Back to Journals » Drug Design, Development and Therapy » Volume 8
Combining structure-based pharmacophore modeling, virtual screening, and in silico ADMET analysis to discover novel tetrahydro-quinoline based pyruvate kinase isozyme M2 activators with antitumor activity
Authors Chen C, Wang T, Wu F, Huang W, He G ![]() , Ouyang L, Xiang M, Peng C, Jiang Q
, Ouyang L, Xiang M, Peng C, Jiang Q
Received 23 February 2014
Accepted for publication 9 July 2014
Published 2 September 2014 Volume 2014:8 Pages 1195—1210
DOI https://doi.org/10.2147/DDDT.S62921
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Can Chen,1,2,* Ting Wang,1,3,* Fengbo Wu,1,* Wei Huang,4 Gu He,1 Liang Ouyang,1 Mingli Xiang,1 Cheng Peng,4 Qinglin Jiang1,2
1State Key Laboratory of Biotherapy and Department of Pharmacy, West China Hospital, Sichuan University, Chengdu, 2College of Pharmacy and the First Affiliated Hospital, Chengdu Medical College, Chengdu, 3Department of Cardiology, Genenal Hospital of Chengdu Military Command, Chengdu, 4State Key Laboratory Breeding Base of Systematic Research, Development and Utilization of Chinese Medicine, Chengdu University of Traditional Chinese Medicine, Chengdu, People’s Republic of China
*These authors contributed equally to this work
Abstract: Compared with normal differentiated cells, cancer cells upregulate the expression of pyruvate kinase isozyme M2 (PKM2) to support glycolytic intermediates for anabolic processes, including the synthesis of nucleic acids, amino acids, and lipids. In this study, a combination of the structure-based pharmacophore modeling and a hybrid protocol of virtual screening methods comprised of pharmacophore model-based virtual screening, docking-based virtual screening, and in silico ADMET (absorption, distribution, metabolism, excretion and toxicity) analysis were used to retrieve novel PKM2 activators from commercially available chemical databases. Tetrahydroquinoline derivatives were identified as potential scaffolds of PKM2 activators. Thus, the hybrid virtual screening approach was applied to screen the focused tetrahydroquinoline derivatives embedded in the ZINC database. Six hit compounds were selected from the final hits and experimental studies were then performed. Compound 8 displayed a potent inhibitory effect on human lung cancer cells. Following treatment with Compound 8, cell viability, apoptosis, and reactive oxygen species (ROS) production were examined in A549 cells. Finally, we evaluated the effects of Compound 8 on mice xenograft tumor models in vivo. These results may provide important information for further research on novel PKM2 activators as antitumor agents.
Keywords: pharmacophore, molecular docking, pyruvate kinase, virtual screening
Introduction
Altered metabolism is one the earliest observed differences between most adult healthy tissues and rapidly growing tissues, such as fetal tissues and tumors. Increased cell proliferation, which characterizes tumor growth, increases the need for nutrients and maintains a balance between the utilization of nutrients for energy production and anabolic processes.1 Cancer cells have a higher nutrient uptake to support the production of new cells and to satisfy the increased biosynthetic demand compared with normal tissues, which require a lower nutrient uptake and use most of the available nutrients for energy production rather than for protein, lipid, and nucleic acid syntheses.2–4 In the 1920s, Warburg reported that cancer cells have increased rates of lactate production even in the presence of oxygen.5 This effect is known as aerobic glycolysis or the Warburg effect and might be due to the reprogramming of metabolic genes to allow cancer cells to achieve the nutrient requirements for cell proliferation.5 Recently, the Warburg effect has been explored for the common clinical detection of tumors via fluorodeoxyglucose positron emission tomography. Although less efficient in ATP production, aerobic glycolysis is commonly enhanced in cancer cells through deregulation pathways, including the activation of the PI3K-AKT-mTOR pathway and the upregulation of hypoxia-inducible factor-1α (HIF-1α) and c-Myc.6–8 Several metabolic enzymes, such as hexokinase 2 (HK2), lactate dehydrogenase A, and pyruvate dehydrogenase kinase 1, are direct targets of oncogenic transcription factors, such as HIF-1α and c-Myc.9
The delicate balance required for tumor cells to endure unfavorable conditions, such as hypoxia and low glucose supply, remains unclear. Pyruvate kinase catalyzes the last step in glycolysis to convert the substrate phosphoenolpyruvate into pyruvate and produce one molecule of ATP per reaction per cycle.10 Takenaka et al discovered the alternative RNA splicing that yields the M1 and M2 forms of pyruvate kinase isozyme (PKM1 and PKM2).11 PKM2 is extensively expressed in undifferentiated embryonic tissues, and there is a switch to express PKM1 during the development of many differentiated tissues. Most cancer cell lines have been studied to determine the exclusive expression of PKM2.12 PKM2 has been found in some normal tissues that require high rates of nucleic acid synthesis. The downregulation of PKM2 activity provides a purposeful divergence from catabolic metabolism toward an anabolic state to provide the necessary resources for rapid cell proliferation.13 PKM2 exists in equilibrium between a low activity T-state and a high activity R-state. The active forms of PKM2 are tetrameric and allosterically activated by FBP through binding at the flexible loop region near the dimer–dimer interface of the tetramer.14–16 Small molecule PKM2 activators stabilize the tetramer form, affect cancer metabolism, and provide a novel antitumor therapeutic method. Recently, Thomas et al reported the first small molecule PKM2 activators, which include a series of diarylsulfoamides (Figure 1).15–17 Additional PKM2 activator chemotypes, such as tetrahydro-quinolines, have been reported since then.18,19 A relatively different structural class with pyrido[1,2-a]pyrimidin moieties was identified in a recent study through an in silico high-throughput screen (Figure 1).20 Computational research should be done to develop a possible antitumor agent design and to further explore the binding mode between the synthesized ligands and PKM2.
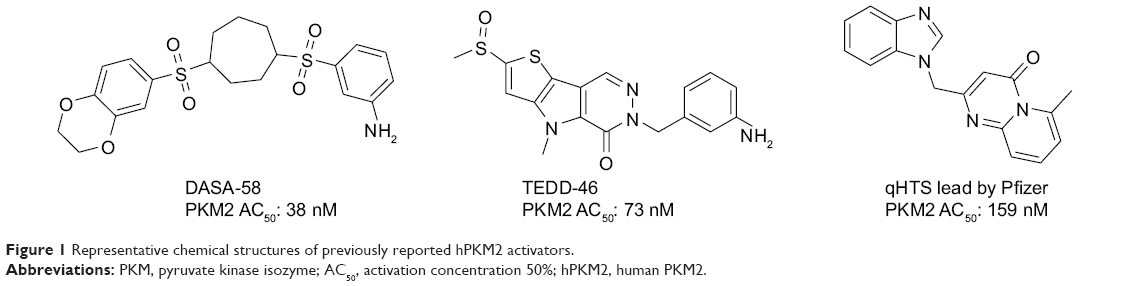 | Figure 1 Representative chemical structures of previously reported hPKM2 activators. |
Our ongoing research aimed to search specific PKM2 activators21 and to explore techniques to generate more accurate and reasonable structure-based computer-aided drug design methods.22–29 Structure-based pharmacophore (SBP) design and hybrid protocol of virtual screening can be used to detect novel tetrahydroquinoline-based lead compounds based on changes in the chemical scaffold and can provide useful references for the design and preliminary evaluation of PKM2 activators. The combination of SBP models and subsequent drug design strategies can provide insights into the required interaction patterns for PKM2 protein allosteric regulation. Moreover, the developed SBP model can be used for virtual screening to discover novel potential lead compounds. These results may provide significant information for further research on novel PKM2 activators as antitumor agents.
Materials and methods
Generation of structure-based pharmacophore models
A set of seven crystal structures of PKM2 in complex with diverse ligands (Table 1) were obtained from the Protein Data Bank (PDB).30 The crystal structures with FBP, the natural ligand of PKM2, were not used in the analyses in order to avoid the unnecessary noise likely to be introduced into the pharmacophore model.31 The generation procedures of the structure-based pharmacophore models were referenced to our previous reports.22–26 The whole process of generation and utilization of the structure-based pharmacophore models were illustrated in Figure 2 and detailed as follows. Briefly, the coordinates of all PKM2-ligand X-ray crystal structures were transformed into a common reference frame by using “Multiple Structure Alignment (Modeller)” module within Discovery Studio (DS).32 The complex-based pharmacophore generation module embedded in DS was used to generate seven individual complex-based pharmacophore models based on the previously aligned structures. All pharmacophore features identified by DS were clustered according to their interaction pattern with the PKM2 protein. The obtained model was further refined by the modification of the constraint tolerance of the spheres in accordance with the default values of pharmacophore modules embedded in DS.
