Back to Journals » Drug Design, Development and Therapy » Volume 15
Combination of Sodium Cantharidinate with Cisplatin Synergistically Hampers Growth of Cervical Cancer
Authors Chen X, Zhou M, Fan W, Yang M, Yang L
Received 18 September 2020
Accepted for publication 16 December 2020
Published 13 January 2021 Volume 2021:15 Pages 171—183
DOI https://doi.org/10.2147/DDDT.S282777
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Xiangxun Chen, Mengxi Zhou, Wenjie Fan, Mingwei Yang, Lin Yang
Department of Radiation Oncology, The First Affiliated Hospital of Anhui Medical University, Hefei, Anhui 230022, People’s Republic of China
Correspondence: Lin Yang
Department of Radiation Oncology, The First Affiliated Hospital of Anhui Medical University, Hefei, Anhui 230022, People’s Republic of China
Tel/Fax +86-0551-65908783
Email [email protected]
Background: Sodium cantharidinate (SC) has been broadly applied in lung cancer treatment in China, while its specific function in cervical cancer (CC), a great contributor to death of female reproductive system cancers, remains unclear. Our research evaluated the anti-tumor effects of SC in CC and the mechanism involved.
Methods: First, cisplatin (DDP)-resistant Caski-1 and ME180 cell lines were developed and treated with SC. The effects of SC on CC cell growth were then evaluated. Subsequently, the genes targeted by SC were predicted via the bioinformatics website. The correlations between PTPN1 expression and tumor stage, lymph node metastasis and tumor differentiation were examined. We further conducted rescue experiments by overexpressing PTPN1 in CC cells, followed by SC and cisplatin treatments. The activation of the PI3K/AKT pathway in CC cells, and the effect of SC on the growth and drug resistance of Caski-1 cells in vivo were investigated.
Results: The sensitivity of Caski-1 and ME180 cells to DDP was increased after SC treatment, which also enhanced the inhibitory effect of DDP on the cell growth. By prediction, we found that SC could target PTPN1. Patients with high expression of PTPN1 had higher clinical stage, lymph node metastasis and lower tumor differentiation. SC inhibited PTPN1 expression. Overexpression of PTPN1 attenuated the effect of SC. Furthermore, PTPN1 activated the PI3K/AKT pathway. Moreover, SC treatment inhibited the growth and drug resistance of Caski-1 cells in vivo.
Conclusion: SC promotes drug sensitivity of CC cells to DDP by targeting PTPN1, thereby impairing the PI3K/AKT pathway.
Keywords: cervical cancer, sodium cantharidinate, cisplatin, PTPN1, the PI3K/AKT pathway
Introduction
According to global cancer statistics in 2018, cervical cancer (CC) ranks the fourth place for both incidence (3.2% for all cases) and mortality (3.3% for all cases).1 Despite progresses in screening, vaccination, and management of CC at early stage, a proportion of patients are diagnosed at advanced stage, or with recurrent CC, for whom systemic chemotherapy represents the cornerstone of treatment regimens.2 As one of the most powerful anticancer drugs, cisplatin (DDP) is a small-molecule platinum that was accidentally exposed to constrain the growth of Escherichia coli and later was discovered to kill tumor cells.3 However, resistance to DDP may develop, which considerably compromised the effectiveness of DDP to treat advanced or recurrent CC.4 As a consequence, exploring the theory and mechanism behind the chemosensitivity of CC cells to DDP is an urgency.
Cantharidin is a terpenoid extracted from Chinese blister beetles that illustrates substantial toxicity against cancer cells, and its pharmacological function includes the suppression of xanthine oxides.5 Cantharidin, the inhibitor of protein phosphatases type 1 and type 2A, has been exhibited to alter cell cycles, including enhancing protein synthesis by facilitating into S phase and cell apoptosis.6 For instance, sodium cantharidinate (SC) has the potency to induce HepG2 cell apoptosis via the LC3 autophagy pathway.7 Nevertheless, no study exists regarding the role of SC in CC to date. Protein-tyrosine phosphatases (PTPs) eliminate phosphate groups from tyrosine residues, thus promoting or repressing signal transduction to impact cellular processes such as cell proliferation.8 Moreover, the significance of PTPs has been underscored in the mediation of human health, and PTP dysfunction is causal to various human diseases, including cancers.9 Among them, PTP nonreceptor type 1 (PTPN1, also known as PTP1B) has been revealed to be overexpressed in esophageal carcinoma, which was closely associated with incidence.10 PTPN1 has been reported to be strongly associated with the development of various tumors, such as non-small cell lung cancer,11 breast cancer12 and glioma.13 However, it has not been intensively studied in CC. Furthermore, it has been noted that overexpression of PTPN1 was associated with Erlotinib resistance in lung adenocarcinoma.14 Therefore, we wondered if PTPN1 was similarly associated with drug resistance in CC. The present study investigated whether PTPN1 also exerts important functions in CC and whether SC may be used to inhibit the expression of PTPN1. The protein and mRNA expression of PTPN1 in the CC cell lines (Caski-1 and ME180) were examined using Western blot and reverse transcription‑quantitative polymerase chain reaction analysis (RT-qPCR), respectively. The function of SC in inducing the chemosensitivity of CC cells to DDP was also probed. This research may provide attracting targets for the treatment of CC and further improve the outcomes of patient with CC.
Materials and Methods
Clinical Sample Collection
The tissue collection and analyses were approved by the Ethics Committee of The First Affiliated Hospital of Anhui Medical University, and all the participants provided written informed consent. The studies were performed as per the Declaration of Helsinki. A total of 59 pairs of CC tissues and adjacent tissues were harvested from January 2016 to January 2019 in The First Affiliated Hospital of Anhui Medical University. All patients had not received radiotherapy or chemotherapy before surgery. Samples from patients with other major diseases were excluded. Samples were selected based on the diagnosis of CC determined by at least two pathologists. The clinical characteristics of the patients were obtained from medical records. Clinical staging and clinicopathological classification were determined in accordance with the International Federation of Gynecology and Obstetrics (FIGO). Tissues were collected at the time of surgery and immediately frozen in liquid nitrogen until use.
