Back to Journals » Neuropsychiatric Disease and Treatment » Volume 10
Combotherapy and current concepts as well as future strategies for the treatment of Alzheimer's disease
Received 2 December 2013
Accepted for publication 14 January 2014
Published 11 March 2014 Volume 2014:10 Pages 439—451
DOI https://doi.org/10.2147/NDT.S45143
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Ling-Yun Fan,1,2 Ming-Jang Chiu2–4
1Department of Neurology, En Chu Kong Hospital, 2Department of Neurology, College of Medicine, Graduate Institute of Brain and Mind Sciences, 3Graduate Institute of Psychology, 4Graduate Institute of Biomedical Engineering and Bioinformatics, National Taiwan University Hospital, Taipei, Taiwan
Abstract: It has been estimated that 35.6 million people globally had dementia in 2010 and the prevalence of dementia has been predicted to double every 20 years. Thus, 115.4 million people may be living with dementia in 2050. Alzheimer's disease (AD) is the leading cause of dementia and is present in 60%–70% of people with dementia. Unfortunately, there are few approved drugs that can alleviate the cognitive or behavioral symptoms of AD dementia. Recent studies have revealed that pathophysiological changes related to AD occur decades before the appearance of clinical symptoms of dementia. This extended preclinical phase of AD provides a critical chance for disease-modifying agents to halt or delay the relentless process of AD. Although several trials targeting various pathological processes are ongoing, the examination of the combined use of different approaches to combat AD seems warranted. In this article, we will review current therapies, future strategies, and ongoing clinical trials for the treatment of AD with a special focus on combination therapies. Furthermore, preventive strategies for cognitively normal subjects in the presymptomatic stages of AD will also be addressed. In this review, we discuss current hypotheses of the disease process. In the decades since the approval of cholinesterase inhibitors, no new drug has ultimately demonstrated clear success in clinical trials. Given the difficulties that have been encountered in attempts to identify a single drug that can treat AD, we must pursue effective multi-target strategies, ie, combination therapies. The combination of cholinesterase inhibitors and memantine is considered well tolerated and safe, and this combination benefits patients with moderate-to-severe AD. In contrast, with the exception of adjuvant therapies of conventional drugs, combinations of different disease-modifying agents with different mechanisms may have promising synergic effects and benefit cognition, behavior, and daily living function.
Keywords: disease-modifying agent, combination therapy, Alzheimer's disease
Introduction
It has been estimated that 35.6 million people globally were living with dementia in 2010, and the prevalence of dementia has been predicted to double every 20 years. Thus, 65.7 million people in 2030 and 115.4 million people in 2050 may be living with dementia.1 Alzheimer’s disease (AD) is the leading cause of dementia and accounts for 60%–70% of the people living with dementia.2 The prevalence of AD dementia exhibits some geographical, ethnic, cultural, and societal variations. For example, rural living is associated with an increased incidence of AD dementia.3 One systematic review estimated that the overall prevalence of AD dementia in developing countries is 3.4%,4 whereas in most countries, the prevalence rates are between 5% and 8% among those over the age of 60 years. Recognized risk factors for AD dementia are older age, the ε4 allele apolipoprotein E (APOE) genotype, family history, illiteracy, cardiovascular risk factors (eg, smoking, diabetes, obesity), and lifestyle and psychosocial factors.5 The course of this devastating and irreversible neurodegeneration process includes progressive cognitive dysfunction, which not only leads to increased mortality rates, enormous societal impacts, and grave caregiver burdens but also exacts a significant worldwide economic burden. Unfortunately, only a few approved drugs can help to alleviate the cognitive or behavioral symptoms of people living with AD dementia.
Intracellular neurofibrillary tangles and extracellular amyloid plaques are the hallmarks of the pathology of AD.6 Many strategies targeting proteinopathies have successfully improved cognitive function in animal models. However, when these approaches have been subjected to clinical trials, most have shown no clinical benefit, regardless of whether decrements in amyloid burden have been shown by biomarker or postmortem pathological studies.
Recent studies have revealed that the pathophysiological changes of AD take place decades before the appearance of clinical symptoms of dementa.7 This extended preclinical phase of AD provides a critical opportunity for disease-modifying agents to halt or to slow the progression of AD.8 Although several trials targeting various pathological processes are ongoing, the examination of the combined use of different approaches that target different mechanisms of AD seems warranted. In this article, we aim to review current therapies, future strategies, and ongoing clinical trials for AD treatment with a special focus on combination therapy. Furthermore, preventive strategies for cognitively normal subjects with presymptomatic AD are also discussed.
Search strategies and terms
A literature search was conducted using the electronic databases of MEDLINE, PubMed, and Cochrane library from the dates of January 2000 to November 2013. In addition, bibliographic references of the selected articles were also evaluated to obtain possible articles not found by search engines. Peer-reviewed studies, thesis dissertations, or gray literature journals were also included. Reviews without original data, conference proceedings, editorial material, meeting abstracts, or language other than English were excluded. For issues related to combination therapies and further treatment strategies, search terms included: cholinesterase inhibitor (ChEI)/donepezil/galantamine/rivastigmine/tacrine, secretase inhibitors, amyloid beta (Aβ) monoclonal antibodies (bapineuzumab, solanezumab, gantenerumab, crenezumab), tauopathy, methylene blue or MB, latrepirdine or Dimebon, omega-3 polyunsaturated fatty acid, vitamin E, statin, Ginkgo biloba, vitamin B12, folate or folic acid, curcumin, phospodiesterase inhibitors or PDEI, thiazolidinedione, nerve growth factors, histamine or 5-HT6 antagonist, transcranial magnetic stimulation or rTMS/TMS, cognitive training or COG. The abovementioned therapeutic interventions were also combined with memantine or N-methyl-D-aspartate (NMDA) receptor antagonist and Alzheimer’s disease or AD to search for combination strategies. Clinicaltrials.gov was also searched to find any ongoing trials.
Study eligibility regarding combination therapies
Studies using ChEIs and memantine combination therapies, systematic reviews, blind randomized controlled trials (RCTs), and cohort studies were included in this review article. Open-label studies (RCTs and non-RCTs) were excluded. Other combination therapies, due to the relative paucity of research in this area, non-RCTs, open-label, pilot studies, or ongoing RCT trials were all included. These studies were designed to compare the combination of two intervention strategies (eg, memantine with ChEI), with either kind of monotherapy (eg, memantine or ChEI), or with a non-treatment group (control group).