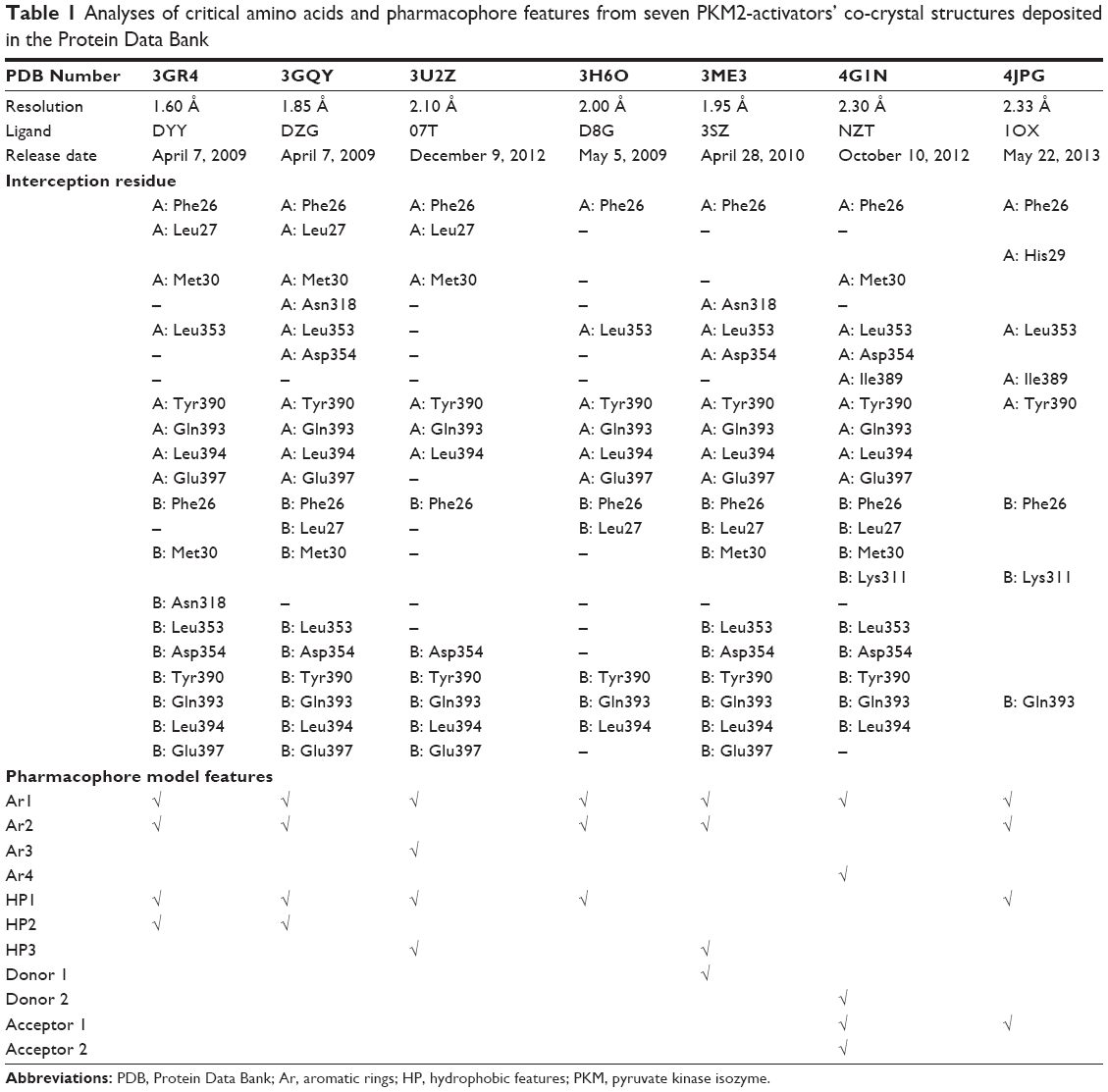 | Table 1 Analyses of critical amino acids and pharmacophore features from seven PKM2-activators’ co-crystal structures deposited in the Protein Data Bank |
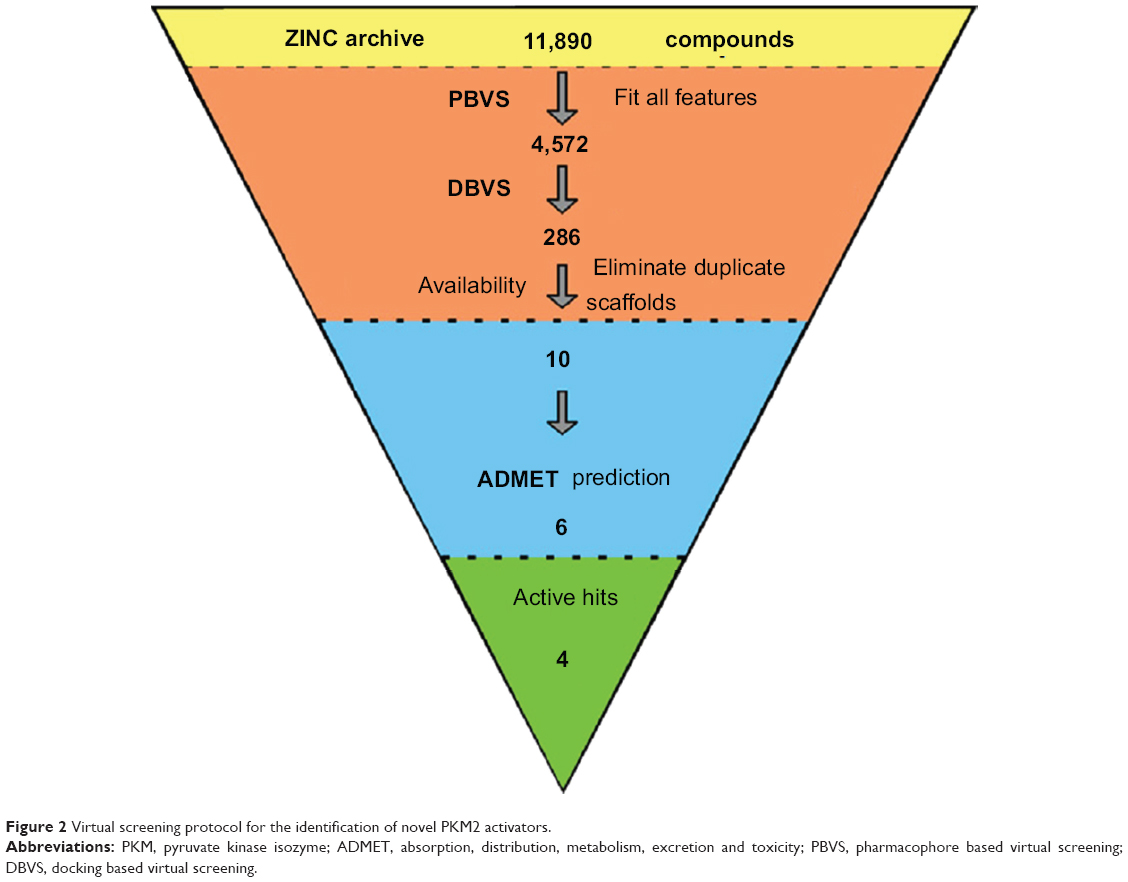 | Figure 2 Virtual screening protocol for the identification of novel PKM2 activators. |
Pharmacophore-based virtual screening
The ligand-based procedure was applied to the ZINC database supplied via the worldwide web, which contained about 35 million commercially available compounds.33,34 The ZINC database provides files corresponding to substances distributed from different vendors, and the entire database was processed by using the SMILES compounds’ codes. Among this large set of compounds the tetrahydro-quinoline scaffold was found only in approximately 12,000 compounds, which were further investigated. In the present study, structure-based pharmacophore model comprising of six chemical features was used as a query for searching tetrahydro-quinoline based chemical library. We performed all database searching experiments using the “Best” conformation generation option and the “Flexible” fitting method option in the “pharmacophore” protocols of DS software.35–37 The molecules in the tetrahydro-quinoline based chemical library which fit with all the features of the pharmacophore model were retained as hits. The criterion for screening for further validation was high fit values, which indicate good matches.
