Back to Journals » Cancer Management and Research » Volume 18

Colorectal Cancer Progression and Bone Metastasis: Molecular Mechanisms, Tumor Microenvironment, and Tumor-Bone Crosstalk
Authors Qi X, Hao S, Sun Y, Li M, Yang J, Kang L, Zhang L, Wang H
Received 6 November 2025
Accepted for publication 18 June 2026
Published 26 June 2026 Volume 2026:18 579585
DOI https://doi.org/10.2147/CMAR.S579585
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Javier-David Benitez-Fuentes
Xiang Qi,1,* Shuai Hao,1,* Yue Sun,2,* Manglai Li,2 Jingyuan Yang,2 Lei Kang,3 Liansheng Zhang,2 Hairui Wang2
1Graduate School, Inner Mongolia Medical University, Hohhot, Inner Mongolia Autonomous Region, People’s Republic of China; 2Department of Bone and Soft Tissue Tumor Surgery, Peking University Cancer Hospital (Inner Mongolia Campus)/Affiliated Cancer Hospital of Inner Mongolia Medical University, Inner Mongolia Cancer Center, Hohhot, Inner Mongolia Autonomous Region, People’s Republic of China; 3Department of Intensive Care Unit, Peking University Cancer Hospital (Inner Mongolia Campus)/Affiliated Cancer Hospital of Inner Mongolia Medical University, Inner Mongolia Cancer Center, Hohhot, Inner Mongolia Autonomous Region, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Liansheng Zhang, Email [email protected] Hairui Wang, Email [email protected]
Abstract: Colorectal cancer (CRC) ranks as the third most common malignancy globally and represents one of the main causes of cancer-related death. This narrative review provides a comprehensive synthesis of recent advances in the molecular mechanisms underlying CRC bone metastasis, with an emphasis on key signaling pathways, tumor microenvironment interactions, and potential therapeutic targets. Bone is a relatively uncommon metastatic site in advanced CRC (1.2– 12% of patients), but it frequently coexists with hepatic or pulmonary metastases and triggers skeletal-related events (SREs) such as pathological fractures, spinal cord compression, and hypercalcemia, which severely compromise patients’ quality of life and survival. Although the molecular mechanisms of CRC and its bone metastasis have been extensively studied, the exact mechanisms underlying its initiation and progression remain incompletely elucidated. Here, we review recent progress focusing on TGF-β signaling, epithelial-mesenchymal transition (EMT), the tumor microenvironment (TME), the Wnt/β-catenin pathway, chemokine regulation, and immune cell interactions within the bone niche. Unlike previous reviews, this article critically distinguishes CRC-specific evidence from data extrapolated from other cancers and provides an evidence-level table to guide clinical translation. By integrating clinical, translational, and preclinical evidence, we aim to present a theoretical basis for understanding CRC bone metastasis and for developing targeted therapeutic strategies.
Keywords: colorectal cancer, bone metastasis, tumor microenvironment, tumor-bone crosstalk, TGF-β signaling, Wnt/β-catenin pathway
Introduction
Colorectal cancer (CRC) is one of the most common malignancies worldwide and ranks third in incidence and second in cancer-related mortality, posing a major threat to human health.1
The reasons for the rising incidence of CRC are unknown, and suspected risk factors include antibiotic use, obesity, lack of physical activity, and changes in dietary patterns, which can affect the gut microbiota.2 In addition, colorectal carcinogenesis and metastasis are associated with numerous genetic and molecular alterations, including epigenetic changes, genetic mutations, microsatellite instability, and chromosomal instability, which disrupt key signaling pathways and deregulate cell and tumor growth.3 Notably, the clinicopathological stratification of cancers of unknown primary (CUP) has recently been further delineated, and the newly recognized favorable subsets now include a CRC-like CUP subtype.4 Because this clinical entity is generally managed according to CRC treatment paradigms, changes in its recognition and classification likely account in part for the rising incidence trends observed in CRC.
The pathogenesis of CRC begins with the malignant transformation of normal intestinal mucosal epithelium. These aberrant intestinal epithelial cells (IECs) lose their inherent structural arrangement and organizational continuity, thereby gaining the potential to develop into adenomas.5 As the disease progresses, tumor cells penetrate the basement membrane and the intestinal mucosal barrier, infiltrating adjacent vascular or lymphatic systems. Through this multistep process, tumor cells gradually acquire the capacity for distant metastasis, which in advanced disease may involve organs such as bone. Despite advances in early detection and treatment, lymphatic and distant metastasis are still the primary causes of mortality in individuals with a recent diagnosis of CRC, and the prognosis of advanced CRC is poor.6 It should be pointed out that among patients undergoing surgical treatment, elderly individuals are more likely to develop severe postoperative complications, although there is no consensus that age itself affects survival outcomes.7 This may be confounded by differences in stage at presentation, tumor site, preexisting comorbidities, and the type of treatment received.
As one of the metastasis sites for advanced CRC, bone metastasis is detected in 1.2–12% of individuals with CRC, and often occurs simultaneously with liver or lung metastasis.8 Once bone metastasis occurs in individuals with advanced CRC, not only do they have to face the adverse effects associated with anti-tumor therapy, but they are also prone to several skeletal-related events (SREs), such as severe bone pain, pathological fractures, spinal cord compression, and hypercalcemia, which seriously reduce the quality of life and significantly shorten the survival period.9 However, the molecular basis of CRC bone metastasis remains incompletely understood, particularly the mechanisms governing bone homing, colonization of the bone niche, and dynamic interactions between tumor cells and the bone microenvironment. This incomplete understanding of CRC bone metastasis limits insight into its underlying biological processes and hampers further refinement of potential biomarkers, risk stratification, and therapeutic strategies targeting molecular mechanisms.
This narrative review provides a comprehensive synthesis of recent advances in the molecular drivers of CRC progression and the specific mechanisms of bone metastasis, with a focus on TGF-β signaling, epithelial-mesenchymal transition (EMT), the tumor microenvironment (TME), the Wnt/β-catenin pathway, chemokine regulation, and immune cell interactions within the bone niche. By integrating clinical, translational, and preclinical evidence, we aim to present a theoretical basis for understanding CRC bone metastasis and for developing targeted therapeutic strategies.
Molecular Drivers of CRC
CRC is a leading global cause of cancer-related deaths. Its pathogenesis originates from progressive accumulation of histopathological, morphological, and epigenetic changes in healthy colonic epithelium, resulting in colorectal adenomas and invasive adenocarcinomas.10 In recent years, deepening research on molecular mechanisms and signaling pathways has provided novel insights into CRC initiation, progression, and metastasis. Several molecular mechanisms, including EMT, apoptosis, chromosomal instability, immune evasion, and TME, are involved in the initiation and onset of tumors.11 These discoveries have also promoted the development of biomarker-driven and histology-agnostic therapeutic strategies in metastatic CRC. However, their clinical implementation remains limited by inter-platform variability in molecular testing, intratumoral heterogeneity, adaptive resistance mechanisms, high testing costs, and unequal access to comprehensive genomic profiling.12
Herein, we delineate the context-dependent roles of TGF-β, EMT, TME, and Wnt signaling in mediating CRC cell proliferation, invasive capacity, and metastatic competence, with an emphasis on their relevance to bone metastasis.
TGF-β: The Paradoxical Shift from Tumor Suppressor to Promoter
TGF-β is a multifunctional cytokine participating in cell growth, differentiation, embryonic development, wound healing, immune regulation, tissue fibrosis, and tumorigenesis.13 In CRC, TGF-β signaling exhibits a context-dependent dual role: it can restrain early tumor growth by inducing cell-cycle arrest, whereas in advanced disease it facilitates malignant progression through EMT induction, invasion, and immune evasion.14 These pro-metastatic effects are highly influenced by tumor stage, cell type, and the surrounding tissue microenvironment. In the context of bone metastasis, TGF-β has been extensively implicated in breast and prostate cancer models, particularly through its role in coupling tumor growth with bone remodeling.15 However, CRC-specific evidence linking TGF-β signaling to bone tropism or bone colonization remains limited, highlighting an important knowledge gap. TGF-β can also suppress immune responses in cancer patients while stimulating angiogenesis under hypoxic conditions. Overexpression of TGF-β induces EMT, extracellular matrix (ECM) deposition, and the formation of cancer-associated fibroblasts (CAFs), ultimately leading to cancer progression.16
The TGF-β/SMAD transduction pathway is commonly mutated in CRC. The SMAD4 protein serves as a key transcription factor in the TGF-β pathway, which modulates cellular proliferation, differentiation, and apoptosis; its aberrant alteration is strongly correlated with poor prognosis in CRC patients.17 SMAD proteins are intracellular molecules that transmit TGF-β signals from the cell membrane to the nucleus to control target gene expression.18 Functional loss of SMAD4 due to genetic mutations hinders its binding to TGF-β receptors, inhibiting normal signal transduction and resulting in dysregulation of cell cycle regulation and apoptosis.19 This leads to CRC cells being susceptible to lymph node metastasis and stimulating tumor angiogenesis, which accelerates the progression of the disease and contributes to a poor prognosis.20 CRC cells evade the tumor-suppressive effects of the TGF-β pathway via SMAD4 loss and disruption of TGF-β receptor-mediated cell cycle regulation. The TGF-β pathway modulates tumor metastasis by influencing vascular endothelial growth factor (VEGF). In the HCT-116 CRC cell line, VEGF overexpression due to SMAD4 loss improves malignant cell migration.21 Conversely, SMAD4 overexpression inhibits CRC growth by suppressing VEGF-A and VEGF-C expression and induces tumor cell apoptosis in xenograft models.22
Elevated TGF-β levels within the CRC TME exert immunosuppressive effects. Various cellular components, including stromal cells, vascular endothelial cells, and macrophages, produce elevated TGF-β, restricting infiltration of CD4⁺ and CD8⁺ T cells into tumor tissue. TGF-β also inhibits T-cell activity and impedes acquisition of the TH1-effector phenotype, conferring immune escape capabilities to CRC cells.23 In the presence of IL-6, TGF-β drives differentiation of CD4⁺ T cells into Th17 cells, although Th17 cells play a limited role in anti-tumor immunity.24 Beyond T lymphocytes, TGF-β regulates innate immune components within the CRC TME. Elevated TGF-β signaling suppresses tumor infiltration and effector functions of natural killer (NK) cells and interferes with maturation and antigen-presenting capacity of dendritic cells (DCs), weakening innate immune surveillance.19 Moreover, TGF-β promotes polarization of macrophages from the M1 phenotype toward the tumor-supportive M2 phenotype, establishing an immunosuppressive microenvironment that favors tumor proliferation and immune evasion.
