Back to Journals » Drug Design, Development and Therapy » Volume 12
Comparative pharmacokinetics of an adalimumab biosimilar SB5 administered via autoinjector or prefilled syringe in healthy subjects
Authors Shin D, Lee Y, Jeong D, Ellis-Pegler R ![]()
Received 14 April 2018
Accepted for publication 1 September 2018
Published 5 November 2018 Volume 2018:12 Pages 3799—3805
DOI https://doi.org/10.2147/DDDT.S169082
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Manfred Ogris
Donghoon Shin,1 Younju Lee,1 Deokyoon Jeong,1 Rod Ellis-Pegler2
1Samsung Bioepis Co. Ltd, Incheon, Republic of Korea; 2Auckland Clinical Studies, Ltd., Auckland, New Zealand
Purpose: The objective of this study was to demonstrate comparable pharmacokinetic (PK), safety, and tolerability parameters of the adalimumab biosimilar SB5 administered via autoinjector (AI) pen or prefilled syringe (PFS).
Patients and methods: In this phase 1, randomized, open-label, single-dose, parallel-group study, healthy subjects aged 18–55 years were randomized 1:1 to a single dose of 40 mg SB5 delivered subcutaneously via AI or PFS. PK parameters, safety, and tolerability were assessed for 57 days post-dose. The primary endpoint was area under the curve (AUC) of the concentration-time curve from zero to infinity (AUCinf) and from zero to last quantifiable concentration (AUClast) and maximum serum concentration (Cmax). Equivalence was determined using predefined margins of 0.80–1.25 for the 90% CI for the ratio of SB5 AI to SB5 PFS.
Results: Ninety-five subjects were randomized to each group. Mean serum concentration-time profiles were superimposable between groups. Mean values for AUCinf, AUClast, and Cmax were similar between the SB5 AI and SB5 PFS groups. For the primary endpoints, the 90% CIs for the ratio of geometric least squares means for SB5 AI to SB5 PFS ranged between 0.9503 and 1.2240, which were all within the equivalence margin of 0.80–1.25. Incidence of treatment-emergent adverse events and injection site reactions was similar between groups.
Conclusion: In healthy subjects receiving a single dose of SB5 via AI or PFS, PK parameters and corresponding 90% CIs were within the predefined margins, showing bioequivalence between the two delivery methods. Safety and tolerability assessments were also similar between groups.
ClinicalTrials.gov identifier: NCT02326233.
EudraCT number: 2014-005178-12.
Keywords: bioequivalence, rheumatoid arthritis, clinical trial, safety, TNF-alpha inhibitor
Introduction
Adalimumab is a recombinant, fully humanized monoclonal antibody that binds specifically to inflammatory cytokine TNF, thereby neutralizing the biological function of TNF. Adalimumab is approved for treatment of rheumatoid arthritis and several other inflammatory diseases.1 Biologic agents including adalimumab are recognized treatment options for rheumatoid arthritis.2,3 However, treatment with biologic agents is often associated with high costs.4 Biosimilars are highly similar to and demonstrate no clinically meaningful differences from existing approved biologic agents and offer the potential to reduce health care expenditure for patients.5 SB5 (Imraldi®; Samsung Bioepis Co., Ltd., Incheon, Republic of Korea) is a biosimilar with adalimumab as the active substance. SB5 has been demonstrated to be highly similar to reference adalimumab (Humira®; AbbVie Deutschland GmbH & Co. KG, Ludwigshafen, Germany)1 and has received regulatory approval for all indications that are authorized for adalimumab in the European Union (EU).6
In a previous phase 1 study in healthy volunteers,7 SB5 demonstrated comparable pharmacokinetic (PK) parameters to both EU-sourced and US-sourced adalimumab, with all primary PK outcomes, test-to-reference ratios, and respective 90% CIs within the predefined limits for equivalence; SB5 also demonstrated comparable safety and immunogenicity with EU- and US-sourced adalimumab. In a phase 3, randomized study,8 SB5 demonstrated comparable efficacy with adalimumab – as well as comparable PKs, safety, and immunogenicity – in patients with moderate to severe rheumatoid arthritis; efficacy results were also comparable with historical studies. Based on results from these studies, the European Commission granted marketing authorization for SB5 in August 2017.