Molecular docking study
Molecular docking studies were performed to generate the bioactive binding poses of PKM2 activators in the active site of proteins by using the LibDock module in DS software. LibDock uses protein site features, referred to as hot spots, consisting of two types (polar and apolar). In the current study, the CHARMm Force Field was used for energy minimization of the ligands. The binding sphere was primarily defined as all residues of the target within 10 Å from the ligand binding site. Conformer Algorithm based on Energy Screening and Recursive build-up (CAESAR) was used for generating conformations. Then, the smart minimizer was used for in situ ligand minimization. All other parameters used were utilized default settings. To better understand the interactions between protein residues and bound ligands, the Ligplot program38 was used for generating the 2D diagrams that helped to identify the binding site residue, including amino acid residues, waters, and ligand compounds.
Fingerprint based similarity analysis
The chemical similarity search was conducted with DS using the standard protocol. The MACCS structural keys were utilized with the Tanimoto Coefficient (Tc) as the similarity metric.39
In silico ADMET analysis
ADMET refers to the absorption, distribution, metabolism, excretion, and toxicity properties of a molecule within an organism, which were predicted using the ADMET module and the TOPKAT module embedded in DS software.40 In the ADMET module, six mathematical models (aqueous solubility, blood–brain barrier penetration, CYP2D6 inhibition, hepatotoxicity, human intestinal absorption, and plasma protein binding) were used to quantitatively predict properties of a set of rules that specify ADMET characteristics of the chemical structure of the molecules. The hit compounds possessing satisfactory ADMET properties were chosen according to TOPKAT calculation. The TOPKAT predictions help in optimizing therapeutic ratios of lead compounds for further development and assessing their potential safety concerns. They will help in evaluating intermediates, metabolites, and pollutants, along with setting dose range for animal assays.36
In vitro pyruvate kinase assay
An in vitro pyruvate kinase enzymatic assay was performed using the Kinase-Glo Plus Assay kit (Promega Corporation, Fitchburg, WI, USA), using methods referred to in the literature.15–18 All reactions were carried out at room temperature for 1 hour in a 50 μL mixture containing PK assay buffer, 0.1 mM ADP (Sigma-Aldrich Co., St Louis, MO, USA), 0.5 mM PEP (Sigma-Aldrich Co.), 0.9 nM PK enzymes (BPS Bioscience, San Diego, CA, USA), and the test compounds. FBP (Sigma-Aldrich Co.) was used as a positive control for activation at concentration of 0.5 mM. At the end of enzymatic reactions, 50 μL of Glo Plus reagent was added to each well and luminescence was measured using a BioTek SynergyTM microplate reader. In the absence of the compound, the luminescence was defined as 0% activity. The percent activity in the presence of each compound was calculated according to the following equation:
% activity = [(L – Lb)/(Lt – Lb) – 1] ×100%, | (1) |
where L = the luminescence in the presence of the compound, Lb = the luminescence in the absence of PKM2, and Lt = the luminescence intensity in the absence of the compound. The values of % activity versus a series of compound concentrations were plotted using non-linear regression analysis of sigmoidal dose-response curve generated with the equation:
Y = B + (T – B)/1 + 10((logEC50–X) × hill slope), | (2) |
where Y = percent activity, B = minimum percent activity, T = maximum percent activity, EC50 = effective concentration 50%, X = logarithm of compound, and Hill Slope = slope factor or Hill coefficient.
Cell lines and animals
A549 (human lung cancer cell line) was obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA). Cells were propagated in RPMI 1,640 media containing Gibco 10% fetal bovine serum (FBS; Thermo Fisher Scientific, Waltham, MA, USA) and 1% antibiotics (penicillin and streptomycin) in 5% CO2 at 37°C. Female BALB/c mice used in this study were obtained from Beijing HFK bioscience CO., Ltd. (Beijing, People’s Republic of China).
Cell proliferation assay
Cells (3–5×103/well) were seeded in 96-well culture plates. After 24 hours of incubation, the cells were treated with vehicle (0.1% DMSO [dimethyl sulfoxide]) and various concentrations of Compound 8 for 72 hours in normoxia (21% O2) or hypoxia (1% O2), respectively. Then, 20 μL of 5 mg/mL MTT was added to each well and the plates were incubated for an additional 2 hours at 37°C. The medium was subsequently discarded, and 150 μL DMSO was added to dissolve the formazan. Absorbance was measured at 570 nM using a Spectra MAX M5 microplate spectrophotometer (Molecular Devices LLC, Sunnyvale, CA, USA) and the IC50 (inhibitory concentration 50%) values were calculated.
Colony formation assay
Briefly, A549 cells (~400–500 cells/well) were seeded in a 6-well plate. After 24 hours of incubation, the cells were treated with various concentrations of Compound 8 and then cultured for another 10 days in hypoxia (1% O2), respectively. After washing by phosphate buffer saline (PBS), colonies were fixed with 4% paraformaldehyde and stained with a 0.5% crystal violet solution. Finally, the colonies with >50 cells were counted under an inverted microscope.
Morphological analysis by EdU staining
Briefly, A549 cells were plated in a 6-well plate for 24 hours. The cells were treated with Compound 8 for another 48 hours in normoxia (21% O2) or hypoxia (1% O2) and then washed with cold PBS. Finally, cells were labeled with 30 mmol/L 5-ethenyl-20-deoxyuridine (EdU; Invitrogen; Thermo Fisher Scientific) solutions according to the manufacturer’s instructions. Then nuclear morphology of cells was examined under an inverted fluorescence microscopy (Zeiss Axiovert 200; Zeiss, Jena, Germany).
Apoptosis analysis by flow cytometry (FCM)
After 15 μM Compound 8 treatment for 48 hours in normoxia (21% O2) or hypoxia (1% O2) the cells were harvested and washed with cold PBS twice. Then the level of apoptosis was determined using the apoptosis detection kit according to manufacturer’s instructions.
Subcutaneous tumor mouse model
To investigate the anti-tumor activity of Compound 8, a subcutaneous A549 tumor mouse model was used. 100 μL of A549 cell suspension (5×105 cells) were injected subcutaneously into the right flank of mice at day 0, and when the tumors were palpable at day 4, mice were randomized into four groups (12 mice per group). At day 4, 9, and 14, mice were injected intravenously with 100 μL of NS (control), low dose Compound 8 (25 mg/kg), or high dose Compound 8 (75 mg/kg), respectively. Mice in each group were used for tumor growth inhibition assay, and tumor size was measured every three days using calipers. Tumor volume was calculated by the equation:
Vol = 0.52 × (a × b2), | (3) |
where vol is tumor volume, a is the length of the major axis, and b is the length of the minor axis.
Results and discussion
SBP models
In our previous reports, the SBP model of PKM2 was generated using six PKM2 activator co-crystal complexes, and pharmacophore-based QSAR was performed for a series of Aryl-sulfonamide derivatives. Recently, a novel series of pyrido[1,2-a]pyrimidin derivatives as PKM2 activators and the corresponding PKM2 co-crystal complexes were resolved simultaneously. In this study, the seven X-ray crystallography structures of PKM2 in complex with small molecular activators were used to construct the SBP. The results of molecular superposition based on Modeller module in the DS software were reported (see Figure S1). The detected pharmacophore features, which determine the amount of complexes in a given feature, are shown in Table 1. The eleven pharmacophore features included two hydrogen bond acceptors (A1–A2), two hydrogen bond donors (D1–D2), three hydrophobic features (HP1–HP3), and four aromatic rings (Ar1–Ar4). In these features, three (Ar1, Ar2, and HP1) were found as common in the seven complexes. The comprehensive pharmacophore map and the ligand-binding conformation at the binding site of PKM2 are shown in Figure 3A. The obtained comprehensive pharmacophore map was initially too restrictive and inappropriate for virtual screening because of the large number of chemical features, and the fit of a molecule to such a pharmacophore was still unattainable for today’s state of the art computational tools.41 A correctly reduced pharmacophore model is more preferable for practical applications.31 According to our previous results and experience, the top-ranked three features (Ar1, Ar2, and HP1) were selected from the comprehensive pharmacophore map and merged to generate the SBP (Figure 3B).