Platelet-derived growth factor D (PDGF-D), a part of the PDGF family (PDGF-A–D), is overexpressed in multiple cancers, including breast cancer, CRC, hepatocellular carcinoma, and lung cancer.25 PDGF-D exerts its biological functions through binding to platelet-derived growth factor receptor-β (PDGFR-β), inducing rapid receptor phosphorylation and activation of downstream signaling pathways.26 Previous studies have demonstrated that serum PDGF-D levels are elevated in patients with CRC and are closely associated with tumor progression.27 In CRC, PDGF-D is overexpressed and regulated by TGF-β through the Notch1/Twist1 pathway, promoting tumor growth, invasion, and angiogenesis.28 Through its involvement in tumor vascular remodeling, PDGF-D supports an adequate supply of blood and nutrients required for sustained tumor mass proliferation. Despite its critical role, no specific inhibitors targeting PDGF-D are currently available, underscoring the need for further in-depth investigation. Targeting the multiple signaling pathways that regulate PDGF-D expression, such as Notch, PI3K/AKT, and NF-κB, may provide alternative therapeutic approaches worthy of future exploration.
TGF-β also drives the conversion of fibroblasts into CAFs, accelerating CRC progression and metastasis.29 CAFs-derived stromal-derived factor-1 (SDF-1) enhances CRC cell migration and invasion via paracrine signaling.30 And CAFs-secreted interleukin-11 (IL-11) activates the GP130/STAT3 pathway in CRC cells, conferring a survival advantage to metastatic cells.31 Elevated TGF-β expression further strengthens the adhesion between CAFs and CRC cells, promoting proliferation, invasion, and hepatic metastasis in vivo.32
TGF-β plays a complex molecular role in maintaining tissue homeostasis, tumorigenesis, and immune regulation (Figure 1). The dual nature of TGF-β presents significant challenges for the development of targeted therapies. Nevertheless, targeting the TGF-β pathway remains highly valuable, particularly when combined with immunotherapeutic agents. Integration of next-generation sequencing (NGS) and genomic profiling in clinical trials will enable molecular subtyping of TGF-β signaling in CRC patients, guiding development of personalized combination therapies.
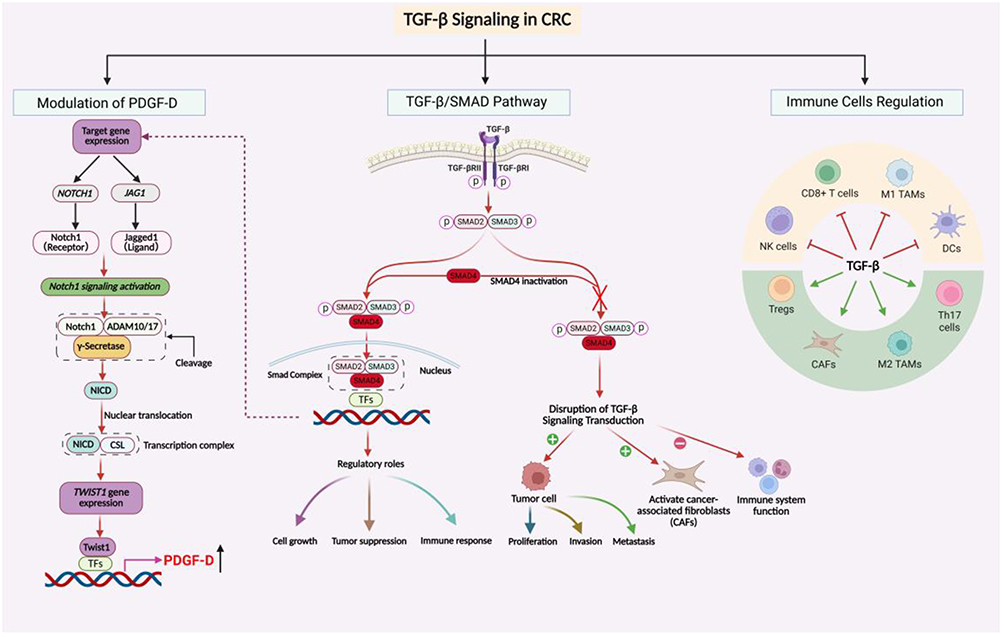 |
Figure 1 TGF-β Signaling in CRC. The schematic illustrates three major aspects of TGF-β signaling in CRC: PDGF-D regulation through the SMAD4–Notch–TWIST1 axis, canonical TGF-β/SMAD signaling, and immune cell modulation. SMAD4 inactivation may disrupt canonical TGF-β signaling and promote tumor cell proliferation, invasion, metastasis, CAF activation, and immune suppression. Green and red arrows indicate stimulatory and inhibitory effects, respectively. Abbreviations: NICD, Notch intracellular domain; TFs, transcription factors; CAFs, cancer-associated fibroblasts; NK, natural killer; Tregs, regulatory T cells; TAMs, tumor-associated macrophages; DCs, dendritic cells. |
EMT: The Molecular Key for Cell Migration and Colonization
EMT is a cellular program in which cells lose epithelial features and acquire mesenchymal traits, playing vital roles in embryonic development, wound healing, and cancer metastasis.33 EMT drives the invasion and metastasis of epithelial-derived carcinomas by the disruption of cell-cell adhesion, reduction of polarity, and reorganization of the cytoskeleton toward a mesenchymal phenotype.34 This multistep process occurs within the dynamic TME and represents a key adaptive response of cancer cells.35 Cells undergoing EMT are characterized by reduced expression of epithelial markers (E-cadherin, cytokeratin, occludin) and upregulation of mesenchymal markers (N-cadherin, vimentin, fibronectin), exhibiting enhanced cell motility and invasive properties.36 Epithelial cells lose their polygonal and cobblestone morphology, acquiring a spindle-shaped mesenchymal appearance that allows the acquisition of metastatic properties.37 This phenotypic transformation favors malignant cells to break away from the primary foci, enter the surrounding tissues, and cross the vascular barrier to enter the systemic circulation, resulting in the generation of metastases in distant organs.
EMT plays a crucial role in the invasion and metastasis of many malignancies, including CRC, and is highly associated with poor prognosis.38 CRC cells exposed to the changing TME can lose their epithelial phenotype and convert to mesenchymal phenotypes, significantly enhancing migration and invasion potential. Throughout EMT, tumor cells undergo tight junction dissolution, loss of apical-basal polarity, and cytoskeletal reorganization, enabling acquisition of an invasive phenotype.39 EMT programming further enables tumor cells to adapt to the dynamic TME, thereby facilitating migration to distant tissue sites. Identification of definitive EMT-associated biomarkers may help predict bone metastatic potential in CRC patients. However, whether EMT markers such as circulating vimentin⁺ or TWIST1⁺ cells specifically enrich for bone-homing capacity remains to be established in CRC cohorts.
Wei et al discovered that tumor-associated macrophages (TAMs) can regulate the JAK2/STAT3/miR-506-3p/FoxQ1 signaling axis to trigger EMT program, enhance invasion and metastasis of CRC cells, and increase expression of chemokine C-C motif ligand 2 (CCL2).40 CRC cell-derived CCL2 binds to and activates endothelial cells via chemokine C-C motif receptor 2 (CCR2), then activates JAK2-Stat5 and p38MAPK pathways, increasing blood vessel permeability and benefiting cancer cell intravasation and extravasation, thereby promoting metastasis.41,42
Runt-related transcription factor 1 (RUNX1), a part of the RUNX family, participates in embryonic development, tumorigenesis, and inflammatory response.43 RNA sequencing of 217 CRC patients showed significant RUNX1 mRNA upregulation in tumor tissues.44 TGF-β upregulates RUNX1, promoting migration and EMT in CRC cells; knockdown of RUNX1 reverses these phenomena.45 These findings establish RUNX1 as a prospective therapeutic target for inhibiting CRC metastasis.
Pre-B-cell leukemia transcription factor 3 (PBX3), a highly conserved member of the PBX protein family, exhibits oncogenic activity in hematological cancers and multiple solid tumors.46 PBX3 is essential for the full EMT phenotype in colon malignant cells, as its genetic knockdown partially blocks ZEB1- and SNAIL-induced EMT while upregulating E-cadherin.47 Additionally, PBX3 promotes proliferation and confers resistance to apoptosis in CRC cells by directly binding to and repressing the p53 promoter, modulating the p53/p21 signaling axis.48 Elevated PBX3 expression correlates significantly with accelerated tumor progression and reduced survival in CRC patients, a promising prognostic predictor that may aid in treatment decision-making for patients with CRC.
