Back to Journals » Drug Design, Development and Therapy » Volume 8
Combined isosorbide dinitrate and ibuprofen as a novel therapy for muscular dystrophies: evidence from Phase I studies in healthy volunteers
Authors Cossu MV, Cattaneo D, Fucile S, Pellegrino P, Baldelli S, Cozzi V, Capetti A, Clementi E
Received 6 December 2013
Accepted for publication 15 January 2014
Published 2 May 2014 Volume 2014:8 Pages 411—419
DOI https://doi.org/10.2147/DDDT.S58803
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Maria Vittoria Cossu,1 Dario Cattaneo,1 Serena Fucile,1 Paolo Pellegrino,1 Sara Baldelli,1 Valeria Cozzi,1 Amedeo Capetti,2 Emilio Clementi1,3
1Unit of Clinical Pharmacology, Consiglio Nazionale delle Ricerche Institute of Neuroscience, Department of Biomedical and Clinical Sciences, University Hospital “Luigi Sacco”, Università di Milano, Milan, Italy; 2Unit of Infectious Diseases, University Hospital “Luigi Sacco”, Milan, Italy; 3Scientific Institute, IRCCS Eugenio Medea, Bosisio Parini, Lecco, Italy
Abstract: We designed two Phase I studies that assessed healthy volunteers in order to evaluate the safety and to optimize the dosing of the combination of the drugs isosorbide dinitrate, a nitric oxide donor, and ibuprofen, a nonsteroidal antiinflammatory drug. We designed these studies with the aim of designing a Phase II trial to evaluate the drugs’ efficacy in patients affected by Duchenne muscular dystrophy. For the first trial, ISOFEN1, a single-dose, randomized-sequence, open-label, active control, three-treatment cross-over study, was aimed at comparing the pharmacokinetics of ibuprofen 200 mg and isosorbide dinitrate 20 mg when given alone and concomitantly. The pharmacokinetics of ibuprofen given alone versus ibuprofen given concomitantly with isosorbide dinitrate were similar, as documented by the lack of statistically significant differences in the main drug’s pharmacokinetic parameters (time to maximal concentration [Tmax], maximal concentration [Cmax], area under the curve [AUC]0–t, and AUC0–∞). Similarly, we found that the coadministration of ibuprofen did not significantly affect the pharmacokinetics of isosorbide dinitrate. No issues of safety were detected. The second trial, ISOFEN2, was a single-site, dose titration study that was designed to select the maximum tolerated dose for isosorbide dinitrate when coadministered with ibuprofen. Eighteen out of the 19 enrolled subjects tolerated the treatment well, and they completed the study at the highest dose of isosorbide dinitrate applied (80 mg/day). One subject voluntarily decided to reduce the dose of isosorbide dinitrate from 80 mg to 60 mg. The treatment-related adverse events recorded during the study were, for the large majority, episodes of headache that remitted spontaneously in 0.5–1 hour – a known side effect of isosorbide dinitrate. These studies demonstrate that the combination of isosorbide dinitrate and ibuprofen does not lead to pharmacokinetic interactions between the two drugs; they also demonstrate that the combination of isosorbide dinitrate and ibuprofen has optimal tolerability and safety profiles that are similar to those previously reported for isosorbide dinitrate and ibuprofen given alone.
Keywords: coadministration, pharmacokinetic profile, adverse events, ibuprofen, isosorbide dinitrate
Introduction
Duchenne muscular dystrophy (DMD) is a severe generic disease that causes progressive wasting of the skeletal muscle accompanied by severe local inflammation, and this invariably leads to death in early adulthood.1 At present, there is no resolutive therapy, and the current therapeutic protocols are still based on the administration of corticosteroids, which provide some delays in the progression of the disease, but they are also associated with severe side effects.1 Therapies that substitute corticosteroids, or that at least may act as corticosteroid-sparing drugs, are thus being actively pursued.2,3
While genetic and stem cell approaches appear to offer significant steps toward a resolute therapy, they are expensive and are still not readily available, as the results of the last clinical trials indicate.4–6 In addition, they target only specific subsets of patients (for example, about 13% in the case of exon 51 skipping).7 “Classical” pharmacological approaches, while not aiming to cure the disease, do appear to be attractive options and are of significant value for a variety of reasons. Drugs may slow disease progression and they may have broad applicability, as they are not selective for a specific gene mutation. In addition, they may be used in combined approaches with gene/cell therapies to enhance the overall therapeutic efficacy, and they may possibly be substituted for corticosteroids, or they may at least lead to corticosteroid-sparing regimens.