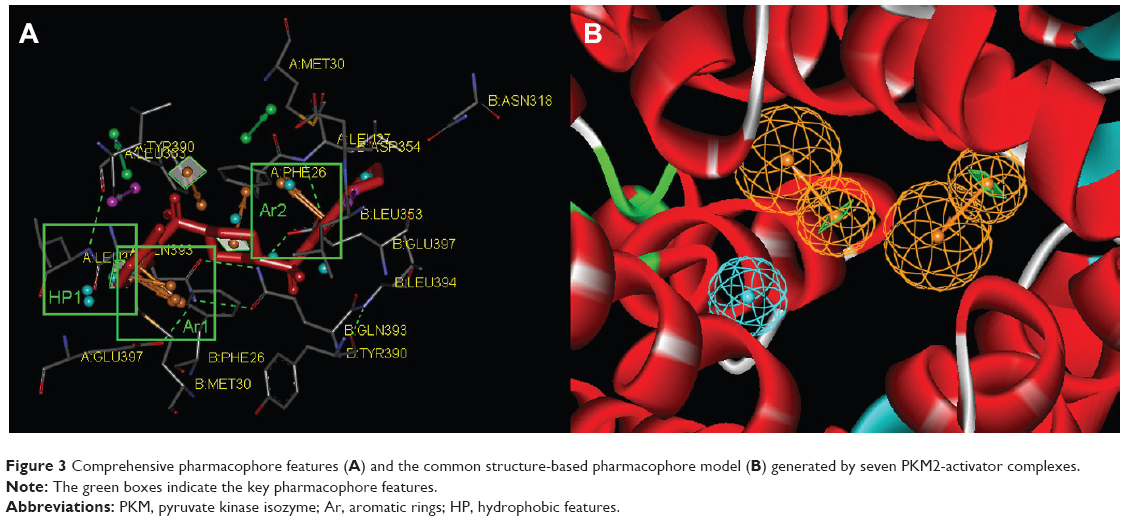 | Figure 3 Comprehensive pharmacophore features (A) and the common structure-based pharmacophore model (B) generated by seven PKM2-activator complexes. |
Virtual screening
Virtual screening of chemical libraries may facilitate the search for novel lead compounds suitable for further research. Compared with other de novo design methods, virtual screening can easily detect the desired compounds from commercial sources for biological activity assay.25 In the present study, we performed the focused tetrahydroquinoline library search by using the Best and Flexible search options. Virtual screening through SBP was performed to assess the commercially available focused tetrahydroquinoline library (11,890 compounds). A hit list of 4,572 compounds that matched the pharmacophore model was obtained. Moreover, the docking program produced several positions with different orientations within the binding site. Each position produced a different LibDock scores. The best score was considered for further studies. The compounds bearing >100 LibDock scores were considered as active new hits and were used to sample a sufficient chemical space and to increase the hit rate. A set of 286 hits satisfied the specified cut-off score and were then used for further evaluations. Compounds with similar structures were rejected for retrieving new scaffolds. Finally, a small set of 10 hits with favorable drug-like properties, high docking score, and preferred docking position were obtained (Table 2). A representative compound with the highest docking score and fitness values, and its interactions with the binding site are shown in Figure S2. The analysis of the protein–ligand complexes revealed the binding site residue, including amino acid residues, water, and ligands. A 2D diagram showing various interactions, such as hydrogen bonds, atomic charge interactions, and Pi-sigma interactions between the surrounding residues and the ligand, was also presented. The high structural diversity of the final hits demonstrated that hits with similar chemical features and novel scaffolds could be retrieved. The pairwise similarity was assessed by Tanimoto coefficients (Tc)42 between VS hits and each hit’s nearest neighbor from the active compounds in the known compounds set. The majority of these virtual hits were found to be less similar to the compounds already known, so it might be interesting to test these hits experimentally. To verify the diversity of those virtual hits, pairwise similarity calculations were performed.
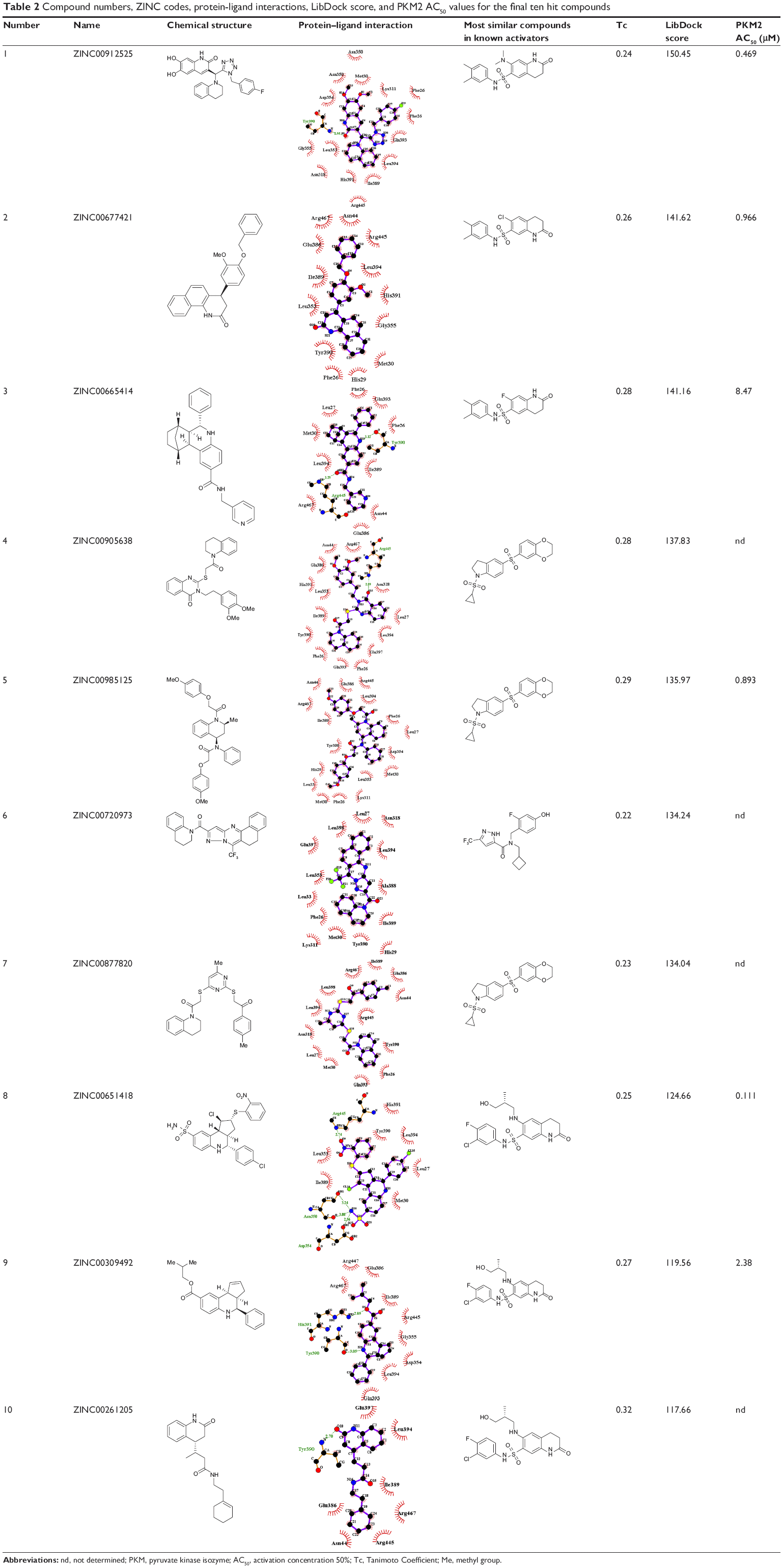 | Table 2 Compound numbers, ZINC codes, protein-ligand interactions, LibDock score, and PKM2 AC50 values for the final ten hit compounds |
In silico ADMET analysis
The docking results suggested that the ten hits had a good docking score (Table 2). The docking studies were promising; thus, the chemical descriptors for the pharmacokinetic properties were calculated. Pharmacokinetic properties, including aqueous solubility, blood–brain barrier penetration, CYP2D6 binding, hepatotoxicity, intestinal absorption, and plasma–protein binding, were determined. The calculation was designated as the first step toward analyzing the novel chemical entities and checking the failure of lead candidates. The failed lead candidates might cause toxicity or might be metabolized by the body into an inactive form or in a form without the ability to cross membranes. The results of this analysis are presented in Figure 4. The biplot figure shows the two analogous 95% and 99% confidence ellipses for the blood–brain barrier penetration and human intestinal absorption models, respectively. The detailed results of pharmacokinetic properties and toxicity analysis for the six selected compounds are shown in Table 3. The solubility and partition coefficient were calculated to determine pharmacokinetic properties, whereas the toxicity, mutagenicity, tumorigenicity, irritation effect, and risk of reproductive effect were predicted. The logP was predicted to determine the hydrophilicity of both compounds. A high logP was associated with poor absorption or permeation; thus, the value should be less than 5. The results suggested that the six compounds were within this limit, and Compound 6 had the best logP. Likewise, skin irritancy was not observed in all compounds except in Compound 9, which showed mild irritancy. Other properties such as rat oral LD50 (lethal dose 50%), Ames mutagenicity, developmental toxicity potential, rat inhalational LC50 (lethal concentration 50%), rat maximum tolerated dose, fathead minnow LC50, and aerobic biodegradability are also provided in Table 4. These results further demonstrate the need to discover new PKM2 activators for cancer treatments.
