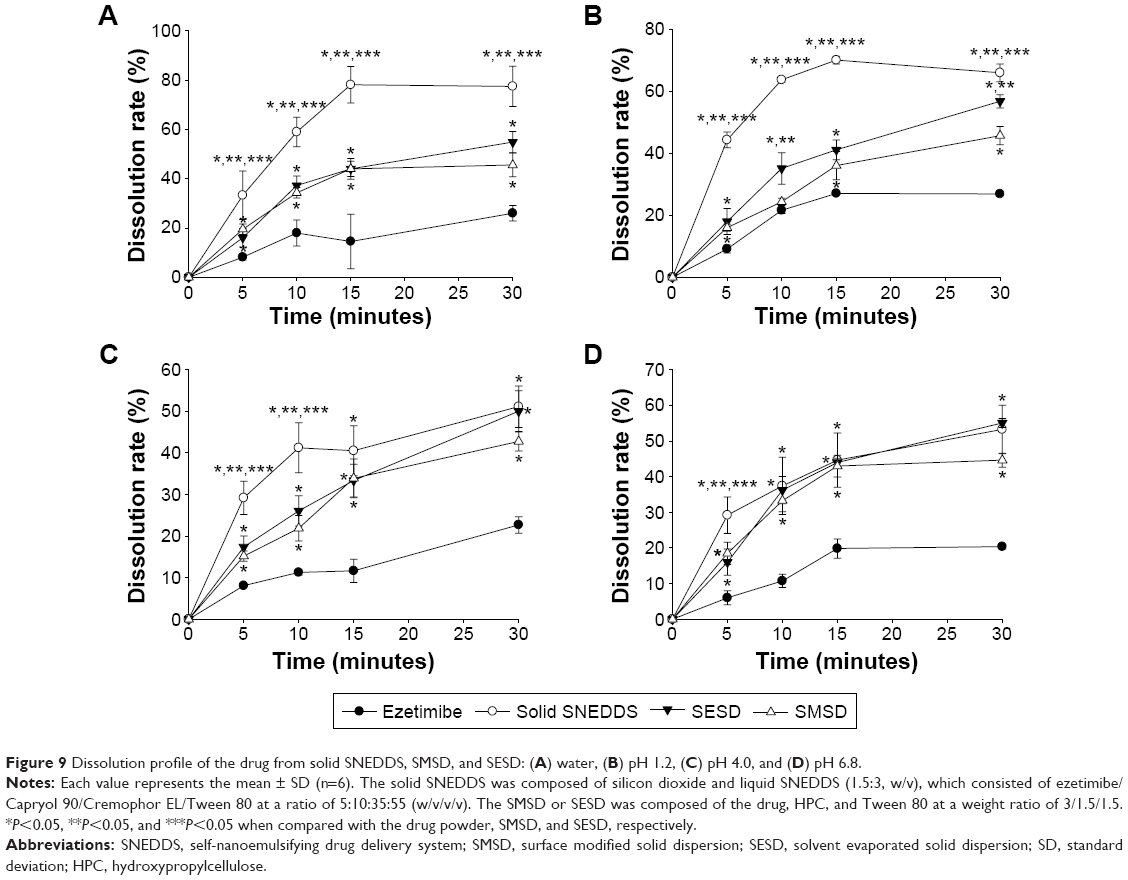 | Figure 9 Dissolution profile of the drug from solid SNEDDS, SMSD, and SESD: (A) water, (B) pH 1.2, (C) pH 4.0, and (D) pH 6.8. |
A pharmacokinetic study was performed to quantify the amount of ezetimibe in solid SNEDDS, SMSD, and SESD (equivalent to 3 mg/kg drug dose) orally administered to rats (Figure 10). Generally, the formulations with equivalent average doses of the drug were administered in the comparative pharmacokinetic study.21 All of the formulations showed a higher plasma concentration of the drug as compared with the drug powder.3 In particular, the total plasma concentrations with SESD were significantly increased as compared with the drug powder in the initial time period (0.5–1.0 hours). Moreover, there were no significant differences in the plasma concentration at each time point among solid SNEDDS, SMSD, and SESD. The AUC values of the drug in solid SNEDDS, SMSD, and SESD were 5.05±0.90, 4.96±0.73, and 5.58±1.25 h·μg/mL, respectively (Table 2). Therefore, SESD gave higher AUC than did other preparations, even if they were not significantly different. Moreover, there were no significant differences in the Tmax and t1/2 values among all preparations.