Possible successful pharmacological strategies are those based on either individual drugs or drug combinations that address the downstream pathogenic effects of dystrophin deficiency, which ultimately leads to muscle loss.
Two key pathological events in DMD appear to be particularly susceptible to pharmacological interventions: local inflammation, which is recognized as playing a role in fiber destruction and in the progression of the disease,8 hence the beneficial effect of corticosteroids; and reductions in the generation of nitric oxide (NO), which leads to muscle fatigue and a reduced bioenergetics capacity, and which plays a role in the neurogenic defects associated with the disease.9–11 Indeed, endogenous NO generation protects muscle from damage during contractile activity12–14 and it enhances its repair.15–19
Studies in the mdx and α-sarcoglycan null mouse models of muscular dystrophy showed that exposure to a combination of NO donation and nonsteroidal antiinflammatory activities has long-term therapeutic effects on disease progression and muscle function.20–26 Among the possibilities we tested, the combination of isosorbide dinitrate (ISO), a NO donating molecule, and ibuprofen, a nonsteroidal antiinflammatory drug – which are licensed for use in humans – was found to be of significant therapeutic effect.25,26 In particular, this therapy reduced muscle inflammation, necrosis, and fibrosis, and ameliorated mouse performance in the free wheel and treadmill tests that measure voluntary movement and resistance to exercise, respectively.25,26 Both the morphostructural and functional beneficial effects of the therapy persisted over the 12 months of experimental treatment.
The safety and tolerability of the coadministration of ISO and ibuprofen in dystrophic patients were confirmed in an open-label, single-centre pilot study.27
These data open novel perspectives for the use of NO-based approaches in DMD and support the design of a new clinical trial to evaluate their efficacy.
Based on the above premises, we decided to design two Phase I studies conducted with healthy volunteers to optimize dosing of the combination of ISO and ibuprofen to be subsequently used in efficacy trials. The first trial (ISOFEN1) was designed to define, through a pharmacokinetic approach, the drug combination’s bioavailability in healthy volunteers when compared with that of the single active principles.
The second trial (ISOFEN2) was designed to define the maximum tolerated dose of ISO in combination with ibuprofen, and to assess the safety of the combined treatment in terms of adverse events (AEs) and variations of blood pressure (BP) during the trial.
Materials and methods
ISOFEN – study planning
ISOFEN1 was a single-dose, randomized-sequence, open-label, active control, three-treatment cross-over study aimed at comparing the pharmacokinetics of ibuprofen (200 mg) and ISO (20 mg) when given alone and concomitantly in healthy volunteers (EudraCT number 2011-002881-19). ISOFEN2 was a single-site, dose titration study carried out in healthy male volunteers, which was designed to select the maximum tolerated dose for ISO when coadministered with ibuprofen (EudraCT number 2012-001193-29).
Both trials were approved by the Competent Authorities (Istituto Superiore di Sanità, Rome, Italy and the Ethical Committee of the Luigi Sacco University Hospital, Milan, Italy) and performed at the Center for Phase I Clinical Studies of the Luigi Sacco University Hospital in accordance with the current revision of the Declaration of Helsinki. All of the subjects were recruited from the Healthy-Volunteer’s Database of the Luigi Sacco University Hospital, and each gave their written informed consent to participate in the study.
ISOFEN1 – study design
For the screening visit (V1), subjects were admitted into the hospital at 7 am in a fasting condition, and they had been previously instructed not to eat or drink anything (except water, which was allowed up to 1 hour before the study initiation) for approximately 8 hours prior to each scheduled visit. They were then allowed to eat standard meals 4 hours after drug administration during the pharmacokinetic visit.
