Back to Journals » Drug Design, Development and Therapy » Volume 14
Colon Targeting of Naringin for Enhanced Cytoprotection Against Indomethacin-Induced Colitis in Rabbits
Authors El Naggar EE, Mohamed EA ![]() , Borg TM, El-Sheakh AR
, Borg TM, El-Sheakh AR ![]() , Hamed MF
, Hamed MF
Received 5 June 2019
Accepted for publication 7 November 2019
Published 19 February 2020 Volume 2020:14 Pages 677—696
DOI https://doi.org/10.2147/DDDT.S218357
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Tuo Deng
Eman Ebrahim El Naggar,1 Elham Abdelmonem Mohamed,2 Thanaa Mohamed Borg,2 Ahmed Ramadan El-Sheakh,3 Mohammed Fawzy Hamed4
1Department of Pharmaceutical Technology, Faculty of Pharmacy, Horus University, New Damietta, Eygpt; 2Department of Pharmaceutics, Faculty of Pharmacy, Mansoura University, Mansoura, Dakahlia, Egypt; 3Department of Pharmacology and Toxicology, Faculty of Pharmacy, Mansoura University, Mansoura, Dakahlia, Egypt; 4Department of Pathology, Faculty of Veterinary Medicine, Mansoura University, Mansoura, Dakahlia, Egypt
Correspondence: Elham Abdelmonem Mohamed
Department of Pharmaceutics, Faculty of Pharmacy, Mansoura University, EL Gomhoreyaha St., Mansoura City 35516, Egypt
Tel +201065690987
Fax +20502247496
Email [email protected]
Background: Naringin is a promising anti-inflammatory drug against various disorders including ulcerative colitis. However, its oral bioavailability is low (8%) possibly due to cleavage at the upper gut. Consequently, colon targeting would be necessary for drug protection at the upper gut, enhanced oral bioavailability and potentiated cytoprotection against colitis.
Methodology: This study involved the formulation of compression-coated tablets of naringin employing mixtures of pH-sensitive Eudragit L100-55 (EUD-L100-55) and different time-dependent polymers including ethyl cellulose (EC), sodium alginate (ALG) and sodium carboxymethyl cellulose (SCMC). Drug–polymer interaction during release was assessed using Fourier transform-infrared spectroscopy (FT-IR) and differential scanning calorimetry (DSC). Tablets were evaluated in vitro. Surface morphology of the optimized tablets either before or after exposure to the different release media was examined employing scanning electron microscopy (SEM). Cytoprotection potential of the optimized tablets against indomethacin-induced colitis in rabbits was screened and compared to core tablets through a histopathological examination of colon, measurement of serum perinuclear antineutrophil cytoplasmic antibodies (pANCA) and immunohistochemical localization of tumor necrosis factor-alpha (TNF-α).
Results: FT-IR and DSC results may indicate drug–polymers interaction during release. Release retardation could be related to polymer swelling that was in the order of SCMC > ALG > EC. SEM examination indicated more porous coats at the buffers relative to the acidic medium. Colon targeting was expected in case of coats of 5% ALG, 5% SCMC and 10% EC (w/w) in combination with EUD-L100-55; thus, they were selected for in vivo evaluation. Effective cytoprotection of selected tablets against indomethacin-induced colitis was indicated by a significant (P< 0.05) reduction in mucosal damage, serum levels of pANCA and TNF-α expression compared to untreated colitis and core-pretreated groups. Compared to EC, higher cytoprotection potential of ALG- and SCMC-based tablets was reflected by lower concentration (5% w/w) to provide cytoprotection against indomethacin-induced colitis.