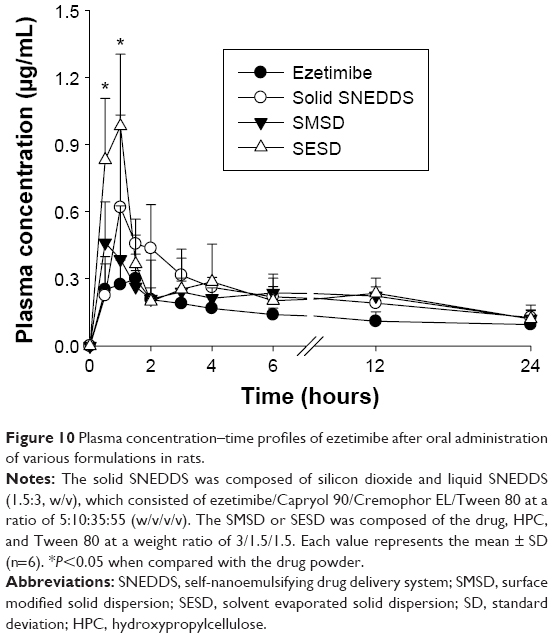 | Figure 10 Plasma concentration–time profiles of ezetimibe after oral administration of various formulations in rats. |
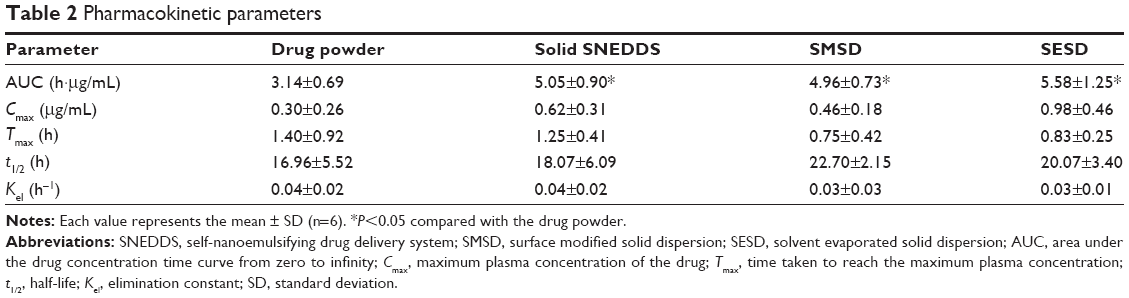 | Table 2 Pharmacokinetic parameters |
Discussion
Ezetimibe has a highly irregular and very slow dissolution rate in GI fluids due to its hydrophobic character,5 resulting in reduced and highly unpredictable bioavailability.6,7,35 In this study, the solubility, dissolution, and bioavailability of ezetimibe-loaded solid SNEDDS, SMSD, and SESD were compared, leading to the selection of the best drug delivery system with the highest oral bioavailability.
The optimized liquid SNEDDS formulation (drug/Tween 80/Cremophor EL/Capryol 90 [5/10/35/55, w/v/v/v]) was spray dried with silicon dioxide to obtain solid SNEDDS. Solid SNEDDS appeared as relatively smooth and round particles of silicon dioxide, whose pores offered space to adsorb the liquid SNEDDS coating. When solid SNEDDS encountered the aqueous atmosphere of the GI tract, the nanoemulsion was formed in the presence of free energy, which was really low in the case of self-emulsifying systems; thus, the spontaneous formation of an interface between water and oil droplets was possible.15 The optimized SMSD or SESD formulations were prepared using the spray drying technique with the formulation ezetimibe/HPC/Tween 80 (3/1.5/1.5, w/w/w) in water as a dispersed form or in a 50% ethanol solution as a clear solution form, respectively.36 This SMSD, prepared using the spray drying procedure, contained relatively coarse and fractured particles, indicating that the hydrophilic polymer and surfactant might be bound on the surface of the hydrophobic crystalline drug, resulting in modified hydrophobicity of the drug to the hydrophilic properties of the polymer and surfactant.37 However, this SESD produced relatively smooth and round nanoparticles and contained the drug in an amorphous form.30
Solid SNEDDS, SESD, and SMSD had particle sizes of approximately 11, 6, and 24 μm, respectively. The aqueous solubility of ezetimibe increased in the order of solid SNEDDS ≥ SESD > SMSD in water and at pH 1.2, pH 4.0, and pH 6.8 because of the reduced particle size and amorphous form of the drug in solid SNEDDS and SESD; these factors modified the drug hydrophobicity to hydrophilicity in SMSD.37 On the other hand, SNEDDS with a relatively larger particle size of approximately 11 μm gave more drug solubility than did the SESD with a smaller particle size of approximately 6 μm because the SNEDDS formed the nanoemulsion with a very small droplet size of approximately 200 nm in the aqueous solutions.10,23 Solid SNEDDS, SMSD, and SESD improved the drug solubility by approximately 200-, 80-, and 110-fold, respectively. Moreover, the drug solubility in each formulation was not dependent upon pH.