At visit two (V2), subjects satisfying all the inclusion/exclusion criteria were randomized to one of the three treatment sequences. Trial treatment medications were ibuprofen (200 mg) or ISO (20 mg), or ibuprofen (200 mg) plus ISO (20 mg) given concomitantly. All treatments were given as single oral doses. Other treatments were given in visits three and four (V3 and V4), according to the sequence randomization scheme with a wash out of at least 3 days between the periods, according to the reported half-lives of the study drugs.28,29 Detailed pharmacokinetic evaluations were performed during each of the treatment visits (V2–V4), together with the assessment of vital signs, pulse rate, and BP.
ISOFEN1 – tolerability assessments
AEs were evaluated before the administration of each dose of the study drug and at the time of each blood draw, based on direct observation and spontaneous reports. Vital signs (BP and heart rate) were measured 1 hour and 3 hours after the study drug intake. All undesirable signs, symptoms, or medical conditions occurring after the start of the study were recorded individually on case report forms by the investigators, separately. BP was measured with an aneroid sphygmomanometer in the right arm, and heart rate was measured manually on the radial artery.
ISOFEN1 – pharmacokinetic analysis
During each pharmacokinetic assessment, subjects had free access to water. Standard meals were provided 4 hours after drug administration. A venous cannula was placed in a forearm vein, and a 4 mL blood sample was drawn into lithium heparin tubes. Blood samples were drawn immediately before the study drug intake (time 0), and at 30 minutes, 60 minutes, 90 minutes, 120 minutes, 150 minutes, 180 minutes, 210 minutes, 240 minutes, and 5 hours, 6 hours, 8 hours, 10 hours, and 12 hours after drug administration. Blood samples were centrifuged within 15 minutes of collection at 2,500× g at 4°C for 10 minutes. Plasma samples were then stored at –20°C in stopped polypropylene tubes until analysis.
Plasma ibuprofen concentrations were assessed using a high-performance liquid chromatography method with diode array detection. The method is linear from 0.5–100 mg/L, with imprecision and inaccuracy below 10%. Plasma isosorbide concentrations were quantified by high-performance liquid chromatography coupled with tandem mass spectrometry. The method is linear over the concentration range of 1.0–1,000 ng/mL, with imprecision and inaccuracy below 15%. Both chromatographic methods have been previously developed and validated30–33 according to the United States Food and Drug Administration’s guidelines for bioanalytical method validation.34
The following drug pharmacokinetic parameters were determined for each subject: maximal concentration (Cmax) and its time (Tmax), as read directly from the data; the area under the curve (AUC)0–12, as determined by the trapezoidal rule; elimination rate (kelimin), as calculated by least squares regression analysis of terminal log–linear portions of the plasma concentration–time profile; t1/2 as Ln2/kelimin; and AUC0–∞ as the sum of AUC0–12 and the extrapolated area given by the quotient of the last measured concentration and kelimin.
ISOFEN2 – study design
The study consisted of a screening visit (V1), a treatment phase (V2–V5), and a final visit (V6). At V2, the subjects satisfying the inclusion/exclusion criteria started treatment with the study drugs at the lowest dose, which was 200 mg once daily (qd) of ibuprofen and 20 mg qd of ISO for 1 week. ISO was titrated up to 80 mg qd (with 20 mg increment per visit) in subjects with diastolic BP (DBP) >60 mmHg; the dose of ibuprofen was maintained at 200 mg qd throughout the duration of the study. In subjects with DBP <60 mmHg or symptomatic hypotension, the dose of ISO was maintained at the current tolerated dosage and up-titrated at the following visit or maintained as such, depending on the DBP values (Figure 1).
 | Figure 1 Flow chart of the ISOFEN2 study. |
The subjects arrived at the Center for Phase I clinical studies in the morning between 7 am and 10 am after fasting for 8 hours overnight, and before the administration of the study drugs. The subjects were confined in the center for at least 3 hours following drug intake. In order to limit potential ibuprofen-related gastrointestinal complications, the proton pump inhibitor pantoprazole 20 mg/day was added throughout the study period. A complete physical examination with the assessment of vital signs, pulse rate, and BP, with particular attention to the DBP, was performed during each visit, before the choice of the new treatment dosage according to the protocol.
At the end of the visit, the drugs were dispensed in blisters containing the number of tablets to be used until the next visit, and instructions were provided to take the study drugs and the rescue therapy every day in the morning.