Establishment of DDP-Resistant Cell Lines
Caski-1 (#CDC0111, China Center for Type Culture Collection, Wuhan, Hubei, China) and ME180 cells (#HTB-33, American Type Culture Collection, Manassas, VA, USA) resistant to DDP were established by sustained exposure to DDP (#P4394, Sigma-Aldrich Chemical Company, St Louis, MO, USA). Briefly, Caski-1 and ME180 cells were exposed to increasing DDP concentrations for 48 hours, once a week. The initial DDP dose was 1.52 μM, which was the 80% inhibition concentration (IC80) for the sensitive cell lines. Once the cells recovered and grew in the exponential growth phase, the DDP concentration gradually increased to as high as 15.20 µM. After 6 cycles of DDP treatment for 6 months, the resulting resistant lines Caski-1/R and ME180/R were sub-cultured and treated monthly with 15.20 μM DDP to maintain chemoresistance. Caski-1/R and ME180/R cells were cultured without DDP for approximately 14 d prior to subsequent experiments.
Cell Survival Assay
Cells were plated at 5 × 103 cells/well in 96-well microtiter plates and allowed to adhere overnight. Then, a gradient concentration of DDP (0.001–100 nmol/mL) was added and incubated for another 72 h. Cell viability was tested using the CellTiter-Glo® (CTG) assay. Each value was normalized to cells treated with dimethylsulfoxide, and IC50 values were calculated using Graphpad Prism software.
5-Ethynyl-2ʹ-Deoxyuridine (EdU) Labeling
EdU was performed to assess the proliferation of Caski-1/R and ME180/R cells. Caski-1/R and ME180/R cells were treated with SC for 24 h prior to a 24-h DDP treatment. Subsequently, the cells were fixed in the mixture of cold ethanol and acetic acid (2:1) for 10 min at ambient temperature and rinsed with phosphate-buffered saline (PBS, pH = 7.4) supplemented with 1% Triton X-100. The fixed cells were treated with 4 mM HCl at 37°C for 15 min to denature the DNA and neutralized with 0.1 M sodium borate (pH = 8.5). The cells were rinsed three times with 0.1% NP-40 (Beyotime Biotechnology Institute, Shanghai, China) in PBS and sealed for 0.5 h at ambient temperature. Finally, cells were treated with 10 μM EdU (#900584, Sigma-Aldrich) for 6 h. Cells were stained with 4ʹ,6-diamidino-2-phenylindole (#D8417, Sigma-Aldrich; Merck KGaA). Staining images were captured using an Axiovert 200 fluorescence microscope (×100, Carl Zeiss, Oberkochen, Germany).
Colony Formation Assay
Cells were plated into 12-well plates at 1 × 103 cells/well. The next day, the cells were incubated with added drugs for 12 d. The medium was renewed every 3 days. Afterward, the cells were fixed with methanol and stained with 0.1% crystal violet (both for 10 min at room temperature) and photographed.
Flow Cytometry
The percentage of apoptosis was determined using the Annexin V/propidium iodide (PI) kit (#C1062, Beyotime Biotechnology Co., Ltd., Shanghai, China). Cells were resuspended in 1 × binding buffer. The cell suspension (1 × 105 cells in 100 μL) was transferred to a 5-mL culture tube and mixed with 5 μL Annexin V-fluorescein isothiocyanate (FITC) and 5 μL PI. The mixture was gently vortexed and incubated at 25°C in darkness for 15 min. Then, 400 μL 1 × binding buffer was loaded to each tube. Samples were analyzed by a flow cytometer within 1 h. The green fluorescence of Annexin V-FITC was measured at 530 nm, and red fluorescence of PI was measured at 585 nm. The results were analyzed using FlowJo software (version 7.6.1, BD Biosciences, San Jose, CA, USA).
Hoechst 33258 Staining
The morphological characteristics of apoptotic cells were detected by Hoechst 33258 staining (#C1017, Beyotime). Following fixation with 4% paraformaldehyde for 0.5 h at ambient temperature, the cells were stained with Hoechst 33258 (5 µg/mL) for 20 min at 37°C in darkness. Images of apoptotic cells exhibiting Hoechst 33258 fluorescence were taken under a fluorescence microscope (Nikon Instruments Inc., Melville, NY, USA). The percentage of apoptotic cells was semi-quantified based on Image-Pro Plus software 6.0 (Media Cybernetics, Bethesda, MD, USA).
Immunofluorescence Staining
Antibodies against CD133 (ab216323, Abcam), CD44 (ab51037, Abcam), and SOX2 (MAB4423, Millipore Corp, Billerica, MA, USA) were applied to stain the transfected cells. Secondary antibodies were Alexa Fluor 488 goat anti-rabbit IgG antibody and coverslips (Invitrogen Inc., Carlsbad, CA, USA). Microscopic observations were conducted under a confocal laser scanning microscope (Leica Microsystems, Bensheim, Germany). Intensity of fluorescence was assessed in five fields of views with 300 cells per coverslip and analyzed using ImageJ 1.37v software (http://rsb.info.nih.gov/ij/index.html).
Sphere Formation Assay
Caski-1/R and ME180/R (1 × 103 cells/mL) were seeded into 6-well Corning ultra-low attachment plates. Cells were cultured in Dulbecco’s modified Eagle’s/F12 medium supplemented with epidermal growth factor (20 ng/mL, PeproTech Inc., Rocky Hill, NJ, USA), basic fibroblast growth factor (20 ng/mL, PeproTech) and B27 (Gibco, Carlsbad, CA, USA). After 12 d, the total number of spheres was counted.
