Back to Journals » Drug Design, Development and Therapy » Volume 8
Comparative steady-state pharmacokinetic study of an extended-release formulation of itopride and its immediate-release reference formulation in healthy volunteers
Authors Yoon S ![]() , Lee H
, Lee H ![]() , Kim T, Lee S
, Kim T, Lee S ![]() , Chee D, Cho J
, Chee D, Cho J ![]() , Yu K
, Yu K ![]() , Jang I
, Jang I ![]()
Received 15 August 2013
Accepted for publication 3 October 2013
Published 15 January 2014 Volume 2014:8 Pages 123—128
DOI https://doi.org/10.2147/DDDT.S53027
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Seonghae Yoon,1,* Howard Lee,2,* Tae-Eun Kim,1 SeungHwan Lee,1 Dong-Hyun Chee,3 Joo-Youn Cho,1 Kyung-Sang Yu,1 In-Jin Jang1
1Department of Clinical Pharmacology and Therapeutics, Seoul National University College of Medicine and Hospital, 2Clinical Trials Center, Seoul National University Hospital, 3AbbVie Ltd., Seoul, Republic of Korea
*These authors contributed equally to this work
Background: This study was conducted to compare the oral bioavailability of an itopride extended-release (ER) formulation with that of the reference immediate-release (IR) formulation in the fasting state. The effect of food on the bioavailability of itopride ER was also assessed.
Methods: A single-center, open-label, randomized, multiple-dose, three-treatment, three-sequence, crossover study was performed in 24 healthy male subjects, aged 22–48 years, who randomly received one of the following treatments for 4 days in each period: itopride 150 mg ER once daily under fasting or fed conditions, or itopride 50 mg IR three times daily in the fasting state. Steady-state pharmacokinetic parameters of itopride, including peak plasma concentration (Cmax) and area under the plasma concentration versus time curve over 24 hours after dosing (AUC0–24h), were determined by noncompartmental analysis. The geometric mean ratio of the pharmacokinetic parameters was derived using an analysis of variance model.
Results: A total of 24 healthy Korean subjects participated, 23 of whom completed the study. The geometric mean ratio and its 90% confidence interval of once-daily ER itopride versus IR itopride three times a day for AUC0–24h were contained within the conventional bioequivalence range of 0.80–1.25 (0.94 [0.88–1.01]), although Cmax was reached more slowly and was lower for itopride ER than for the IR formulation. Food delayed the time taken to reach Cmax for itopride ER, but AUC0–24h was not affected. There were no serious adverse events and both formulations were generally well tolerated.
Conclusion: At steady state, once-daily itopride ER at 150 mg has a bioavailability comparable with that of itopride IR at 50 mg given three times a day under fasting conditions. Food delayed the absorption of itopride ER, with no marked change in its oral bioavailability.
Keywords: itopride, extended-release, immediate-release, bioavailability, pharmacokinetics
Introduction
Itopride, a benzamide derivative, has both acetylcholine esterase inhibitory and dopamine D2 receptor antagonist effects.1–3 These combined effects make itopride a useful gastroprokinetic agent for the symptomatic treatment of various gastrointestinal disorders, including functional dyspepsia4–7 and gastroesophageal reflux disease.2 After oral administration, itopride is rapidly absorbed, and peak plasma levels are reached within half an hour.8 Furthermore, itopride is metabolized by flavin-containing monooxygenase in the liver, unlike cisapride and mosapride, which are metabolized by cytochrome P450 (CYP) enzymes. In addition, itopride did not inhibit five CYP enzymes, including CYP2C19 and CYP3A4, in a human hepatic microsomal study.9 Collectively, these results suggest that itopride is less likely to have drug-drug interactions.
Itopride has been approved to treat functional dyspepsia in South Korea. The current labeled dosing regimen of itopride is oral administration three times a day at 50 mg. Because many gastroprokinetic agents, including itopride, exert their effects through antidopaminergic action,10,11 maintaining a certain therapeutic level over a sustained period becomes important.12 An extended-release (ER) formulation is a good therapeutic option in this regard because it reduces fluctuation in the plasma concentration, while less frequent dosing enhances patient convenience and compliance. Based on this understanding, an ER formulation of itopride at 150 mg was developed, which was intended to be given once daily. The ER formulation of itopride is a capsule, which contains a large number of coated pellets that are released slowly but continuously at different times in the gastrointestinal tract, depending on the composition and amount of coating applied. Therefore, this ER formulation is expected to provide a more steady and sustained level of itopride in the body.