ISOFEN2 – tolerability assessments
The subjects were continuously monitored by the investigators for AEs. These were evaluated before the administration of each dose of the study drug, based on direct observation and spontaneous reports. Vital signs (BP and heart rate) were measured at predose, 30 minutes, 1 hour, and 3 hours post-drug intake, and all undesirable signs, symptoms, or medical conditions occurring after the start of the study were recorded individually on case report forms by the investigators, separately. We considered the maximum tolerated ISO dose as the one that, when given with ibuprofen at 200 mg qd, was well tolerated by at least 85% of the enrolled subjects.
Statistical analysis
Demographic characteristics were described according to qualitative or quantitative data. The assessment of safety was based primarily on the frequency of AEs, laboratory abnormalities, and serious AEs. In agreement with the European Medicines Agency’s (EMA’s) Guideline on the Investigation of Bioequivalence,35 the assessment of bioequivalence was based upon 90% confidence intervals (CIs) for the ratio of the population geometric means (drug alone/combined treatment). The pharmacokinetic parameters under consideration (ie, AUC0–t, AUC0–∞, Cmax, Tmax) were analyzed using analysis of variance (ANOVA), and data were transformed prior to analysis using a logarithmic transformation. A CI for the difference between treatments on the log-transformed scale was obtained from the ANOVA model. The CI was then back-transformed to obtain the desired CI for the ratio on the original scale. For these parameters, the 90% CI for the ratio of the test and reference products should be contained within the acceptance interval of 0.80–1.25.
Results
ISOFEN1 – subjects’ characteristics
Twelve healthy Caucasian male volunteers were screened, randomly assigned to one of the three sequence treatments, and included in the pharmacokinetics study. Baseline characteristics of the enrolled subjects are given in Table 1. All subjects were Caucasians and the large majority were nonsmokers. The mean age was 23±2 years, and the mean body mass index was 22.62±2.58 kg/m2.
 | Table 1 Demographic and baseline characteristics of the subjects of the ISOFEN1 study |
ISOFEN1 – tolerability
All enrolled subjects completed the study. Safety assessments, including hematochemical analyses, vital signs, and cardiac and respiratory evaluations showed no differences between the beginning and the end of the study (Table 2). A total of 15 AEs were reported during the study. Of these, 13 were assessed as treatment-related. In particular, all subjects experienced episodes of mild headache, remitting spontaneously in 0.5–1 hours – a known side effect of ISO. No AEs led to discontinuation, and no AEs that were defined as serious occurred during the study.
 | Table 2 Variations of the laboratory analyses and vital signs between the screening and the final visit (ISOFEN1) |
ISOFEN1 – pharmacokinetics
The mean plasma drug concentration–time profiles after the administration of ibuprofen (200 mg) given alone or in combination with ISO are given in Figure 2A. The temporal pharmacokinetic profiles of ibuprofen alone or in combination with ISO were comparable. The lack of effect of ISO on ibuprofen exposure was also confirmed by the nonsignificant difference in the ibuprofen AUCt–∞ (5.9±8.4 mg*hour/L versus 6.0±6.6 mg*hour/L; P=0.08). The same result was also confirmed by the other main pharmacokinetic parameters (Cmax, Tmax, t1/2; Table 3).
 | Figure 2 Pharmacokinetic profiles of isosorbide and ibuprofen after the administration of ISO, ibuprofen, or their combination. |
 | Table 3 Main isosorbide and ibuprofen pharmacokinetic parameters (following the administration of ISO and ibuprofen alone or in combination) |
The mean plasma drug concentration–time profile after the administration of ISO, given alone or in combination with ibuprofen, are shown in Figure 2B. No significant differences were observed in the Tmax and Cmax when comparing ISO given alone versus ISO given concomitantly with ibuprofen. A significant difference was found in the AUC0–t (1,155±201 ng*hour/mL versus 1,270±234 ng*hour/mL; P=0.019), but not confirmed when the AUC0–∞ was considered (1,478±266 ng*hour/mL versus 1,533±249 ng*hour/mL; P=0.241; Table 3).