In the phase 1 and phase 3 studies,7,8 40 mg SB5 was administered subcutaneously via a prefilled syringe (PFS). Although PFSs increase the likelihood that patients will be adherent to therapy and offer greater patient control compared with vials or products that must be reconstituted, they may be limited by patients’ fear of needles, confidence in using the device, or reactions at the injection site.9 Additionally, PFSs may be difficult to use in patients with advanced stages of rheumatoid arthritis and in those who lack dexterity of the hand.9,10 To overcome these limitations, autoinjector (AI) pens were developed and are becoming increasingly acceptable, and may be useful in patients with late-stage rheumatoid arthritis. In a phase 2 study of patient preferences for PFS vs AI, 88.5% of patients reported preferring the AI compared with 5.8% of patients who preferred the PFS.10 Of the patients who preferred the AI, ease of use, less pain, and convenience were common reasons for preferring this delivery method.
An SB5 AI has been developed to address some of the limitations mentioned previously that may be encountered in patients with rheumatoid arthritis. The SB5 AI contains the same dose as the SB5 PFS and is a single-use, disposable combination product that incorporates not only an ergonomic design for greater handling, flexibility, and control, but also safety features to prevent needle-stick injuries before, during, and after subcutaneous injection. The objective of the current bridging study was to demonstrate PK equivalence and comparable safety and tolerability of subcutaneous injection of SB5 via AI or PFS.
Material and methods
Study design
This was a phase 1, randomized, open-label, single-dose, parallel-group study conducted at one site in Belgium and one in New Zealand (EudraCT number: 2014-005178-12; ClinicalTrials.gov identifier: NCT02326233). The objectives were to investigate and compare the PK profiles of the SB5 AI and PFS in healthy subjects and to investigate safety and tolerability. Subjects were randomized 1:1 to receive a single dose of 40 mg SB5 subcutaneously via AI or PFS and were observed for 57 days, during which PK parameters, safety, and tolerability were assessed. The study was approved by independent ethics committees in both locations (Ziekenhuis Netwerk Antwerpen, Commissie voor Medische Ethiek, Belgium; Ministry of Health, Health and Disability Ethics Committees, New Zealand) and was conducted in accordance with the Declaration of Helsinki, International Conference on Harmonisation Good Clinical Practice Guidelines, and local regulatory requirements and laws. All subjects provided written informed consent before entering the study.
Subjects
Healthy men and women aged 18 through 55 years were enrolled in the study. Women had to be of non-childbearing potential, and all subjects were required to have electrocardiogram (ECG), vital signs, physical examination, and clinical laboratory tests (measured at screening and day 1) within the normal range or, if outside the normal range, assessed as not clinically significant. Additionally, subjects had to have a body weight between 65.0 and 90.0 kg and body mass index between 20.0 and 29.9 kg/m2 at screening and day 1, inclusive. Subjects were excluded if they met any of the following criteria: history and/or presence of atopic allergy, hypersensitivity, or allergic reaction to any component of the test or reference study drug formulation or comparable drug; active or latent tuberculosis; history of systemic or local infection, known risk for developing sepsis, or active inflammatory process within 6 months before study drug administration; serious infection requiring intravenous antibiotics or hospitalization within 6 months before study drug administration; previous treatment with adalimumab or receipt of adalimumab for investigational purpose; or history of cancer, impaired liver function, or immunodeficiency.
Study assessments
PKs
Blood samples (approximately 3.5 mL) for PK analysis were collected at pre-dose (0 hours) and at 24, 48, 96, 120, 144, 168, 216, 264, 336, 408, 504, 600, 696, 840, 1,008, and 1,344 hours post-dose. The serum concentration of adalimumab was measured using an ELISA specific for the detection and quantification of adalimumab. The lower limit of quantitation was 0.05 μg/mL, and the upper limit of quantitation was 1.20 μg/mL. Intra- and inter-assay precision and accuracy displayed a coefficient of variation (CV%) of 7.8% and 16.7%, respectively. PKs were assessed in the PK population, which included all subjects who were randomized, received the study drug, and had evaluable primary PK parameters without major protocol deviations.