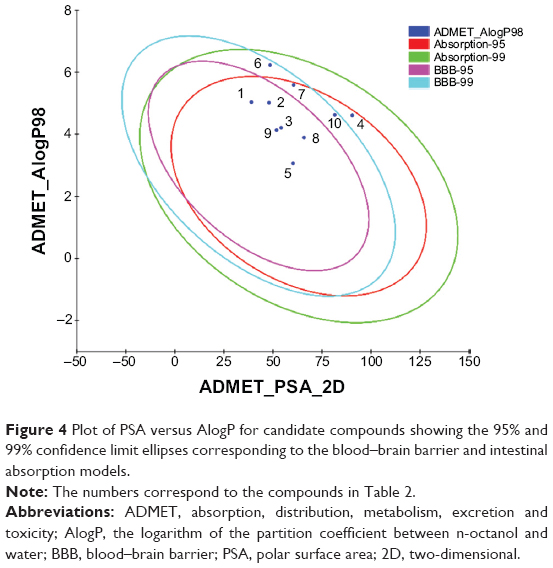 | Figure 4 Plot of PSA versus AlogP for candidate compounds showing the 95% and 99% confidence limit ellipses corresponding to the blood–brain barrier and intestinal absorption models. |
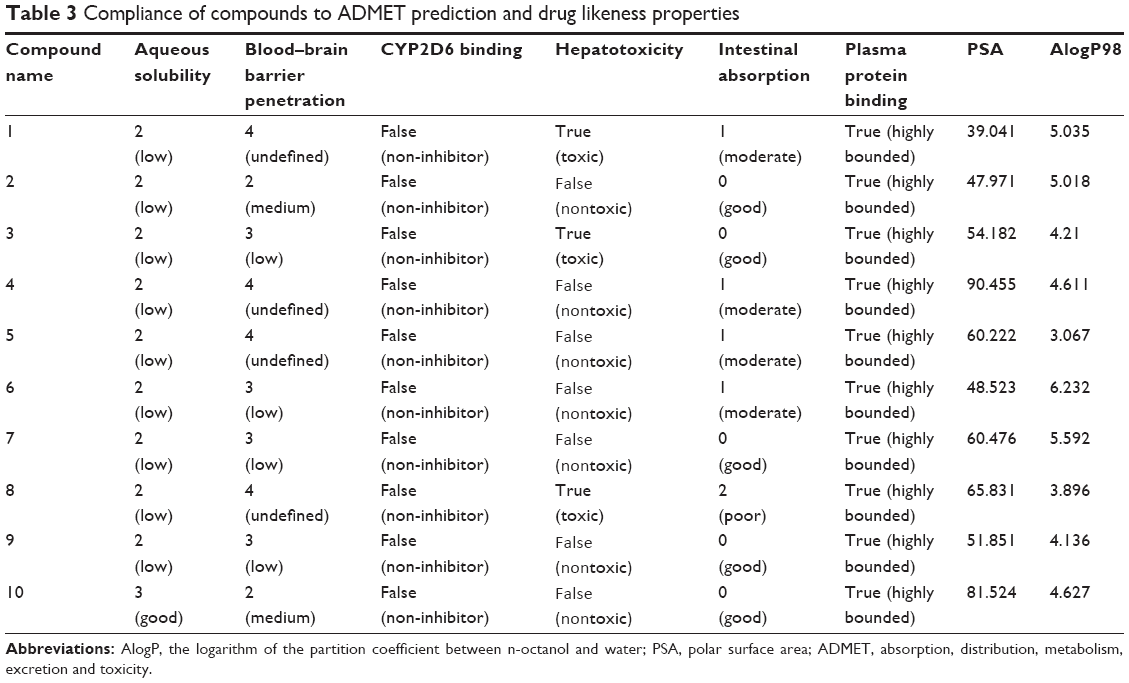 | Table 3 Compliance of compounds to ADMET prediction and drug likeness properties |
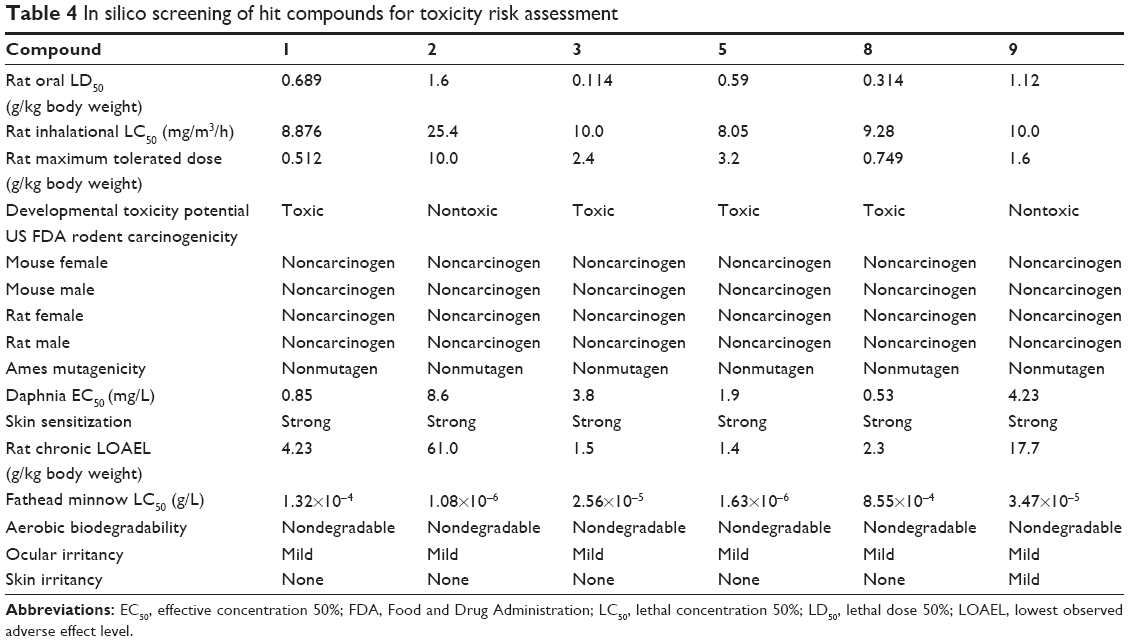 | Table 4 In silico screening of hit compounds for toxicity risk assessment |
In vitro pyruvate kinase assay
An in vitro pyruvate kinase enzymatic assay was performed. Hit Compound 8, which was identified from the virtual screen, possessed the highest AC50 (activation concentration 50%) value versus 0.111 μM PKM2. However, Compounds 3 and 9 can activate PKM2 with AC50 values of 8.47 and 2.38 μM, respectively. Compounds 3, 8, and 9 had a similar scaffold, and the main difference between these compounds was the fusion ring at the 3,4 position of the tetrahydroquinoline scaffold. The other three compounds activated the PKM2 with AC50 values from 0.469 μM to 0.966 μM. This finding suggests that the tetrahydroquinoline core structure is a potent moiety of hit compounds to interact with PKM2. Thus, modifying the core structure might result in determining more potent PKM2 activators.
The anti-proliferative effects of Compound 8
We investigated the proliferation inhibition caused by Compound 8 treatment on A549 human lung cancer cells. After exposure to Compound 8 for 72 hours, only a few decreases in the cellular viability were observed. Moreover, exposure of A549 cells to hypoxia, or hypoxia combined with Compound 8, for 24 hours, 48 hours and 72 hours, respectively, caused a marked decrease in the viability (Figure 5A). These results demonstrated that Compound 8 inhibited the proliferation of A549 cells in a hypoxic environment in a time- and concentration-dependent manner.