MicroRNAs (miRNAs) are small non-coding RNAs whose dysregulation is closely related to cancer initiation, progression, metastasis, and treatment resistance. miR-452-5p is upregulated in early-stage CRC and triggers cell growth via ERK pathway activation while suppressing EMT by inhibiting the transcription factor Slug and upregulating E-cadherin, reducing cancer cell invasiveness.49 Furthermore, specific miRNAs directly target mesenchymal genes, exemplified by miR-17-5p, which controls EMT by targeting vimentin in CRC.50 Beyond their role in EMT regulation, circulating and tissue miRNAs have also been investigated as predictive biomarkers of treatment response in advanced CRC. For example, upregulation of serum miR-19a has been associated with resistance to FOLFOX chemotherapy, while circulating miR-126 dynamics have been linked to response or resistance to bevacizumab-containing regimens in metastatic CRC. In the anti-EGFR setting, dysregulation of miR-31, miR-100, miR-125b, and miR-7 has been implicated in cetuximab resistance, suggesting that miRNA profiles may help connect EMT-related tumor plasticity with therapeutic tolerance in metastatic disease.51
The central role of EMT in CRC progression, metastasis, and tumor resistance has been confirmed in preclinical and early clinical investigations.52 Its molecular markers may act as outcome indicators and possible therapeutic targets in CRC. Identification of definitive EMT-associated biomarkers will present a theoretical basis for developing individualized treatment approaches for cancer patients, which deserves further investigation.
TME: Indispensable “Soil” and Conspirators
Genetic changes affect the malignant potential of cancer cells, as does the TME, a necessary prerequisite for cancer progression and metastasis. Tumors are complex accumulations of malignant cells and simultaneously constitute intricate ecosystems composed of heterogeneous cancer cells, immune cells, stromal cells, blood vessels, and lymphatic vessels, with their composition differing according to the tumor type.53 The TME is characterized by hypoxia, acidity, and chronic inflammation, providing favorable conditions for tumor cell survival, proliferation, and invasion. Concurrently, the TME accelerates tumor progression by inhibiting effector T cell (Teff) function and activating immunosuppressive cells, establishing immune escape mechanisms.
The Inflammatory Microenvironment Within the TME
Rudolf Virchow first documented leukocytic infiltrates within tumor tissues in the 19th century, showing a prospective correlation between inflammation and malignancies.54 Inflammatory responses play roles at several stages of tumor aggressiveness, malignant cell survival, immune adaptation and escape, and altered responses to hormones and chemotherapy. Acute inflammation participates in injured tissue restitution through leukocyte recruitment, neovascularization, and ECM remodeling.55 Conversely, chronic inflammation drives dysregulation of tissue repair responses, fostering a tumor-permissive microenvironment conducive to oncogenesis. In tumors, which resemble “wounds that never heal”, a wide variety of immune and inflammatory cells enter the tissue, favoring cell renewal, proliferation, and invasion.56 Inflammation can improve the cross-presentation of tumor antigens and trigger subsequent anti-tumor immune responses, while, on the other hand, it can promote malignant tumor behavior.57
Epidemiological investigations indicate that chronic inflammation predisposes individuals to various malignancies, and underlying infections and inflammatory responses are associated with 15–20% of global malignancy deaths.58 The inflammatory microenvironment is a fundamental component of all tumors. Chronic inflammation and increased epithelial turnover promote progression from low- and high-grade dysplasia to CRC. Individuals with inflammatory bowel disease (IBD), such as ulcerative colitis (UC) and Crohn’s disease (CD), have significantly elevated CRC risk.59 Patients with long-standing UC or CD have an approximately two- to three-fold elevated risk, particularly with extensive colonic involvement.60,61
Inflammasomes are protein complexes critical for innate immunity, regulating caspase-1 (CASP1) stimulation and subsequent maturation and release of IL-1β and IL-18, triggering inflammatory responses. The NLRC4 inflammasome serves as a tumor inhibitor in CRC, while NLRC4 variants can cause recessive immune dysregulation or serve as risk factors for UC, increasing CRC susceptibility.62 Furthermore, inflammation affects key cytokine receptor-mediated signaling pathways that control tumor-initiating and tumor-promoting processes in CRC, such as activation of nuclear factor-κB (NF-κB) downstream of tumor necrosis factor (TNF) receptor and IL-1 receptor signaling, as well as gp130-dependent activation of STAT3 downstream of IL-6- and/or IL-11-induced signaling.63
Inflammation can epigenetically modulate cancer-related genes, silencing key tumor suppressors.64 Prostaglandin E2 (PGE2), abundant in human gastrointestinal cancers including CRC, binds to its receptors (EP1-EP4) to suppress tumor suppressor and DNA repair genes by inducing DNA methyltransferases DNMT1 and DNMT3B.65 Additionally, PGE2 triggers cancer stem cell (CSC) formation and expansion by stimulating NF-κB through EP4-dependent PI3K/MAPK signaling.
Immunoregulation Within the TME
The immune system suppresses tumorigenesis via immune surveillance, recognizing and eliminating malignant cells. Tumor development disrupts immune homeostasis, impairing immune cell function within the TME. Infiltrating immune cells often lose anti-tumor activity and instead interact with tumor cells to trigger oncogenesis and progression.66
The tumor-infiltrating immune cells within the TME comprise DCs, macrophages, NK cells, T cells, B cells, and other innate immune cells such as neutrophils and mast cells. The immune response has a complicated function in the early phases of tumor development, which is related to cancer development and, more specifically, the CRC progression. In CRC, mismatch repair (MMR) status is closely associated with anti-tumor immunity and immune checkpoint expression. Immune cell programmed death-ligand 1 (PD-L1) expression has been reported to be higher in microsatellite instability-high (MSI-H) CRC than in MMR-proficient or MSI-low tumors, with no marked differences among different MSI-H molecular subtypes.67 Recommended screening for defective DNA MMR includes immunohistochemistry (IHC) for MMR proteins and/or MSI testing; however, biological and technical heterogeneity may complicate clinical interpretation.68 For example, discordant MMR IHC results may occur in some cases, potentially because of additional somatic alterations.
As components of the cellular adaptive immune response, T helper cells (CD4+ T cells), cytotoxic T lymphocytes (CTLs), and M1-phenotype TAMs inhibit tumor growth and development, while regulatory T cells (Tregs) and M2 phenotype TAMs promote tumor progression.69 CD4⁺ helper T cells activate CTLs, which directly eliminate tumor cells by the recognition of tumor antigen peptides provided by antigen-presenting cells, like DCs.70 The spatial density of CTLs within cancer cell nests shows a significant positive correlation with patient survival.71,72 However, with disease progression, prolonged antigen exposure triggers CTL exhaustion, characterized by impaired cytotoxic function and overexpression of inhibitory receptors such as programmed death-1 (PD-1), cytotoxic T lymphocyte antigen-4 (CTLA-4), and lymphocyte-activation gene-3 (LAG-3), enabling tumor immune evasion.73
Tregs, an immunosuppressive CD4⁺ T-cell subset defined by forkhead box protein P3 (FOXP3) expression, maintain immune tolerance but become pro-inflammatory and tumor-promoting in CRC.74 Tregs mediate tumor immune escape by suppressing growth, stimulation, and cytokine generation of CD4⁺ and CD8⁺ T cells. Increased intratumoral Treg infiltration is related to enhanced tumor progression, immunotherapy failure, poorer prognosis, and therapeutic resistance in CRC.75 CRC cell-derived CCL20 recruits Tregs via the FOXO1/CEBPB/NF-κB pathway, and Tregs subsequently promote chemoresistance.76
TAMs represent 30–50% of cellular constituents within the TME and are crucial in fostering a permissive niche for tumor progression. They are categorized into anti-tumor M1 and pro-tumor M2 subtypes.77 The transition between M1 and M2 polarization states is largely governed by cytokines present in the TME, such as IL-4, M-CSF/CSF-1, IL-10, IL-33, IL-21, and TGF-β, allowing tumor cells to utilize macrophage plasticity for their own benefit.78 During advanced tumor progression, M2-polarized macrophages promote tumor cell proliferation, angiogenesis, invasion, and metastasis by upregulating angiogenesis-related genes, activating endothelial growth factor signaling, and expressing matrix proteases.79 Studies have found that TAMs produce TGF-β to support hypoxia-inducible factor 1α (HIF-1α) expression, upregulating Tribbles pseudokinase 3 (TRIB3) in tumor cells, leading to activation of the β-catenin/Wnt signaling pathway and ultimately enhancing stem cell-like phenotype and cell invasion in CRC.80
WNT: Guardian of Intestinal Tract Cell Homeostasis and Instigator of Cancerous Progression
The Wnt pathway constitutes a highly conserved and complex network of protein interactions that regulates embryonic development, cell growth, differentiation, apoptosis, and malignancies correlated to inflammation.81 This pathway mainly includes classical (Wnt/β-catenin) and noncanonical (β-catenin-independent) pathways. Current research on the Wnt pathway is mostly focused on the Wnt/β-catenin branch.