The primary PK endpoints were area under the curve (AUC) of the concentration-time curve from zero to infinity (AUCinf), AUC from zero to last quantifiable concentration (AUClast), and maximum serum concentration (Cmax; obtained directly from concentration-time curve). Secondary endpoints included time to Cmax (Tmax), apparent volume of distribution based on terminal phase (Vz/F), terminal rate constant (λz), terminal half-life (t1/2), apparent total body clearance (CL/F), and AUC extrapolated from time t to infinity as a percentage of total AUC (%AUCextrap). Linear regression after loge-transformation using the last three (or more) non-zero concentrations was used to calculate λz. CL/F was determined as dose/AUCinf, and Vz/F was estimated as CL/F/λz. t1/2 was calculated by ln(2)/λz, and %AUCextrap was calculated as (1–[AUClast/AUCinf])×100.
Safety
Safety was assessed through adverse event (AE) reporting, with all reported terms for AEs coded using the Medical Dictionary for Regulatory Activities (MedDRA v17.0). Safety parameters included treatment-emergent adverse events (TEAEs), vital signs, clinical laboratory tests, 12-lead ECGs, physical examinations, and injection site assessments. Specifically, injection sites were assessed for redness, bruising, swelling, itching, and pain using a numeric rating scale from 0 to 3 (0= none, 3= severe). The total score was the sum of these points, ranging from 0 to 15; an injection site reaction with a total score of ≥2 according to the rating scale was documented as an AE. Safety was assessed in the safety population, which included all subjects who received the study drug.
Statistical analyses
It was determined that a sample size of 85 completing subjects from each group would provide 90% power to detect a 20% difference in PK parameters between the test (AI) and reference (PFS) study drug. This was based on an assumption of 5% difference in true geometric least squares (LS) means between groups and inter-subject CV% of 39.5% (CV% of primary PK endpoints of adalimumab [PFS] ranged from 29.1% to 39.5% based on the results of a prior study with SB5 [SB5-G11-NHV]).7 With AUCinf, AUClast, and Cmax as the primary endpoints for this study, the 39.5% inter-subject CV% was used as a conservative approach. This was a one-sided t-test with a 5% significance level. Assuming a 10% dropout rate consistent with SB5-G11-NHV study results, a total of 188 subjects (94 in each group) were needed for the study.
Demographics, PKs, and safety parameters were summarized by treatment group using descriptive statistics. For comparison of primary PK parameters an ANOVA model with treatment as fixed effect was performed based on non-compartmental analysis methods using WinNonlin Phoenix 6.2 (Pharsight Corporation, Palo Alto, CA, USA). The difference in geometric LS means of primary endpoints between the SB5 AI and SB5 PFS and the associated 90% CIs were determined. The ratio of geometric LS means and 90% CIs for these ratios were determined by back transformation. Equivalence of the primary endpoints was determined if the 90% CI for the ratio of geometric LS means of the SB5 AI to SB5 PFS was within the acceptance interval of 0.80–1.25.
Results
Subject disposition
A total of 307 subjects were screened, and 190 were randomized (SB5 AI, n=95; SB5 PFS, n=95) (Figure 1). One subject from the SB5 PFS group did not meet all exclusion criteria and did not receive study drug; all other subjects completed the study. One subject (SB5 AI) who did not receive the full amount of the study drug because of failure of the AI pen was not included in the PK analysis. Baseline demographic characteristics were well-balanced between the two groups (Table 1). The overall mean age was 30.8 years, and most subjects were male (90.0%) and white (83.2%).