Lactate Dehydrogenase (LDH) Release Assay
Parental or drug-resistant Caski-1 and ME180 cells (1.5 × 105 cell/well) were cultured overnight in 24-well cell culture plates. The supernatant was collected and added into a 96-well black culture plate (200 μL/well). LDH release levels were measured using the LDH cytotoxicity assay kit (#C0016, Beyotime) as per the manufacturer’s instructions. The optical density (OD) value at 450 nm in each well was read using a microplate reader.
Caspase-3 Activity Assay
Parental or drug-resistant Caski-1 and ME180 cells were cultured with SC or DDP for 72 h. The Caspase-Glo 3 kit (#G8090, Promega Corporation, Madison, WI, USA) was used to assess the Caspase-3 activity in cells in 96-well plates (Promega, Madison, WI, USA) following the manufacturer’s protocol.
RT-qPCR
After transfection, cells or spheres were harvested and RNA was isolated using TRIZOL reagent (Sigma-Aldrich). RNA (10 ng) was used for reverse transcription with the TaqMan RNA RT kit (Applied Biosystems, Life Technologies, Madison, WI, USA). PCR was performed in a quadruple reaction in Rotor-Gene Corbett Q 6000 PCR. Data were collected and analyzed using Rotor-Gene quantitation software. RT-PCR primers (Cat no 204337) were purchased from Exiqon (Woburn, MA, USA).
Western Blot
Treated cells or tissues were ice-bathed in radio immunoprecipitation assay buffer containing 1 mmol/L fluoride. Cell lysates or tissue samples containing 20 μg protein were subjected to 8–12% SDS-PAGE, and proteins were transferred to polyvinylidene difluoride membranes (Millipore). The membranes were sealed with 5% skim milk for 90 min at ambient temperature, followed by incubation with primary antibodies (1:1000 dilution) to PTPN1 (MABS197, Millipore), phos-PI3K (ab182651, Abcam), and phos-AKT1 (#bs-1448R, Bioss Biotech, Beijing, China) at 4°C overnight and with appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies (1:10,000 dilution; Proteintech, Chicago, IL, USA) for 60 min at ambient temperature. Enhanced chemiluminescence reagent (Beyotime) was used for chemiluminescence.
Xenograft Assay in Nude Mice
All animal experiments were approved by the Research Animal Resource Center of The First Affiliated Hospital of Anhui Medical University. All animal procedures were implemented according to the NIH guidelines. Caski-1 and Caski-1/R cells (3 × 106 cells/mL) were injected subcutaneously into the axilla of 5-week-old female nude mice (n = 6, 20–22g, Hunan SJA Laboratory Animal Co., Ltd., Changsha, Hunan, China). The mice were treated by gavage with SC or PBS at a dose of 100 mg/kg per day. Meanwhile, the mice were also treated with DDP every 5 days. The volume of tumors formed by Caski-1 cells was calculated every five days starting from the fifth day after cell implantation and determined by the formula volume = (length × width2)/2. All procedures were performed under intravenous anesthesia with pentobarbital sodium at a concentration of 100 mg/kg, and every effort was made to minimize the suffering of mice.
Immunohistochemistry
For immunohistochemistry, tissue samples were incubated with antibodies to KI67 (1:400, #bs-2007R, Bioss Biotech, Beijing, China), PTPN1 (1:500, MABS197, Millipore), phos-PI3K (1:400, ab182651, Abcam), and phos-AKT1 (1:400, #bs-1448R, Bioss) at 4°C overnight. The sections were treated with HRP at 37°C for 60 min, colored with 3,3-diaminobenzidine (DAB) and then stored at ambient temperature without light for 10 min. Staining degree scores were determined as 0 point: 0–5% stained cells, 1 point: 6–25% stained cells, 2 points: 26–50% stained cells, 3 points: 51–75% stained cells, and 4 points: > 75% stained cells. Staining intensity scores were assessed as 0–1: negative, 2–3: weakly positive, 4–5: moderately positive, and 6–7: strongly positive. The sum of the degree and intensity scores was defined as the final staining score.
TUNEL Assay
The percentage of apoptosis in tissues was measured using the TUNEL kit (#G7360, Promega). Tissue sections were waxed and rehydrated. Following antigen retrieval with proteinase K working solution and permeabilization, the slides were incubated at 37°C for 2 h with a mixture of TdT and dUTP (1:9). Then, the activity of endogenous peroxidase was inactivated. The tissue was covered with reagent 3 (converter-peroxidase), reacted with DAB color development reagent and counterstained with hematoxylin for 3 min. Finally, the tissues were continuously dehydrated in 70%, 80%, 95%, and 100% ethanol, cleared with xylene, followed by resin mounting. Nuclei stained with hematoxylin were in blue, and positive cells were in brownish yellow. Viewed under a microscope, the tissues were analyzed using Image-Pro Plus 6.0 (Media Cybernetics).
Statistical Analysis
All the experiments were performed with at least three repetitions. The data were analyzed with SPSS 20.0 (IBM Corp. Armonk, N.Y., USA) and GraphPad Prism Software 8.0 (GraphPad Software, San Diego, CA, USA). Statistical analysis was performed utilizing the one-way or two-way ANOVA test combined with Tukey’s test or t-test. Data are displayed as mean ± standard deviation (SD). Differences between means were considered significant for p < 0.05.