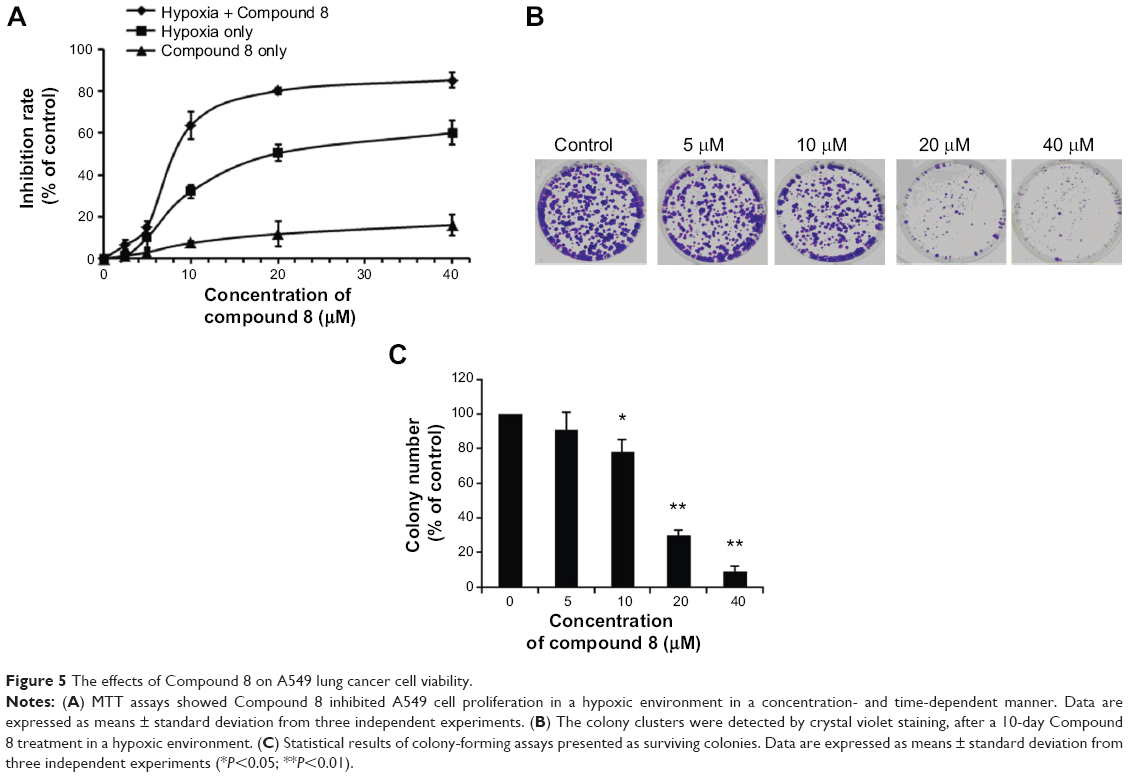 | Figure 5 The effects of Compound 8 on A549 lung cancer cell viability. |
Moreover, to further determine whether Compound 8 could inhibit the proliferation of A549 cells, we conducted clonogenic assay under hypoxia. A clonogenic assay clearly showed that Compound 8 treatment significantly decreased the colony size and number of colonies in a concentration-dependent manner (Figure 5B, C). The results from the clonogenic assay were in accordance with the MTT assay. Taken together, these results indicated that A549 could inhibit the proliferation of A549 cells in a hypoxic environment.
Morphological changes and the proliferation capacity of A549 cells treated by Compound 8 were investigated through EdU staining (Figure 6). In the A549 cells, the combined treatment of compound 8 and hypoxia resulting high bright-blue fluorescent (DAPI stained) and low red fluorescent (EdU stained), and the difference between each groups suggested that Compound 8 inhibited the proliferation of A549 cells in a hypoxic environment.
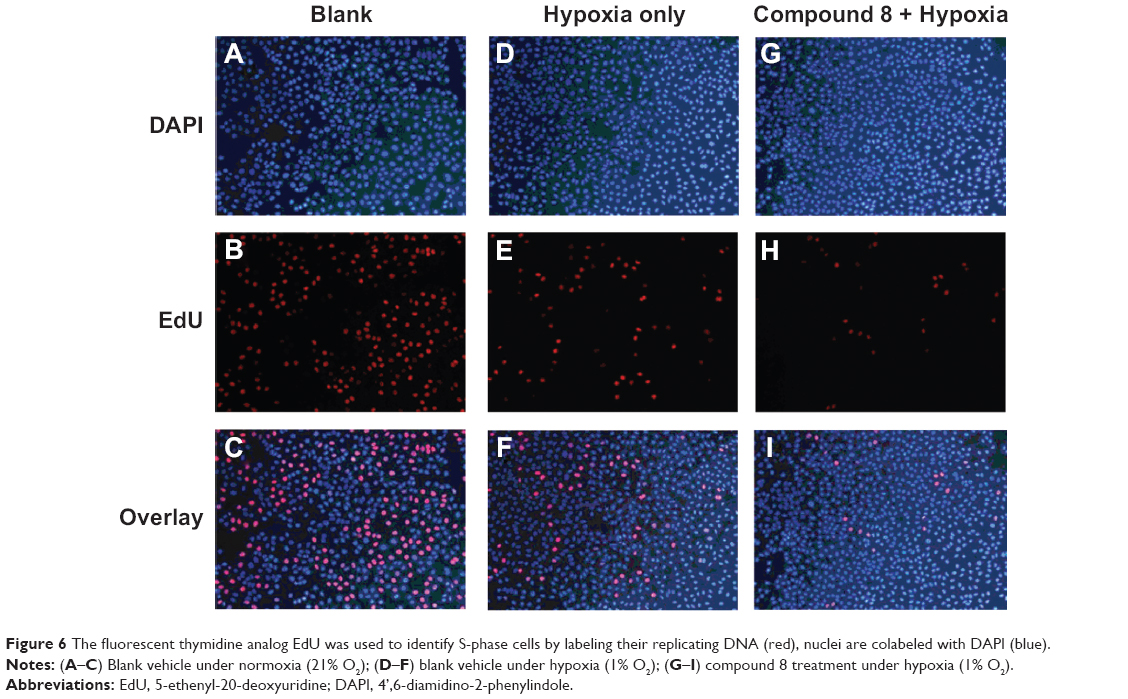 | Figure 6 The fluorescent thymidine analog EdU was used to identify S-phase cells by labeling their replicating DNA (red), nuclei are colabeled with DAPI (blue). |
Inducing apoptosis by Compound 8 under hypoxia
To investigate the underlying mechanism of anti-proliferative activity, DCFH-DA (2′,7′-Dichlorofluorescin diacetate) was applied to detect the change of intracellular reactive oxygen species (ROS) level after Compound 8 treatment. As compared with control cells, Compound 8 combined with hypoxia caused a marked increase of ROS levels in A549 cells, enhancing ROS to almost triple these levels (Figure 7A).