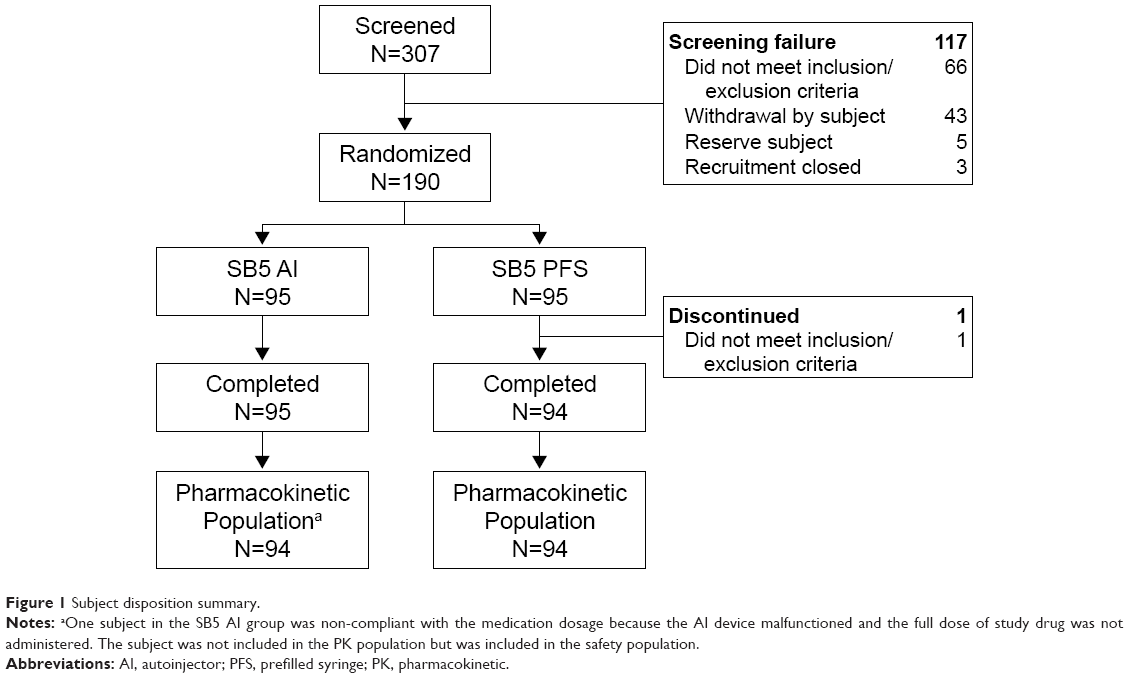 | Figure 1 Subject disposition summary. |
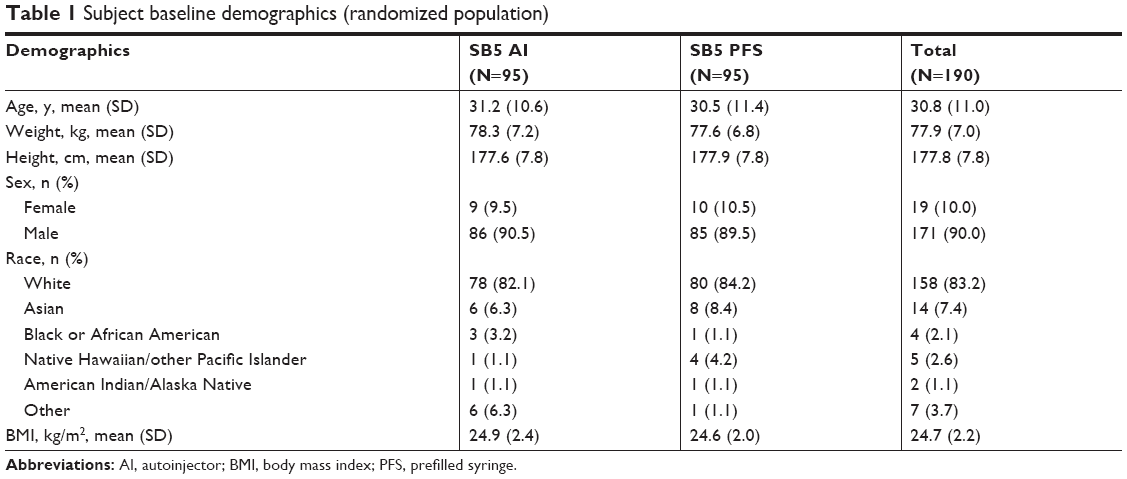 | Table 1 Subject baseline demographics (randomized population) |
Pharmacokinetics
The PK population included 94 subjects in each group. The mean serum concentration-time profiles were superimposable between the two groups with a rapid increase in serum concentrations after injection followed by a slow elimination phase (Figure 2). The mean values of PK parameters were similar and are summarized in Table 2. The comparative bioequivalence between SB5 AI and SB5 PFS is presented in Table 3. The 90% CIs for the geometric LS mean ratios of AUCinf, AUClast, and Cmax ranged from 0.9503 to 1.2240 and were within the predefined equivalence margin of 0.8–1.25 (Table 3).