In the canonical Wnt/β-catenin pathway, Wnt ligands bind the low-density lipoprotein receptor-related protein 5/6 (LRP5/6) and Frizzled (Fzd) receptor complex to send transmembrane signaling.82 This results in cytoplasmic β-catenin overload, nuclear translocation, and interaction with the transcription factor T cell factor/lymph enhancer factor (TCF/LEF), leading to the activation of transcription of proliferation-related genes. Without Wnt signaling activation, β-catenin is phosphorylated by the axis inhibition protein (Axin)/adenomatous polyposis coli (APC)/casein kinase 1α (CK1α)/glycogen synthase kinase-3β (GSK-3β) destruction complex and then ubiquitinated and degraded by the proteasome, maintaining low pathway activity.83 Figure 2 presents a schematic diagram illustrating the key mechanisms governing activation and inhibition of the Wnt/β-catenin signaling pathway.
 |
Figure 2 Wnt/β-catenin pathway ON and OFF. In the Wnt-ON state, Wnt binding to Frizzled/LRP activates Disheveled and inhibits the β-catenin destruction complex, allowing β-catenin to accumulate, translocate into the nucleus, and activate TCF/LEF-dependent target gene transcription. In the Wnt-OFF state, the Axin/APC/GSK-3β/CK1α destruction complex phosphorylates β-catenin, leading to ubiquitin-mediated proteasomal degradation and suppression of target gene expression. Abbreviations: LRP, low-density lipoprotein receptor-related protein; APC, adenomatous polyposis coli; GSK-3β, glycogen synthase kinase-3β; CK1α, casein kinase 1α; TCF/LEF, T-cell factor/lymphoid enhancer factor. |
The canonical Wnt/β-catenin pathway is vital for keeping normal intestinal epithelial architecture and is the most important signaling pathway for keeping the self-renewal of intestinal stem cells (ISCs). This pathway can drive the continuous proliferation of ISCs and inhibit their premature differentiation, ensuring the stable existence of the ISC niche positioned at the crypt base.84 Given the fundamental function of the Wnt pathway in maintaining homeostasis, it is unsurprising that its overactivation with the mutation of downstream genes leads to multiple cancer diseases, including CRC.85 Several investigations have illustrated a direct causal relationship between Wnt signaling abnormalities and human CRC. Eto et al demonstrated that RNF43 mutations promote tumor growth and increase recurrence rates in CRC patients by activating Wnt signaling.86 Separately, experimental evidence revealed that POLE2 expression is upregulated in CRC, where it facilitates EMT through modulating the Wnt/β-catenin signaling pathway.87 Wnt2 is an evolutionarily conserved secreted-type glycoprotein in the Wnt signaling pathway. miR-627-5p suppresses the classical Wnt/β-catenin pathway and its downstream target gene expression by directly suppressing Wnt2, thereby reducing the malignant tendency of CRC cells.88
Aberrant Wnt/β-catenin signaling can transform ISCs into CSCs, and these cancer cells with high Wnt activity exhibit sustained self-renewal and heightened tumorigenic potential.89 In mediating this malignant transformation, mutations in the APC gene play a central driving role. APC mutations were initially detected in familial adenomatous polyposis (FAP), and biallelic APC inactivation initiates approximately 75%–80% of CRC cases.90 APC is considered a critical negative regulator by mediating β-catenin phosphorylation and ubiquitin-mediated degradation. Mutational inactivation of APC results in aberrant stabilization of β-catenin and activation of the Wnt signaling cascade, thereby upregulating key downstream target genes, including Cyclin D1 and C-Myc, and facilitating CRC tumorigenesis and disease progression.
Axin1 and Axin2 mutations are also found in a subset of CRC. These proteins serve as critical molecular scaffolds for assembling the β-catenin destruction complex. Inactivating mutations in Axin genes impair the assembly efficiency of the destruction complex, causing aberrant β-catenin accumulation and contributing to CRC tumorigenesis.91,92
Investigating the mechanisms of the Wnt pathway in cell growth, differentiation, and maintenance and regeneration of tissue homeostasis is of great value for understanding CRC pathology and developing diagnosis and treatment strategies. In particular, abnormalities such as overactivation of signaling pathways or mutations in key components can significantly elevate the CRC susceptibility in humans, which has a critical function in CRC occurrence and progression.93 Topical application of Wnt modulators may be a key direction for future research, which is believed to lay a novel cornerstone for the precise treatment of Wnt-driven malignancies such as CRC.
Bone Metastasis in CRC
Metastasis is a hallmark of advanced cancer and a key therapeutic challenge. Metastatic dissemination arises when genetically unstable tumor cells adapt to microenvironments at sites distant from the primary lesion.94
Bone is the third most predominant location of distant metastasis after the lung and liver. Bone metastases can cause SREs, including spinal cord compression, pathological fractures, and severe pain, thereby markedly impairing prognosis and quality of life.95 Most Individuals with bone metastases present with pathologic T3/4 disease at the time of CRC diagnosis. Bone metastasis in CRC typically occurs via dissemination from the colorectal site through the Batson vertebral plexus, most frequently affecting the spine, though it may also involve the sacroiliac region, pelvis, long bones of the extremities, and thoracic skeletal structures.96,97
General Patterns of Bone Metastasis in CRC
Among cancer-related causes of death, CRC ranks as the second leading cause of cancer mortality among both males and females.98 Bone is a predominant location for metastasis of malignant solid tumors, with metastatic bone disorder most prevalently originates from pulmonary (24.8%), prostatic (19.4%), breast (19.3%), gastrointestinal (9.4%), and urological (6.5%) cancers.99 Among common malignancies with bone metastasis, prostate cancer typically presents with osteoblastic lesions, whereas breast cancer and CRC predominantly develop osteolytic metastases, the latter being prone to causing structural damage.100
Although isolated bone metastasis is comparatively uncommon in CRC patients, it frequently co-occurs with hepatic and pulmonary metastases. The spine is the most commonly involved location. Right colon cancer correlated significantly with long bone metastasis, while left colon cancer correlated significantly with spinal bone metastasis.101 CRC exhibits a pronounced propensity for hepatic and pulmonary metastasis, with lung metastases predicted to have a more prospective progression to bone than liver metastases.102
Mechanism of CRC Bone Metastasis
Bone Remodeling and Chemokine Regulation in Bone Metastasis
The basic multicellular unit (BMU), comprising osteoblasts, osteoclasts, and osteocytes, serves as a key structural and functional entity in bone and is necessary for maintaining bone homeostasis. Pathological fractures and hypercalcemia resulting from bone metastasis disrupt this homeostatic balance, triggering aberrant osteoclast activation and leading to excessive bone resorption at tumor sites.103 Tumor-induced osteoclast overactivation is a primary driver of osteolytic bone lesions and cancer-associated pain.104 In the TME, reciprocal interactions between osteoclasts and tumor cells create a permissive niche that promotes bone metastasis, forming a “vicious cycle” of bone destruction and tumor bone metastasis.105
Chemokines play critical roles in regulating tumor growth, invasion, and metastasis.106 Among them, chemokine C-C motif ligand 7 (CCL7) is ubiquitously elevated in expression within the TME of multiple malignancies, such as CRC, breast, renal, and gastric cancers.107,108 CCL7 has a key function in recruiting osteoclast precursors (OCPs) during CRC bone metastasis, promoting their migration to metastatic sites and enhancing the bone-resorptive capacity of CRC cells.109 Inhibition of CCL7 significantly reduces osteolysis and preserves trabecular bone architecture in CRC bone metastasis. Additionally, CCL3 is expressed in CRC tissues and metastatic lesions, with expression relating to clinical stage. In general, bone marrow-derived monocytes/macrophages (BMMs) differentiate into osteoclasts.110 In the microenvironment of bone metastasis in CRC, CCL3 promotes osteoclastogenesis by enhancing bone marrow-derived BMM infiltration.102 Further investigation illustrated that the ERK/CREB pathway can be stimulated in BMMs by CRC cell-derived epidermal growth factor (EGF) and promotes CCL3 generation.111 Inhibition of either EGF or CCL3 effectively suppresses osteoclastogenesis and attenuates bone destruction.
The Bone Microenvironment: A “Fertile Soil” for Bone Metastasis
The “seed and soil” hypothesis posits that the selective growth advantage of cancer cells is determined by the dynamic interplay between their inherent properties and the specific organ microenvironment. Upon reaching a distant organ, disseminated tumor cells “seed” into a friendly “soil” and finally turn into metastatic lesions.112 Within the bone microenvironment, diverse cell types release signaling molecules that collectively establish a privileged site for metastatic cancer cell colonization. After cancer cells metastasize to bone, osteoclast-stimulating factors, such as parathyroid hormone-related protein (PTHrP) secreted by metastatic cancer cells, activate bone destruction, leading to increased release of bone-derived growth factors, including insulin-like growth factors (IGFs) and TGF-β.113
The physiological state of the bone microenvironment is characterized by hypoxic conditions, with bone exhibiting significantly lower oxygen partial pressure than other tissues.114 Hypoxia stimulates osteoclast production by inducing pro-osteoclastogenic cytokines such as M-CSF, receptor activator of nuclear factor-κB ligand (RANKL), VEGF, insulin-like growth factor 2 (IGF-2), and growth differentiation factor-15 (GDF-15), while suppressing osteoprotegerin (OPG) generation.115,116 Hypoxia also induces the expression of HIF-1α, and its elevated expression triggers bone metastasis in tumor cells.113
Immune Cells in the Bone Metastasis
The skeletal system is a distinct organ that functions as an integral component of both the musculoskeletal and immune systems.117 Bone metastasis development is often accompanied by bone destruction and alterations in immune composition, involving key players including Tregs, myeloid-derived suppressor cells (MDSCs), and TAMs.
Tregs are potent immunosuppressive cells that weaken anti-tumor immunity within metastatic lesions. Preclinical and clinical investigations illustrate that Tregs impair immune surveillance, inhibit effective anti-tumor immunity, and promote tumor progression. Tregs can dampen Teff responses through immunosuppressive cytokines, including IL-10, IL-35, and TGF-β, and through direct cell–cell interactions. In a murine model of bone metastasis from breast cancer, the overexpression of cyclooxygenase-2 (COX2) in tumor cells enhanced Tregs recruitment and increased apoptosis of intratumoral CD8⁺ T cells.118 Furthermore, in the immune microenvironment of prostate cancer bone metastasis, the population of Tregs is increased, and Tregs transfer to bone marrow via the C-X-C motif receptor 4 (CXCR4)/ C-X-C motif chemokine ligand 12 (CXCL12) pathway.119 Although direct CRC-specific evidence remains limited, these findings indicate that the bone marrow can serve as a reservoir for activated Tregs and may provide an immunosuppressive niche that facilitates metastatic colonization.