In water, at pH 1.2, pH 4.0, and pH 6.8, all formulations significantly increased the dissolution frequency as compared with the drug powder. Similar to the order of aqueous solubility, the dissolution rate increased in the same order of solid SNEDDS ≥ SESD > SMSD. When solid SNEDDS came into contact with the aqueous solution in the dissolution medium, the free energy needed to form a nanoemulsion was very low, as is the case in self-emulsifying systems; hence, the extemporaneous formation of an interface between water and oil droplets was possible, leading to the greatly improved dissolution rate of ezetimibe.38 The drug was in an amorphous form in the SESD system. However, in the SMSD system, the hydrophilic polymer and surfactants covered the surface of the drug crystals without altering the crystallinity. Thus, the surface of the drug crystals was converted from hydrophobic to hydrophilic. Upon contacting water, these drug crystals interacted with the aqueous microenvironment, thereby dispersing the drug into the supersaturated solution. Tween 80 obstructed recrystallization, owing to a higher level of supersaturation due to the interfacial tension between the small drug particles.11,31 These results suggest that SESD, solid SNEDDS, and SMSD significantly improved the dissolution of ezetimibe.37
All of the formulations led to higher total ezetimibe plasma concentrations as compared with the drug powder. There were no significant differences in the plasma concentrations among the three formulations. Unlike the order of aqueous solubility, the AUC increased in the order of SESD > solid SNEDDS = SMSD, with significantly higher AUC values as compared with the drug powder (P<0.05). In our study, solid SNEDDS showed greater solubility but lower oral bioavailability of ezetimibe as compared with SESD. Irrespective of the high solubility and dissolution of the drug, the relatively lower bioavailability of solid SNEDDS might be due to the presence of Cremophor EL used as a carrier.39 Ezetimibe had a rapid first-pass metabolism and P-glycoprotein efflux.6,7 Furthermore, this drug is mainly conjugated by the act of uridine diphosphate (UDP) glucuronosyltransferases in the small intestine and remains biologically active as free ezetimibe and an ezetimibe-glucuronide conjugate.34,40 Cremophor EL might interact with P-glycoprotein40–42 and serum protein,43,44 leading to a hindrance of the absorption of ezetimibe in the GI tract. Such enhanced oral bioavailability of ezetimibe in SESD was due to the amorphous nature and small particle size of the drug, which ultimately improved solubility and dissolution, resulting in increased drug absorption in the GI tract.30 Among these formulations tested in this study, SESD most improved the oral bioavailability of ezetimibe, even though they were not significantly different. In addition, SESD is easier and more simply manufactured than are solid SNEDDS and SMSD.10,31 Thus, SESD is strongly recommended as a drug delivery system for the oral administration of poorly water-soluble ezetimibe.
Conclusion
Ezetimibe-loaded SMSD, SESD, and solid SNEDDS enhanced apparent drug solubility and oral bioavailability. SESD gave higher AUC than did SNEDDS and SMSD, even if they were not significantly different, suggesting more improved oral bioavailability. In particular, SESD improved drug solubility by approximately 110-fold and oral bioavailability by twofold because of the reduced particle size and change in crystallinity. Thus, among the various formulations tested in this study, the SESD system would be strongly recommended as a drug delivery system for the oral administration of ezetimibe with its poor water solubility.
Acknowledgment
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (Korean Ministry of Education, Science and Technology) (No.2015R1A2A2A05027872) and the Medical Research Center Program (No.2015R1A5A2009124).
Disclosure
The authors report no conflicts of interest in this work.