Results
DDP-Resistant CC Cell Lines are Successfully Developed
We first used gradient concentrations of DDP to treat Caski-1 and ME180 cells for the development of DDP-resistant CC cell lines (Caski-1/R and ME180/R). To determine the success of resistant cell line construction, we examined the survival of parental and resistant Caski-1 and ME180 cells at various concentrations of DDP using the CTG kit, and we observed that Caski-1/R and ME180/R cells had significantly higher IC50 values for DDP (Figure 1A). A dose of 10 μM DDP was used to treat parental and drug-resistant cells for 6 h. Apoptosis rate and activity were measured using flow cytometry and EdU staining. We found that the apoptosis rate of parental cells was significantly higher and the number of proliferating cells was significantly reduced after 10 μM DDP treatment compared to drug-resistant cells (Figure 1B and C). Moreover, we further used immunofluorescence staining to detect the expression of stem cell markers CD133, CD44, and SOX2 in the cells. The expression of stem cell markers was significantly increased in drug-resistant cells (Figure 1D). Moreover, the size of the tumor spheres formed by DDP-resistant cells was also significantly larger than that of the parental cells (Figure 1E). The above results indicate that DDP-resistant CC cell lines were successfully constructed and that the resistant cells exhibited stem cell properties.
 |
Figure 1 DDP-resistant CC cell lines are successfully induced. (A) cell survival detection by CTG kit; (B) proliferative activity of cells determined by EdU staining assay; (C) proportions of apoptotic cells evaluated by flow cytometry; (D) immunofluorescence detection of expression of stem cell markers in cells; (E) the size and number of spheres formed by cells assessed by sphere formation assay. The experiments were repeated at least three times. The data are displayed as the means ± SD of three experiments. Statistical analysis was performed utilizing the one-way (panel B, C and E) or two-way ANOVA (panel A and D) test combined with Tukey’s test. **p < 0.01 vs Caski-1 cells; ##p < 0.01 vs ME180 cells. |
SC Inhibits the Growth of CC Cells
SC was found to cause apoptosis in hepatocellular carcinoma cells in a previous study.7 We anticipated that SC may have a similar role in CC cells. Thus, we treated parental and DDP-resistant Caski-1 and ME180 cells with different concentrations of SC, and assayed cell activity using the CTG kit. It has been revealed that as the concentration of SC increased, the activity of Caski-1 and ME180 cells decreased significantly (Figure 2A and B), and the apoptosis rate increased significantly (Figure 2C). We subsequently used immunofluorescence to assess the changes in the expression of stem cell markers in resistant cells in the presence of SC. SC significantly inhibited the expression of CD133, CD44, and SOX2 in the cells (Figure 2D). Sphere formation assay also showed a significant decline in the size and number of spheres formed by Caski-1/R and ME180/R cells after SC treatment (Figure 2E).
 |
Figure 2 SC inhibits the growth of CC cells. CC cells were treated with SC alone. (A) cell survival detection by CTG kit; (B) proliferative activity of cells determined by EdU staining assay; (C) proportions of apoptotic cells evaluated by flow cytometry; (D) immunofluorescence detection of expression of stem cell markers in cells; (E) the size and number of spheres formed by cells assessed by sphere formation assay. The experiments were repeated at least three times. The data are expressed as the means ± SD of three experiments. Statistical analysis was performed utilizing the unpaired t-test (panel E), one-way (panel B and C) or two-way ANOVA (panel A and D) test combined with Tukey’s test. **p < 0.01 vs Caski-1 + PBS; ##p < 0.01 vs Caski-1/R + PBS; @@p < 0.01 vs ME180 + PBS; &&p < 0.01 vs ME180/R + PBS. |
SC Promotes Sensitivity of CC Cells to DDP
Now that we established that SC could inhibit the growth of CC cells and the stem cell properties of drug-resistant cells, we hypothesized that SC treatment promotes drug sensitivity of cells to DDP. To validate our conjecture, we treated parental and resistant Caski-1 and ME180 cells with 50 mM SC and with a gradient concentration of DDP for the IC50 value assessment. We were surprised to find that the IC50 values of Caski-1 and ME180 parental and resistant cells to DDP were significantly reduced after SC treatment (Figure 3A). Moreover, our treatment of Caski-1 and ME180 cells with SC significantly augmented the inhibitory effect of DDP on the growth of both parental and drug-resistant cells (Figure 3B and C), and the proportion of apoptosis was significantly higher (Figure 3D and E). We further examined the release of LDH from Caski-1 and ME180 cells, where SC treatment significantly promoted the release of LDH from both parental and drug-resistant cells after DDP treatment (Figure 3F), and the activity of caspase-3 was similarly increased (Figure 3G).
 |
Figure 3 SC promotes drug sensitivity of CC cells to DDP. CC cells were treated with DDP in the presence of 50 mM SC. (A) cell survival detection by CTG kit; (B) proliferative activity of cells determined by EdU staining assay; (C) colony formation of cell tested by colony formation assay; (D) proportions of apoptotic cells evaluated by flow cytometry; (E) the percentage of apoptosis in cells determined by Hoechst 33258 staining; (F) the LDH release of CC cells assessed by LDH kits; (G) the activity of caspase-3 in cells examined by Caspase-3 kits. The experiments were repeated at least three times. The data are expressed as the means ± SD of three experiments. Statistical analysis was performed utilizing the one-way (panel B–G) or two-way ANOVA (panel A) test combined with Tukey’s test. **p < 0.01 vs Caski-1 + PBS; ##p < 0.01 vs Caski-1/R + PBS; @@p < 0.01 vs ME180 + PBS; &&p < 0.01 vs ME180/R + PBS. |
SC Promotes Drug Sensitivity of CC Cells to DDP via Inhibition of PTPN1
To clarify the molecular mechanism of SC, we used the SWISS Prediction (http://www.swisstargetprediction.ch/) website to predict the target of SC, and we noted that the targets of SC are distributed in the phosphatase class (Figure 4A and B). We found that SC can target PTPN1 (Supplementary Table 1). Thus, we determined PTPN1 expression in Caski-1 and ME180 cells and found that the expression of PTPN1 was significantly increased in DDP-resistant cells, but had a significant decrease after further treatment with SC (Figure 4C and D). Moreover, we further analyzed the correlation between PTPN1 expression and the clinical stage, lymph node metastasis, and tumor differentiation of the 59 CC patients and found that patients with overexpression of PTPN1 had higher clinical stage and higher risks for lymph node metastasis, and the poorer tumor differentiation. However, PTPN1 expression was independent of the age and tumor size of patients (Table 1).