Keywords: naringin, colon targeting, compression-coated tablets, Eudragit L, release retardants, colitis
Introduction
Inflammatory bowel diseases including ulcerative colitis and Crohn's disease are among the most challenging human illness. Etiology of ulcerative colitis is largely unknown, yet multiple immune and genetic factors have been suggested to affect its initiation and progression.1,2 Most of the current treatment regimens of ulcerative colitis involve treatment with glucocorticosteroids and 5-aminosalicylic acid.1,2 Severe ulcerative colitis is associated with a sustained massive production of cytokines such as TNF-α.3–5 Therefore, immunosuppressive drugs have been also used to control severe colitis; however, they may cause serious complications and side effects.4 There is a great need for new therapeutic approaches permitting efficacy and safety because many patients either do not respond to the currently available therapies or suffer from side effects possibly due to the prolonged use.2
It has been documented that naringin can influence the inflammatory process through its effects on the expression and activity of inducible nitric oxide synthetase and cyclooxygenase-2 (COX-2).6 Naringin has exhibited an activity against colitis in mice.7 However, it has been reported that this drug undergoes cleavage in the lumen or in the cell gut at the harsh pH (1–2.5) and enzymatic conditions of the upper gastrointestinal tract (GIT); thus, its absorption following oral administration is low and irregular.8–10 Consequently, the oral bioavailability of naringin is low (about 8%) and its half-life is short (2.6 hrs).11 The formulation of naringin gastro-resistant micro-particles has been attempted by applying cellulose acetate phthalate as a coating material using the spray-drying technique to obtain a controlled drug release to the intestine and to prevent the drug degradation in the gastric medium.10 This study did not involve in vivo evaluation of the prepared formulations. Furthermore, naringin is mainly hydrolyzed by the microflora in the distal part of the small intestine and colon into its aglycone, naringenin.12 It was reported that naringenin treatment significantly improved colitis through down-regulating the mRNA expression of several pro-inflammatory mediators, including TNF-α.13 Thus, it would be of medical importance to formulate naringin in drug delivery systems for colon targeting to attain high drug concentrations in the colon and allow the hydrolysis to its aglycone naringenin for providing cytoprotection against colitis and overcoming its instability at the upper GIT. Site-specific drug delivery systems for colon targeting have been successfully used to deliver drugs for local colon diseases such as Crohn’s disease, ulcerative colitis, irritable bowel syndrome and constipation. Colon targeted drug delivery system should hinder drug release and absorption in the stomach as well as the small intestine but allow its release and absorption once the system reaches the colon.14 Colon as a delivery site offers several advantages including nearly neutral pH, long transit time, reduced digestive enzymatic activity and great responsiveness to absorption enhancers.15 Local treatment of inflammatory bowel diseases, such as ulcerative colitis, is highly challenging since it requires minimal release in the upper GIT.16
Different colonic delivery systems including pH-, time-, bacterial- and pressure-responsive systems have been employed to overcome this restriction.17,18 However, tablets coated with pH-sensitive polymers, such as Eudragits (EUDs) S and L, hinder the drugs release mainly in the gastric fluid and possibly the proximal part of the small intestine. As well, time-responsive systems have been used due to the simple preparation, yet variable physiological conditions along GIT made their potential as colonic delivery systems doubtful.19 Variation of gastric emptying time (less than 2 hrs under fasting and 2–12 hrs under fed conditions in humans) is an example of critical physiological variation that may lead to the failure of colon targeting.20 Therefore, colon targeted systems based on combinations of pH-sensitive and time-dependent polymers were successfully used.21,22 For example, the addition of water-insoluble polymer as ethyl cellulose (EC) has been utilized to overcome the short gastric resistance and premature release of pH-dependent colon targeted delivery systems.22 Sodium alginate (ALG) has been successfully used in compression coating along with EUD-S100 to retard flurbiprofen release in the upper GIT but progressively release the drug in the colon.23 Sodium carboxymethyl cellulose (SCMC) was used in combination with EUD-L to provide tablets for colonic delivery of albendazole.24
Direct compression coating of tablets for colon targeting is more convenient particularly for large-scale production when compared to the traditional liquid coating processes due to several reasons.25 Among these, direct compression coating is solvent-free, less time-consuming and less expensive because no special coating equipment is required. As well, it provides thicker and more stable coatings. EUD-L is a spray-dried methacrylic acid-ethyl acrylate copolymer, which is advantageous to EUD-S particularly for direct compression coating because of its good flowability and compressibility.22
To the best of our knowledge, no study was reported about colon-targeting of naringin in the form of coated tablets employing the simple direct compression technique that would encourage the large-scale production. Therefore, colon-targeted tablets of this drug based on coats of EUD L100-55 combined with hydroxypropyl methylcellulose were successfully prepared and evaluated in our laboratory.26 In addition, our previous study indicated that naringin colon targeting was attained due to the drug release retardation that was related to the swelling rates of the coats that depended on the release retardant concentration. Thus, it was worth to prepare directly compressed coated tablets for colonic delivery of the naturally occurring naringin employing mixtures of pH-sensitive EUD-L and time-dependent polymers other than hydroxypropyl methylcellulose including EC, ALG and SCMC as compression coating. The possibility of drug interaction with EUD-L or any of the release retardants used following exposure to the different release media employed was assessed. SEM examination was performed to examine the effects of the release media used on the porosity of the swollen coats of the selected tablets and hence the drug release. The prepared tablets were evaluated in vitro, compared and optimized. In comparison with the core tablets, the selected tablets were examined for their cytoprotective activity against indomethacin-induced colitis in rabbits based on histopathological examination of colon, measurement of serum pANCA and immunohistochemical localization of TNF-α.