References
Clader JW. The discovery of ezetimibe: a view from outside the receptor. J Med Chem. 2004;4:1–9. | ||
Basha SJ, Naveed SA, Tiwari NK, et al. Concurrent determination of ezetimibe and its phase-I and II metabolites by HPLC with UV detection: quantitative application to various in vitro metabolic stability studies and for qualitative estimation in bile. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;853:88–96. | ||
Karanam SR, Katakam P, Chandu BR, Hwisa NT, Adiki SK. Simultaneous determination of ezetimibe and simvastatin in rat plasma by stable-isotope dilution LC-ESI–MS/MS and its application to a pharmacokinetic study. J Pharm Anal. 2013;4:286–294. | ||
Gulsun T, Gursoy RN, Oner L. Design and characterization of nanocrystal formulations containing ezetimibe. Chem Pharm Bull (Tokyo). 2011;59:41–45. | ||
Patel R, Bhimani D, Patel J, Patel D. Solid-state characterization and dissolution properties of ezetimibe–cyclodextrins inclusion complexes. J Incl Phenom Macrocycl Chem. 2008;60:241–251. | ||
Ezzet F, Krishna G, Wexler DB, Statkevich P, Kosoglou T, Batra VK. A population pharmacokinetic model that describes multiple peaks due to enterohepatic recirculation of ezetimibe. Clin Ther. 2001;23:871–885. | ||
Kosoglou T, Statkevich P, Johnson-Levonas A, Paolini JF, Bergman AJ, Alton KB. Ezetimibe: a review of its metabolism, pharmacokinetics and drug interactions. Clin Pharmacokinet. 2005;44:467–494. | ||
Mandawgade SD, Sharma S, Pathak S, Patravale VB. Development of SMEDDS using natural lipophile: application to β-artemether delivery. Int J Pharm. 2008;362:179–183. | ||
Balakrishnan P, Lee BJ, Oh DH, et al. Enhanced oral bioavailability of dexibuprofen by a novel solid self-emulsifying drug delivery system (SEDDS). Eur J Pharm Biopharm. 2009;72:539–545. | ||
Patel AR, Vavia PR. Preparation and in vivo evaluation of SMEDDS (self-microemulsifying drug delivery system) containing fenofibrate. AAPS J. 2007;9:E344–E352. | ||
Nazzal S, Khan MA. Controlled release of a self-emulsifying formulation from a tablet dosage form: stability assessment and optimization of some processing parameters. Int J Pharm. 2006;315:110–121. | ||
Bandyopadhyay S, Katare O, Singh B. Development of optimized supersaturable self-nanoemulsifying systems of ezetimibe: effect of polymers and efflux transporters. Expert Opin Drug Deliv. 2014;11:479–492. | ||
Tang B, Cheng G, Gu JC, Xu CH. Development of solid self-emulsifying drug delivery systems: preparation techniques and dosage forms. Drug Discov Today. 2008;13:606–612. | ||
Yan YD, Sung JH, Kim KK, et al. Novel valsartan-loaded solid dispersion with enhanced bioavailability and no crystalline changes. Int J Pharm. 2012;422:202–210. | ||
Oh DH, Park YJ, Kang JH, Yong CS, Choi HG. Physicochemical characterization and in vivo evaluation of flurbiprofen-loaded solid dispersion without crystalline change. Drug Deliv. 2011;18:46–53. | ||
Rashid R, Kim DW, Din FU, et al. Effect of hydroxypropylcellulose and Tween 80 on physicochemical properties and bioavailability of ezetimibe-loaded solid dispersion. Carbohydr Polym. 2015;130:26–31. | ||
Park YJ, Ryu DS, Li DX, et al. Physicochemical characterization of tacrolimus-loaded solid dispersion with sodium carboxylmethyl cellulose and sodium lauryl sulfate. Arch Pharm Res. 2009;32:893–898. | ||
Tran TH, Poudel BK, Marasini N, et al. Preparation and evaluation of raloxifene-loaded solid dispersion nanoparticle by spray drying technique without an organic solvent. Int J Pharm. 2013;44:50–57. | ||
Bae JW, Choi CI, Park SH, Jang CG, Lee SY. Analytical LC-MS/MS method for ezetimibe and its application for pharmacokinetic study. J Liq Chromatogr Relat Technol. 2012;35:141–152. | ||
Kang BK, Lee JS, Chon SK, et al. Development of self-microemulsifying drug delivery systems (SMEDDS) for oral bioavailability enhancement of simvastatin in beagle dogs. Int J Pharm. 2004;274:65–73. | ||
Singh AK, Chaurasiya A, Singh M, Upadhyay SC, Mukherjee R, Khar RK. Exemestane loaded self-microemulsifying drug delivery system (SMEDDS): development and optimization. AAPS Pharm Sci Tech. 2008;9:628–634. | ||