 |
Table 1 Correlation Analysis Between PTPN1 Expression and Clinical Features (N = 59) |
 |
Figure 4 SC promotes drug sensitivity of CC cells to DDP via inhibition of PTPN1. (A) chemical structure of SC; (B) the targets of SC predicted by SWISS Prediction (http://www.swisstargetprediction.ch/) website; (C) mRNA expression of PTPN1 in Caski-1 and ME180 cells determined by RT-qPCR; (D) protein expression of PTPN1 in Caski-1 and ME180 cells determined by Western blot. The experiments were repeated at least three times. The data are expressed as the means ± SD of three experiments. Statistical analysis was performed utilizing the one-way ANOVA (panel C–D) test combined with Tukey’s test. **p < 0.01 vs Caski-1; ##p < 0.01 vs Caski-1 + PBS; @@p < 0.01 vs Caski-1/R + PBS; &&p < 0.01 vs ME180; $$p < 0.01 vs ME180 + PBS; %%p < 0.01 vs ME180/R + PBS. |
Overexpression of PTPN1 Reverses the Enhancer Effect of SC on DDP Sensitivity of CC Cells
To further validate that SC promotes DDP drug sensitivity in CC cells through targeting PTPN1, we transfected PTPN1 overexpression plasmids in parental and drug-resistant Caski-1 and ME180 cells and verified successful transfection by RT-qPCR and Western blot (Figure 5A and B). We found that after overexpression of PTPN1 in cells, the IC50 values of DDP were significantly increased in parental and drug-resistant Caski-1 and ME180 cells (Figure 5C). Besides, the inhibition of cell growth by DDP and SC (Figure 5D and E) was significantly alleviated, and the percentage of apoptosis after DDP and SC treatments was also inhibited (Figure 5F). Similarly, the release of LDH from cells as well as the activity of caspase-3 was significantly inhibited following PTPN1 upregulation (Figure 5G–I).
 |
Figure 5 Overexpression of PTPN1 reverses the promoting effects of SC on DDP sensitivity of CC cells. The PTPN1 overexpression plasmids were transfected into parental and drug-resistant Caski-1 and ME180 cells treated with DDP and SC. (A) mRNA expression of PTPN1 in Caski-1 and ME180 cells determined by RT-qPCR; (B) protein expression of PTPN1 in Caski-1 and ME180 cells determined by Western blot; (C) cell survival detection by CTG kit; (D) proliferative activity of cells determined by EdU staining assay; (E) colony formation of cell tested by colony formation assay; (F) proportions of apoptotic cells evaluated by flow cytometry; (G) the percentage of apoptosis in cells determined by Hoechst 33258 staining; (H) the LDH release of CC cells assessed by LDH kits; (I) the activity of caspase-3 in cells examined by Caspase-3 kits. The experiments were repeated at least three times. The data are expressed as the means ± SD of three experiments. Statistical analysis was performed utilizing the one-way (panel A, B, D, E, F, G, H and I) or two-way ANOVA (panel C) test combined with Tukey’s test. **p < 0.01 vs Caski-1 + oe-NC; ##p < 0.01 vs Caski-1/R + oe-NC; @@p < 0.01 vs ME180 + oe-NC, &&p < 0.01 vs ME180/R + oe-NC. |
PTPN1 Activates the PI3K/AKT Pathway in CC Cells
In a study by Jin et al, it was noted that PTPN1 promotes the malignant biological behavior of gliomas through activation of the PI3K/AKT pathway.15 Thus, we examined the extents of PI3K and AKT phosphorylation in Caski-1 and ME180 cells by Western blot. The extents of PI3K and AKT phosphorylation were significantly increased in DDP-resistant cells but were significantly decreased after further treatment with SC. In addition, overexpression of PTPN1 partially counteracted the inhibitory effect of SC on the extents of PI3K and AKT phosphorylation (Figure 6A and B). Subsequently, we treated both parental and drug-resistant Caski-1 and ME180 cells with the PI3K/AKT-specific agonist Recilisib in combination with SC. After activation of PI3K/AKT, we found a significant increase in IC50 values for DDP in both parental and drug-resistant Caski-1 and ME180 cells, suggesting that CC cells were less sensitive to DDP after activation of the PI3K/AKT signaling pathway (Figure 6C). Moreover, the inhibitory effect of DDP on CC cell growth promoted by SC was largely counteracted by Recilisib, which mainly manifested by the potentiated apoptosis and suppressed proliferation of CC cells caused by SC treatment was significantly attenuated after activation of the PI3K/AKT signaling pathway (Figure 6D–H).