Materials and Methods
Materials
Naringin and sodium carboxymethylcellulose (SCMC) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Eudragit® L100-55 (EUD-L) was obtained as a gift from Evonik Rhom GmbH (Darmstadt, Germany). Sodium Alginate (ALG) was supplied by AppliChem GmbH (Germany). Ethyl cellulose (EC) was purchased from ACROS (New Jersy, USA). Indomethacin was obtained from Kahira Pharmaceuticals & Chemical Industries Co. (Cairo, Egypt). Rabbit antineutrophilic perinuclear antibodies (pANCA) Kit was purchased from MyBioSource (San Diego, CA, USA). Avicel pH 101 was obtained from Eipico Pharmaceutical Chemicals Co. (Cairo, Egypt). Magnesium stearate and talc were provided as a gift by Amriya Pharmaceutical Industries Co. (Alexandria, Egypt). All other chemicals were of fine analytical grade.
Preparation of Naringin Core Tablets
Naringin at a dose of 15.8 mg/kg has been reported to protect against colitis in mice.7 This dose was converted to that of human according to a reported method.27 According to this method, naringin dose expected to exert cytoprotection against colitis in an adult of average body weight equal to 70 kg was 125 mg. Consequently, core tablets containing 125 mg naringin were prepared by direct compression. Naringin was passed through a 200-µm sieve (sieve No. 70), mixed with Avicel pH 101 using a mortar and a pestle for 10 mins and then lubricated with a 2:1 mixture of talc (2% w/w) and magnesium stearate (1% w/w). Finally, an amount equal to 180 mg of the blend was fed into a die of a single punch tableting press (Type EKO, Erweka Apparatebau, GmbH, Germany), equipped with flat-faced punches. The diameter of the compression punch used for the preparation of naringin core tablets was 9 mm. Tablet hardness between 5 and 6 kg/cm2 was obtained by adjusting compression pressure.
Preparation of Naringin Compression-Coated Tablets
Drug cores were compression-coated with EUD-L either alone or combined with 5% and 10% w/w of each of EC, ALG and SCMC maintaining core:coat weight ratio at 1:2 (Table 1). Polymers were passed through a 200-µm sieve (sieve No. 70), mixed and lubricated using a 2:1 talc–magnesium stearate blend. Compression-coated tablets were prepared according to a reported method.23 In brief, the tablets were prepared by first filling one-half (180 mg) of the coat mixture in the die cavity, positioning the core tablet centrally on the powder bed and finally filling the remaining half (180 mg) of the coat mixture on the top to be directly compressed (Type EKO, Erweka Apparatebau). The diameter of the compression punch used for the preparation of naringin coated tablets was 12 mm. The compression pressure was adjusted to give tablet hardness ranging from 7 to 10 kg/cm2 to decrease coat porosity for lag time prolongation.23
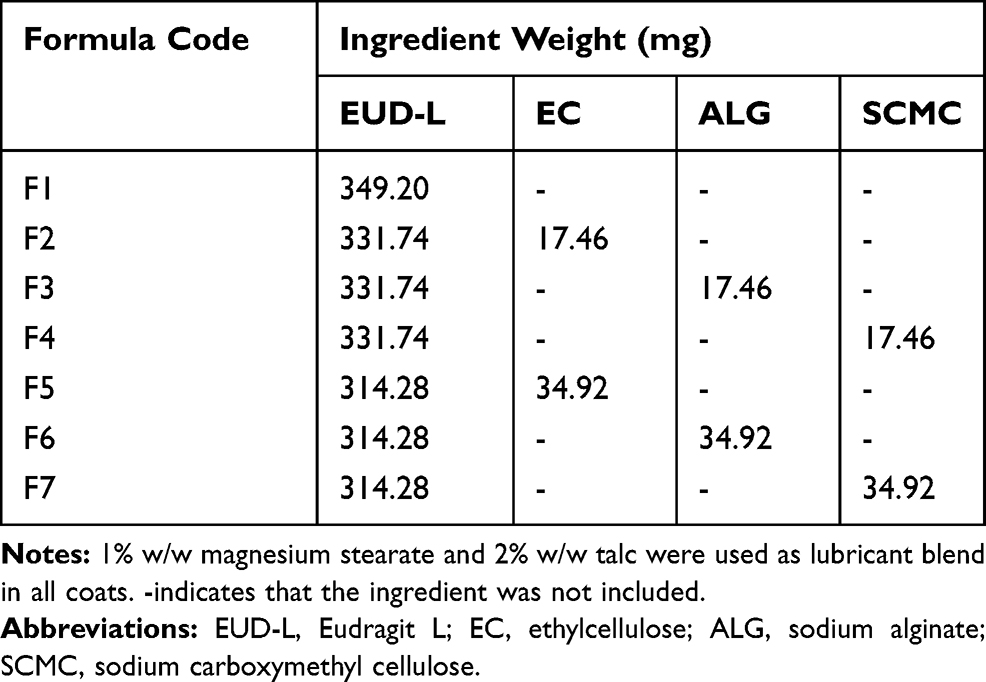 |
Table 1 Composition of Compression Coats |
Characterization of Core and Compression-Coated Tablets
The prepared cores and coated tablets were evaluated regarding weight variation, thickness, diameter, hardness, friability and drug content uniformity according to the procedures stated in US pharmacopoeia.28 Randomly picked 20 tablets of each formulation were weighed using an electronic balance (Mettler Toledo, Switzerland), and the average weight was calculated to assess the weight variation. Tablet hardness was measured using the randomly selected 10 tablets (Erweka-Apparatebau). Ten tablets of each formulation were accurately weighed (W1) and placed in the drum of Roche friabilator (Erweka-Apparatebau) to be rotated at 25 rpm for 4 mins (100 revolutions). Afterwards, they were taken out of the drum and the dust, if any, was removed by a brush to be reweighed (W2) to calculate friability percent according to the relation, Friability% = [(W1−W2)/W1] ×100.