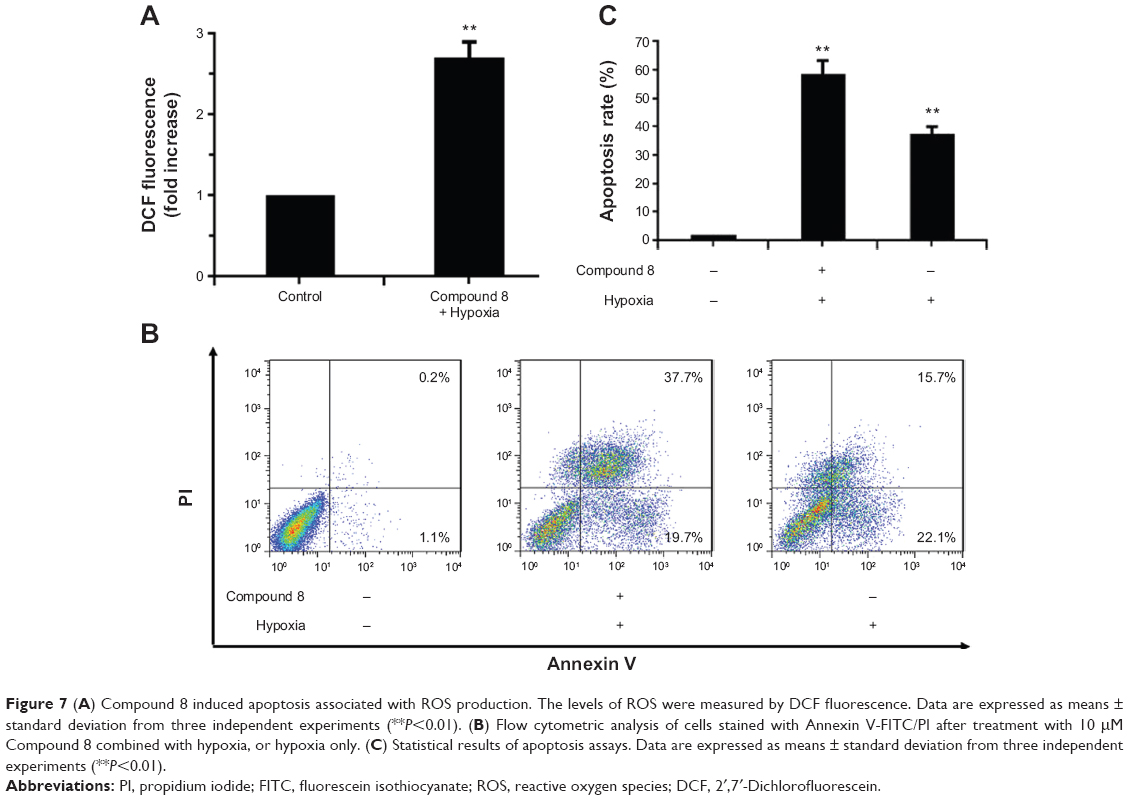 | Figure 7 (A) Compound 8 induced apoptosis associated with ROS production. The levels of ROS were measured by DCF fluorescence. Data are expressed as means ± standard deviation from three independent experiments (**P<0.01). (B) Flow cytometric analysis of cells stained with Annexin V-FITC/PI after treatment with 10 μM Compound 8 combined with hypoxia, or hypoxia only. (C) Statistical results of apoptosis assays. Data are expressed as means ± standard deviation from three independent experiments (**P<0.01). |
To determine whether hypoxia played an important role in the induction of apoptosis following Compound 8 treatment in A549 cells, an Annexin V assay was conducted. After a 48 hours treatment of hypoxia with or without the combination of Compound 8 (20 μM), the level of apoptosis was determined by FCM. The apoptosis rates in A549 cells of control and Compound 8 combined hypoxia treatment were 1.3% and 57.4%, respectively (Figure 7B, C). The data indicated that treatment with Compound 8 significantly increased the rate of apoptosis of A549 cells in a hypoxic environment compared with the controls, which were consistent with the results of Hoechst staining. Moreover, single treatment with hypoxia could significantly reduce the rate of apoptosis (Figure 7B, C). Taken together, these data indicated that the hypoxic environment is critical for the observed Compound 8-induced apoptosis.
In vivo antitumor effect in subcutaneous tumor model
To test the antitumor effect of Compound 8 in vivo, female BALB/c mice were inoculated subcutaneously with A549 cells. Mice then received an intraperitoneal injection (ip) of Compound 8 (25 mg/kg and 75 mg/kg, respectively) or vehicle every 3 days for 21 days. The results showed that the tumor growth of the Compound 8 treatment group become significant slowed after 6 days treatment. Compound 8 substantially inhibited tumor growth in a dose-dependent manner compared with the control after 21 days treatment (Figure 8A). Moreover, Compound 8 treatment was well tolerated and did not cause significant loss in body weight compared with the vehicle group (Figure 8B). The above results demonstrated that Compound 8 could efficiently suppress tumor growth in vivo.
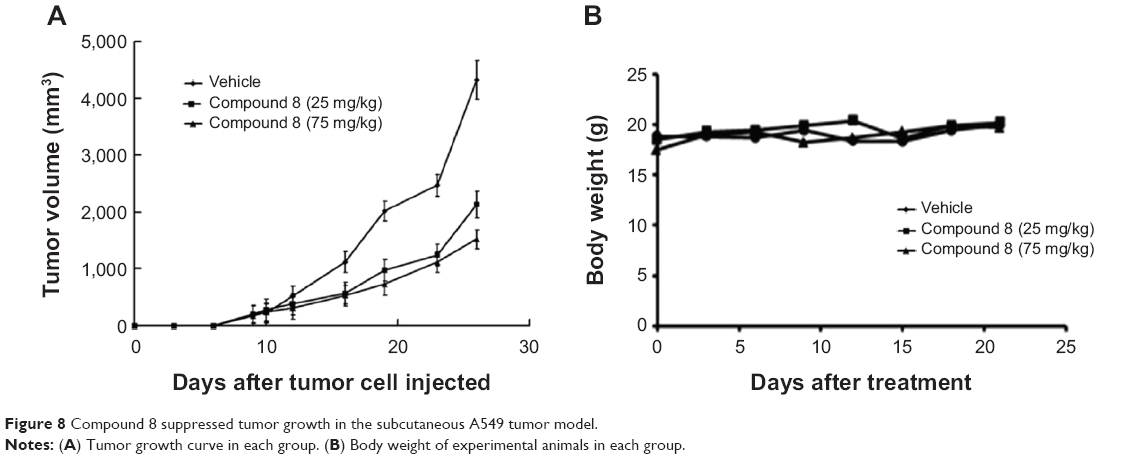 | Figure 8 Compound 8 suppressed tumor growth in the subcutaneous A549 tumor model. |
Conclusion
In this study, we attempted to obtain PKM2 activators with novel scaffolds via the combined pharmacophore-based virtual screening with molecular docking and in silico ADMET analysis. The SBP model was established using the seven co-crystal complexes of PKM2 and activators. The model was further used as a 3D search query to screen the focused tetrahydroquinoline chemical library. About 286 compounds were selected based on the fit value of more than 4,486 for molecular docking study, which was performed to improve the reliability and accuracy of virtual screening. The 10 preferable hits based on the best docking score were selected for in silico ADMET analysis. The results of in silico ADMET study showed that the six hit compounds possessed good ADMET properties. These compounds were shifted to in vitro pyruvate kinase assay. The results showed that four out of the six hit compounds could activate the recombined PKM2 with AC50 values lower than 1.0 μM. Hence, we successfully identified several potent PKM2 activators with novel scaffolds via a combination of various in silico techniques. These compounds had simultaneous binding ability with the binding site as well as in vitro pyruvate kinase activation. Compound 8 inhibited the proliferation of A549 human lung cancer cells and induced cellular apoptosis via the ROS-mediated-mitochondrial pathway. Moreover, Compound 8 could significantly inhibit A549 xenograft tumor growth in vivo. Therefore, our studies provided strong evidence that PKM2 activators may be a novel lead compound as lung cancer drug candidate.
Acknowledgments
We gratefully acknowledge the support from the National Natural Science Foundation of China (No. 30901837, 81001357 and 81273471), the Fund of the Health Department of Sichuan Province (No.120487), and the Open Research Fund of State Key Laboratory Breeding Base of Systematic Research, Development and Utilization of Chinese Medicine.
Disclosure
The authors report no conflicts of interest in this work.