The lack of a mutual pharmacokinetic drug-to-drug interaction between ibuprofen and ISO was also confirmed when applying the more stringent criteria dictated by the EMA’s Guideline on the Investigation of Bioequivalence.35 These guidelines require that the 90% CIs for the ratio of the test and reference products are contained within the acceptance interval of 0.80–1.25.35 Indeed, the ratios of the AUC0–∞ between each drug when administered alone versus when combined with the other were 0.99 (90% CI: 0.92–1.06) and 1.06 (90% CI: 0.98–1.14) for ibuprofen and ISO, respectively.
ISOFEN2 – subjects’ characteristics
Nineteen healthy Caucasian male volunteers participated in the ISOFEN2 study. Baseline characteristics of the enrolled subjects are given in Table 4. All subjects were Caucasian and the majority were nonsmokers. The mean age was 24±5 years, and the mean body mass index was 22±2 kg/m2.
 | Table 4 Demographic and baseline characteristics of the subjects of the ISOFEN2 study |
ISOFEN2 – tolerability
All the enrolled subjects completed the study, being treated with the study drugs for a median of 27 days (range: 24–28 days). Safety assessments, including hematochemical analyses, vital signs, and cardiac and respiratory evaluations, showed no differences between the beginning and the end of the study (Table 5).
 | Table 5 Variations of the laboratory analyses and vital signs between the screening visit and the final visit (ISOFEN2) |
Eighteen out of the 19 enrolled subjects tolerated the treatment well and completed the study, reaching the highest dose of ISO (80 mg/day). Only one subject voluntarily decided to reduce the dose of ISO from 80 mg to 60 mg. Overall, 31 treatment-related AEs were recorded during the study, the large majority being episodes of headache (Table 6). Such episodes did not persist, and they gradually decreased in intensity during the first week of the treatment period. Only one subject reported persistent, albeit tolerable, headaches up to the end of the study. The time course of BP variations determined at the different visits is graphically represented in Figures 3 and 4. No significant differences were observed in the diastolic or systolic BP measured at 0 minutes, 30 minutes, 60 minutes, 120 minutes, and 180 minutes following drug administration between the different ISO dosages.
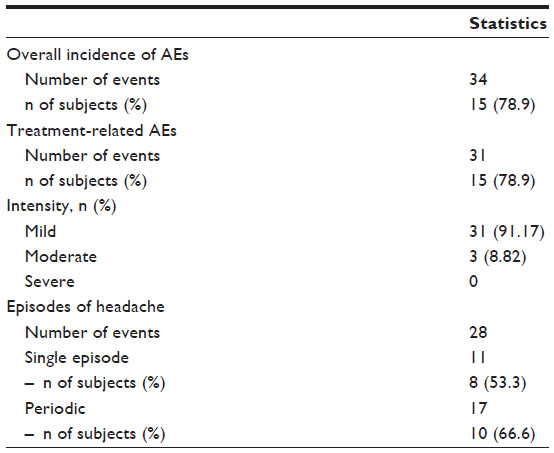 | Table 6 Overview of adverse events recorded during the ISOFEN2 trial |
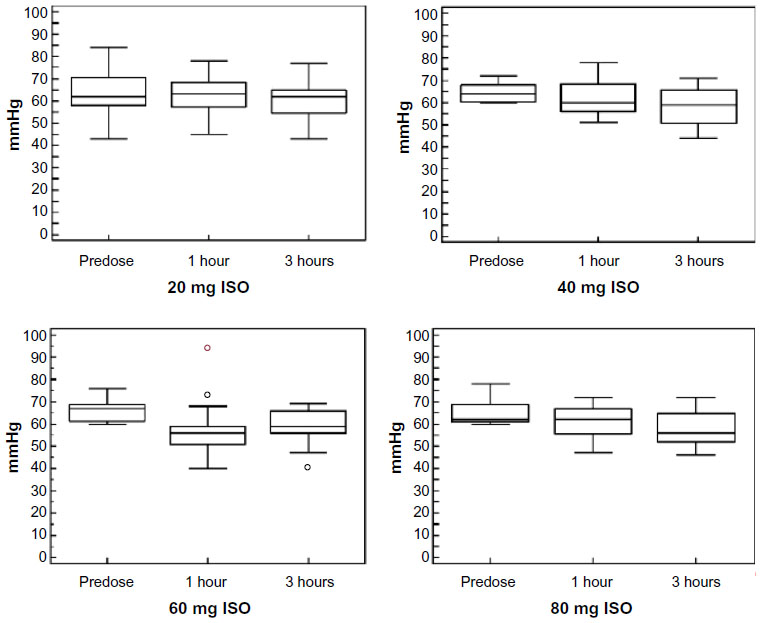 | Figure 3 Time course of diastolic blood pressure variations during the study. |
 | Figure 4 Time course of systolic blood pressure variations during the study. |
Discussion
In the ISOFEN1 study, we demonstrated that the pharmacokinetics of ibuprofen given alone versus ibuprofen given concomitantly with ISO were similar, as documented by the lack of statistically significant differences in the main drug pharmacokinetic parameters (Tmax, Cmax, AUC0–t, and AUC0–∞). Similarly, we found that the coadministration of ibuprofen did not significantly affect the pharmacokinetics of ISO. Indeed, only a minor increase in the AUC0–12 of ISO was observed when ibuprofen was added concomitantly. This increase, which