Naringin cores and coated tablets were evaluated for their drug content in triplicate. Ten tablets of each formulation were finely powdered and an amount equivalent to 125 mg of naringin was accurately weighed and thoroughly mixed with phosphate buffer (PB) pH 7.4, filtered through a 45-µm membrane filter, properly diluted and analyzed spectrophotometrically (UV/VIS spectrophotometer, V-550, Jasco, Japan) for drug content at 282 nm. All results were expressed as the mean of three measurements ± standard deviation (SD).
Study of Drug–Polymers Interaction
The possibility of drug interaction with EUD-L or any of the release retardants used following exposure to the different release media employed was assessed. Kneaded mixtures (KMs) of the drug with each of these polymers were prepared by kneading corresponding physical mixtures (PMs) at a weight ratio of 1:1 with each of the release media used to be then dried and ground. PMs were prepared at a weight ratio of 1:1 to maximize the probability of detecting any interaction, if any, and minimize the dilution effect.29 The prepared KMs were then examined employing Fourier transform-infrared (FT-IR) spectroscopy and differential scanning calorimetry (DSC) in comparison with the pure drug and the corresponding 1:1 PMs.
Fourier Transform-Infrared (FT-IR) Spectroscopy
Fourier transform-infrared (FT-IR) spectra of the drug, each polymer, 1:1 drug–polymer PM and the corresponding KM were recorded using Fourier transform-infrared spectrophotometer (Thermo Fisher Scientific, Inc., Waltham, MA, USA). Each sample (2 mg) was ground with potassium bromide (200 mg) and compressed into discs by a hydraulic press to be finally scanned over a wavenumber ranged from 500 to 4000 cm−1.
Differential Scanning Calorimetry (DSC)
DSC runs of the drug, each polymer, 1:1 drug–polymer PM and the corresponding KM were performed using a differential scanning calorimeter (PerkinElmer, model DSC-4, New York, USA). In aluminum crimped pans, samples (4 mg) were heated at a rate of 10°C/min over a temperature range of 35–400°C under the flow of nitrogen gas. Indium (99.99% purity, m.p. 156.6°C) was utilized as a standard in DSC runs to attain temperature calibration.
Swelling Studies
Swelling of tablets coated with EUD-L combined with 5% and 10% w/w of each release retardant was studied according to the method previously adopted.30,31 Swelling studies were carried out to screen the effects of the natural release retardant and concentration as well as the release medium pH on the coat swelling; hence, the drug release at the three media was used.
Briefly, each tablet was individually weighed (W1) and transferred into a beaker containing 200 mL of 0.1N HCl and maintained in a water bath at 37 ± 0.5°C. At regular time intervals (0.5, 1 and 2 hrs), each tablet was removed from the beaker, excess surface liquid was carefully removed using a filter paper and the swollen tablet was reweighed (W2). The process was repeated in each of PBs at pH 6.8 and 7.4 using the tablet previously placed in 0.1N HCl. Regarding buffers, the tablets were reweighed at time intervals of 0.5, 1, 2 and 3 hrs. The process duration and sequence of medium change resembled that followed during in vitro release study to screen the effects of natural release retardant and concentration as well as the release medium pH on coat swelling and hence drug release at the three-release media. The percent swelling was calculated according to the following equation: Percent swelling = ((W2−W1)/W1) × 100. The experiment was carried out in triplicate, and the average percent swelling of each coat at each time interval was calculated.