Combination therapies of cholinesterase inhibitor and memantine
Three ChEIs (donepezil, rivastigmine, galantamine) are US Food and Drug Administration (FDA)-approved for the treatment of mild-to-moderate AD. The efficacies of these drugs have been broadly reviewed and documented.9 These therapeutic approaches are based on enhancing the attention system by delaying the reduction of acetylcholine level in the synaptic clefts. Stimulation of synaptic NMDA receptors, acting mainly through Ca2+ signals, leads to neuroprotection with increased neuronal survival reservoir. Meanwhile, excessive tonic activation of extra-synaptic NMDA receptors leads to mitochondrial dysfunction and neuronal apoptosis. These changes are closely related to AD pathology.6,10 Imbalance of synaptic and extra-synaptic signaling may be due to synaptic loss, resting potential alternation, or redistributions of NMDA receptors. The redistribution of NMDA receptors from synaptic to extra-synaptic sites may contribute to cell death and neurodegeneration.11 Memantine is an open-channel blocker with a fast off-rate, and is voltage-dependent, with an uncompetitive nature.12 Low-dose memantine results in an effective blockade of the chronic extra-synaptic NMDA receptor activity through antagonizing NR2B subunits, consequently halting downstream cell death signaling.11 In addition, it is proposed that in the CA1 region of AD hippocampus, the excitation–inhibition balance regulated by glutaminergic pyramidal cells (PCs) and GABAergic interneurons (Ints) is altered toward over-excitation states. Ints dysfunction is likely to increase the synchrony of excitatory PCs, thus destabilizing neuronal regulation. Memantine is relatively selective for NR2B and NR2A, which are subunits more predominant in PC NMDA receptors, whereas NR2C and NR2D are more expressed in Int NMDA receptors. Therefore, memantine antagonism is more robust in PCs than in Ints. The memantine antagonism might attenuate the over-excitation of PCs without alleviating the inhibition of interneurons to obtain a neuroprotective effect.13 Memantine has been approved by the FDA, the European Agency for the Evaluation of Medicinal Products (EMEA), and the UK National Institute for Health and Clinical Excellence (NICE) for the treatment of patients with moderate-to-severe AD dementia.12
Due to the nature of the neurodegenerative process, the efficacies of these approved medications are modest. A combination of these aforementioned drugs has been put into clinical practice for a long time. There are several completed clinical trials comparing the efficacy of combination therapy with ChEI monotherapy.14,15 One observational cohort study investigated these efficacies and predicted the long-term clinical outcomes of 4 years of combination therapy (COMBO), ChEI monotherapy, and no therapy among 328 AD patients. These patients were assessed with the Blessed Dementia Scale (BDS) and the Weintraub Activities of Daily Living Scale (ADL) and had a mean cumulative medication prescription interval of 22.5 months.16 The COMBO group exhibited a significantly lower mean annual rate of deterioration compared to the ChEI alone and no treatment groups as measured by BDS and ADL scores. The long-term course of the COMBO group exhibited increasing effect size benefits over treatment time, which implies a synergistic effect. Another observational study compared AD patients receiving combination therapy with those receiving ChEI monotherapy and those who had never received a cognitive enhancer, using nursing home admission or death as an endpoint.17 Results from this cohort study showed that the addition of memantine to ChEI significantly altered the history of AD patients with only monotherapy or those without medication by extending the time to nursing home admission. Three RCTs have been completed (Table 1). Porsteinsson et al evaluated the efficacy and tolerability of adjuvant therapy with memantine and placebo in 433 patients with mild-to-moderate AD dementia already receiving ChEI.18 The results revealed no significant differences in cognitive, functional, or behavioral outcomes at the end of the 24th week. Tariot et al evaluated the efficacies of adjuvant therapy with memantine and placebos in 404 patients with moderate-to-severe AD dementia (Mini Mental State Examination [MMSE]: 5–14).19 The results revealed a significant benefit of combination therapy in terms of cognitive, functional, behavioral, and global outcomes at the 24th week of treatment. The third RCT was conducted by Haward et al.20 Authors designed a 2 × 2 factorial comparison and divided moderate-to-severe (MMSE of 5–13) AD patients who had received donepezil for at least 3 months into four study groups: those who continued with donepezil and a memantine placebo, those who continued with donepezil with memantine, those who discontinued donepezil and received both donepezil and memantine placebos, and those who discontinued donepezil and received a donepezil placebo and memantine. The patients were followed for 52 weeks, and MMSE and Bristol Activities of Daily Living Scale (BADLS) were evaluated for use as the co-primary outcomes. Further secondary outcomes were the Neuropsychiatry Inventory (NPI), Measurement of health-related Quality of Life for people with Dementia (DEMQOL)-proxy, and General Health Questionnaire (GHQ)-12. The results revealed modest cognitive and functional benefits of continuing donepezil over the time period of the study’s observation. Memantine did not exhibit significant clinical efficacy regardless of whether the patients discontinued donepezil. The authors drew the conclusion that combined donepezil and memantine treatment was not significantly superior to treatment with donepezil alone based on the primary or secondary outcomes. A recent systemic review conducted by Muayqil and Camicioli pooled these three double-blind RCTs and included patients with mild-to-severe AD for meta-analysis.21 The results favored combination therapy in terms of cognitive, functional, and behavioral outcomes with a small but significant effect size among the moderate-to-severe AD patients. In the Tariot et al study, the use of Severe Impairment Battery (SIB) might help to avoid the floor effect, which would be encountered in the clinical trials on moderate-to-severe AD patients.19 Notably, this combination therapy is generally considered safe, and adverse effects are infrequent.
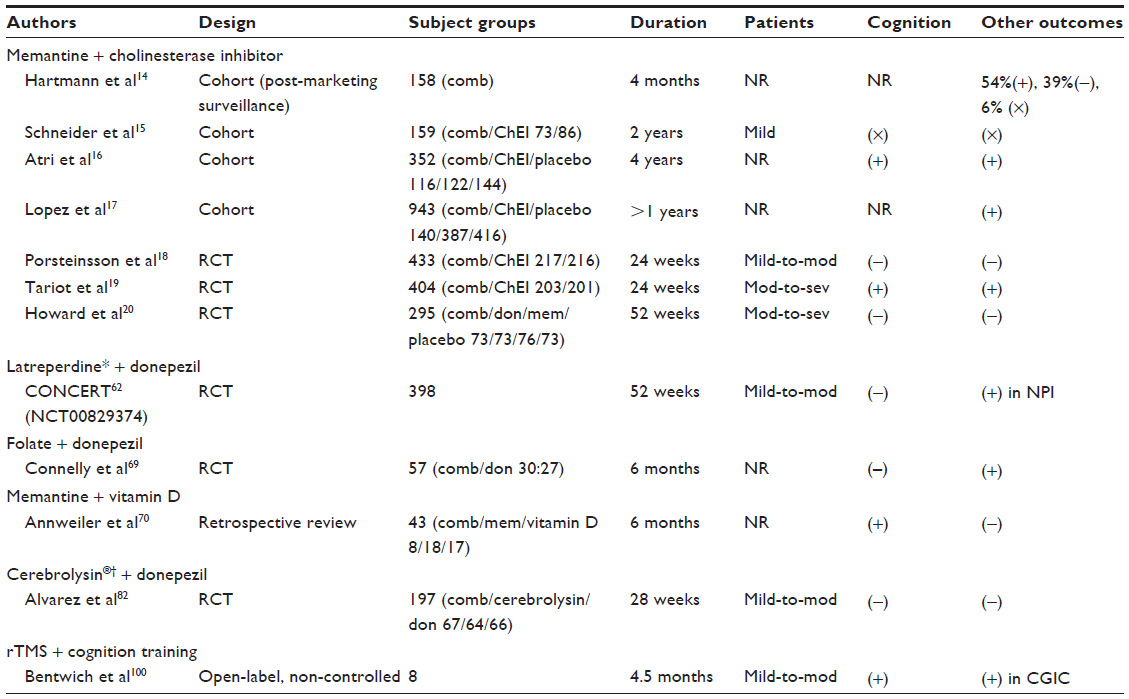 | Table 1 Characteristics and main outcomes of published studies with combination therapies |
Amyloid cascade
The amyloid cascade is one of the most well-known hypotheses of AD. This cascade begins with the deregulation of the intramembrane amyloid precursor protein (APP), which is catalyzed sequentially by beta and gamma secretases to form Aβ peptides. The Aβ peptides are mainly composed of 39–43 residues. Aβ 40 is the major component, and Aβ 42 tends to aggregate to form oligomers. The oligomers, globulomers, protofibrils, and other Aβ-aggregate intermediates are considered neurotoxic and pathophysiologically relevant because these intermediates lead to NMDA receptor dysfunction, neuronal synaptic dysfunction, oxidative injury, and neuronal loss. The amyloid fibrillar plaques are relatively inert.22 Early onset familial AD patients identified with pathogenic mutations of APP, presenilin 1 (PSEN1), or presenilin 2 (PSEN2) genes exhibit elevated production of Aβ 42. Carriers of the APOE ε4 genotype also have increased amyloid deposition in the neocortex via amyloid imaging scan. It is noteworthy that the degree of amyloid deposition in these carriers parallels with their genetic loads, and amyloid deposition begins in the preclinical stage of AD.23 All these findings support the amyloid cascade hypothesis.