 | Figure 2 Mean serum concentration profiles (pharmacokinetic population). |
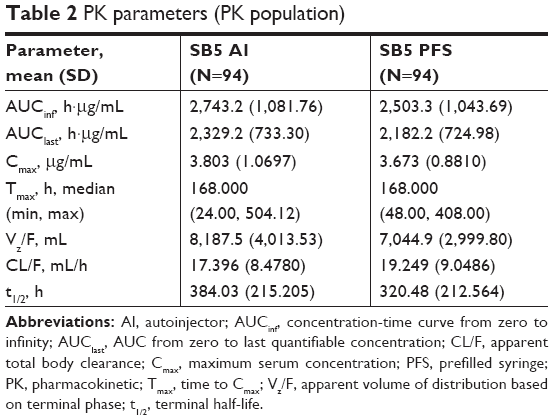 | Table 2 PK parameters (PK population) |
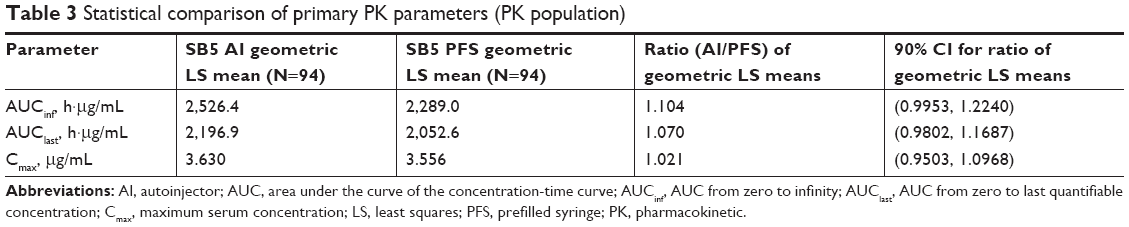 | Table 3 Statistical comparison of primary PK parameters (PK population) |
Safety
The safety population included 95 subjects from the SB5 AI group and 94 subjects from the SB5 PFS group. The percentage of subjects who experienced TEAEs was similar between groups (SB5 AI, 68.4%; SB5 PFS, 60.6%) (Table 4). Most TEAEs were mild in severity (SB5 AI, 96.6%; SP5 PFS, 94.2%), and most were considered unrelated to treatment (SB5 AI, 94.8%; SP5 PFS, 92.3%). The most frequently reported TEAE was upper respiratory tract infection, which occurred in 17.5% (33/189) of subjects overall. The incidence of fatigue, back pain, and vessel puncture site bruise was numerically higher in the SB5 AI group compared to SB5 PFS group. However when assessed by their relatedness with study drug the incidence was comparable. No deaths or discontinuations due to TEAEs occurred during the study. A serious adverse event (SAE) was reported in one subject (1.1%) in the SB5 AI group (schizophrenia) and in two subjects (2.1%) in the SB5 PFS group (clavicle fracture and hand fracture); none of the SAEs were considered related to treatment by the investigator.