The US Food and Drug Administration recommends that bioavailability studies of an orally administered ER formulation adequately characterize its absorption profile, which may be compared with that of the immediate-release (IR) formulation.13 Likewise, the Korean regulatory guidance14 states that a bioequivalence study or comparative pharmacokinetic study can be conducted to evaluate the pharmacokinetic profile of a newly developed formulation.
The objective of this study was to evaluate the steady-state systemic exposure to an itopride ER formulation, and to compare it with that of itopride IR in the fasting state. In addition, the effect of food on the bioavailability of itopride ER was assessed.
Materials and methods
Study participants
Healthy Korean volunteers aged 22–55 years with a body mass index of 18–27 kg/m2 were enrolled if they had no clinically significant abnormalities based on medical history, physical examination, vital signs, 12-lead electrocardiography, and routine clinical laboratory tests (clinical chemistry, hematology, and urinalysis) performed within 3 weeks before enrollment. Subjects were excluded if they had any of the following: a history of clinically significant cardiovascular, respiratory, renal, hepatic, or gastrointestinal disease; a history of alcohol or drug abuse; a history of drug allergy or known hypersensitivity to itopride; or any clinical condition which might affect the absorption, distribution, biotransformation, or excretion of the study drug. Subjects were also excluded if they took any prescription drug, over-the-counter medication, or herbal medications within 14 days before scheduled study drug administration, or smoked more than ten cigarettes per day.
Study design and procedures
This study was conducted using a randomized, open-label, single-center, multiple-dose, three-treatment, three-sequence, crossover design. The following three oral regimens were compared: a single ER tablet of itopride hydrochloride 150 mg given every 24 hours under fasting (regimen A) or fed (regimen B) conditions, and a single IR tablet of itopride hydrochloride 50 mg given 30 minutes before each meal and administered at around 9 am, 2 pm, and 7 pm (regimen C). Eligible subjects randomly received one of the three itopride regimens for 4 days in each period according to one of the following sequences: ABC, BCA, or CAB. There was a 4-day washout between adjacent periods. The subjects were admitted to the Clinical Trials Center at Seoul National University Hospital, Seoul, Korea, on the night before the first day of each period and were confined to the center throughout the whole study period, during which they were discouraged from undertaking any strenuous or athletic activity. A standard diet (~20%–25% of calories from fat) was provided, and subjects were instructed to sit upright for 4 (regimens A and B) or 2 (regimen C) hours after each administration of the study drug.
Steady-state pharmacokinetic blood samples were obtained on day 4 of each period for regimens A and B at 0 (ie, predose), and 1, 2, 3, 4, 5, 6, 7, 8, 10, 12, 16, and 24 hours after dosing. Because regimen C was administered three times a day, the sampling scheme was slightly modified, ie, 0, 0.5, 1, 1.5, 2, 3 and 5 hours after the first and second doses of day 4, and 0, 0.5, 1, 1.5, 2, 3, 5, 7, and 14 hours after the third dose of the day. Blood samples were centrifuged using a refrigerated centrifuge to separate the plasma within one hour of collection, were frozen within 2 hours of collection, and maintained at ≤−20°C until analysis of itopride concentration. Physical examination, vital signs, a 12-lead electrocardiogram, and laboratory tests were conducted throughout the study. Adverse events were captured using a structured questionnaire and spontaneous reports by study participants.
The study protocol and an informed consent form were reviewed and approved by the institutional review board of Seoul National University Hospital. Further, all procedures of the study were conducted in accordance with the principles of the Declaration of Helsinki and Korean Good Clinical Practice Guidelines.
Determination of itopride plasma concentrations
Plasma concentrations of itopride were determined using high-performance liquid chromatography (1100 series, Agilent Technologies, Santa Clara, CA, USA) coupled with tandem mass spectrometry (API3000, Applied Biosystems/MDS Sciex, Toronto, Canada). After adding 200 μL of glipizide (internal standard) in acetonitrile solution (50 ng/mL) to 50 μL of plasma, the mixture was stirred immediately for 30 seconds and then centrifuged for 5 minutes at 13,000 g, after which 150 μL of 10 mM ammonium acetate with 0.1% formic acid (mobile phase) was added to 50 μL of supernatant. Throughout this process, reconstitution was completed and 10 μL of the reconstituted solution was injected into the liquid chromatography-tandem mass spectrometry system. Chromatographic separation was performed using a Luna C8 column (3 μm, 4 × 2 mm, Phenomenex, Torrance, CA, USA) and the column temperature was maintained at 25°C. All separations were achieved at a flow rate of 0.3 mL per minute.