Strategies attenuating the amyloidogenic pathway by β-secretase inhibitor (BACEI) and γ-secretase inhibitors have been under investigation for decades. However, the development of BACEI turned out to be very challenging due to problems of brain access, cell penetration, and oral bioavailability. In addition, BACEI is also involved in myelination. Many small molecules targeting BCAEI, either anti-β site antibodies or anti-β-secretase antibodies showed efficacy of reducing brain Aβ deposits as well as enhancing cognition in animal models.24 LY2811376, an orally available non-peptide BACEI produced profound long-lasting cerebrospinal fluid (CSF) Aβ reduction in healthy volunteers.25 However, these small molecules did not show their efficacy in cognitive improvement for AD patients in early clinical trials and this study was halted due to liver toxicity.26 MK-8919 is another BACEI currently being tested in Phase III clinical trials on patients with mild-to-moderate AD dementia. One subgroup study includes evaluation with amyloid positron emission tomography (PET) and CSF biomarkers and is currently recruiting (NCT01739348).27 Regarding γ-secretase inhibitors, several collateral side effects were encountered, such as hematological and gastrointestinal toxicity and skin reactions caused by inhibition of the Notch signal pathway. The result of a recent Phase III trial of semagacestat did not improve cognitive status. Patients receiving a higher dose had significant worsening of functions and had higher risk of skin cancers and infections.28
The proposed mechanisms of passive immunization include microglia-mediated phagocytosis, alternation of Aβ aggregation, neutralization of Aβ toxicity, and intracerebral sequestration with peripheral sink.29 However, amyloid-related imaging abnormalities were revealed while patients were receiving bapineuzumab in Phase II trials. The prevalence of abnormalities was higher in those patients who received a higher dose and those who carried ApoE4 alleles. Amyloid-related imaging abnormalities are due to the activation of antibody-induced complement systems and pre-existing amyloid angiopathy with altered vascular permeability.30 A Phase III trial of bapineuzumab announced disappointing results for patients with mild-to-moderate AD, although the positive effects of reduced cerebral amyloid deposition and decreased phosphorylated tau in the CSF were observed. Solanezumab is another antibody that recognizes a liner epitope in the center of Aβ, but this antibody also failed to improve cognitive and functional outcomes among mild-to-moderate AD patients in the Phase III EXPEDITION study. However, subgroup analyses revealed cognitive benefits in mild AD, and analyses of serum amyloid suggested that this antibody may promote the removal of amyloid from the brain to the blood.31 Gantenerumab is a fully human monoclonal antibody that is capable of neutralizing amyloid monomers, oligomers, and fibrils. Gantenerumab has been proven to reduce cerebral amyloid and is currently in Phase III studies (NCT01760005).32 Crenezumab is another humanized antibody that targets protofibrillar epitopes of Aβ and inhibits Aβ aggregation and is currently undergoing a Phase II trial (NCT01723826).33
Tau-centered treatments
Tau is a cytoplasmic protein that binds to tubulin and stabilizes microtubules, which are responsible for the neuronal transport of vesicles and cargo to the synapse. Neurofibrillary tangles were found to be a hallmark of the pathological findings in AD brains, which made tauopathy receive extensive attention in AD research. Hyperphosphorylated tau with incompletely understood mechanisms are prone to aggregate into oligomers and fibrils, which exhibit decreased binding ability to microtubules. Microtubule destabilization arrests anterograde axonal transportation resulting in neuronal damage and loss of neuronal function.34 On the other hand, degradation systems including proteasomal and autophagic processes contribute to the turnover of tau. However, while tauopathy accumulates, the “gain of toxicity” such as disrupting mitochondrial morphological alternation and inducing mitochondrial mis-localization takes place.35 Animal models have revealed that tau may mediate Aβ-induced excitotoxicity.36 Through a series of mutant-animal studies, Lors et al proposed a hypothesis that postsynaptic toxicity of Aβ is tau-dependent.37 Hyperphosphorylated tau could sensitize NMDA receptors, which makes neurons more vulnerable to Aβ toxicity. On the other hand, Aβ progressively triggers tau hyperphosphorylation, though the exact mechanism is still unknown.
It is well known that medial temporal lobe tauopathy is a feature of aging. Autopsy studies of individuals under 30 years of age have revealed that tauopathy precedes deposition.38 In vivo observations of the brains of AD animal models have revealed that these deregulated proteins can be transmitted across synapses and spread from brainstem nuclei to the entorhinal cortex through diverse connective networks.39 Price and Morris hypothesized that the turning point of the acceleration of the spread of antecedent subcortical tauopathy to the neocortex is the key that leads to further neurodegeneration.40 Evidence has shown that an increase in CSF total or phosphorylated tau, a decrease in regional metabolism on 18F-fluoro-deoxyglucose PET (FDG-PET), and brain atrophy on conventional neuroimages correlated more with neurodegeneration and functional loss.41,42 Nevertheless, amyloid deposition in brain areas correlates fairly well with a disruption of functional networks (ie, default mode network).43 The relationships, dependencies, and pathological synergies between the amyloid cascade and tauopathy are not yet clarified. The core causative pathophysiology of AD remains to be elucidated. While nearly all anti-amyloid therapies have confronted obstacles in clinical trials with symptomatic AD patients, therapeutics targeting tau must be pursued.
There are several possible therapeutic targets to alleviate tau-related toxicity, which are currently undergoing preclinical studies: inhibition of tau hyperphosphorylation (kinase inhibition or phosphatase activation), promoting misfolded tau degradation, inducing active immunization by utilizing various tau epitopes, and using antibodies targeting prefilament oligomeric species.44 Methylthioninium chloride, also called methylene blue, is one of the phenothiazines that was initially proved to prevent tau–tau aggregation in vitro.45 The ability of this drug to attenuate tauopathy-related neuronal dysfunction and improve behavior in animal tauopathy models by promoting autophagy and proteosomal degradation, preventing Aβ aggregation, and possibly protecting the mitochondria via an antioxidant effect has been established.46 A randomized, double-blind, placebo-controlled, Phase II trial of methylene blue (Rember®, TauRx Therapeutics Ltd., Singapore) that included 332 mild-to-moderate AD patients and utilized an endpoint evaluation time of 24 weeks, showed that it improved cognitive function in a moderate AD group at a dose of 60 mg per day. Further, Rember stabilized progression in both mild and moderate AD groups over 50 weeks. In the same study, a comparison was made in 18 patients between FDG-PET studies at baseline and 24 weeks later. The results revealed that patients who received Rember exhibited increased glucose metabolism in the medial temporal area, while the placebo group showed no changes in signal. A Phase III trial is recruiting mild-to-moderate AD patients to investigate the safety and efficacy of this drug (NCT01689246).47
Mitochondrial-targeted therapy and antioxidants
APP, PSEN1, and PSEN2 mutant gene carriers account for only 1%–5% of all AD patients. With the exception of the carriers of these familial causal genes and other identified genes associated with increased risks of late onset sporadic AD, the risks for AD dementia are now considered to be multi-factorial. The most prominent observation is that the risk for AD dementia increases with age. From the biochemical perspective, this effect of aging can be explained by the accumulation of structural and functional defects in the mitochondria, increases in the production of reactive oxygen species, and the acceleration of cell death.48 Decreases in mitochondrial function and synapse damage occur during the early stage of AD. Studies of hybrid cell lines, transgenic animals, and postmortem studies have determined that mitochondrial defects are closely linked to AD.49–51 It has been proposed that the activities of mitochondrial enzymes in the AD brain are defective. Decreases in cyclooxygenase (COX) activity result in increased levels of reactive oxygen species, and biogenetic stress leads to further activation of the cell apoptosis cascade. The apoptosis cascade is a vicious cycle of abnormal expression of mitochondrial DNA, beta-secretase activation, and tau hyperphosphorylation and aggregation. Mitochondrial Aβ decreases cyclooxygenase activities, induces free radicals, enhances the activity of the mitochondrial-related cell apoptosis pathway through membrane permeability transition pores, interacts with mitochondrial protein to cause morphological and functional abnormalities, and disturbs mitochondrial trafficking. Finally, insufficient adenosine triphosphate at the synapse causes synaptic degeneration and cognitive decline. Additionally, Aβ decreases superoxidase dismutase activity, which serves as a defense mechanism against oxidative stress in animal models.35,52,53 The mitochondrial cascade was proposed by Swerdlow.54 In this cascade, variable baseline mitochondrial vulnerability is determined individually by genes, and critical changes that exceed a threshold in the mitochondria due to aging lead to vicious cycles and irreversible pathological AD changes.