MDSCs represent a heterogeneous population of myeloid-lineage cells endowed with immunosuppressive capacity. MDSCs can inhibit NK cell function through upregulation of inhibitory receptors such as PD-1 and by secreting immunosuppressive factors including arginase-1 (Arg-1), reactive oxygen species (ROS), and nitric oxide (NO).120 MDSCs also hinder T cell activation by inducing inducible nitric oxide synthase (iNOS) expression.121 In CRC, elevated circulating MDSC levels have been correlated with shorter progression-free interval (PFI) and accelerated tumor metastasis and progression.27,122 In breast cancer bone metastasis models, MDSCs expand within metastatic lesions and differentiate into osteoclasts. The expanded MDSC population exhibits increased TGF-β and IL-17 expression, enhancing PTHrP expression and driving RANKL-dependent osteoclast activation, thereby further activating osteoclast-mediated bone destruction.123,124 These mechanisms are biologically plausible in CRC bone metastasis but require further CRC-specific validation.
TAMs constitute a major cellular component of the TME and drive tumor development and dissemination via phagocytosis and the production of diverse cytokines and chemokines.125 In CRC, TAM-derived cytokines, including TGF-β, IL-6, and IL-1β, can promote EMT, stemness, and invasive behavior in tumor cells.126,127 Correlative clinical data have shown that elevated TAM infiltration is linked to unfavourable outcomes in over 80% of human malignancies, including colorectal, breast, ovarian, and non-small cell lung cancer.128 TAMs predominantly exhibit an M2-like pro-tumor phenotype, contributing to cancer cell proliferation, immune evasion, and reduced sensitivity to anti-cancer drugs.129 Current studies have shown a pro-metastatic role for macrophages in bone, suggesting their potential as a novel target for immunotherapy in bone metastases.130 Tumor cells that reach bone tissue can induce macrophages in the bone microenvironment to increase C-X-C motif chemokine ligand 10 (CXCL10) expression, thereby promoting bone metastasis through CXCL10 binding to C-X-C motif receptor 3 (CXCR3) on tumor cells, whereas CXCL10 neutralization reduces bone metastatic burden.131,132 Macrophage depletion has also been shown to restrict tumor growth and improve responses to chemotherapy or anti-angiogenic therapy in experimental models.127 Therapeutically, targeting the CSF-1/CSF-1R axis or reprogramming M2-like TAMs toward an anti-tumor M1-like phenotype may represent promising strategies.127,129,133 However, the high phenotypic plasticity of TAMs remains a major barrier to clinical translation.
Together, these findings indicate that CRC bone metastasis is shaped by coordinated interactions among tumor-intrinsic signaling pathways, osteoclast-mediated bone remodeling, chemokine-driven cellular recruitment, and immune suppression within the bone niche. However, the strength of evidence varies substantially across mechanisms. Some pathways, such as CCL7-mediated osteoclast precursor recruitment and the EGF/CCL3/ERK/CREB axis, have direct CRC-specific preclinical support, whereas others, including TGF-β-driven tumor-bone crosstalk, Treg accumulation, and MDSC-mediated osteoclastogenesis, are partly inferred from other osteotropic cancers. To clarify these distinctions and improve readability, the major molecules, pathways, and cellular mediators discussed in this review are summarized in Table 1 according to their proposed relevance to CRC bone metastasis and the corresponding level of evidence.
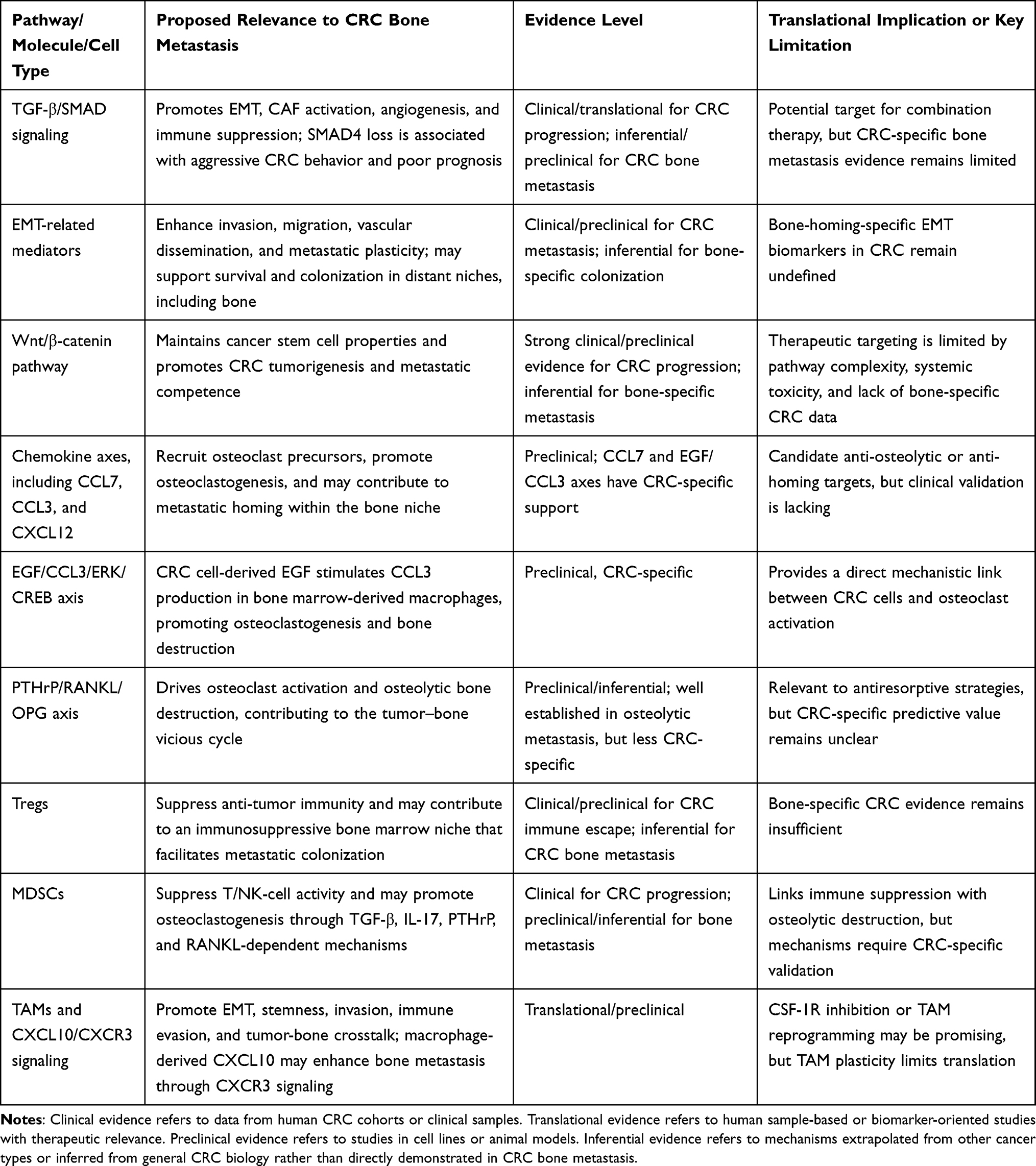 |
Table 1 Major Molecular Pathways and Cellular Mediators Implicated in CRC Bone Metastasis |
Conclusions
Bone metastasis in colorectal cancer, though less frequent than hepatic or pulmonary spread, represents a critical transition to late-stage disease with profound effects on patient mobility, independence, and survival. This narrative review distills several bone-metastasis-specific mechanistic themes:
First, the TGF-β/EMT/Wnt axis forms a core signaling network that not only drives primary tumor invasion but also primes CRC cells for bone colonization, in part by enhancing chemokine production (CCL7, CCL3) that recruits osteoclast precursors. Second, the bone microenvironment —with its unique hypoxia, mineralized matrix, and resident osteoblasts/osteoclasts — acts as a fertile “soil” that actively reprograms disseminated tumor cells via growth factors (TGF-β, IGFs) and metabolic signals. Third, immune components within the bone metastatic niche, particularly Tregs, M2-polarized TAMs, and MDSCs, create a profoundly immunosuppressive and osteoclastogenic environment that distinguishes bone metastases from primary tumors and other metastatic sites.
The most robust mechanistic themes supported by CRC-specific evidence include: (i) CCL7/EGF/CCL3-driven osteoclast recruitment; (ii) APC/β-catenin mutations as drivers of both tumorigenesis and bone tropism; and (iii) TAM-derived TGF-β/HIF-1α/TRIB3 signaling that enhances stemness. In contrast, pathways such as non-canonical Wnt signaling and several MDSC mechanisms remain largely extrapolated from breast or prostate cancer models and require CRC-specific validation.