accounted for less than 10% of the daily drug exposure, was considered not clinically relevant. Interestingly, no significant differences were observed when comparing ISO alone versus the drug combination for the other pharmacokinetic parameters. The lack of a mutual pharmacokinetic drug-to-drug interaction between ibuprofen and ISO was confirmed also when applying the more stringent criteria dictated by the EMA’s Guideline on the Investigation of Bioequivalence.35 For the application of these guidelines, we considered the AUC of each drug given alone as the “reference” and the AUC of each drug given in combination as the “test” product. In every instance, we found that the ratio of the AUCs from zero to infinity were within the window of acceptance of 0.80%–1.25%.
Concerning the safety profile of the drugs given alone or combined, no serious AEs occurred during the study, with no meaningful effects on vital signs, electrocardiograms, or laboratory parameters. The majority of the AEs recorded during the study were scored as treatment-related. In particular, all subjects experienced mild-to-moderate headache, remitting spontaneously in 0.5–1 hour – a known side effect of ISO. The results of the ISOFEN1 study confirmed the optimal tolerability of the combined therapy with ibuprofen and ISO previously documented in a pilot study in dystrophic patients.23 In mouse models of DMD, the effect of ISO plus ibuprofen is endowed with a synergistic therapeutic effect.25,26 The similar pharmacokinetic profiles of ibuprofen and ISO, when given alone or combined, allow us to exclude that the synergism between these two drugs is eventually related to an unmasked pharmacokinetic drug-to-drug interaction.
The main result of the ISOFEN2 study was the definition of the maximum tolerated dose of ISO in combination with ibuprofen, and the further assessment of the safety of the combined treatment in terms of AEs and variations in BP. We found that the combination of ibuprofen (200 mg) with ISO (up to 80 mg/day) was well tolerated by nearly 95% of the subjects, thus largely exceeding the 85% cutoff fixed as the primary end point. No serious AEs were recorded during the study with respect to vital signs, electrocardiograms, or laboratory parameters. The majority of the AEs were judged as treatment-related and scored as mild to moderate in intensity. Also, in this study, headache was experienced at the highest concentrations of ISO, and it was moderate and remitted spontaneously within 1 hour following drug intake. All AEs resolved spontaneously without any concomitant treatment.
In a previous study conducted in patients, the combination of ibuprofen (200 mg twice a day) in combination with ISO at 20 mg/day was found to be well tolerated.27 The results of the ISOFEN2 study also confirmed the optimal tolerability of the combined therapy when ISO is given at higher concentrations, up to 80 mg/day. Accordingly, it can be reasonably speculated that DMD patients with a poor response to the combined treatment at low doses (20 mg twice a day) of ISO may eventually benefit from progressive up-titration of drug doses up to 80 mg/day, provided that strict monitoring of the patient’s BP is performed.