The targeting of dysfunctional mitochondria is a new approach that has been shown to improve the clinical courses of mild-to-moderate AD patients in a single Phase II study.55 However, this result was not confirmed in a subsequent Phase III study (CONNECTION).56 Latrepirdine has been reported to block NMDA receptors and block L-type Ca2+ channels selectively to prevent cell death.57 In addition, it inhibits acetyl cholinesterase, a-adrenergic receptors, histamine H1 and H2 receptors, and serotonin 5-HT2c, 5-HT5A, and 5-HT6 receptors with high affinity. H1 inhibition is much related to its cognition-enhancing effect. It also has significant effect on dopamine D1, D2s, and D3 receptors, Imidazole I2 receptors, and serotonin 5-HT2 and 5-HT2B receptors. It further enhances mitochondrial function and inhibits mitochondrial permeability transition pores.57–60 It has been proposed that latrepirdine decreases amyloid burden and improves cognition in animal models of AD by enhancing autophagy and postponing the neurodegenerative process.61 One meta-analysis pooled the five currently available RCTs to ascertain the effect of latrepirdine on cognitive function. This meta-analysis included a recent CONCERT trial that evaluated the efficacy of combined latrepirdine and donepezil in mild-to-moderate AD patients. This study demonstrated that latrepirdine improved cognitive scores, but only the neuropsychiatric benefit was significant.62
There are other potential strategies that employ antioxidants, including omega-3 polyunsaturated fatty acids, vitamin E, statins, G. biloba, vitamin B12, folate, and curcumin. All of these strategies have produced inconsistent results across the studies that have been completed, and no evidence from systemic reviews suggests that these antioxidants improve cognition.63–68 The efficacy of statins has been tested in a Phase III clinical trial with negative results.56 Other combination therapies are being tested; for example, a pilot RCT of the combinations of vitamin D and memantine or folate with CI has shown significant cognitive benefits (Table 2).69,70 Another Phase III RCT testing the combination of vitamin D and memantine for the treatment of AD (AD-IDEA) is ongoing.71
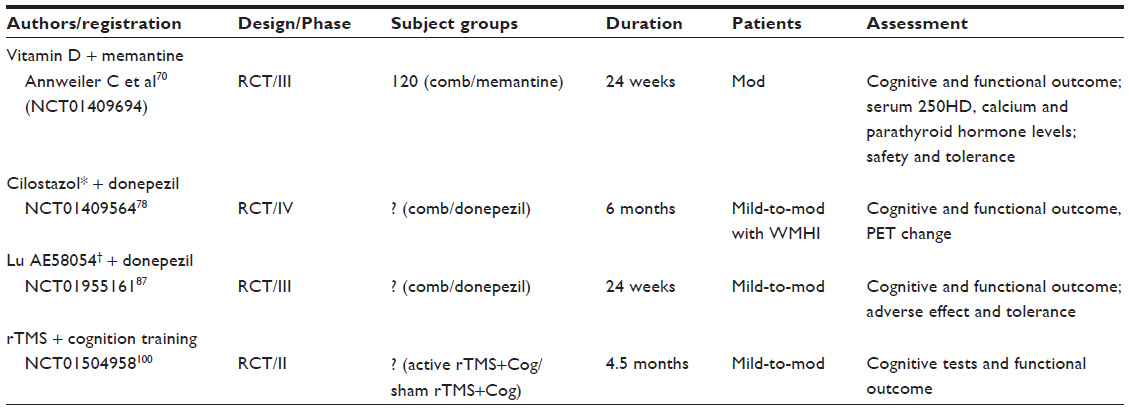 | Table 2 List of ongoing clinical trials with combination therapies |
Other potential agents and combination strategies
Phosphodiesterase inhibitors
Phosphodiesterase inhibitors regulate signal transduction pathways by elevating cyclic guanosine monophosphate (cGMP) or cyclic adenosine monophosphate (cAMP) levels, both of which ultimately promote cAMP response element-binding (CREB)-dependent gene expression and thus enhance long-term potentiation, synapse plasticity, long-term memory formation, and neuroprotection. Human neurons and vascular endothelial cells contain diverse phosphodiesterase inhibitors.72 In animal models, various phosphodiesterase inibitors have been proven to reverse memory impairments via synapse remolding or prevention of Aβ aggregation.73,74 The administration of PDE3 inhibitors has proven efficacious in counteracting Aβ-induced alterations of the expression of phosphorylated CREB.75 In AD mice, PDE5 inhibitors improve memory consolidation by decreasing the levels of hyperphosphorylated tau in brain regions involved in memory via the elevation of cGMP and the deactivation of GSK3β.76 One randomized, open-label pilot study showed that cilostazol, a PDE3 inhibitor, can increase regional cerebral blood flow in the right anterior cingulate gyrus and prevent cognitive decline in patients with AD dementia and vascular dementia.77 Another Phase IV study seeks to evaluate the augmentation of subcortical white matter hyperintensities by cilostazol in mild-to-moderate AD patients who are receiving donepezil using a cognitive task and PET neuroimaging at the expected endpoint of 6 months. This Phase IV clinical trial is currently recruiting subjects (NCT01409564).78
Thiazolidinedione
Thiazolidinediones stimulate the nuclear peroxisome proliferator-activated receptor γ and thus suppress β-secretase and promote APP ubiquitination. Pioglitazone is a member of the thiazolidinedione group of oral hypoglycemic agents.79 Although a Phase II study of this drug failed to produce results,80 pioglitazone is now being tested in a Phase III trial to evaluate its ability to postpone progression in high-risk patients with mild cognitive impairment due to AD (NCT01931566).81
Nerve growth factors
The loss of cholinergic neurons in the basal forebrain is strongly correlated with the clinical severity of AD. In in vitro and in vivo experiments, nerve growth factors (NGFs) have been demonstrated to be effective in producing neuronal protection, augmenting cholinergic function via the enhancement of choline acetyltransferase, and facilitating the differentiation of neuron progenitor cells into cholinergic neurons. Cerebrolysin® consists of low-molecular-weight peptides and free amino acids, and mimics the actions of endogenous neurotrophic factors. Cerebrolysin has demonstrated nerve growth factor (NGF)-like effects on cholinergic cells. Therefore, it has been proposed that Cerebrolysin and ChEIs have a synergistic effect. In a Phase II study, the combination of intravenous Cerebrolysin and oral donepezil significantly benefited the global outcomes of mild-to-moderate AD patients and was well tolerated.82 NGF has also been delivered via the stereotactic injection of viral vectors into the nucleus basalis of Meynert in mild-to-moderate AD patients, showing benefit on cognition and increased cerebral metabolism in a Phase I trial.83 Studies of this delivery method are ongoing (CERE-110; NCT00087789).84
5-HT6 receptor antagonists
Serotoninergic drugs represent another potential therapeutic approach. 5-HT6 receptor antagonists can enhance cholinergic release and transmission, improve learning and memory, and may augment monoaminergic neurotransmission both in vivo and in vitro.85 These antagonists were found to be well tolerated and efficacious in improving the cognition of patients with mild-to-moderate AD in a Phase II trial.86 The benefit of 5-HT6 receptor antagonists as an adjuvant therapy for patients with mild-to-moderate AD who are already taking donepezil is currently being assessed in a randomized, double-blind, placebo-controlled Phase III study (NCT01955161).87
Transcranial magnetic stimulation
Transcranial magnetic stimulation (TMS) is a noninvasive technique for the stimulation of the human brain. Based on its electromagnetic induction, TMS has been used in research to examine the brain–behavior relationship, map sensory, motor, and cognitive functions, and evaluate cortical excitability. The abilities of TMS to modulate brain activity and plasticity in many neurological and psychiatric diseases have been investigated.88 The TMS technique has been demonstrated to reduce short-latency afferent inhibition and resting motor thresholds (eg, enhance motor cortex excitability) in AD patients.89 The actual neurophysiological mechanisms that mediate the modulation of cognition by repetitive TMS (rTMS) remain unknown. Some researchers have proposed that these mechanisms are due to long-term potentiation and long-term depression effects mediated by alterations in synapse plasticity and depolarization.90 Moreover, TMS’s ability to enhance or interfere with cognition and behavior depends on the status of the underlying cortical region and the timing of different stimulating parameters. In a systematic review, Guse et al reported that high-frequency stimulation can significantly enhance cognition.91 Another study showed that high-frequency rTMS also induces local increases in cerebral blood flow.92 One early clinical study showed that rTMS can transiently enhance associative memory in elders with subjective memory complaints. Based on functional magnetic resonance imaging (fMRI) studies, this effect has been attributed to the compensatory recruitment of activation of the right prefrontal and bilateral posterior cortical regions.93 Recent preliminary studies have utilized active high-frequency rTMS in different cortical regions and assessed the corresponding cognitive improvement in AD patients and produced promising results.94–96 These studies have demonstrated that rTMS applied to the left or right dorsolateral prefrontal cortex can enhance action-naming accuracy, object-naming accuracy, and sentence comprehension. One study applied rTMS to the bilateral dorsolateral prefrontal cortex and found that this treatment produced long-term positive effects on cognition (as measured with the MMSE), global functioning, and depression scale scores that lasted as long as 3 months in patients with mild-to-moderate AD.97