Priority areas for future research include: (1) developing circulating or imaging biomarkers for early detection of CRC bone metastasis; (2) designing CRC-specific clinical trials that target the bone–tumor–immune interface (eg, CSF-1R inhibitors, TGF-β blockade combined with immune checkpoint inhibitors); and (3) overcoming the high phenotypic plasticity of TAMs and MDSCs that currently limits translation of macrophage-targeting therapies. Addressing these priorities will be essential to move from descriptive mechanisms to clinically actionable strategies for patients with CRC bone metastasis.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
Inner Mongolia Autonomous Region Public Hospitals High-Level Clinical Specialty Construction Science and Technology Project. Project number: 2023SGGZ071-02. Inner Mongolia Autonomous Region Science and Technology Plan Project. Project number: 2026SYFSH0023. Science and Technology Program of the Joint Fund of Scientific Research for the Public Hospitals of Inner Mongolia Academy of Medical Sciences. Project number: 2025GLLH0267.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Bray F, Laversanne M, Sung H, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA a Cancer J Clin. 2024;74(3):229–16. doi:10.3322/caac.21834
2. Spaander MCW, Zauber AG, Syngal S, et al. Young-onset colorectal cancer. Nat Rev Dis Primers. 2023;9(1):21. doi:10.1038/s41572-023-00432-7
3. Dekker E, Tanis PJ, Vleugels JLA, et al. Colorectal cancer. Lancet. 2019;394(10207):1467–1480. doi:10.1016/S0140-6736(19)32319-0
4. Mathew BG, Aliyuda F, Taiwo D, et al. From biology to diagnosis and treatment: the ariadne’s thread in cancer of unknown primary. Int J Mol Sci. 2023;24(6):5588. doi:10.3390/ijms24065588
5. Lucas C, Barnich N, Nguyen HTT. Microbiota, inflammation and colorectal cancer. Int J Mol Sci. 2017;18(6):1310. doi:10.3390/ijms18061310
6. Huang D, Sun W, Zhou Y, et al. Mutations of key driver genes in colorectal cancer progression and metastasis. Cancer Metastasis Rev. 2018;37(1):173–187. doi:10.1007/s10555-017-9726-5
7. Osseis M, Nehmeh WA, Rassy N, et al. Surgery for T4 colorectal cancer in older patients: determinants of outcomes. J Personalized Med. 2022;12(9):1534. doi:10.3390/jpm12091534
8. Park HS, Chun YJ, Kim HS, et al. Clinical features and KRAS mutation in colorectal cancer with bone metastasis. Sci Rep. 2020;10(1):21180. doi:10.1038/s41598-020-78253-x
9. Hong S, Youk T, Lee SJ, et al. Bone metastasis and skeletal-related events in patients with solid cancer: a Korean nationwide health insurance database study. PLoS One. 2020;15(7):e0234927. doi:10.1371/journal.pone.0234927
10. Jung G, Hernández-Illán E, Moreira L, et al. Epigenetics of colorectal cancer: biomarker and therapeutic potential. Nat Rev Gastroenterol Hepatol. 2020;17(2):111. doi:10.1038/s41575-019-0230-y
11. Liu L, Yan Q, Chen Z, et al. Overview of research progress and application of experimental models of colorectal cancer. Front Pharmacol. 2023;14:1193213. doi:10.3389/fphar.2023.1193213
12. Sartore-Bianchi A, Agostara AG, Patelli G, et al. Application of histology-agnostic treatments in metastatic colorectal cancer. Dig Liver Dis. 2022;54(10):1291–1303. doi:10.1016/j.dld.2022.05.013
13. Deng Z, Fan T, Xiao C, et al. TGF-β signaling in health, disease, and therapeutics. Signal Transd Target Ther. 2024;9(1):61. doi:10.1038/s41392-024-01764-w
14. Chen J, Ji C, Liu S, et al. Transforming growth factor-β (TGF-β) signaling pathway-related genes in predicting the prognosis of colon cancer and guiding immunotherapy. Cancer Pathogenesis Ther. 2024;2(4):299–313. doi:10.1016/j.cpt.2023.12.002
15. Trivedi T, Pagnotti GM, Guise TA, et al. The role of TGF-β in bone metastases. Biomolecules. 2021;11(11):1643. doi:10.3390/biom11111643
16. Peng D, Fu M, Wang M, et al. Targeting TGF-β signal transduction for fibrosis and cancer therapy. Mol Cancer. 2022;21(1):104. doi:10.1186/s12943-022-01569-x
17. Shan H, Tian G, Zhang Y, et al. Exploring the molecular mechanisms and therapeutic potential of SMAD4 in colorectal cancer. Cancer Biol Ther. 2024;25(1):2392341. doi:10.1080/15384047.2024.2392341
18. Fasano M, Pirozzi M, Miceli CC, et al. TGF-β modulated pathways in colorectal cancer: new potential therapeutic opportunities. Int J Mol Sci. 2024;25(13):7400. doi:10.3390/ijms25137400
19. Singh S, Gouri V, Samant M. TGF-β in correlation with tumor progression, immunosuppression and targeted therapy in colorectal cancer. Med Oncol. 2023;40(11):335. doi:10.1007/s12032-023-02204-5
20. Li X, Wu Y, Tian T. TGF-β signaling in metastatic colorectal cancer (mCRC): from underlying mechanism to potential applications in clinical development. Int J Mol Sci. 2022;23(22):14436. doi:10.3390/ijms232214436
21. Papageorgis P, Cheng K, Ozturk S, et al. Smad4 inactivation promotes malignancy and drug resistance of colon cancer. Cancer Res. 2011;71(3):998–1008. doi:10.1158/0008-5472.CAN-09-3269
22. Li X, Li X, Lv X, et al. Smad4 inhibits VEGF-a and VEGF-C expressions via enhancing Smad3 phosphorylation in colon cancer. Anatomical Record. 2017;300(9):1560–1569. doi:10.1002/ar.23610
23. Tauriello DVF, Palomo-Ponce S, Stork D, et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. 2018;554(7693):538–543. doi:10.1038/nature25492
24. Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*). Ann Rev Immunol. 2010;28(1):445–489. doi:10.1146/annurev-immunol-030409-101212
25. Wu Q, Hou X, Xia J, et al. Emerging roles of PDGF-D in EMT progression during tumorigenesis. Cancer Treat Rev. 2013;39(6):640–646. doi:10.1016/j.ctrv.2012.11.006
26. Raica M, Cimpean AM. Platelet-derived growth factor (PDGF)/PDGF receptors (PDGFR) axis as target for antitumor and antiangiogenic therapy. Pharmaceuticals. 2010;3(3):572–599. doi:10.3390/ph3030572
27. Tada K, Kitano S, Shoji H, et al. Pretreatment immune status correlates with progression-free survival in chemotherapy-treated metastatic colorectal cancer patients. Cancer Immunol Res. 2016;4(7):592–599. doi:10.1158/2326-6066.CIR-15-0298
28. Chen J, Yuan W, Wu L, et al. PDGF-D promotes cell growth, aggressiveness, angiogenesis and EMT transformation of colorectal cancer by activation of Notch1/Twist1 pathway. Oncotarget. 2017;8(6):9961–9973. doi:10.18632/oncotarget.14283
29. Liu J, Huang Z, Chen H-N, et al. ZNF37A promotes tumor metastasis through transcriptional control of THSD4/TGF-β axis in colorectal cancer. Oncogene. 2021;40(19):3394–3407. doi:10.1038/s41388-021-01713-9
30. Chen C-Y, Yang S-H, Chang P-Y, et al. Cancer-associated-fibroblast-mediated paracrine and autocrine SDF-1/CXCR4 signaling promotes stemness and aggressiveness of colorectal cancers. Cells. 2024;13(16):1334. doi:10.3390/cells13161334
31. Calon A, Espinet E, Palomo-Ponce S, et al. Dependency of colorectal cancer on a TGF-β-driven program in stromal cells for metastasis initiation. Cancer Cell. 2012;22(5):571–584. doi:10.1016/j.ccr.2012.08.013
32. Gonzalez-Zubeldia I, Dotor J, Redrado M, et al. Co-migration of colon cancer cells and CAFs induced by TGFβ1 enhances liver metastasis. Cell Tissue Res. 2015;359(3):829–839. doi:10.1007/s00441-014-2075-6
33. Huang Z, Zhang Z, Zhou C, et al. Epithelial–mesenchymal transition: the history, regulatory mechanism, and cancer therapeutic opportunities. MedComm. 2022;3(2):e144. doi:10.1002/mco2.144
34. Jiang L, Yang Y, Feng H, et al. Pinocembrin inhibits the proliferation, migration, invasiveness, and epithelial-mesenchymal transition of colorectal cancer cells by regulating LACTB. Cancer Biother Radiopharm. 2022;37(7):527–536. doi:10.1089/cbr.2020.4052
35. Pastushenko I, Brisebarre A, Sifrim A, et al. Identification of the tumour transition states occurring during EMT. Nature. 2018;556(7702):463–468. doi:10.1038/s41586-018-0040-3
36. Seoane J, Gomis RR. TGF-β family signaling in tumor suppression and cancer progression. Cold Spring Harbor Perspect Biol. 2017;9(12):a022277. doi:10.1101/cshperspect.a022277
37. Nie F, Sun X, Sun J, Zhang J, Wang Y. Epithelial-mesenchymal transition in colorectal cancer metastasis and progression: molecular mechanisms and therapeutic strategies. Cell Death Discov. 2025;11(1). doi:10.1038/s41420-025-02593-8
38. Roepman P, Schlicker A, Tabernero J, et al. Colorectal cancer intrinsic subtypes predict chemotherapy benefit, deficient mismatch repair and epithelial-to-mesenchymal transition. Int J Cancer. 2014;134(3):552–562. doi:10.1002/ijc.28387
39. Vu T, Datta PK. Regulation of EMT in colorectal cancer: a culprit in metastasis. Cancers. 2017;9(12):171. doi:10.3390/cancers9120171
40. Wei C, Yang C, Wang S, et al. Crosstalk between cancer cells and tumor associated macrophages is required for mesenchymal circulating tumor cell-mediated colorectal cancer metastasis. Mol Cancer. 2019;18(1):64. doi:10.1186/s12943-019-0976-4
41. Wolf MJ, Hoos A, Bauer J, et al. Endothelial CCR2 signaling induced by colon carcinoma cells enables extravasation via the JAK2-Stat5 and p38MAPK pathway. Cancer Cell. 2012;22(1):91–105. doi:10.1016/j.ccr.2012.05.023
42. Roblek M, Protsyuk D, Becker PF, et al. CCL2 is a vascular permeability factor inducing CCR2-dependent endothelial retraction during lung metastasis. Mol Cancer Res. 2019;17(3):783–793. doi:10.1158/1541-7786.MCR-18-0530
43. Tang X, Sun L, Wang G, et al. RUNX1: a regulator of NF-kB signaling in pulmonary diseases. Curr Protein Pept Sci. 2018;19(2):172–178. doi:10.2174/1389203718666171009111835
44. Slattery ML, Herrick JS, Mullany LE, et al. The co-regulatory networks of tumor suppressor genes, oncogenes, and miRNAs in colorectal cancer. Genes Chromosomes Cancer. 2017;56(11):769–787. doi:10.1002/gcc.22481
45. Lu C, Yang Z, Yu D, et al. RUNX1 regulates TGF-β induced migration and EMT in colorectal cancer. Pathol Res Pract. 2020;216(11):153142. doi:10.1016/j.prp.2020.153142
46. Luo X, Wei M, Li W, et al. PBX3 promotes pentose phosphate pathway and colorectal cancer progression by enhancing G6PD expression. Int J Bio Sci. 2023;19(14):4525–4538. doi:10.7150/ijbs.86279
47. Lamprecht S, Kaller M, Schmidt EM, et al. PBX3 is part of an EMT regulatory network and indicates poor outcome in colorectal cancer. Clin Cancer Res. 2018;24(8):1974–1986. doi:10.1158/1078-0432.CCR-17-2572
48. Li WF, Herkilini A, Tang Y, et al. The transcription factor PBX3 promotes tumor cell growth through transcriptional suppression of the tumor suppressor p53. Acta Pharmacol Sin. 2021;42(11):1888–1899. doi:10.1038/s41401-020-00599-9
49. Koyamo Y, Fujihara S, Chiyo T, et al. Role of mir-452-5p overexpression in epithelial-mesenchymal transition (EMT) in early-stage colorectal cancer. In Vivo. 2023;37(5):1980–1990.