The main results of the two studies (ie, the safety and absence of pharmacokinetic interactions) indicate that from a therapeutic perspective, the combination of ISO and ibuprofen is highly manageable. This information can be used to design novel studies conducted with the aim of evaluating the safety and efficacy of this combined treatment in patients affected by DMD. Several acquired and inherited neuromuscular disorders are associated with a loss of functional NO signaling and inflammation, including muscular dystrophies other than the Duchenne form and myopathies.36 The high clinical manageability of the combination of ISO and ibuprofen encourages preclinical studies to verify its beneficial effect in these other pathologies.
Acknowledgments
The financial support of Parent Project Italia Onlus is gratefully acknowledged. We wish to thank Filippo Buccella of the same charity for his advice and support during this study.
Disclosure
The authors report no conflicts of interest in this work.
References
Dubowitz V. Prednisone for Duchenne muscular dystrophy. Lancet Neurol. 2005;4(5):264. | |
Ruegg UT. Pharmacological prospects in the treatment of Duchenne muscular dystrophy. Curr Opin Neurol. 2013;26(5):577–584 | |
Jarmin S, Kymalainen H, Popplewell L, Dickson G. New developments in the use of gene therapy to treat Duchenne muscular dystrophy. Expert Opin Biol Ther. 2014;14(2):209–230. | |
Pichavant C, Aartsma-Rus A, Clemens PR, et al. Current status of pharmaceutical and genetic therapeutic approaches to treat DMD. Mol Ther. 2011;19(5):830–840. | |
Liu N, Williams AH, Maxeiner JM, et al. microRNA-206 promotes skeletal muscle regeneration and delays progression of Duchenne muscular dystrophy in mice. J Clin Invest. 2012;122(6):2054–2065. | |
Wang L, Zhou L, Jiang P, et al. Loss of miR-29 in myoblasts contributes to dystrophic muscle pathogenesis. Mol Ther. 2012;20(6):1222–1233. | |
Malik V, Rodino-Klapac LR, Viollet L, et al. Gentamicin-induced readthrough of stop codons in Duchenne muscular dystrophy. Ann Neurol. 2010;67(6):771–780. | |
Wehling M, Spencer MJ, Tidball JG. A nitric oxide synthase transgene ameliorates muscular dystrophy in mdx mice. J Cell Biol. 2001;155(1):123–131. | |
Rando TA. Role of nitric oxide in the pathogenesis of muscular dystrophies: a “two hit” hypothesis of the cause of muscle necrosis. Microsc Res Tech. 2001;55(4):223–235. | |
Kobayashi YM, Rader EP, Crawford RW, et al. Sarcolemma-localized nNOS is required to maintain activity after mild exercise. Nature. 2008;456(7221):511–515. | |
Lai Y, Thomas GD, Yue Y, et al. Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J Clin Invest. 2009;119(3):624–635. | |
Stamler JS, Meissner G. Physiology of nitric oxide in skeletal muscle. Physiol Rev. 2001;81(1):209–237. | |
Nisoli E, Clementi E, Paolucci C, et al. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science. 2003;299(5608):896–899. | |
Wehling-Henricks M, Oltmann M, Rinaldi C, Myung KH, Tidball JG. Loss of positive allosteric interactions between neuronal nitric oxide synthase and phosphofructokinase contributes to defects in glycolysis and increased fatigability in muscular dystrophy. Hum Mol Genet. 2009;18(18):3439–3451. | |
Anderson JE. A role for nitric oxide in muscle repair: nitric oxide-mediated activation of muscle satellite cells. Mol Biol Cell. 2000;11(5):1859–1874. | |
Cazzato D, Assi E, Moscheni C, et al. Nitric oxide drives embryonic myogenesis in chicken through the upregulation of myogenic differentiation factors. Exp Cell Res. 2014;320(2):269–280. | |
Colussi C, Mozzetta C, Gurtner A, et al. HDAC2 blockade by nitric oxide and histone deacetylase inhibitors reveals a common target in Duchenne muscular dystrophy treatment. Proc Natl Acad Sci U S A. 2008;105(49):19183–19187. | |
Percival JM, Anderson KN, Gregorevic P, Chamberlain JS, Froehner SC. Functional deficits in nNOSmu-deficient skeletal muscle: myopathy in nNOS knockout mice. PLoS One. 2008;3(10):e3387. | |