Cognitive training (COG) and rehabilitation may improve the cognitive function of patients with AD dementia. However, a meta-analysis and a recent Cochrane systemic review found that the efficacy of COG is limited, although the level of evidence was inadequate.98,99 One study examined the possible synergic effects of a combination of the COG and high-frequency rTMS. The treatment protocol for this study included daily rTMS-COG sessions (5 per week) for 6 weeks that were followed by maintenance sessions (2 per week) for an additional 3 months. rTMS was applied to six different cortical regions, and the stimulation was interlaced with various cognitive trainings that corresponded to these cortical areas. In this preliminary trial, the seven patients who completed the study exhibited significant improvements in cognition as demonstrated by decreases of approximately 4 points on the Alzheimer’s Disease Assessment Scale – Cognitive subscale (ADAS-Cog) that were persistent after 4.5 months.100 Neither rTMS nor rTMS-COG produced adverse effects, and both were well tolerated. This combination therapy seems promising, and this approach will be evaluated in a double-blind, randomized, placebo-controlled Phase II trial of patients with mild-to-moderate AD in the near future (NCT01504958).101
Presymptomatic AD treatment
Recent studies have connected biomarkers with the pathophysiological process of AD by showing decreased CSF Aβ42 levels and increased brain amyloid deposition in amyloid PET. These are the earliest detectable signs in the preclinical stage of AD.41 One prospective longitudinal study on 128 participants with known genetic mutations for autosomal dominant AD found that CSF concentrations of Aβ42 appeared to decline 25 years prior to the expected onset of symptoms of dementia and that Aβ42 deposition could be detected 15 years before.102 The expected onsets of symptoms were defined by the ages at which the participants’ parents were clinically diagnosed with AD dementia. Through autopsy studies, AD patients and mutant carriers have gradually elucidated the correlated sequential dynamic changes in these biomarkers.103 Failure of disease-modifying drugs targeting Aβ at Phase III trials may indicate that our intervention is too late for the pathogenic proteinopathy.
Consequently, the reasons for initiating the treatment of presymptomatic subjects who are at the highest imminent risk of developing symptomatic AD are compelling. The Dominantly Inherited Alzheimer’s Network (DIAN) has cooperated internationally to recruit participants who are at risk of ADAD (autosomal dominant AD).104 The DIAN consists of 242 members who are the adult biological children of patients with any causative gene for ADAD. This population is composed of both symptomatic and asymptomatic mutant gene carriers and non-carriers. In a proposed Phase I trial, all of these participants will undergo a series of biomarker analyses (structural, functional, and amyloid imaging, blood and CSF amyloid and tau),103 APOE genotyping, and cognitive function assessment with a neuropsychological test battery at baseline and will be followed longitudinally (with the exception of the genetic studies). This trial will determine and describe the pathochronological changes in various biomarkers of AD with the aim of extending our knowledge of which AD-slowing treatments that target biomarkers can best predict clinically meaningful outcomes. Thus, the second prevention RCT will investigate two scheduled immunotherapies (ie, solanezumab and gentanezumab) via the measurement of downstream AD biomarkers and the examination of clinical assessments. The non-carriers will only receive placebos. Currently, the DIAN is enrolling willing individuals with the goal of reaching a sample size of 400 participants. The Alzheimer’s Prevention Initiative (API) trial seeks to enroll cognitively normal PSEN1 E280A mutant carrier at ages within 10 years of the estimated mean age of dementia onset from Antioquia, Colombia, which is currently the area with the highest incidence of early-onset ADAD.105 The trial will examine the efficacy of crenezumab therapy. The 100 mutation carriers engaged in this study will be randomly assigned to active treatment or placebo groups, and the noncarriers will be assigned to a placebo group. This study is designed to last for at least 5 years and seeks to determine the effects of crenezumab on biomarkers and the cognitive endpoints of the subjects. Another trial will focus on AD dementia prevention in cognitively normal participants aged 60–80 years with the APOE ε4 homozygous genotype.106 At this age, the participants will be approaching the estimated median age of the dementia onset. The bioactive drug to be examined in this trial has not yet been chosen. The study will be a 24-month, double-blind, RCT investigating biomarkers and the cognitive endpoints of subjects at high risk for the development of sporadic AD.
The DIAN and API studies will both focus on the efficacy of anti-amyloid treatments in people who are at risk for the development of AD due to underlying causative genes. However, the majority of AD patients worldwide will not be subjects. Therefore, the Anti-amyloid Treatment in Asymptomatic Alzheimer’s Disease (A4) trial will bridge the gap between familial AD and sporadic AD to determine the generalizability of preventive treatments administered at the preclinical stage. This study is designed to enroll 1,000 people who are positive for amyloid (as measured by PET) over the age of 70 years without dementia. The subjects will be randomly assigned to receive either solanezumab or placebo.106 Moreover, 500 PET-negative amyloid participants over the age of 70 years will participate in this trial as controls. This trial will last at least 3 years and use measures of cognition as outcomes, biomarkers, and functional networks as well. It is reasonable to speculate that this drug may have greater positive effects in participants with AD who are in the preclinical stage to prevent these patients from progressing to AD dementia.
Regarding combination therapies of AD, most clinical trials are add-on designs (eg, ChEI added on memantine vs ChEI added on placebo) thus lacking a true placebo arm. The rationale is mainly an ethical reason because most of the clinical trials on AD last for at least 6 months and it might take up to 2 years to obtain a significant difference between two treatment groups for the slowly progressive course of AD. It would be unethical if a patient with AD was put in a true placebo arm for such a long period of time, as it would expose individuals in the control group to unprotected neurodegeneration.
Psychoactive drugs are another important issue that may pose a significant confounding effect on the performance of cognitive tests. Most clinical trials used cognitive efficacy as the primary endpoint and behavioral or functional effects as secondary outcomes. Typically, psychoactive drugs must be maintained on a stable dose or adjusted within a narrow range of dose to avoid this confounding.
Conclusion
AD has multiple risk factors and is likely to have multiple pathogenic pathways. In this review, we discuss various current hypotheses of AD. Recently, the understanding of this disease has moved from proteins to organelles (eg, the mitochondria), at least from the perspective of some researchers. In contrast, in the era of the genome, some of the pathognomonic genes for AD have been identified by epidemiological studies and risk factor analyses. Briefly, AD is not a one-gene, one-protein disease and should be attributed to a network of interactions between genes, proteins, organelles, cells, neurotransmitters, and the environment. Those disease-modifying agents currently being developed typically target one hypothesis and one protein. Thus, it is clear that a single drug for the successful treatment of AD is not yet available. It is reasonable to explore multi-target strategies and combination therapies. Based on RCTs and meta-analyses, the combination of ChEI and NMDA antagonists is well tolerated and safe, and may benefit patients with moderate-to-severe AD dementia. In addition, different disease-modifying agents targeting different pathogenic pathways could be used together, for example, monoclonal antibody targeting Aβ combined with substance-inhibiting tau aggregation or BACEI added with mitochondrial protectors. A better chance of fighting against this relentless disease would then be possible.