The plasma concentration of itopride was determined from the standard curve using the ratio between the peak area of itopride and that of the internal standard. We used 1, 4, 20, 100, 200, 500, and 100 ng/mL of itopride solution to construct the calibration curve. The regression correlation coefficients of the calibration curves were >0.9982. The lowest limit of quantification of itopride was 1 ng/mL. The performance of the assay during sample analysis was evaluated for independently prepared quality control samples at four concentrations (1, 3.2, 200, and 800 ng/mL) in each analytical batch. From these quality control samples, interbatch accuracy ranged from 97.7% to 103.3%, and interbatch precision, expressed as percent coefficient of variation (%CV), ranged from 4.0% to 13.2%. In addition, intrabatch accuracy and precision (%CV) ranged from 86.4% to 101.2% and from 2.8% to 4.1%, respectively.
Pharmacokinetic analysis
A noncompartmental analysis was performed using WinNonlin® (Profession Network Version 5.2.1, Pharsight Corporation, Mountain View, CA, USA). The actual sampling time of each subject was used in pharmacokinetic analysis. The following steady-state pharmacokinetic parameters were obtained for each itopride formulation: peak plasma drug concentrations (Cmax,ss), time to reach Cmax,ss (Tmax,ss), area under the plasma concentration-time curve over 24 hours post dose (AUC0–24,ss), and elimination half-life (t1/2,ss). Cmax,ss and Tmax,ss were obtained directly from the observed values. The terminal elimination rate constant (λz) was estimated by linear regression of the log-linear decline portion of the individual plasma concentration-time data. Next, t1/2,ss was calculated as the natural logarithm of 2 divided by λz. AUC0–24,ss was derived using the trapezoidal rule from 0 to 24 hours post dose for regimens A and B, whereas partial AUCs for 0–5 hours, 5–10 hours, and 10–24 hours post dose was summed for regimen C.
Statistical analysis
Given the exploratory nature of the present study, sample size was not based on formal statistical methods, but was considered a reasonable size to address the study objectives. Statistical analysis was performed using Statistical Package for the Social Sciences software (SPSS Korea, Seoul, Korea). Descriptive statistics were used to summarize baseline demographics such as age, body mass index, and body weight, and pharmacokinetic parameters including AUC0–24,ss, Cmax,ss, Tmax,ss, and t1/2. Between-regimen comparison was performed using the geometric mean ratio of AUC0–24,ss and Cmax,ss, where regimen C (ie, IR formulation under fasting) was the reference. Likewise, the effect of food on the ER formulation was examined using the geometric mean ratio of AUC0–24,ss and Cmax,ss with (regimen B) and without (regimen A) food. To this end, Cmax,ss and AUC0–24,ss were log-transformed and entered into a mixed-effects analysis of variance model, where period and treatment were fixed effects.
Results
Study participants
This study enrolled a total of 24 healthy Korean subjects, whose mean ± standard deviation age, weight, and body mass index were 30.8±7.4 years, 67.3±9.6 kg, and 22.7±2.0 kg/m2, respectively (Table 1). One participant withdrew consent on day 4 due to abdominal pain, and was included only in the tolerability analysis data set.
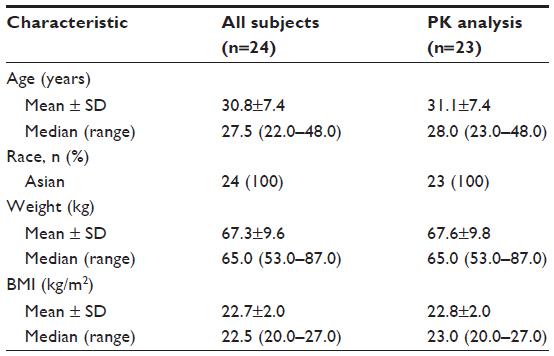 | Table 1 Demographic characteristics of participants |
Pharmacokinetic analysis
Itopride ER showed the typical pharmacokinetic profile of an extended-release formulation, particularly in the fasting state (Figure 1), ie, Cmax,ss was reached more slowly and was lower for the ER formulation than for the IR formulation (median 3.1 hours versus 0.8 hours for Tmax,ss; 244.4±94.4 ng/mL versus 416.2±141.3 ng/mL for Cmax, ss, Table 2). However, in the fasting state, once-daily ER itopride had a pharmacokinetic profile comparable with that of IR itopride given three times a day in terms of the extent of total systemic exposure. For example, AUC0–24,ss under fasting conditions was similar between the two formulations, leading to a geometric mean ratio for ER to IR of 0.94, and its 90% confidence interval, ie, 0.88–1.01 (Table 3), fell entirely within the conventional bioequivalence range of 0.8–1.25.