References
Christofk HR, Vander Heiden MG, Wu N, et al. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature. 2008;452:181–186. | ||
Chaneton B, Gottlieb E. Rocking cell metabolism: revised functions of the key glycolytic regulator PKM2 in cancer. Trends Biochem Sci. 2012;37:309–316. | ||
Christofk HR, Vander Heiden MG, Harris MH, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230–233. | ||
Altenberg B, Greulich KO. Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics. 2004;84:1014–1020. | ||
Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. | ||
Hsu PP, Sabatini DM. Cancer cell metabolism: Warburg and beyond. Cell. 2008;134:703–707. | ||
Tamada M, Suematsu M, Saya H. Pyruvate Kinase M2: Multiple Faces for Conferring Benefits on Cancer Cells. Clin Cancer Res. 2012;18:5554–5561. | ||
Ye J, Mancuso A, Tong X. et al. Pyruvate kinase M2 promotes de novo serine synthesis to sustain mTORC1 activity and cell proliferation. Proc Natl Acad Sci. 2012;109:6904–6909. | ||
Lv L, Li D, Zhao D, et al. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol Cell. 2011;42:719–730. | ||
Dombrauckas JD, Santarsiero BD, Mesecar AD. Structural basis for tumor pyruvate kinase M2 allosteric regulation and catalysis. Biochemistry. 2005;44:9417–9429. | ||
Takenaka M, Noguchi T, Sadahiro S, et al. Isolation and characterization of the human pyruvate kinase M gene. Eur J Biochem. 1991;198:101–106. | ||
Cheong H, Lu C, Lindsten T, et al. Therapeutic targets in cancer cell metabolism and autophagy. Nat Biotech. 2012;30:1–8. | ||
Yamada K, Noguchi T. Regulation of pyruvate kinase M gene expression. Biochem Biophys Res Commun. 1999;256:257–262. | ||
DeBerardinis RJ, Sayed N, Ditsworth D, et al. Brick by brick: metabolism and tumor cell growth. Curr Opin Genet Dev. 2008;18:54–61. | ||
Anastasiou D, Yu Y, Israelsen WJ, et al. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat Chem Biol. 2012;8:839–847. | ||
Boxer MB, Jiang JK, Vander Heiden MG, et al. Evaluation of substituted N,N’-diarylsulfonamides as activators of the tumor cell specific M2 isoform of pyruvate kinase. J Med Chem. 2010;53:1048–1055. | ||
Jiang JK, Boxer MB, Vander Heiden MG, et al. Evaluation of thieno[3,2-b]pyrrole[3,2-d]pyridazinones as activators of the tumor cell specific M2 isoform of pyruvate kinase. Bioorg Med Chem Lett. 2010;20:3387–3393. | ||
Walsh MJ, Brimacombe KR, Veith H, et al. 2-Oxo-N-aryl-1,2,3,4-tetrahydroquinoline-6-sulfonamides as activators of the tumor cell specific M2 isoform of pyruvate kinase. Bioorg Med Chem Lett. 2011;21:6322–6327. | ||
Xu Y, Liu XH, Saunders M, et al. Discovery of 3-(trifluoromethyl)-1H-pyrazole-5-carboxamide activators of the M2 isoform of pyruvate kinase (PKM2). Bioorg Med Chem Lett. 2014;24:515–519. | ||
Guo C, Linton A, Jalaie M, et al. Discovery of 2-((1H-benzo[d]imidazol-1-yl)methyl)-4H-pyrido[1,2-a]pyrimidin-4-ones as novel PKM2 activators. Bioorg Med Chem Lett. 2013;23:3358–3363. | ||
Chen ZJ, Jiang QL, He G, et al. Multicomplex-Based Pharmacophore and QSAR of Aryl-Sulfamides as Pyruvate Kinase M2 Activators. Acta Phys Chim Sin. 2013;29:1793–1803. | ||
He G, Qiu M, Li R, et al. Multicomplex-based pharmacophore-guided 3D-QSAR studies of N-substituted 2′-(aminoaryl)benzothiazoles as Aurora-A inhibitors. Chem Biol Drug Des. 2012;79:960–971. | ||
Ouyang L, He G, Huang W, et al. Combined Structure-Based Pharmacophore and 3D-QSAR Studies on Phenylalanine Series Compounds as TPH1 Inhibitors. Int J Mol Sci. 2012;13:5348–5363. | ||
Li B, Zhou R, He G, et al. Molecular Docking, QSAR and Molecular Dynamics Simulation on Spiro-oxindoles as MDM2 Inhibitors. Acta Chim Sin. 2013;71:1396–1403. | ||
Wu F, Xu T, He G, et al. Discovery of novel focal adhesion kinase inhibitors using a hybrid protocol of virtual screening approach based on multicomplex-based pharmacophore and molecular docking. Int J Mol Sci. 2012;13:15668–15678. | ||
Peng F, Peng A, Luo Y, et al. Combined structure-based pharmacophore, virtual screening, and 3D-QSAR studies of structural diverse dehydrosqualene synthase inhibitors. Med Chem Res. 2013;22:4858–4866. | ||
Liu C, He G, Jiang Q, et al. Novel Hybrid Virtual Screening Protocol Based on Molecular Docking and Structure-Based Pharmacophore for Discovery of Methionyl-tRNA Synthetase Inhibitors as Antibacterial Agents. Int J Mol Sci. 2013;14:14225–14239. | ||
Ouyang L, Huang Y, Zhao Y, et al. Preparation, antibacterial evaluation and preliminary structure-activity relationship (SAR) study of benzothiazol- and benzoxazol-2-amine derivatives. Bioorg Med Chem Lett. 2012;22:3044–3049. | ||
Zhong H, Huang W, He G, et al. Molecular dynamics simulation of tryptophan hydroxylase-1: binding modes and free energy analysis to phenylalanine derivative inhibitors. Int J Mol Sci. 2013;14:9947–9962. | ||
Berman HM, Westbrook J, Feng Z, et al. The protein data bank. Nucl Acid Res. 2000;28:235–242. | ||
Zou J, Xie HZ, Yang SY, et al. Towards more accurate pharmacophore modeling: multicomplex-based comprehensive pharmacophore map and most frequent-feature pharmacophore model of CDK2. J Mol Graph Model. 2008;27:430–438. | ||
Discovery Studio, version 3.5; Accelrys Inc.: San Diego, CA, USA, 2012. | ||
Irwin JJ, Sterling T, Mysinger MM, et al. ZINC: A Free Tool to Discover Chemistry for Biology. J Chem Inf Model. 2012;52:1757–1768. | ||
Irwin JJ, Shoichet BK. ZINC-a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45:177–182. | ||
Xiang ML, Lei K, Fan WJ, et al. In silico identification of EGFR-T790M inhibitors with novel scaffolds: start with extraction of common features. Drug Des Devel Ther. 2013;7:789–839. | ||
Alam S, Khan F. QSAR and docking studies on xanthone derivatives for anticancer activity targeting DNA topoisomerase IIα. Drug Des Devel Ther. 2014;8:183–195. | ||
Yadav DK, Meena A, Srivastava A, et al. Development of QSAR model for immunomodulatory activity of natural coumarinolignoids. Drug Des Devel Ther. 2010;4:173–186. | ||
Laskowski RA, Swindells MB. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51:2778–2786. | ||
Luo M, Wang XS, Roth BL, Golbraikh A, Tropsha A. Application of Quantitative Structure–Activity Relationship Models of 5-HT1A Receptor Binding to Virtual Screening Identifies Novel and Potent 5-HT1A Ligands. J Chem Inf Model. 2014;54:634–647. | ||
Enslein K, Blake BW, Borgstedt HH. Prediction of probability of carcinogenicity for a set of ongoing NTP bioassays. Mutagenesis. 1990;5:305–306. | ||
Chaudhaery SS, Roy KK, Saxena AK. Consensus Superiority of the Pharmacophore-Based Alignment, Over Maximum Common Substructure (MCS): 3D-QSAR Studies on Carbamates as Acetylcholinesterase Inhibitors. J Chem Inf Model. 2009;49:1590–1601. | ||
Roth BL, Baner K, Westkaemper R, et al. Salvinorin A: a potent naturally occurring nonnitrogenous kappa opioid selective agonist. Proc Natl Acad Sci U S A. 2002;99:11934–11939. |
Supplementary materials
 | Figure S1 Superimposition of seven PKM2-activator complexes. |
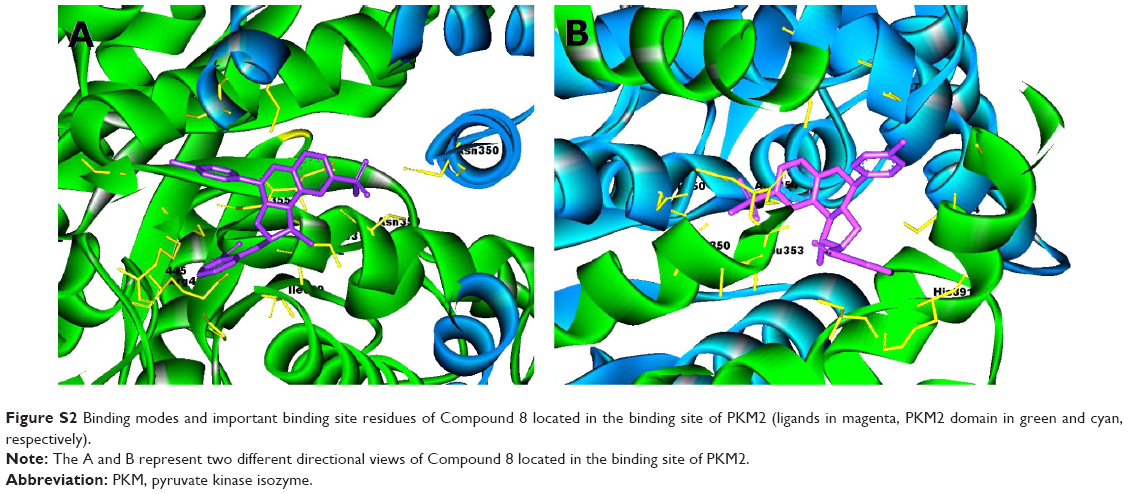 | Figure S2 Binding modes and important binding site residues of Compound 8 located in the binding site of PKM2 (ligands in magenta, PKM2 domain in green and cyan, respectively). |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.