Scanning Electron Microscope (SEM) Examination
The surface morphological features of the optimized naringin tablets coated with the different release retardants combined with EUD-L were analyzed before and after in vitro release at different media (pH 1.2, 6.8 and 7.4) using scanning electron microscope (SEM; JSM 5500 LV; JEOL, Tokyo, Japan). SEM examination was performed to assess the effects of the different release media used on the porosity of the different coats of the studied tablets and hence the drug release. Following the exposure to the different release media, the tablets were dried at room temperature overnight. The examined tablets were split by a sharp knife and then coated with gold employing a sputter coater to be imaged using a 15-kV electron beam.
In vitro Drug Release
Drug release from the core and coated tablets was studied using USP apparatus I (Release Apparatus USP Standards, Scientific, DA-6D, Bombay, India). The release media of 0.1N HCl (pH 1.2), PB pH 6.8 and finally PB pH 7.4 were used to simulate pH values pertaining to stomach, small intestine and colon, respectively.32 It has been documented that the average residence time of a formulation in stomach is 2 hrs, and that in small intestine is 3 hrs.33,34 Consequently, the release medium was changed during the study as follows: 0.1N HCl (pH 1.2) for 2 hrs, PB pH 6.8 for 3 hrs, and finally PB pH 7.4 up to 3 hrs to examine the ability of the compression-coated tablets to protect the drug against early release in stomach and small intestine before reaching colon. The release medium (500 mL) was maintained at 37 ± 0.5°C and stirred at 100 rpm. Three milliliters were withdrawn at predetermined time intervals and replaced by fresh release medium. The samples were filtered through a 45-µm membrane filter, properly diluted with the respective release medium and analyzed spectrophotometrically at 282 nm (UV/VIS spectrophotometer, V-550, Jasco, Japan). Plots of mean cumulative percent drug released versus time were constructed to evaluate in vitro drug release.
Release Kinetics
To elucidate the mechanism of drug release, in vitro release data at each of the three release media used were analyzed according to zero-order, first-order,35 and diffusion-controlled release mechanism.36 Korsmeyer–Peppas kinetic model was also employed to identify the release mechanism applying the equation mt/m∞=ktn, where mt/m∞ is the fraction of drug released, k is the kinetic constant, t is the release time and n is the diffusional exponent for drug release and it equals the slope of log mt/m∞ vs log time curve.37 The model with the highest correlation coefficient (r2) was considered to describe naringin release from the prepared tablet formulations.
In vivo Evaluation of the Optimized Naringin Tablets
Induction of Colitis in Rabbits
Male New Zealand rabbits weighing ≈2.5 kg were used in this study because the pharmacological responses of rabbit colonic smooth muscles to inflammatory mediators closely resemble those of human colon.38 Indomethacin has been effectively used as a single intragastric administration of 10 mg/kg to induce intestinal injury and Caco-2 cells damage in rats.39 Firstly, human dose of indomethacin (1.62 mg/kg) was calculated through the relation (human dose (mg/kg) = rat dose (10 mg/kg) × (Km for rat/Km for human), where Km is the correction factor estimated by dividing the average body weight of the species (kg) by its average body surface area (m2), being 6 and 37 for rat and human, respectively.40,41 Then, indomethacin dose (≈8 mg/kg) required to induce intestinal injury in rabbits was calculated by multiplying the average human dose (113.51 mg/70kg) by 0.07.27,42
The experimental procedures were performed according to the ethical principles of the scientific committee of Faculty of Pharmacy, Mansoura University, Egypt, for the use of experimental animals (Approval No. 2013–64 on December 3, 2013). Based on the results of in vitro release and swelling studies as discussed later, the selected tablet formulations to perform in vivo study were those coated with combinations of EUD-L with each of 5% w/w ALG (F3), 5% w/w SCMC (F4) and 10% w/w EC (F5) in comparison with the core tablets (Table 1).