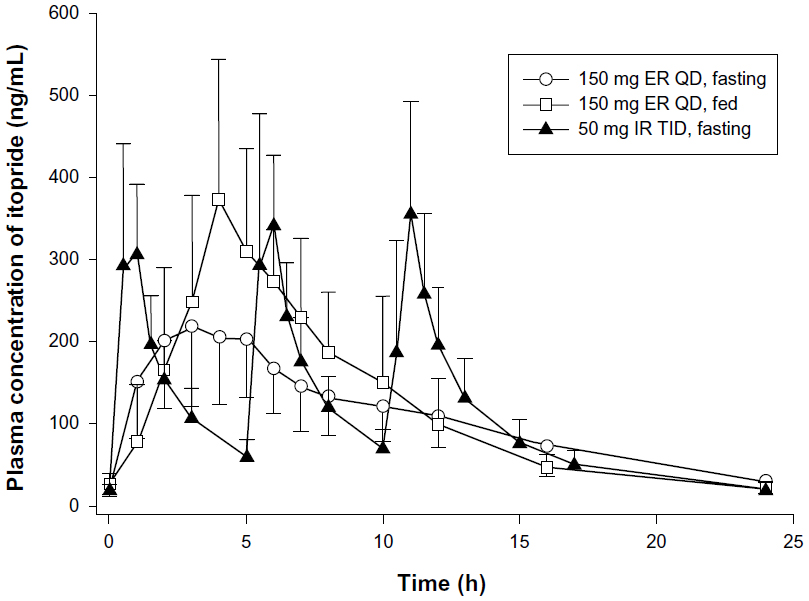 | Figure 1 Mean steady-state plasma concentration-time profiles for oral itopride after QD dosing with the ER formulation under fasting or fed conditions, and after TID dosing with the IR formulation in the fasting state. The error bars denote the standard deviation. |
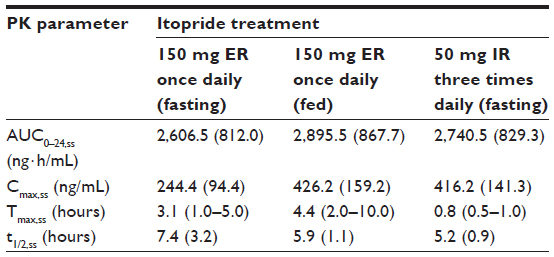 | Table 2 Steady-state pharmacokinetic parameters of oral itopride after once-daily administration of 150 mg of the ER formulation under fasting or fed conditions and after 50 mg of the IR formulation given three times daily in the fasting state |
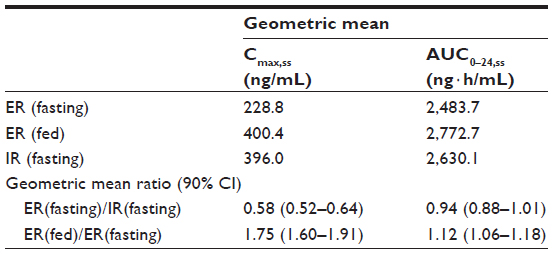 | Table 3 Geometric mean and ratio of pharmacokinetic parameters for itopride in the ER and IR formulations |
Food delayed the time taken to reach peak concentration with ER itopride (4.4 hours versus 3.1 hours for Tmax,ss with and without food, respectively, Table 2 and Figure 1). In addition, Cmax,ss was increased when ER itopride was given with food, reaching close to the value seen with IR itopride given without food (Table 2). However, food did not affect the extent of absorption, ie, the geometric mean ratio (90% confidence interval) of AUC0–24,ss for ER itopride with and without food was 1.12 (1.06–1.18) as shown in Table 3.