 |
Figure 6 PTPN1 activates the PI3K/AKT pathway in CC cells. (A) the extents of PI3K and AKT phosphorylation in parental and drug-resistant Caski-1 cells determined by Western blot (**p < 0.01 vs Caski-1 cells; ##p < 0.01 vs Caski-1 + PBS; @@p < 0.01 vs Caski-1/R + PBS; &&p < 0.01 vs Caski-1 + oe-NC; $$p < 0.01 vs Caski-1/R + oe-NC); (B) the extents of PI3K and AKT phosphorylation in parental and drug-resistant ME180 cells determined by Western blot (**p < 0.01 vs ME180 cells; ##p < 0.01 vs ME180 + PBS; @@p < 0.01 vs ME180/R + PBS; &&p < 0.01 vs ME180 + oe-NC; $$p < 0.01 vs ME180/R + oe-NC). Parental and drug-resistant Caski-1 and ME180 cells were treated with the PI3K/AKT-specific agonist Recilisib in combination with SC and DDP. (C) cell survival detection by CTG kit; (D) proliferative activity of cells determined by EdU staining assay (**p < 0.01 vs Caski-1 + DMSO; ##p < 0.01 vs Caski-1/R + DMSO; @@p < 0.01 vs ME180 + DMSO; &&p < 0.01 vs ME180/R + DMSO); (E) colony formation of cell tested by colony formation assay (**p < 0.01 vs Caski-1 + DMSO; ##p < 0.01 vs Caski-1/R + DMSO; @@p < 0.01 vs ME180 + DMSO; &&p < 0.01 vs ME180/R + DMSO); (F) proportions of apoptotic cells evaluated by flow cytometry (**p < 0.01 vs Caski-1 + DMSO; ##p < 0.01 vs Caski-1/R + DMSO; @@p < 0.01 vs ME180 + DMSO; &&p < 0.01 vs ME180/R + DMSO); (G) the LDH release of CC cells assessed by LDH kits (**p < 0.01 vs Caski-1 + DMSO; ##p < 0.01 vs Caski-1/R + DMSO; @@p < 0.01 vs ME180 + DMSO; &&p < 0.01 vs ME180/R + DMSO); (H) the activity of caspase-3 in cells examined by Caspase-3 kits (**p < 0.01 vs Caski-1 + DMSO; ##p < 0.01 vs Caski-1/R + DMSO; @@p < 0.01 vs ME180 + DMSO; and &&p < 0.01 vs ME180/R + DMSO). The experiments were repeated at least three times. The data are expressed as the means ± SD of three experiments. Statistical analysis was performed utilizing the one-way (panel D, E, F, G and H) or two-way ANOVA (panel A, B and C) test combined with Tukey’s test. |
SC Inhibits the Growth and Drug Resistance of CC Cells in vivo
To determine the effect of SC on the growth and drug resistance of CC cells in vivo, we implanted parental and resistant Caski-1 cells into the axilla of nude mice and treated the mice with SC or PBS by gavage at a dose of 100 mg/kg per day and DDP every 5 days (Figure 7A). The volume of xenograft tumors formed by Caski-1 cells was calculated every five days starting from the fifth day after inoculation. SC treatment significantly inhibited the growth and resistance of Caski-1 cells to DDP in vivo (Figure 7B and C). Moreover, we further used immunohistochemistry to detect the number of positive cells for KI67, PTPN1, phos-PI3K, and phos-AKT1 in tumor tissues and found that the staining intensities of KI67, PTPN1, phos-PI3K, and phos-AKT1 in tumor tissues were significantly reduced (Figure 7D–G) and the number of TUNEL-positive cells was remarkably enhanced (Figure 7H) after SC treatment. In summary, SC can promote the drug sensitivity of CC cells to DDP by targeting PTPN1 and inhibiting the activation of the PI3K/AKT pathway.
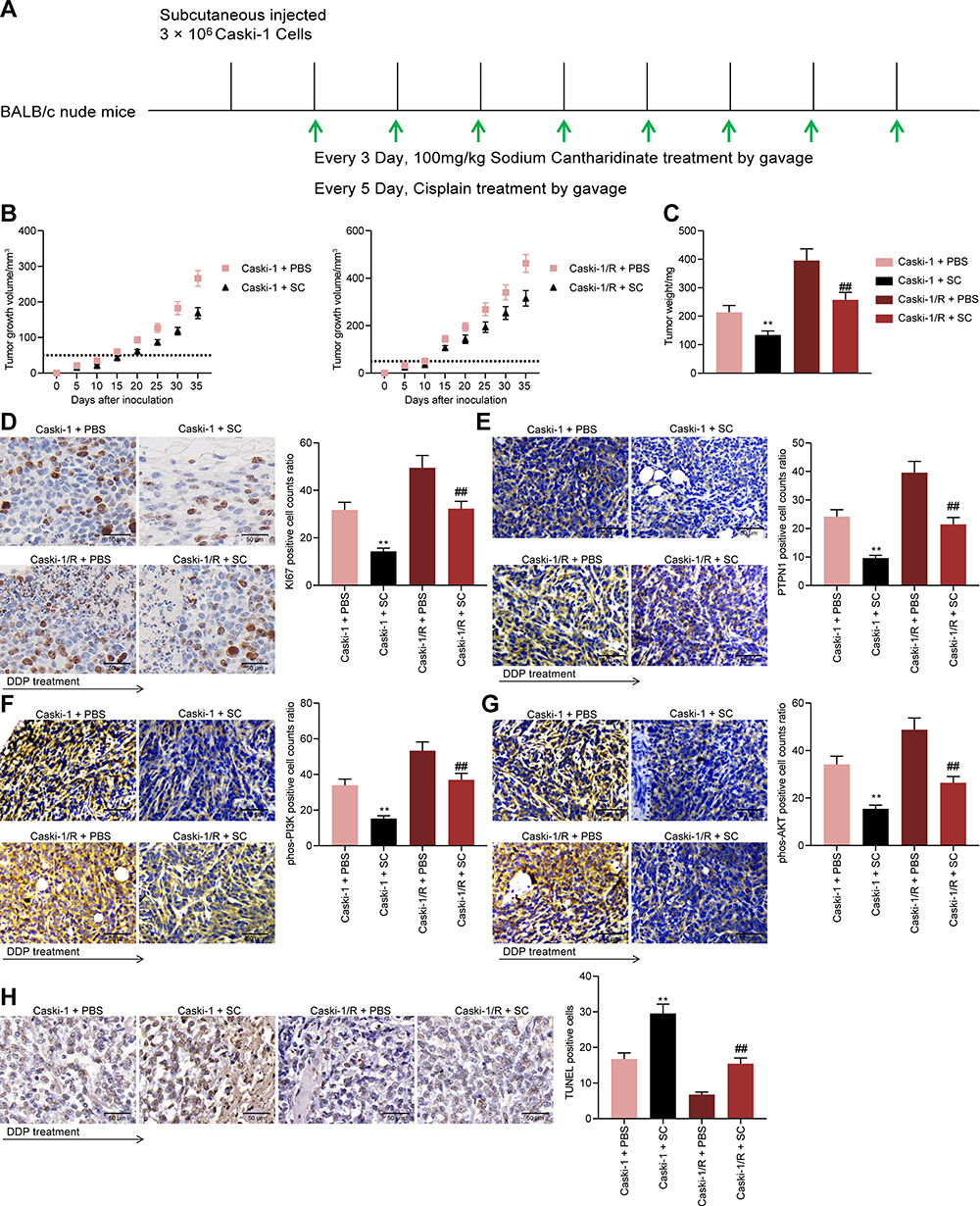 |
Figure 7 SC inhibits the growth as well as drug resistance of CC cells in vivo. (A) process diagram; (B) growth curves of tumors; (C) weight of tumors; (D–G) immunohistochemical staining of the number of positive cells for KI67 (D), PTPN1 (E), phos-PI3K (F), and phos-AKT1 (G) in xenograft tumors; (H), the proportion of apoptotic cells in tumors determined by TUNEL staining. The experiments were repeated at least three times. N = 6. The data are expressed as the means ± SD of three experiments. Statistical analysis was performed utilizing the one-way (panel C–H) or two-way ANOVA (panel B) test combined with Tukey’s test. **p < 0.01 vs mice injected with Caski-1 + PBS; ##p < 0.01 vs mice injected with Caski-1/R + PBS. |
Discussion
DDP, a renowned chemotherapeutic drug, is widely used for management of plentiful human malignancies, involving bladder, head and neck, lung, ovarian, as well as testicular cancers.16 A major contributor of the DPP treatment failure is drug resistance, which may ultimately affect treatment efficacy, survival and prognosis of CC patients and lead to distant metastasis and relapse.17 In this investigation, we showed that SC treatment hampered the growth of CC cells and further reinforced the growth inhibitory effect of DDP to CC cells. Mechanistically, SC significantly repressed PTPN1 expression in CC cells, while restoration of PTPN1 expression conferred chemoresistance to CC cell. Finally, PTPN1-mediated PI3K/AKT pathway activation was accomplished to suppress chemosensitivity in CC.