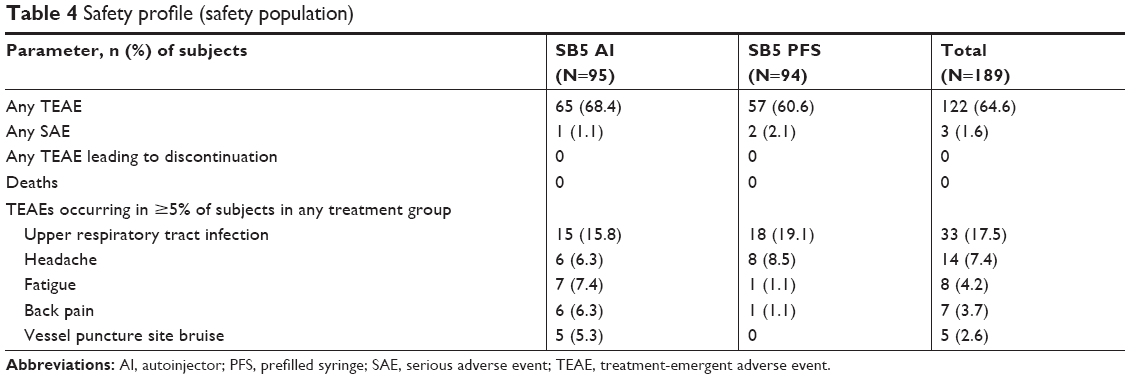 | Table 4 Safety profile (safety population) |
When assessed for redness, swelling, bruising, itching, and pain, few local injection site reactions were observed in either group, and all were mild in severity except for one subject in the SB5 PFS group who reported moderate redness on day 1. The most frequently reported injection site reaction was redness on day 1 (SB5 AI, 40.0%; SB5 PFS, 39.4%). In the SB5 PFS group, redness was observed in four subjects (4.3%) on day 2 and in one subject (1.1%) on day 5; no additional redness was reported in either group at other time points. On day 1, one subject (1.1%) in the SB5 PFS group had a total injection site reaction score of 3; no other subject had a total sum of injection site reaction score of ≥2. Laboratory data, vital signs, and ECG parameters did not show any relevant changes over time that might be considered related to study drug.
Discussion
This randomized, open-label, two-arm, parallel-group, bridging study was designed to compare the PKs and safety of SB5 administered via AI and PFS, which is part of the required information for new devices to be used in patients.11 Results from this study demonstrated that the two delivery methods display comparable PK parameters. The safety profiles of the SB5 AI and SB5 PFS were also comparable, with similar incidences of TEAEs and injection site reactions.
The mean serum concentration-time profiles for SB5 AI and SB5 PFS were superimposable, displaying a rapid increase in serum concentration followed by a slow elimination phase. Primary PK parameters (AUCinf, AUClast, and Cmax) and their 90% CIs were within the predefined equivalence margins, indicating that the two delivery methods are bioequivalent. These results suggest that the SB5 AI would likely be equally efficacious to the SB5 PFS, as well as to the reference adalimumab PFS.
In a previous study designed to show acceptability of switching from an adalimumab PFS to AI, patients considered the AI to be easier to use than the PFS.9 Similarly, in a study of patients with multiple sclerosis receiving IFNβ-1a, 94% of patients preferred a prefilled AI pen over a PFS, with preferences related to an easier injection process (eg, holding, gripping, and injecting).12 Use of an AI pen has also been associated with high levels of confidence in being able to perform self-injections in the future.13 Although the current study did not assess subject preferences or perceived ease of use, results from these previous trials10,12,13 suggest that patients would likely prefer to use the AI compared to the PFS for similar reasons. As patients with moderate to severe rheumatoid arthritis may have limited dexterity, the ease of use associated with AIs may provide a better option for self-injection of medication.
Treatment with biologic agents may be associated with administration-related reactions, and the incidence of pain9 and bruising10 has been reported to differ between delivery devices. In the present study, injection site assessments – including redness, bruising, swelling, itching, and pain – were used to evaluate tolerability of the delivery methods. Results from both the SB5 AI and SB5 PFS groups suggest that both delivery methods are associated with few injection site reactions and are well tolerated, as only one subject who received SB5 via PFS had a total sum of injection site reactions >2.
Limitations of the present study include factors related to the study design and population. As it was impossible to blind the delivery method, this may have affected subjects’ beliefs regarding expected TEAEs including pain associated with injection. Healthy subjects were selected for inclusion in the study because they were considered to be a more homogeneous and therefore a more sensitive population to compare PK characteristics than patients with various disease-related factors. A separate clinical study has been conducted to compare device usability and preference in patients with rheumatoid arthritis, and data are currently in press.
Conclusion
In healthy subjects receiving a single dose of the adalimumab biosimilar SB5, the AI and PFS demonstrated similar PK parameters, safety, and injection site assessments. These results suggest that there would be no clinical differences between the SB5 AI and SB5 PFS in terms of safety and efficacy and demonstrate that the SB5 AI represents an alternative delivery option to PFSs.