Tolerability
Eight subjects experienced a total of 16 adverse events (Table 4), among which four events in three subjects were considered “possibly related” to the study drug, ie, abdominal pain (n=2), diarrhea (n=1), and dizziness (n=1). All adverse events resolved without treatment, except in one subject who needed intravenous hydration to manage vasovagal syncope, which was not considered related to the study drug. No serious adverse event occurred during the entire course of the study. Likewise, there were no clinically significant abnormalities in laboratory tests, physical examinations, or electrocardiograms.
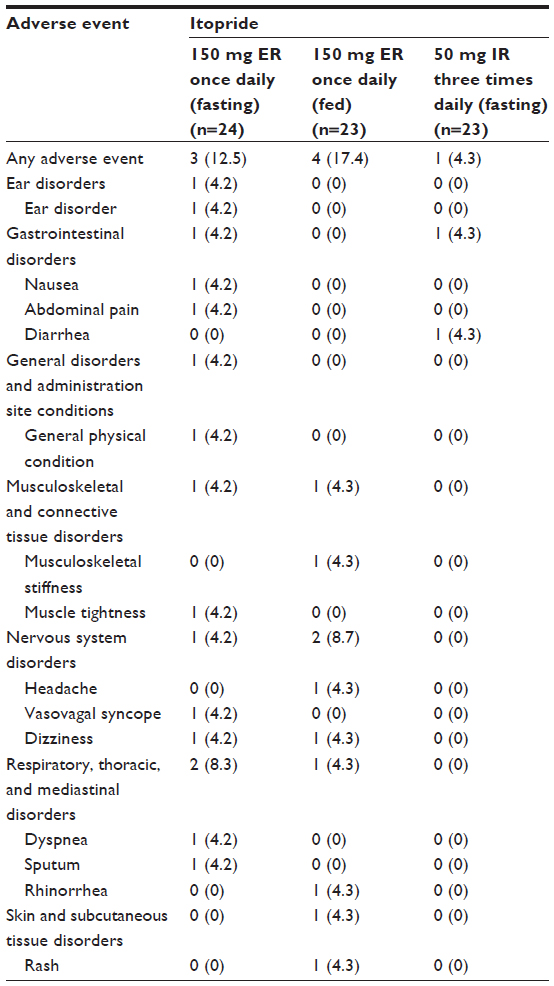 | Table 4 Treatment-emergent adverse events after treatment with itopride |
Discussion
The present study shows that, at steady state under fasting conditions, the ER formulation of itopride has a pharmacokinetic profile comparable with that of the IR formulation in terms of the extent of absorption. For example, AUC0–24,ss was similar between the two formulations, and the geometric mean ratio of ER to IR was 0.94 with a 90% confidence interval of 0.88–1.01 (Table 3), falling entirely within the conventional bioequivalence range of 0.8–1.25. As expected, the peak plasma concentration was 42% lower and delayed by 2.3 hours after administration of itopride ER compared with IR (Table 3). However, peak-to-trough variability in concentration was much smaller for the ER formulation; its pharmacokinetic profile was sustained and smoothened out (Figure 1). These pharmacokinetic properties of itopride ER would enable therapeutic itopride concentrations to be maintained over a longer period of time, contributing to the antidopaminergic effect of itopride.12 Collectively, the once-daily ER formulation of itopride will help patients to comply with the regimen while ensuring a systemic exposure comparable with that of the IR formulation.13,15
Our results show that food increased the peak concentration of itopride ER by 75% and delayed the time taken for it to reach peak concentration, while AUC0–24,ss was increased by 12%, leading to a bioequivalent geometric mean ratio of 1.12 (90% confidence interval 1.06–1.18, Table 3). Because food may affect gastric emptying, and thereby drug absorption,15 a change in gastric emptying might have contributed to the delay in time taken to reach the peak concentration of itopride. In addition, itopride can alter intestinal motility, which may further modify the absorption profile of itopride ER.
Both the ER and IR formulations of itopride were well tolerated in the present study. Further, subjects did not experience any serious adverse events, and all adverse events resolved spontaneously without any intervention, except for one subject who required hydration because of vasovagal syncope that was not related to the study medication.
The present study had several limitations. First, it was conducted using an open-label design, in which subjective assessment of adverse events can be biased. To overcome this potential bias in assessment of adverse events, a standardized structured questionnaire was used in addition to subject self-reporting. Second, because this study was conducted in healthy and relatively young subjects, our results may not be fully representative of the intended patient population for itopride, medical conditions in whom could alter the pharmacokinetic profile of the drug. Therefore, future studies are warranted to adequately characterize the absorption behavior of itopride ER in patients with functional dyspepsia.