50. Kim TW, Lee YS, Yun NH, et al. MicroRNA-17-5p regulates EMT by targeting vimentin in colorectal cancer. Br J Cancer. 2020;123(7):1123–1130.
51. Zhang J, Zhu H, Liu W, et al. Prognostic and predictive molecular biomarkers in colorectal cancer. Front Oncol. 2025;15:1532924. doi:10.3389/fonc.2025.1532924
52. Zhang N, Ng AS, Cai S, et al. Novel therapeutic strategies: targeting epithelial-mesenchymal transition in colorectal cancer. Lancet Oncol. 2021;22(8):e358–e368. doi:10.1016/S1470-2045(21)00343-0
53. Liu Q, Song J, Pan Y, et al. Wnt5a/CaMKII/ERK/CCL2 axis is required for tumor-associated macrophages to promote colorectal cancer progression. Int J Bio Sci. 2020;16(6):1023–1034. doi:10.7150/ijbs.40535
54. Virchow R. An address on the value of pathological experiments. Br Med J. 1881;2(1075):198–203. doi:10.1136/bmj.2.1075.198
55. Maman S, Witz IP. A history of exploring cancer in context. Nat Rev Cancer. 2018;18(6):359–376. doi:10.1038/s41568-018-0006-7
56. Suarez-Carmona M, Lesage J, Cataldo D, et al. EMT and inflammation: inseparable actors of cancer progression. Mol Oncol. 2017;11(7):805–823. doi:10.1002/1878-0261.12095
57. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883–899. doi:10.1016/j.cell.2010.01.025
58. Aggarwal BB, Vijayalekshmi RV, Sung B. Targeting inflammatory pathways for prevention and therapy of cancer: short-term friend, long-term foe. Clin Cancer Res. 2009;15(2):425–430. doi:10.1158/1078-0432.CCR-08-0149
59. Si X, Jia H, Liu N, et al. Alpha-ketoglutarate attenuates colitis in mice by increasing lactobacillus abundance and regulating stem cell proliferation via wnt-hippo signaling. Mol Nutr Food Res. 2022;66(10):e2100955. doi:10.1002/mnfr.202100955
60. Krugliak Cleveland N, Torres J, Rubin DT. What does disease progression look like in ulcerative colitis, and how might it be prevented? Gastroenterology. 2022;162(5):1396–1408. doi:10.1053/j.gastro.2022.01.023
61. Shah SC, Itzkowitz SH. Colorectal cancer in inflammatory bowel disease: mechanisms and management. Gastroenterology. 2022;162(3):715–730.e3. doi:10.1053/j.gastro.2021.10.035
62. Tong G, Shen Y, Li H, et al. NLRC4, inflammation and colorectal cancer (review). Int J Oncol. 2024;65(4). doi:10.3892/ijo.2024.5687
63. Schmitt M, Greten FR. The inflammatory pathogenesis of colorectal cancer. Nat Rev Immunol. 2021;21(10):653–667. doi:10.1038/s41577-021-00534-x
64. Colotta F, Allavena P, Sica A, et al. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 2009;30(7):1073–1081. doi:10.1093/carcin/bgp127
65. Wang D, DuBois RN. Role of prostanoids in gastrointestinal cancer. J Clin Investig. 2018;128(7):2732–2742. doi:10.1172/JCI97953
66. Yin Y, Yao S, Hu Y, et al. The immune-microenvironment confers chemoresistance of colorectal cancer through macrophage-derived IL6. Clin Cancer Res. 2017;23(23):7375–7387. doi:10.1158/1078-0432.CCR-17-1283
67. Boussios S, Ozturk MA, Moschetta M, et al. The developing story of predictive biomarkers in colorectal cancer. J Personalized Med. 2019;9(1):12. doi:10.3390/jpm9010012
68. Gallon R, Herrero-Belmonte P, Phelps R, et al. A novel colorectal cancer test combining microsatellite instability and BRAF/RAS analysis: clinical validation and impact on lynch syndrome screening. BJC Reports. 2024;2(1):48. doi:10.1038/s44276-024-00072-8
69. Makaremi S, Asadzadeh Z, Hemmat N, et al. Immune checkpoint inhibitors in colorectal cancer: challenges and future prospects. Biomedicines. 2021;9(9):1075. doi:10.3390/biomedicines9091075
70. Colangelo T, Polcaro G, Muccillo L, et al. Friend or foe? The tumour microenvironment dilemma in colorectal cancer. Biochim Biophys Acta Rev Cancer. 2017;1867(1):1–18. doi:10.1016/j.bbcan.2016.11.001
71. Deschoolmeester V, Baay M, Van Marck E, et al. Tumor infiltrating lymphocytes: an intriguing player in the survival of colorectal cancer patients. BMC Immunol. 2010;11(1):19. doi:10.1186/1471-2172-11-19
72. Calon A, Lonardo E, Berenguer-Llergo A, et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat Genet. 2015;47(4):320–329. doi:10.1038/ng.3225
73. Thommen DS, Schumacher TN. T cell dysfunction in cancer. Cancer Cell. 2018;33(4):547–562. doi:10.1016/j.ccell.2018.03.012
74. Zhou Y, Xu J, Luo H, et al. Wnt signaling pathway in cancer immunotherapy. Cancer Lett. 2022;525:84–96. doi:10.1016/j.canlet.2021.10.034
75. Markman JL, Shiao SL. Impact of the immune system and immunotherapy in colorectal cancer. J Gastrointestinal Oncol. 2015;6(2):208–223. doi:10.3978/j.issn.2078-6891.2014.077
76. Wang D, Yang L, Yu W, et al. Colorectal cancer cell-derived CCL20 recruits regulatory T cells to promote chemoresistance via FOXO1/CEBPB/NF-κB signaling. J ImmunoTher Cancer. 2019;7(1):215. doi:10.1186/s40425-019-0701-2
77. Sica A, Allavena P, Mantovani A. Cancer related inflammation: the macrophage connection. Cancer Lett. 2008;267(2):204–215. doi:10.1016/j.canlet.2008.03.028
78. Murray PJ, Allen JE, Biswas SK, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41(1):14–20. doi:10.1016/j.immuni.2014.06.008
79. Balkwill F, Mantovani A. Inflammation and cancer: back to virchow? Lancet. 2001;357(9255):539–545. doi:10.1016/S0140-6736(00)04046-0
80. Liu C, Zhang W, Wang J, et al. Tumor-associated macrophage-derived transforming growth factor-β promotes colorectal cancer progression through HIF1-TRIB3 signaling. Cancer Sci. 2021;112(10):4198–4207. doi:10.1111/cas.15101
81. Tewari D, Bawari S, Sharma S, et al. Targeting the crosstalk between canonical wnt/β-catenin and inflammatory signaling cascades: a novel strategy for cancer prevention and therapy. Pharmacol Ther. 2021;227:107876.