Filippin LI, Moreira AJ, Marroni NP, Xavier RM. Nitric oxide and repair of skeletal muscle injury. Nitric Oxide. 2009;21(3–4):157–163. | |
De Palma C, Clementi E. Nitric oxide in myogenesis and therapeutic muscle repair. Mol Neurobiol. 2012;46(3):682–692. | |
Buono R, Vantaggiato C, Pisa V, et al. Nitric oxide sustains long-term skeletal muscle regeneration by regulating fate of satellite cells via signaling pathways requiring Vangl2 and cyclic GMP. Stem Cells. 2012;30(2):197–209. | |
Cordani N, Pisa V, Pozzi L, Sciorati C, Clementi E. Nitric oxide controls fat deposition in dystrophic skeletal muscle by regulating fibro-adipogenic precursor differentiation. Stem Cells. Epub October 29, 2013. | |
Brunelli S, Sciorati C, D’Antona G, et al. Nitric oxide release combined with nonsteroidal antiinflammatory activity prevents muscular dystrophy pathology and enhances stem cell therapy. Proc Natl Acad Sci U S A. 2007;104(1):264–269. | |
Sciorati C, Miglietta D, Buono R, et al. A dual acting compound releasing nitric oxide (NO) and ibuprofen, NCX 320, shows significant therapeutic effects in a mouse model of muscular dystrophy. Pharmacol Res. 2011;64(3):210–217. | |
Sciorati C, Staszewsky L, Zambelli V, et al. Ibuprofen plus isosorbide dinitrate treatment in the mdx mice ameliorates dystrophic heart structure. Pharmacol Res. 2013;73:35–43. | |
Sciorati C, Buono R, Azzoni E, et al. Co-administration of ibuprofen and nitric oxide is an effective experimental therapy for muscular dystrophy, with immediate applicability to humans. Br J Pharmacol. 2010;160(6):1550–1560. | |
D’Angelo MG, Gandossini S, Martinelli Boneschi F, et al. Nitric oxide donor and non steroidal anti inflammatory drugs as a therapy for muscular dystrophies: evidence from a safety study with pilot efficacy measures in adult dystrophic patients. Pharmacol Res. 2012;65(4):472–479. | |
Fung HL, McNiff EF, Ruggirello D, Darke A, Thadani U, Parker JO. Kinetics of isosorbide dinitrate and relationships to pharmacological effects. Br J Clin Pharmacol. 1981;11(6):579–589. | |
Bushra R, Aslam N. An overview of clinical pharmacology of ibuprofen. Oman Med J. 2010;25(3):155–161. | |
Farrar H, Letzig L, Gill M. Validation of a liquid chromatographic method for the determination of ibuprofen in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2002;780(2):341–348. | |
Zhao X, Chen D, Li K, Wang D. Sensitive liquid chromatographic assay for the simultaneous determination of ibuprofen and its prodrug, ibuprofen eugenol ester, in rat plasma. Yakugaku Zasshi. 2005;125(9):733–737. | |
Verma RK, Garg S. A validated high performance liquid chromatographic method for analysis of isosorbide mononitrate in bulk material and extended release formulations. J Pharm Biomed Anal. 2002;30(3):583–591. | |
Stockis A, De Bruyn S, Deroubaix X, et al. Pharmacokinetic profile of a new controlled-release isosorbide-5-mononitrate 60 mg scored tablet (Monoket Multitab). Eur J Pharm Biopharm. 2002;53(1):49–56. | |
US Department of Health and Human Services; Food and Drug Administration; Center for Drug Evaluation and Research (CDER); Center for Veterinary Medicine (CVM). Guidance for Industry: Bioanalytical Method Validation. Rockville, MD: Center for Drug Evaluation and Research (CDER); 2001. Available from: http://www.fda.gov/downloads/Drugs/Guidances/ucm070107.pdf. Accessed January 10, 2013. | |
European Medicines Agency; Committee for Medicinal Products for Human Use (CHMP). Guideline on the Investigation of Bioequivalence. London, UK: European Medicines Agency; 2010. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf. Accessed January 10, 2013. | |
Finanger Hedderick EL, Simmers JL, Soleimani A, et al. Loss of sarcolemmal nNOS is common in acquired and inherited neuromuscular disorders. Neurology. 2011;76(11):960–967. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.