A crucial drawback of the previous clinical trials of AD drugs that has led to their failure is that the patients receive treatment too late. Strategies targeting Aβ to prevent presymptomatic AD from progressing to clinical AD dementia and strategies targeting asymptomatic high-risk individuals at the predicted age of the clinical onset of dementia are being investigated in current clinical trials. Investigations of the correlations of biomarkers and clinical outcomes are also ongoing. These approaches may, on the one hand, test fundamental theories of AD and, on the other hand, identify the proper timing for treatment. The identification of the proper timing for treatment is the most promising approach among all of the methods currently being investigated in clinical trials, as appropriately timed treatment may halt the initiation or slow the evolution of the relentless process of deterioration in AD.
Disclosure
The authors report no conflicts of interest in this work.
References
Sosa-Ortiz AL, Acosta-Castillo I, Prince MJ. Epidemiology of dementias and Alzheimer’s disease. Arch Med Res. 2012;43(8):600–608. | |
Reitz C, Brayne C, Mayeux R. Epidemiology of Alzheimer disease. Nat Rev Neurol. 2011;7(3):137–152. | |
Russ TC, Batty GD, Hearnshaw GF, Fenton C, Starr JM. Geographical variation in dementia: systematic review with meta-analysis. Int J Epidemiol. 2012;41(4):1012–1032. | |
Kalaria RN, Maestre GE, Arizaga R, et al. Alzheimer’s disease and vascular dementia in developing countries: prevalence, management, and risk factors. Lancet Neurol. 2008;7(9):812–826. | |
Daviglus ML, Bell CC, Berrettini W, et al. National Institutes of Health State-of-the-Science Conference statement: preventing alzheimer disease and cognitive decline. Ann Intern Med. 2010;153(3):176–181. | |
Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010;362(4):329–344. | |
Morris JC. Early-stage and preclinical Alzheimer disease. Alzheimer Dis Associ Disord. 2005;19(3):163–165. | |
Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):280–292. | |
Birks J. Cholinesterase inhibitors for Alzheimer’s disease. Cochrane Database Syst Rev. 2006(1):CD005593. | |
Bordji K, Becerril-Ortega J, Buisson A. Synapses, NMDA receptor activity and neuronal Abeta production in Alzheimer’s disease. Rev Neurosci. 2011;22(3):285–294. | |
Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. 2010;11(10):682–696. | |
McShane R, Areosa Sastre A, Minakaran N. Memantine for dementia. Cochrane Database Sys Rev. 2006(2):CD003154. | |
Martina M, Comas T, Mealing GA. Selective pharmacological modulation of pyramidal neurons and interneurons in the CA1 region of the rat hippocampus. Front Pharmacol. 2013;4:24. | |
Hartmann S, Mobius HJ. Tolerability of memantine in combination with cholinesterase inhibitors in dementia therapy. Int Clin Psychopharmacol. 2003;18(2):81–85. | |
Schneider LS, Insel PS, Weiner MW; Alzheimer’s Disease Neuroimaging I. Treatment with cholinesterase inhibitors and memantine of patients in the Alzheimer’s Disease Neuroimaging Initiative. Arch Neurol. 2011;68(1):58–66. | |
Atri A, Shaughnessy LW, Locascio JJ, Growdon JH. Long-term course and effectiveness of combination therapy in Alzheimer disease. Alzheimer Dis Associ Disord. 2008;22(3):209–221. | |
Lopez OL, Becker JT, Wahed AS, et al. Long-term effects of the concomitant use of memantine with cholinesterase inhibition in Alzheimer disease. J Neurol Neurosurg Psychiatry. 2009;80(6):600–607. | |
Porsteinsson AP, Grossberg GT, Mintzer J, Olin JT; Memantine MEM-MD-12 Study Group. Memantine treatment in patients with mild to moderate Alzheimer’s disease already receiving a cholinesterase inhibitor: a randomized, double-blind, placebo-controlled trial. Curr Alzheimer Res. 2008;5(1):83–89. | |
Tariot PN, Farlow MR, Grossberg GT, et al. Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. JAMA. 2004;291(3):317–324. | |
Howard R, McShane R, Lindesay J, et al. Donepezil and memantine for moderate-to-severe Alzheimer’s disease. N Engl J Med. 2012;366(10):893–903. | |
Muayqil T, Camicioli R. Systematic review and meta-analysis of combination therapy with cholinesterase inhibitors and memantine in Alzheimer’s disease and other dementias. Dement Geriat Cogn Disord Extra. 2012;2(1):546–572. | |
Moreth J, Mavoungou C, Schindowski K. Passive anti-amyloid immunotherapy in Alzheimer’s disease: What are the most promising targets? Immun Age. 2013;10(1):18. | |
Ringman JM, Coppola G. New genes and new insights from old genes: update on Alzheimer disease. Continuum (Minneap Minn). 2013; 19(2 Dementia):358–371. | |
Mangialasche F, Solomon A, Winblad B, Mecocci P, Kivipelto M. Alzheimer’s disease: clinical trials and drug development. Lancet Neurol. 2010;9(7):702–716. | |
May PC, Dean RA, Lowe SL, et al. Robust central reduction of amyloid-beta in humans with an orally available, non-peptidic beta-secretase inhibitor. J Neurosci. 2011;31(46):16507–16516. | |
Mikulca JA, Nguyen V, Gajdosik DA, et al. Potential novel targets for Alzheimer pharmacotherapy: II. Update on secretase inhibitors and related approaches. J Clin Pharma Ther. 2014;39(1):25–37. | |
Merck Sharp and Dohme Corp. An Efficacy and Safety Trial of MK-8931 in Mild to Moderate Alzheimer’s Disease (P07738) (EPOCH). In: ClinicalTrials.gov [website on the Internet]. Bethesda, MD: US National Library of Medicine; 2012 [updated January 30, 2014]. Available from http://clinicaltrials.gov/show/NCT01739348. NLM identifier: NCT01739348. Accessed February 2, 2014. | |
Doody RS, Raman R, Farlow M, et al; Semagacestat Study Group. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N Engl J Med. 2013;369(4):341–350. | |
Fu HJ, Liu B, Frost JL, Lemere CA. Amyloid-beta immunotherapy for Alzheimer’s disease. CNS Neurol Disord Drug Targets. 2010;9(2):197–206. | |
Sperling R, Salloway S, Brooks DJ, et al. Amyloid-related imaging abnormalities in patients with Alzheimer’s disease treated with bapineuzumab: a retrospective analysis. Lancet Neurol. 2012;11(3):241–249. | |
Matthews G, Bader V. Independent analysis of solanezumab provides evidence that compound may remove amyloid from brain in Alzheimer’s disease [webpage on the Internet]. Monaco; October 29, 2012. Available from: http://www.fiercebiotech.com/press-releases/independent-analysis-solanezumab-provides-evidence-compound-may-remove-amyl. Accessed January 22, 2014. | |
Ostrowitzki S, Deptula D, Thurfjell L, et al. Mechanism of amyloid removal in patients with Alzheimer disease treated with gantenerumab. Arch Neurol. 2012;69(2):198–207. | |
Genentech. A Long-Term Safety Extension Study of Studies ABE4869g And ABE4955g in Patients With Mild To Moderate Alzheimer’s Disease Treated With Crenezumab. In: ClinicalTrials.gov [website on the Internet]. Bethesda, MD: US National Library of Medicine; 2012 [updated January 21, 2014]. Available from http://clinicaltrials.gov/ct2/show/NCT01723826?term=NCT01723826&rank=1. NLM identifier: NCT01723826. Accessed February 2, 2014. | |
LaPointe NE, Morfini G, Pigino G, et al. The amino terminus of tau inhibits kinesin-dependent axonal transport: implications for filament toxicity. J Neurosci Res. 2009;87(2):440–451. | |