82. Bikle DD. The vitamin D receptor as tumor suppressor in skin. Adv Exp Med Biol. 2020;1268:285–306.
83. Schneikert J, Behrens J. The canonical wnt signalling pathway and its APC partner in colon cancer development. Gut. 2007;56(3):417–425. doi:10.1136/gut.2006.093310
84. Sato T, Van Es JH, Snippert HJ, et al. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature. 2011;469(7330):415–418. doi:10.1038/nature09637
85. Nusse R, Clevers H. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell. 2017;169(6):985–999. doi:10.1016/j.cell.2017.05.016
86. Eto T, Miyake K, Nosho K, et al. Impact of loss-of-function mutations at the RNF43 locus on colorectal cancer development and progression. J Pathol. 2018;245(4):445–455. doi:10.1002/path.5098
87. Jian W, Zhang L. POLE2 silencing inhibits the progression of colorectal carcinoma cells via wnt signaling axis. Cancer Biol Ther. 2024;25(1):2392339. doi:10.1080/15384047.2024.2392339
88. Zhao D-Y, Yin T-F, Sun X-Z, et al. microRNA-627-5p inhibits colorectal cancer cell proliferation, migration and invasion by targeting Wnt2. World J Gastrointestinal Oncol. 2023;15(2):318–331. doi:10.4251/wjgo.v15.i2.318
89. Wend P, Holland JD, Ziebold U, et al. Wnt signaling in stem and cancer stem cells. Semin Cell Dev Biol. 2010;21(8):855–863. doi:10.1016/j.semcdb.2010.09.004
90. Fennell LJ, Kane A, Liu C, et al. APC mutation marks an aggressive subtype of BRAF mutant colorectal cancers. Cancers. 2020;12(5):1171. doi:10.3390/cancers12051171
91. Aghabozorgi AS, Ebrahimi R, Bahiraee A, et al. The genetic factors associated with wnt signaling pathway in colorectal cancer. Life Sci. 2020;256:118006. doi:10.1016/j.lfs.2020.118006
92. Korkmaz G, Horozoglu C, Arıkan S, et al. LGALS3 and AXIN1 gene variants playing role in the wnt/β-catenin signaling pathway are associated with mucinous component and tumor size in colorectal cancer. Bosnian J Basic Med Sci. 2016;16(2):108–113. doi:10.17305/bjbms.2016.721
93. Voloshanenko O, Erdmann G, Dubash TD, et al. Wnt secretion is required to maintain high levels of wnt activity in colon cancer cells. Nat Commun. 2013;4(1):2610. doi:10.1038/ncomms3610
94. Wang Y, Jia J, Wang F, et al. Pre-metastatic niche: formation, characteristics and therapeutic implication. Signal Transd Target Ther. 2024;9(1):236. doi:10.1038/s41392-024-01937-7
95. Takei D, Tagami K. Management of cancer pain due to bone metastasis. J Bone Mineral Metab. 2023;41(3):327–336. doi:10.1007/s00774-022-01382-y
96. Li -A-A, Cao Z-Y, Liu J-M, et al. The risk factors for bone metastases in patients with colorectal cancer. Medicine. 2018;97(40):e12694. doi:10.1097/MD.0000000000012694
97. Santini D, Tampellini M, Vincenzi B, et al. Natural history of bone metastasis in colorectal cancer: final results of a large Italian bone metastases study. Ann Oncol. 2012;23(8):2072–2077. doi:10.1093/annonc/mdr572
98. Siegel RL, Giaquinto AN, Jemal A. Cancer statistics, 2024. CA a Cancer J Clin. 2024;74(1):12–49. doi:10.3322/caac.21820
99. Christ AB, Piple AS, Gettleman BS, et al. Prevalence of primary malignant tumours, rates of pathological fracture, and mortality in the setting of metastatic bone disease. Bone Joint Open. 2023;4(6):424–431. doi:10.1302/2633-1462.46.BJO-2023-0042.R1
100. Kreps LM, Addison CL. Targeting intercellular communication in the bone microenvironment to prevent disseminated tumor cell escape from dormancy and bone metastatic tumor growth. Int J Mol Sci. 2021;22(6):2911. doi:10.3390/ijms22062911
101. Kawamura H, Yamaguchi T, Yano Y, et al. Characteristics and prognostic factors of bone metastasis in patients with colorectal cancer. Dis Colon Rectum. 2018;61(6):673–678. doi:10.1097/DCR.0000000000001071
102. Zhu Z, zhu Z. Retrospective study of predictors of bone metastasis in colorectal cancer patients. J Bone Oncol. 2017;9:25–28. doi:10.1016/j.jbo.2017.10.003
103. Clohisy DR, Ramnaraine ML, Scully S, et al. Osteoprotegerin inhibits tumor-induced osteoclastogenesis and bone tumor growth in osteopetrotic mice. J Orthop Res. 2000;18(6):967–976. doi:10.1002/jor.1100180617
104. Wang K, Donnelly CR, Jiang C, et al. STING suppresses bone cancer pain via immune and neuronal modulation. Nat Commun. 2021;12(1):4558. doi:10.1038/s41467-021-24867-2
105. Yoneda T, Hiraga T. Crosstalk between cancer cells and bone microenvironment in bone metastasis. Biochem Biophys Res Commun. 2005;328(3):679–687. doi:10.1016/j.bbrc.2004.11.070
106. Jia S-N, Han Y-B, Yang R, et al. Chemokines in colon cancer progression. Semi Cancer Biol. 2022;86(Pt 3):400–407. doi:10.1016/j.semcancer.2022.02.007
107. Xie C, Ye F, Zhang N, et al. CCL7 contributes to angiotensin II-induced abdominal aortic aneurysm by promoting macrophage infiltration and pro-inflammatory phenotype. J Cell Mol Med. 2021;25(15):7280–7293. doi:10.1111/jcmm.16757
108. Lee YS, Cho YB. CCL7 signaling in the tumor microenvironment. Adv Exp Med Biol. 2020;1231:33–43.
109. Yang H, Jian L, Jin Q, et al. CCL7 playing a dominant role in recruiting early OCPs to facilitate osteolysis at metastatic site of colorectal cancer. Cell Commun Signal. 2022;20(1):94. doi:10.1186/s12964-022-00867-7
110. Udagawa N, Koide M, Nakamura M, et al. Osteoclast differentiation by RANKL and OPG signaling pathways. J Bone Mineral Metab. 2021;39(1):19–26. doi:10.1007/s00774-020-01162-6
111. Zi-Chen G, Jin Q, Yi-Na Z, et al. Colorectal cancer cells promote osteoclastogenesis and bone destruction through regulating EGF/ERK/CCL3 pathway. Biosci Rep. 2020;40(6):BSR20201175. doi:10.1042/BSR20201175
112. Fornetti J, Welm AL, Stewart SA. Understanding the bone in cancer metastasis. J Bone Miner Res. 2018;33(12):2099–2113. doi:10.1002/jbmr.3618
113. Hiraga T. Hypoxic microenvironment and metastatic bone disease. Int J Mol Sci. 2018;19(11):3523. doi:10.3390/ijms19113523
114. Spencer JA, Ferraro F, Roussakis E, et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature. 2014;508(7495):269–273. doi:10.1038/nature13034
115. Arnett TR, Gibbons DC, Utting JC, et al. Hypoxia is a major stimulator of osteoclast formation and bone resorption. J Cell Physiol. 2003;196(1):2–8. doi:10.1002/jcp.10321
116. Knowles HJ. Hypoxic regulation of osteoclast differentiation and bone resorption activity. Hypoxia. 2015;3:73–82. doi:10.2147/HP.S95960
117. Liu C, Wang M, Xu C, et al. Immune checkpoint inhibitor therapy for bone metastases: specific microenvironment and current situation. J Immunol Res. 2021;2021:8970173. doi:10.1155/2021/8970173
118. Karavitis J, Hix LM, Shi YH, et al. Regulation of COX2 expression in mouse mammary tumor cells controls bone metastasis and PGE2-induction of regulatory T cell migration. PLoS One. 2012;7(9):e46342. doi:10.1371/journal.pone.0046342
119. Zhao E, Wang L, Dai J, et al. Regulatory T cells in the bone marrow microenvironment in patients with prostate cancer. Oncoimmunology. 2012;1(2):152–161. doi:10.4161/onci.1.2.18480
120. Joshi S, Sharabi A. Targeting myeloid-derived suppressor cells to enhance natural killer cell-based immunotherapy. Pharmacol Ther. 2022;235:108114.
121. Wang Y, Ding Y, Guo N, et al. MDSCs: key criminals of tumor pre-metastatic niche formation. Front Immunol. 2019;10:172. doi:10.3389/fimmu.2019.00172
122. Sun H-L, Zhou X, Xue Y-F, et al. Increased frequency and clinical significance of myeloid-derived suppressor cells in human colorectal carcinoma. World J Gastroenterol. 2012;18(25):3303–3309. doi:10.3748/wjg.v18.i25.3303
123. Owen KL, Parker BS. Beyond the vicious cycle: the role of innate osteoimmunity, automimicry and tumor-inherent changes in dictating bone metastasis. Mol Immunol. 2019;110:57–68. doi:10.1016/j.molimm.2017.11.023
124. Danilin S, Merkel AR, Johnson JR, et al. Myeloid-derived suppressor cells expand during breast cancer progression and promote tumor-induced bone destruction. Oncoimmunology. 2012;1(9):1484–1494. doi:10.4161/onci.21990
125. Kielbassa K, Vegna S, Ramirez C, et al. Understanding the origin and diversity of macrophages to tailor their targeting in solid cancers. Front Immunol. 2019;10:2215. doi:10.3389/fimmu.2019.02215
126. Magliacane Trotta S, Adinolfi A, D’Orsi L, et al. Cancer-derived exosomal alu RNA promotes colorectal cancer progression. Exp Mol Med. 2024;56(3):700–710.
127. Allavena P, Mantovani A. Immunology in the clinic review series; focus on cancer: tumour-associated macrophages: undisputed stars of the inflammatory tumour microenvironment. Clin Exp Immunol. 2012;167(2):195–205. doi:10.1111/j.1365-2249.2011.04515.x
128. Guerriero JL. Macrophages: the road less traveled, changing anticancer therapy. Trends Mol Med. 2018;24(5):472–489. doi:10.1016/j.molmed.2018.03.006
129. Umeda H, Shigeyasu K, Takahashi T, et al. ADAR1-high tumor-associated macrophages induce drug resistance and are therapeutic targets in colorectal cancer. Mol Cancer. 2025;24(1):116. doi:10.1186/s12943-025-02312-y
130. Yuan G, Huang Y, Yang S-T, et al. RGS12 inhibits the progression and metastasis of multiple myeloma by driving M1 macrophage polarization and activation in the bone marrow microenvironment. Cancer Commun. 2022;42(1):60–64. doi:10.1002/cac2.12228
131. Lee J-H, Kim H-N, Kim K-O, et al. CXCL10 promotes osteolytic bone metastasis by enhancing cancer outgrowth and osteoclastogenesis. Cancer Res. 2012;72(13):3175–3186. doi:10.1158/0008-5472.CAN-12-0481
132. Xin Z, Qin L, Tang Y, et al. Immune mediated support of metastasis: implication for bone invasion. Cancer Commun. 2024;44(9):967–991. doi:10.1002/cac2.12584
133. Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer Cell. 2015;27(4):462–472. doi:10.1016/j.ccell.2015.02.015
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.