Acknowledgments
The authors thank the volunteers who were involved in this study, the study personnel who made this work possible, and the study investigators. This study was funded by Samsung Bioepis Co., Ltd. (Incheon, Republic of Korea). Medical writing assistance was provided by Krystina Neuman, PhD, and Michael S McNamara, MS, from C4 MedSolutions, LLC (Yardley, PA), a CHC Group company, and was funded by Samsung Bioepis Co., Ltd.
Author contributions
D Shin, Y Lee, and D Jeong contributed to the conception and design of the study and analysis and interpretation of data. R Ellis-Pegler contributed to the conception and design of the study, acquisition of data, and analysis and interpretation of data. All authors were involved in drafting or critically revising the article for intellectual content, provided approval for the final version to be published, and agree to be accountable for all aspects of the work.
Disclosure
D Shin, Y Lee, and D Jeong are employees of Samsung Bioepis Co., Ltd. D Shin reports personal fees from Samsung Bioepis during the conduct of the study. Y Lee reports owning Samsung Bioepis Co., Ltd. stock outside the submitted work. R Ellis-Pegler is a principal investigator and physician consultant for Auckland Clinical Studies (ACS), Auckland, New Zealand. The authors report no other conflicts of interest in this work.
References
Humira® (adalimumab). Summary of product characteristics. Ludwigshafen, Germany: AbbVie Deutschland GmbH & Co. KG; 2018. | ||
Smolen JS, Landewé R, Bijlsma J, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann Rheum Dis. 2017;76(6):960–977. | ||
Singh JA, Saag KG, Bridges SL, et al. 2015 American College of Rheumatology Guideline for the Treatment of Rheumatoid Arthritis. Arthritis Care Res. 2016;68(1):1–25. | ||
Dörner T, Strand V, Castañeda-Hernández G, et al. The role of biosimilars in the treatment of rheumatic diseases. Ann Rheum Dis. 2013;72(3):322–328. | ||
Hirsch BR, Lyman GH. Biosimilars: a cure to the U.S. health care cost conundrum? Blood Rev. 2014;28(6):263–268. | ||
European Medicines Agency. Imraldi: Summary of Opinion (initial authorisation). EMA; 2017. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Summary_of_opinion_-_Initial_authorisation/human/004279/WC500229906.pdf. Accessed September 10, 2018. | ||
Shin D, Lee Y, Kim H, Körnicke T, Fuhr R. A randomized phase I comparative pharmacokinetic study comparing SB5 with reference adalimumab in healthy volunteers. J Clin Pharm Ther. 2017;42(6):672–678. | ||
Weinblatt ME, Baranauskaite A, Niebrzydowski J, et al. Phase III randomized study of SB5, an adalimumab biosimilar, versus reference adalimumab in patients with moderate-to-severe rheumatoid arthritis. Arthritis Rheumatol. 2018;70(1):40–48. | ||
Borrás-Blasco J, Gracia-Pérez A, Rosique-Robles JD, Casterá MD, Abad FJ. Acceptability of switching adalimumab from a prefilled syringe to an autoinjection pen. Expert Opin Biol Ther. 2010;10(3):301–307. | ||
Kivitz A, Cohen S, Dowd JE, et al. Clinical assessment of pain, tolerability, and preference of an autoinjection pen versus a prefilled syringe for patient self-administration of the fully human, monoclonal antibody adalimumab: the TOUCH trial. Clin Ther. 2006;28(10):1619–1629. | ||
US Food and Drug Administration. Guidance for Industry Rheumatoid Arthritis: Developing Drug Products for Treatment. 2013. | ||
Phillips JT, Fox E, Grainger W, Tuccillo D, Liu S, Deykin A. An open-label, multicenter study to evaluate the safe and effective use of the single-use autoinjector with an Avonex® prefilled syringe in multiple sclerosis subjects. BMC Neurol. 2011;11:126. | ||
Kivitz A, Baret-Cormel L, van Hoogstraten H, et al. Usability and Patient Preference Phase 3 Study of the Sarilumab Pen in Patients with Active Moderate-to-Severe Rheumatoid Arthritis. Rheumatol Ther. 2018;5(1):231–242. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.