Although some reports have concentrated on the antitumor activity of SC in clinical practice in recent years, the activity of SC in CC has not been elucidated to date. For instance, SC improved tumor responses and quality of life with reduced risk of hepatotoxicity and gastrointestinal toxicity relative to chemotherapy alone in non-small-cell lung cancer.18 Moreover, SC possesses the therapeutic ability to lower the expression of S100A3 in hepatocellular carcinoma cells and to induce cellular apoptosis.19 In the present work, we firstly treated parental and drug resistance cells with SC and provided evidence that SC could remarkably diminish the expression patterns of cancer stem cell markers and spheres formed. Additional DDP treatment disclosed that SC is able to reduce the IC50 value of CC cells to DPP and to expedite apoptosis and LDH release. Consistently, disodium cantharidinate, which is a ramification of cantharidin, contributed to a significant elevation of caspase-3 and PARP protein expression in dendritic cells of patients with bladder carcinoma.20 In our studies, we confirmed that SC is able to decrease the viability of DPP-resistant CC cells to enhance chemosensitivity.
Mechanistically, we revealed that PTPN1 was targeted and repressed by SC. Also, our study underlined that PTPN1 enhanced the activation of the PI3K/AKT pathway. These findings might offer an explanation for why acquired chemoresistance develops in CC. PTPN1 inhibitors could be regarded a prospective anticancer treatment, no matter as monotherapy and in coordination with other anticancer drugs.21 For instance, PTPN1 protein expression was dramatically elevated in breast cancer tissues and PTPN1 promoted the proliferation, and suppressed the apoptosis of HER2-positive and triple-negative breast cancer cells.22 Our in vitro evidence displayed that overexpression of PTPN1 reduced the LDH release and apoptosis in the presence of SC.
Dysfunction of the PI3K pathway is frequently observed in many different types of malignancies, and the combination of the PI3K inhibitors with chemotherapy and/or other targeted therapy may be beneficial for optimizing the efficacy of therapies.23 Amirani et al also suggested that inhibition of the PI3K/AKT pathway restored sensitivity of cancer cells to hormonal, chemical and radioactive agents.24 In addition, the positive correlation between PTPN1 expression and the PI3K/AKT pathway activation has been corroborated in glioma.15 By contrast, SC suppressed osteosarcoma cell proliferation and stimulated cell cycle arrest by impairing the PI3K/AKT activation.25 In the same vein, we noted that SC reduced the extents of PI3K and AKT phosphorylation in parental and DDP-resistant CC cells, which was partially restored by PTPN1 overexpression. To further substantiate the involvement of the PI3K/AKT pathway deficit in SC-mediated DDP chemosensitivity, we delivered the PI3K/AKT pathway agonist Recilisib into CC cells treated with DDP and SC. Recilisib was found to lead to acquired chemoresistance to DDP by preventing CC cells from apoptosis. Also, in cardiomyocytes, Recilisib-activated PI3K/AKT pathway restrained apoptosis induced by miR-23a-5p overexpression.26 More specifically, KRAS knockdown blocked the activation of PI3K/AKT pathway to inhibit epithelial-mesenchymal transition and proliferation of breast cancer cells.27
Conclusion
In conclusion, using in vitro and in vivo models, we confirmed that SC can decrease the viability of Caski-1 and ME180 CC cells, and the DDP chemoresistance. Our studies also revealed that SC could suppress the expression of PTPN1 and block the induction of the PI3K/AKT pathway. We therefore demonstrated that SC could be regarded as a possible pharmacological agent in the treatment or prevention of CC. A potential pitfall of this work may be the lack of in vivo experiments using the ME180 cell line. Still, we believe that the existing results already demonstrated that SC can inhibit the growth and drug resistance of CC cells in vitro and in vivo.