Thirty-six rabbits were randomly divided into six groups, with each group consisting of six animals. Animals were fasted but allowed free access to water for 24 hrs before the day of the experiment. Rabbits of group I were normalized and did not receive any treatment (normal control). Group II was given indomethacin suspended in 1% w/v CMC orally (8 mg/kg, untreated colitis) as a single intragastric administration on the fifth day. Group III was treated with naringin core tablets. Group IV received 10% w/w EC-coated tablets (F5). Group V administered 5% w/w ALG-coated tablets (F3). Rabbits of group VI were given 5% w/w SCMC-coated tablets (F4). Rabbits of the later four groups (III, IV, V and VI) were given examined tablets for 5 successive days. On the fifth day, these rabbits received indomethacin suspension in 1% w/v CMC (8 mg/kg) as a single intragastric administration 2 hrs after tablets administration. Rabbits were euthanized by an overdose of sodium pentobarbital 24 hrs after indomethacin administration.43 Blood samples were collected and centrifuged for biomarker assay. Sera were separated and stored at −80°C. Portions of colonic specimens were cleaned of fats, washed with 0.9% (w/v) saline solution and kept in 10% (v/v) buffered formalin (pH 7.4) for histopathological examination and immunohistochemical localization of TNF-α.
Histopathological Study
Colonic tissues fixed in 10% (v/v) buffered formalin solution were washed, dehydrated by alcohol, cleared in xylene and embedded in paraffin in hot air oven (56°C) for 24 hrs. Sections of 5 μm thickness were deparaffinized and stained with hematoxylin and eosin (H&E). Histopathological alterations in untreated colitis (II) and tablet-pretreated (III–VI) groups were recorded using a light microscope fitted with a digital camera (Leica Microsystems, Germany) and compared to normal group (I). The mucosal damage was scored based on a reported method.44,45 The loss of epithelial surface, destruction of crypts and inflammatory cell infiltration into mucosa was scored by a blinded pathologist as follows: 0 = no change, 1 = localized and mild, 2 = localized and moderate, 3 = extensive and moderate, and 4 = extensive and severe. The mucosal damage score was estimated as the sum of the scores of the three parameters for each animal.
Measurement of Serum (pANCA)
The primary serologic marker associated with ulcerative colitis is typical perinuclear antineutrophil cytoplasmic antibodies (pANCA).46 This serologic marker has been used to distinguish ulcerative colitis from Crohn's disease since increased levels are more common in patients with the former.47,48 The serum levels of pANCA were estimated using a commercially available kit (MyBioSource). The total protein concentration was determined.
Immunohistochemical Localization of TNF-α
Paraffin blocks from rabbit colon were used for immunohistochemical analysis of TNF-α. Cutting of tissue sections was followed by their mounting on Superfrost Plus microscope slides (Thermo Fisher Scientific). Histostain bulk kit-Invitrogen Lab-SA detection system was employed. Slides were prepared using the manual immunohistochemistry stainer following the previously reported procedure.49,50 Sections were deparaffinized with graded xylene and dehydrated in ethanol. Antigen retrieval was accomplished using citrate buffer pH 6.0. The activity of endogenous peroxidase was blocked by 3% H2O2 for 5 mins at room temperature. Sections were incubated with anti-TNF-α antibody (Boster Biological Technology, CA, USA, dilution 1/100) for 40 mins at room temperature and then incubated with the secondary antibody of Universal HRP Multimer for 8 mins at 37°C. Slides were treated with DAB + H2O2 substrate for 8 mins, followed by hematoxylin and the bluing reagent counterstain at 37°C. Reaction buffer (PBS) was used as the washing solution. The staining intensity of positively stained cells was recorded. Controls were prepared by staining without primary antibody. Staining was recorded with a digital camera (Olympus) mounted on a microscope (Leica Microsystems). Specimen examination was done by a senior pathologist. Immunoreactive cells for TNF-α were counted using ImageJ analysis (NIH, Bethesda, MD, USA). The number of immunopositive cells per 1000 cells was determined in triplicate and the average was calculated.51
Statistical Analysis
The nonparametric mucosal damage scores were statistically analyzed applying Kruskal–Wallis test followed by Dunn's test to compare all pairs. The normality of data distribution of pANCA and TNF-α levels was assessed using Kolmogorov–Smirnov normality test. The statistical analysis of the normally distributed results was accomplished by applying one-way ANOVA followed by Tukey–Kramer multiple comparison test. The statistical analysis was performed employing GraphPad prism software (version 5.00; GraphPad software, San Diego, CA, USA).