In conclusion, at steady state under fasting conditions, comparable oral bioavailability was found between itopride ER 150 mg given once daily and itopride IR 50 mg given three times a day. Food delayed the absorption of itopride ER, with no marked change in oral bioavailability. Itopride ER and IR were well tolerated. The smoothened-out pharmacokinetic profile of the ER formulation of itopride, along with its once-daily regimen, will increase efficacy and compliance with treatment.
Disclosure
This study was supported by a research grant from Abbott Korea Ltd. Dong-Hyun Chee is a former employee of Abbott Korea Ltd, is now an employee of AbbVie Ltd, and may own Abbott and AbbVie stock. The present work was presented as a poster at the 2013 annual meeting of the American Society for Clinical Pharmacology and Therapeutics (March 5–9, 2013, Indianapolis, IN, USA). Otherwise, the authors report no conflicts of interest in this work.
References
Iwanaga Y, Kimura T, Miyashita N, et al. Characterization of acetylcholinesterase-inhibition by itopride. Jpn J Pharmacol. 1994;66(3):317–322. Available from: http://dx.doi.org/10.1254/jjp.66.317. Accessed December 3, 2013. | |
Kim YS, Kim TH, Choi CS, et al. Effect of itopride, a new prokinetic, in patients with mild GERD: a pilot study. World J Gastroenterol. 2005;11(27):4210–4214. | |
Katagiri F, Shiga T, Inoue S, Sato Y, Itoh H, Takeyama M. Effects of itopride hydrochloride on plasma gut-regulatory peptide and stress-related hormone levels in healthy human subjects. Pharmacology. 2006;77(3):115–121. | |
Kreutzkamp B. [Functional dyspepsia. Itopride improves symptoms]. Med Monatsschr Pharm. 2006;29(10):387–388. German. | |
Sun J, Yuan YZ, Holtmann G. Itopride in the treatment of functional dyspepsia in chinese patients: a prospective, multicentre, post-marketing observational study. Clin Drug Investig. 2011;31(12):865–875. | |
Talley NJ, Tack J, Ptak T, Gupta R, Giguere M. Itopride in functional dyspepsia: results of two phase III multicentre, randomised, double-blind, placebo-controlled trials. Gut. 2008;57(6):740–746. | |
Holtmann G, Talley NJ, Liebregts T, Adam B, Parow C. A placebo-controlled trial of itopride in functional dyspepsia. N Engl J Med. 2006;354(8):832–840. | |
Nakajima M, Uematsu T, Nakajima S, Nagata O, Yamaguchi T. Phase 1 study of HSR-803. Jpn Pharmacol Ther. 1993;21(11):4157–4173. | |
Mushiroda T, Douya R, Takahara E, Nagata O. The involvement of flavin-containing monooxygenase but not CYP3A4 in metabolism of itopride hydrochloride, a gastroprokinetic agent: comparison with cisapride and mosapride citrate. Drug Metab Dispos. 2000;28(10):1231–1237. | |
Demol P, Ruoff HJ, Weihrauch TR. Rational pharmacotherapy of gastrointestinal motility disorders. Eur J Pediatr. 1989;148(6):489–495. | |
Keith A Sharkey, Wallace JL. Treatment of disorders of bowel motility and water flux; anti-emetics; agents used in biliary and pancreatic disease. In: Brunton LL, Blumenthal DK, Murri N, Dandan RH, Knollmann BC. Goodman and Gilman’s the Pharmacological Basis of Therapeutics. 12th ed. New York, NY, USA: The McGraw-Hill Companies, Inc; 2011. | |
Tonini M, Cipollina L, Poluzzi E, Crema F, Corazza GR, De Ponti F. Review article: clinical implications of enteric and central D2 receptor blockade by antidopaminergic gastrointestinal prokinetics. Aliment Pharmacol Ther. 2004;19(4):379–390. | |
Malinowski H, Marroum P, Uppoor VR, et al. Draft guidance for industry extended-release solid oral dosage forms. Development, evaluation and application of in vitro-in vivo correlations. Adv Exp Med Biol. 1997;423:269–288. | |
Guidance on Evaluation of the Safety and Efficacy of Medicinal Products. Republic of Korea: The Korean Food and Drug Administration; 2013. | |
Welling PG. Influence of food and diet on gastrointestinal drug absorption: a review. J Pharmacokinet Biopharm. 1977;5(4):291–334. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.