Wang X, Su B, Lee HG, et al. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J Neurosci. 2009;29(28):9090–9103. | |
Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A. Tau is essential to beta-amyloid-induced neurotoxicity. Proc Nat Acad Sci U S A. 2002;99(9):6364–6369. | |
Ittner LM, Gotz J. Amyloid-beta and tau – a toxic pas de deux in Alzheimer’s disease. Nat Rev Neurosci. 2011;12(2):65–72. | |
Braak H, Del Tredici K. The pathological process underlying Alzheimer’s disease in individuals under thirty. Acta Neuropathol. 2011;121(2):171–181. | |
Liu L, Drouet V, Wu JW, et al. Trans-synaptic spread of tau pathology in vivo. PloS One. 2012;7(2):e31302. | |
Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann Neurol. 1999;45(3):358–368. | |
Jack CR Jr, Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12(2):207–216. | |
Musiek ES, Holtzman DM. Origins of Alzheimer’s disease: reconciling cerebrospinal fluid biomarker and neuropathology data regarding the temporal sequence of amyloid-beta and tau involvement. Curr Opin Neurol. 2012;25(6):715–720. | |
Pievani M, de Haan W, Wu T, Seeley WW, Frisoni GB. Functional network disruption in the degenerative dementias. Lancet Neurol. 2011;10(9):829–843. | |
Yoshiyama Y, Lee VM, Trojanowski JQ. Therapeutic strategies for tau mediated neurodegeneration. J Neurol Neurosurg Psychiatry. 2013;84(7):784–795. | |
Wischik CM, Edwards PC, Lai RY, Roth M, Harrington CR. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc Nat Acad Sci U S A. 1996;93(20):11213–11218. | |
Oz M, Lorke DE, Petroianu GA. Methylene blue and Alzheimer’s disease. Biochem Pharmacol. 2009;78(8):927–932. | |
TauRx Therapeutics Ltd. Safety and Efficacy Study Evaluating TRx0237 in Subjects With Mild to Moderate Alzheimer’s Disease. In: ClinicalTrials.gov [website on the Internet]. Bethesda, MD: US National Library of Medicine; 2012 [updated November 24, 2013]. Available from http://clinicaltrials.gov/ct2/show/NCT01689246?term=NCT01689246&rank=1. NLM identifier: NCT01689246. Accessed February 2, 2014. | |
Reddy PH, Reddy TP. Mitochondria as a therapeutic target for aging and neurodegenerative diseases. Curr Alzheimer Res. 2011;8(4):393–409. | |
Ghosh SS, Swerdlow RH, Miller SW, Sheeman B, Parker WD, Jr., Davis RE. Use of cytoplasmic hybrid cell lines for elucidating the role of mitochondrial dysfunction in Alzheimer’s disease and Parkinson’s disease. Ann NY Acad Sci. 1999;893:176–191. | |
Hauptmann S, Scherping I, Drose S, et al. Mitochondrial dysfunction: an early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol Aging. 2009;30(10):1574–1586. | |
Rice AC, Keeney PM, Algarzae NK, Ladd AC, Thomas RR, Bennett Jr JP. Mitochondrial DNA Copy Numbers in Pyramidal Neurons are Decreased and Mitochondrial Biogenesis Transcriptome Signaling is Disrupted in Alzheimer’s Disease Hippocampi. J Alzheimers Dis. Jan 21 2014 [Epub ahead of print]. | |
Swerdlow RH. Alzheimer’s disease pathologic cascades: who comes first, what drives what. Neurotox Res. 2012;22(3):182–194. | |
Eckert GP, Renner K, Eckert SH, et al. Mitochondrial dysfunction – a pharmacological target in Alzheimer’s disease. Mol Neurobiol. 2012;46(1):136–150. | |
Swerdlow RH, Burns JM, Khan SM. The Alzheimer’s disease mitochondrial cascade hypothesis. J Alzheimers Dis. 2010;20 Suppl 2:S265–S279. | |
Doody RS, Gavrilova SI, Sano M, et al. Effect of dimebon on cognition, activities of daily living, behaviour, and global function in patients with mild-to-moderate Alzheimer’s disease: a randomised, double-blind, placebo-controlled study. Lancet. 2008;372(9634):207–215. | |
Dimebon Disappoints in Phase 3 Trial. 04 Mar 2010; http://www.alzforum.org/news/research-news/dimebon-disappoints-phase-3-trial. | |
Lermontova NN, Redkozubov AE, Shevtsova EF, Serkova TP, Kireeva EG, Bachurin SO. Dimebon and tacrine inhibit neurotoxic action of beta-amyloid in culture and block L-type Ca(2+) channels. Bull Exp Biol Med. 2001;132(5):1079–1083. | |
Wu J, Li Q, Bezprozvanny I. Evaluation of Dimebon in cellular model of Huntington’s disease. Mol Neurodegener. 2008;3:15. | |
Grigorev VV, Dranyi OA, Bachurin SO. Comparative study of action mechanisms of dimebon and memantine on AMPA- and NMDA-subtypes glutamate receptors in rat cerebral neurons. Bull Exp Biol Med. 2003;136(5):474–477. | |
Bachurin SO, Shevtsova EP, Kireeva EG, Oxenkrug GF, Sablin SO. Mitochondria as a target for neurotoxins and neuroprotective agents. Ann N Y Acad Sci. 2003;993:334–344; discussion 345–339. | |
Steele JW, Gandy S. Latrepirdine (Dimebon(R)), a potential Alzheimer therapeutic, regulates autophagy and neuropathology in an Alzheimer mouse model. Autophagy. 2013;9(4):617–618. | |
Cano-Cuenca N, Solis-Garcia Del Pozo JE, Jordan J. Evidence for the efficacy of latrepirdine (Dimebon) treatment for improvement of cognitive function: a meta-analysis. J Alzheimers Dis. 2014;38:155–164. | |
McGuinness B, O’Hare J, Craig D, Bullock R, Malouf R, Passmore P. Statins for the treatment of dementia. Cochrane Database Sys Revs. 2010(8):CD007514. | |
Malouf R, Grimley Evans J. The effect of vitamin B6 on cognition. Cochrane Database Sys Rev. 2003(4):CD004393. | |
Farina N, Isaac MG, Clark AR, Rusted J, Tabet N. Vitamin E for Alzheimer’s dementia and mild cognitive impairment. Cochrane Database Sys Revs. 2012;11:CD002854. | |
Malouf R, Grimley Evans J. Folic acid with or without vitamin B12 for the prevention and treatment of healthy elderly and demented people. Cochrane Database Sys Revs. 2008(4):CD004514. | |
Janssen IM, Sturtz S, Skipka G, Zentner A, Velasco Garrido M, Busse R. Ginkgo biloba in Alzheimer’s disease: a systematic review. Wien Med Wochenschr. 2010;160(21–22):539–546. | |
Malouf R, Areosa Sastre A. Vitamin B12 for cognition. Cochrane Database Sys Revs. 2003(3):CD004326. | |
Connelly PJ, Prentice NP, Cousland G, Bonham J. A randomised double-blind placebo-controlled trial of folic acid supplementation of cholinesterase inhibitors in Alzheimer’s disease. Int J Geriat Psychiatry. 2008;23(2):155–160. | |
Annweiler C, Herrmann FR, Fantino B, Brugg B, Beauchet O. Effectiveness of the combination of memantine plus vitamin D on cognition in patients with Alzheimer disease: a pre-post pilot study. Cogn Behav Neurol. 2012;25(3):121–127. | |
Annweiler C, Fantino B, Parot-Schinkel E, Thiery S, Gautier J, Beauchet O. Alzheimer’s disease – input of vitamin D with mEmantine assay (AD-IDEA trial): study protocol for a randomized controlled trial. Trials. 2011;12:230. | |
Garcia-Osta A, Cuadrado-Tejedor M, Garcia-Barroso C, Oyarzabal J, Franco R. Phosphodiesterases as therapeutic targets for Alzheimer’s disease. ACS Chem Neurosci. 2012;3(11):832–844. | |
Hiramatsu M, Takiguchi O, Nishiyama A, Mori H. Cilostazol prevents amyloid beta peptide(25–35)-induced memory impairment and oxidative stress in mice. Br J Pharmacol. 2010;161(8):1899–1912. | |
Gong B, Vitolo OV, Trinchese F, Liu S, Shelanski M, Arancio O. Persistent improvement in synaptic and cognitive functions in an Alzheimer mouse model after rolipram treatment. J Clin Invest. 2004;114(11):1624–1634. | |