Results and Discussion
Characterization of the Prepared Tablets
The characteristics of core and coated tablets are illustrated in Table 2. All tablets showed good weight uniformity with a small deviation from the mean (≤1.76). Average hardness was 5.17 ± 0.60 kg/cm2 for the prepared core and ranged from 7.27 ± 0.51 to 10.05 ± 0.30 kg/cm2 for coated tablets complying with the pharmacopoeial limit of 4–10 kg/cm2.28 In accordance, all tablets were non-friable as indicated by friability% less than 1%. The higher hardness of coated tablets relative to the cores could decrease compression coatings porosity, and hence increase the lag time permitting colon targeting.23 The average thickness and diameter of core tablets (2.87 ± 0.02 and 9.03 ± 0.01 mm, respectively) were increased on applying the compression coatings up to 3.78 ± 0.09 mm and 12.09 ± 0.01 mm, respectively. All tablets exhibited drug content ranging from 99.00 ± 1.70 to 101.00 ± 2.00 in accordance with the pharmacopoeial range (90–110%) of the labeled claim.28
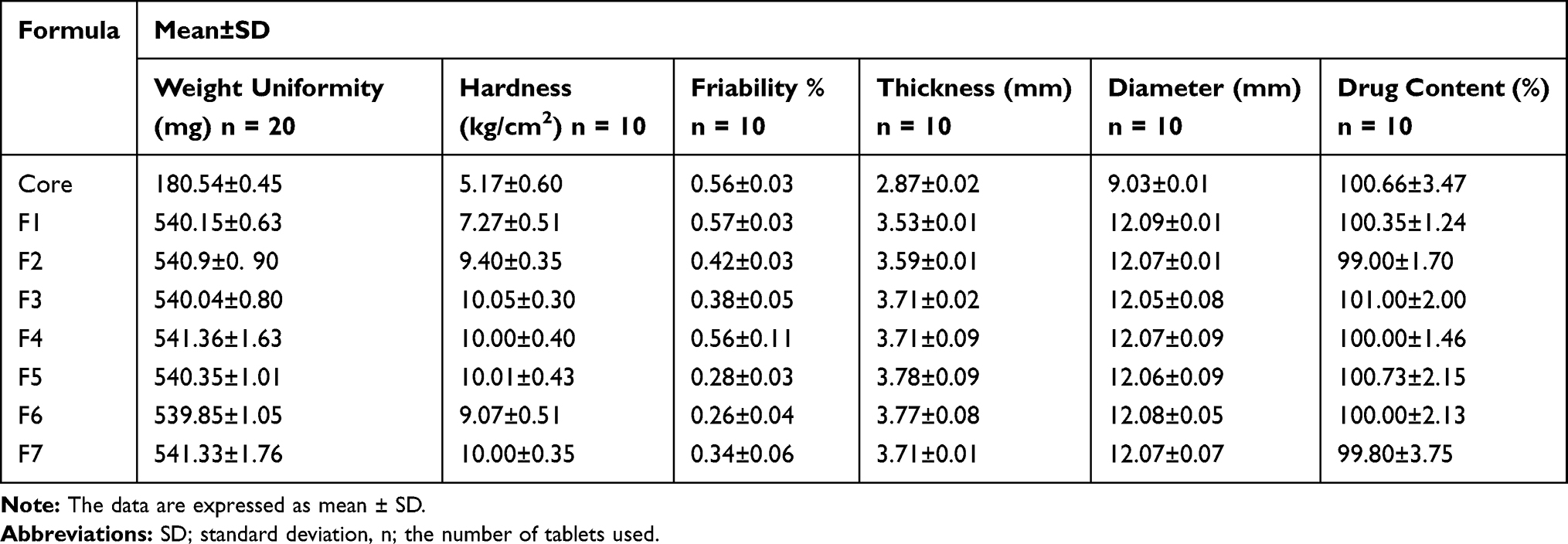 |
Table 2 Characterization of the Prepared Tablets |
Study of Drug–Polymer Interaction
Fourier Transform-Infrared (FT-IR) Spectroscopy
Figure 1 depicts the FT-IR spectra of free naringin, each of the polymers used, drug–polymer 1:1 PM and the corresponding KMs exposed to the different release media. Naringin has polyhydroxy groups; thus O–H bond stretching resulted in a strong band in the range of 3000–3500 cm−1.50,52 Other characteristic bands of naringin included aromatic –OH at 1516 cm−1, phenolic –OH at 1207 and 1362 cm−1, and carbonyl group at 1644 cm−1. Similar data have been reported.53 The band of –OH axial deformation at 1042 cm−1 appeared in the FT-IR spectrum of naringin.54