Funding
There is no funding to report.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Menderes G, Black J, Schwab CL, Santin AD. Immunotherapy and targeted therapy for cervical cancer: an update. Expert Rev Anticancer Ther. 2016;16(1):83–98. doi:10.1586/14737140.2016.1121108
3. Shen DW, Pouliot LM, Hall MD, Gottesman MM. Cisplatin resistance: a cellular self-defense mechanism resulting from multiple epigenetic and genetic changes. Pharmacol Rev. 2012;64(3):706–721. doi:10.1124/pr.111.005637
4. Zhu H, Luo H, Zhang W, et al. Molecular mechanisms of cisplatin resistance in cervical cancer. Drug Des Devel Ther. 2016;10:1885–1895. doi:10.2147/DDDT.S106412
5. Yeh CB, Su CJ, Hwang JM, Chou MC. Therapeutic effects of cantharidin analogues without bridging ether oxygen on human hepatocellular carcinoma cells. Eur J Med Chem. 2010;45(9):3981–3985. doi:10.1016/j.ejmech.2010.05.053
6. Deng LP, Dong J, Cai H, Wang W. Cantharidin as an antitumor agent: a retrospective review. Curr Med Chem. 2013;20(2):159–166. doi:10.2174/092986713804806711
7. Tao R, Sun WY, Yu DH, et al. Sodium cantharidinate induces HepG2 cell apoptosis through LC3 autophagy pathway. Oncol Rep. 2017;38(2):1233–1239. doi:10.3892/or.2017.5779
8. Hale AJ, Ter Steege E, den Hertog J. Recent advances in understanding the role of protein-tyrosine phosphatases in development and disease. Dev Biol. 2017;428(2):283–292. doi:10.1016/j.ydbio.2017.03.023
9. Lee H, Yi JS, Lawan A, et al. Mining the function of protein tyrosine phosphatases in health and disease. Semin Cell Dev Biol. 2015;37:66–72. doi:10.1016/j.semcdb.2014.09.021
10. Chen J, Zhao X, Yuan Y, Jing JJ. The expression patterns and the diagnostic/prognostic roles of PTPN family members in digestive tract cancers. Cancer Cell Int. 2020;20(1):238. doi:10.1186/s12935-020-01315-7
11. Liu H, Wu Y, Zhu S, et al. PTP1B promotes cell proliferation and metastasis through activating src and ERK1/2 in non-small cell lung cancer. Cancer Lett. 2015;359(2):218–225. doi:10.1016/j.canlet.2015.01.020
12. Kuban-Jankowska A, Gorska-Ponikowska M, Sahu KK, et al. Docosahexaenoic acid inhibits PTP1B phosphatase and the viability of MCF-7 breast cancer cells. Nutrients. 2019;11(11):2554. doi:10.3390/nu11112554
13. Li Z, Hu C, Zhen Y, et al. Pristimerin inhibits glioma progression by targeting AGO2 and PTPN1 expression via miR-542-5p. Biosci Rep. 2019;39(5).
14. Chen Y, Tang J, Lu T, Liu F. CAPN1 promotes malignant behavior and erlotinib resistance mediated by phosphorylation of c-Met and PIK3R2 via degrading PTPN1 in lung adenocarcinoma. Thorac Cancer. 2020;11(7):1848–1860. doi:10.1111/1759-7714.13465
15. Jin T, Li D, Yang T, et al. PTPN1 promotes the progression of glioma by activating the MAPK/ERK and PI3K/AKT pathways and is associated with poor patient survival. Oncol Rep. 2019;42(2):717–725. doi:10.3892/or.2019.7180
16. Dasari S, Tchounwou PB. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol. 2014;740:364–378. doi:10.1016/j.ejphar.2014.07.025
17. Fang X, Zhong G, Wang Y, et al. Low GAS5 expression may predict poor survival and cisplatin resistance in cervical cancer. Cell Death Dis. 2020;11(7):531. doi:10.1038/s41419-020-2735-2
18. Xiao Z, Wang C, Tan Z, et al. Clinical efficacy and safety of sodium cantharidinate plus chemotherapy in non-small-cell lung cancer: a systematic review and meta-analysis of 38 randomized controlled trials. J Clin Pharm Ther. 2019;44(1):23–38. doi:10.1111/jcpt.12761
19. Tao R, Wang ZF, Qiu W, et al. Role of S100A3 in human hepatocellular carcinoma and the anticancer effect of sodium cantharidinate. Exp Ther Med. 2017;13(6):2812–2818. doi:10.3892/etm.2017.4294
20. Zang GH, Li R, Zhou RS, et al. Effects of disodium cantharidinate on dendritic cells of patients with bladder carcinoma. Oncol Lett. 2018;15(2):2273–2277. doi:10.3892/ol.2017.7589
21. Kostrzewa T, Styszko J, Gorska-Ponikowska M, et al. Inhibitors of protein tyrosine phosphatase ptp1b with anticancer potential. Anticancer Res. 2019;39(7):3379–3384. doi:10.21873/anticanres.13481
22. Yu M, Liu Z, Liu Y, et al. PTP1B markedly promotes breast cancer progression and is regulated by miR-193a-3p. FEBS J. 2019;286(6):1136–1153. doi:10.1111/febs.14724
23. Bartholomeusz C, Gonzalez-Angulo AM. Targeting the PI3K signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16(1):121–130. doi:10.1517/14728222.2011.644788
24. Amirani E, Hallajzadeh J, Asemi Z, et al. Effects of chitosan and oligochitosans on the phosphatidylinositol 3-kinase-AKT pathway in cancer therapy. Int J Biol Macromol. 2020;164:456–467. doi:10.1016/j.ijbiomac.2020.07.137
25. Kong DL, Liu Y, Wang JY, et al. Sodium cantharidinate suppresses human osteosarcoma MG63 cell proliferation and induces cell cycle arrest by inhibition of PI3K/AKT activation. Oncol Rep. 2019;41(2):1351–1358. doi:10.3892/or.2018.6906
26. Huang J, Jiang R, Chu X, et al. Overexpression of microRNA-23a-5p induces myocardial infarction by promoting cardiomyocyte apoptosis through inhibited of PI3K/AKT signalling pathway. Cell Biochem Funct. 2020;38(8):1047–1055. doi:10.1002/cbf.3536
27. Zhang Y, Liu JL, Wang J. KRAS gene silencing inhibits the activation of PI3K-Akt-mTOR signaling pathway to regulate breast cancer cell epithelial-mesenchymal transition, proliferation and apoptosis. Eur Rev Med Pharmacol Sci. 2020;24(6):3085–3096. doi:10.26355/eurrev_202003_20673
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.