Cheng YF, Wang C, Lin HB, et al. Inhibition of phosphodiesterase-4 reverses memory deficits produced by Abeta25-35 or Abeta1-40 peptide in rats. Psychopharmacology. 2010;212(2):181–191. | |
Cuadrado-Tejedor M, Hervias I, Ricobaraza A, et al. Sildenafil restores cognitive function without affecting beta-amyloid burden in a mouse model of Alzheimer’s disease. Br J Pharmacol. 2011;164(8):2029–2041. | |
Sakurai H, Hanyu H, Sato T, et al. Effects of cilostazol on cognition and regional cerebral blood flow in patients with Alzheimer’s disease and cerebrovascular disease: a pilot study. Geriat Gerontol Int. 2013;13(1):90–97. | |
Seoul National University Hospital. Cilostazol Augmentation Study in Dementia. In: ClinicalTrials.gov [website on the Internet]. Bethesda, MD: US National Library of Medicine; 2011 [updated August 3, 2011]. Available from http://clinicaltrials.gov/ct2/show/NCT01409564?term=NCT01409564&rank=1. NLM identifier: NCT01409564. Accessed February 2, 2014. | |
Landreth G, Jiang Q, Mandrekar S, Heneka M. PPARgamma agonists as therapeutics for the treatment of Alzheimer’s disease. Neurotherapeutics. 2008;5(3):481–489. | |
Geldmacher DS, Fritsch T, McClendon MJ, Landreth G. A randomized pilot clinical trial of the safety of pioglitazone in treatment of patients with Alzheimer disease. Arch Neurol. 2011;68(1):45–50. | |
Takeda. Biomarker Qualification for Risk of Mild Cognitive Impairment (MCI) Due to Alzheimer’s Disease (AD) and Safety and Efficacy Evaluation of Pioglitazone in Delaying Its Onset. In: ClinicalTrials.gov [website on the Internet]. Bethesda, MD: US National Library of Medicine; 2013 [updated November 11, 2013]. Available from http://clinicaltrials.gov/ct2/show/NCT01931566?term=NCT01931566&rank=1. NLM identifier: NCT01931566. Accessed February 2, 2014. | |
Alvarez XA, Cacabelos R, Sampedro C, et al. Combination treatment in Alzheimer’s disease: results of a randomized, controlled trial with cerebrolysin and donepezil. Curr Alzheimer Res. 2011;8(5):583–591. | |
Mandel RJ. CERE-110, an adeno-associated virus-based gene delivery vector expressing human nerve growth factor for the treatment of Alzheimer’s disease. Curr Opin Mol Ther. 2010;12(2):240–247. | |
Ceregene. CERE-110 in Subjects With Mild to Moderate Alzheimer’s Disease. In: ClinicalTrials.gov [website on the Internet]. Bethesda, MD: US National Library of Medicine; 2004[updated June 30, 2010]. Available from http://clinicaltrials.gov/ct2/show/NCT00087789?term=NCT00087789&rank=1. NLM identifier: NCT00087789. Accessed February 2, 2014. | |
Geldenhuys WJ, Van der Schyf CJ. Serotonin 5-HT6 receptor antagonists for the treatment of Alzheimer’s disease. Curr Top Med Chem. 2008;8(12):1035–1048. | |
Maher-Edwards G, Zvartau-Hind M, Hunter AJ, et al. Double-blind, controlled phase II study of a 5-HT6 receptor antagonist, SB-742457, in Alzheimer’s disease. Curr Alzheimer Res. 2010;7(5):374–385. | |
H. Lundbeck A/S. Study of Lu AE58054 in Patients With Mild - Moderate Alzheimer’s Disease Treated With Donepezil (STARSHINE). In: ClinicalTrials.gov [website on the Internet]. Bethesda, MD: US National Library of Medicine; 2013[updated October 10, 2013]. Available from http://clinicaltrials.gov/ct2/show/NCT01955161?term=NCT01955161&rank=1. NLM identifier: NCT01955161. Accessed February 2, 2014. | |
Wagner T, Valero-Cabre A, Pascual-Leone A. Noninvasive human brain stimulation. Ann Rev Biomed Eng. 2007;9:527–565. | |
Freitas C, Mondragon-Llorca H, Pascual-Leone A. Noninvasive brain stimulation in Alzheimer’s disease: systematic review and perspectives for the future. Exp Gerontol. 2011;46(8):611–627. | |
Hoogendam JM, Ramakers GM, Di Lazzaro V. Physiology of repetitive transcranial magnetic stimulation of the human brain. Brain Stimul. 2010;3(2):95–118. | |
Guse B, Falkai P, Wobrock T. Cognitive effects of high-frequency repetitive transcranial magnetic stimulation: a systematic review. J Neural Transm. 2010;117(1):105–122. | |
Bohning DE, Shastri A, Wassermann EM, et al. BOLD-f MRI response to single-pulse transcranial magnetic stimulation (TMS). J Magnetic Resonance Image. 2000;11(6):569–574. | |
Sole-Padulles C, Bartres-Faz D, Junque C, et al. Repetitive transcranial magnetic stimulation effects on brain function and cognition among elders with memory dysfunction. A randomized sham-controlled study. Cereb Cortex. 2006;16(10):1487–1493. | |
Cotelli M, Manenti R, Cappa SF, et al. Effect of transcranial magnetic stimulation on action naming in patients with Alzheimer disease. Arch Neurol. 2006;63(11):1602–1604. | |
Cotelli M, Manenti R, Cappa SF, Zanetti O, Miniussi C. Transcranial magnetic stimulation improves naming in Alzheimer disease patients at different stages of cognitive decline. Euro J Neurol. 2008;15(12):1286–1292. | |
Cotelli M, Calabria M, Manenti R, et al. Improved language performance in Alzheimer disease following brain stimulation. J Neurol Neurosurg Psychiatry. 2011;82(7):794–797. | |
Ahmed MA, Darwish ES, Khedr EM, El Serogy YM, Ali AM. Effects of low versus high frequencies of repetitive transcranial magnetic stimulation on cognitive function and cortical excitability in Alzheimer’s dementia. J Neurol. 2012;259(1):83–92. | |
Sitzer DI, Twamley EW, Jeste DV. Cognitive training in Alzheimer’s disease: a meta-analysis of the literature. Acta Psychiatr Scand. 2006;114(2):75–90. | |
Bahar-Fuchs A, Clare L, Woods B. Cognitive training and cognitive rehabilitation for mild to moderate Alzheimer’s disease and vascular dementia. Cochrane Database Sys Rev. 2013;6:CD003260. | |
Bentwich J, Dobronevsky E, Aichenbaum S, et al. Beneficial effect of repetitive transcranial magnetic stimulation combined with cognitive training for the treatment of Alzheimer’s disease: a proof of concept study. J Neural Transm. 2011;118(3):463–471. | |
Beth Israel Deaconess Medical Center. Effects of a Combined Transcranial Magnetic Stimulation (TMS) and Cognitive Training in Alzheimer Patients. In: ClinicalTrials.gov [website on the Internet]. Bethesda, MD: US National Library of Medicine; 2011 [updated September 9, 2013]. Available from http://clinicaltrials.gov/ct2/show/NCT01504958?term=NCT01504958&rank=1. NLM identifier: NCT01504958. Accessed February 2, 2104. | |
Bateman RJ, Xiong C, Benzinger TL, et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med. 2012;367(9):795–804. | |
Bateman RJ, Aisen PS, De Strooper B, et al. Autosomal-dominant Alzheimer’s disease: a review and proposal for the prevention of Alzheimer’s disease. Alzheimers Res Ther. 2011;3(1):1. | |
Morris JC, Aisen PS, Bateman RJ, et al. Developing an international network for Alzheimer research: The Dominantly Inherited Alzheimer Network. Clin Invest. 2012;2(10):975–984. | |
Reiman EM, Langbaum JB, Fleisher AS, et al. Alzheimer’s Prevention Initiative: a plan to accelerate the evaluation of presymptomatic treatments. J Alzheimers Dis. 2011;26 Suppl 3:321–329. | |
Miller G. Alzheimer’s research. Stopping Alzheimer’s before it starts. Science. 2012;337(6096):790–792. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.