Pouton CW. Lipid formulations for oral administration of drugs: non-emulsifying, self-emulsifying and ‘self-microemulsifying’ drug delivery systems. Eur J Pharm Sci. 2000;11:S93–S98. | ||
Balakrishnan P, Lee BJ, Oh DH, et al. Enhanced oral bioavailability of Coenzyme Q10 by self-emulsifying drug delivery systems. Int J Pharm. 2009;374:66–72. | ||
Constantinides PP, Scalart JP, Lancaster C, et al. Formulation and intestinal absorption enhancement evaluation of water-in-oil microemulsions incorporating medium-chain glycerides. Pharma Res. 1994;11:1385–1390. | ||
Bandyopadhyay S, Katare O, Singh B. Optimized self nano-emulsifying systems of ezetimibe with enhanced bioavailability potential using long chain and medium chain triglycerides. Colloids Surf B Biointerfaces. 2012;100:50–61. | ||
Lewis RJ, editor. Sax’s Dangerous Properties of Industrial Materials. 11th ed. New York, NY: Wiley; 2004:2013. | ||
Cho HJ, Park JW, Yoon IS, Kim DD. Surface-modified solid lipid nanoparticles for oral delivery of docetaxel: enhanced intestinal absorption and lymphatic uptake. Int J Nanomedicine. 2014;9:495–504. | ||
Dannenfelser RM, He H, Joshi Y, Bateman S, Serajuddin A. Development of clinical dosage forms for a poorly water soluble drug I: application of polyethylene glycol–polysorbate 80 solid dispersion carrier system. J Pharm Sci. 2004;93:1165–1175. | ||
Lee SN, Poudel BK, Tran TH, et al. A novel surface-attached carvedilol solid dispersion with enhanced solubility and dissolution. Arch Pharm Res. 2013;36:79–85. | ||
Sethia S, Squillante E. Solid dispersion of carbamazepine in PVP K30 by conventional solvent evaporation and supercritical methods. Int J Pharm. 2004;272:1–10. | ||
Joe JH, Lee WM, Park YJ, et al. Effect of the solid-dispersion method on the solubility and crystalline property of tacrolimus. Int J Pharm. 2010;395:161–166. | ||
Wang X, Michoel A, Van den Mooter G. Solid state characteristics of ternary solid dispersions composed of PVP VA64, Myrj 52 and itraconazole. Int J Pharm. 2005;303:54–61. | ||
Newa M, Bhandari KH, Li DX, et al. Preparation, characterization and in vivo evaluation of ibuprofen binary solid dispersions with poloxamer 188. Int J Pharm. 2007;343:228–237. | ||
Sancheti P, Karekar P, Vyas V, Shah M, Pore Y. Preparation and physicochemical characterization of surfactant based solid dispersions of ezetimibe. Pharmazie. 2009;64:227–231. | ||
Bali V, Ali M, Ali J. Study of surfactant combinations and development of a novel nanoemulsion for minimising variations in bioavailability of ezetimibe. Colloids Surf B Biointerfaces. 2010;76:410–420. | ||
Ming Thau S, Ching Min Y, Sokoloski TD. Characterization and dissolution of fenofibrate solid dispersion systems. Int J Pharm. 1994;103:137–146. | ||
Kim DW, Kwon MS, Yousaf AM, et al. Comparison of a solid SMEDDS and solid dispersion for enhanced stability and bioavailability of clopidogrel napadisilate. Carbohydr Polym. 2014;114:365–374. | ||
Zhang P, Liu Y, Feng N, Xu J. Preparation and evaluation of self-microemulsifying drug delivery system of oridonin. Int J Pharm. 2008;355:269–276. | ||
Gelderblom H, Verweij J, van Zomeren DM, et al. Influence of Cremophor EL on the bioavailability of intraperitoneal paclitaxel. Clin Cancer Res. 2002;8(4):1237–1241. | ||
Woodcock DM, Jefferson S, Linsenmeyer ME, et al. Reversal of the multidrug resistance phenotype with cremophor EL, a common vehicle for water-insoluble vitamins and drugs. Cancer Res. 1990;50:4199–4203. | ||
Schuurhuis G, Broxterman H, Pinedo H, et al. The polyoxyethylene castor oil Cremophor EL modifies multidrug resistance. Br J Cancer. 1990;62:591. | ||
Ross DD, Wooten PJ, Tong Y, et al. Synergistic reversal of multidrug-resistance phenotype in acute myeloid leukemia cells by cyclosporin A and cremophor EL. Blood. 1994;83:1337–1347. | ||
Sykes E, Woodburn K, Decker D, Kessel D. Effects of Cremophor EL on distribution of Taxol to serum lipoproteins. Br J Cancer. 1994;70:401–404. | ||
Woodburn K, Chang C, Lee S, Henderson B, Kessel D. Biodistribution and PDT efficacy of a ketochlorin photosensitizer as a function of the delivery vehicle. Photochem Photobiol. 1994;60:154–159. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.