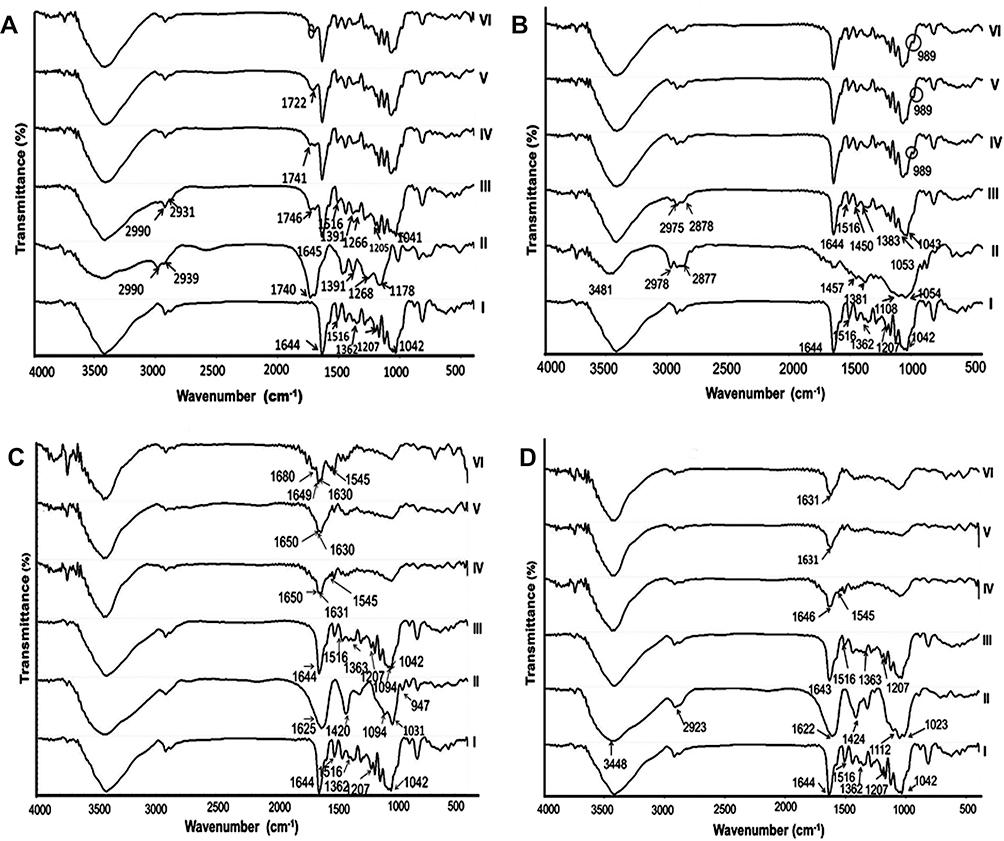 |
Figure 1 FT-IR spectra. Notes: (A) EUD-L, (B) EC, (C) ALG, and (D) SCMC. (I) naringin, (II) polymer, (III) 1:1 PM, and (IV) 1:1 KM at pH 1.2, (V) 1:1 KM at pH 6.8, and (VI) 1:1 KM at pH 7.4. Abbreviations: FT-IR, Fourier transform-infrared; EUD-L, Eudragit L; EC, ethylcellulose; ALG, sodium alginate; SCMC, sodium carboxymethyl cellulose; PM, physical mixture; KM, kneaded mixture. |
The spectrum of EUD-L 100-55 showed a broad band of hydroxyl groups (–OH stretch vibration) in the range of 3000–3600 cm−1, methyl and methylene C–H stretch vibration at 2990 and 2939 cm−1, a strong stretching C=O band at 1740 cm−1, and two bands of ester C–O stretching vibration at 1268 and 1178 cm−1 (Figure 1A).55 FT-IR spectrum of naringin-EUD-L 1:1 PM appeared to be an overlap of that of drug and polymer. In KMs with EUD-L at pH 6.8, there was a shift in C=O stretching peak of EUD-L to 1722 cm−1 versus 1746 cm−1 in corresponding PM. At pH 7.4, it can be said that such a peak was noticeably reduced. An interaction through hydrogen bonding between C=O of EUD-L and –OH groups of naringin in the presence of the buffers may explain these changes. Similar results have been reported for EUD-L 100-55 nanofibers of diclofenac sodium.55
In FT-IR spectrum of EC (Figure 1B), a distinct peak at 3481 cm−1 due to –OH groups, asymmetric peak around 2978–2877 cm−1 describing –CH stretching, a peak of –CH2 bending at 1457 cm−1, a peak at 1381 cm−1 due to –CH3 bending and a broad peak of C–O–C stretch in the cyclic ether at 1108 cm−1 were observed.56 EC-naringin PM showed characteristic bands of both components. A new peak appeared at 989 cm−1 in all KMs of the drug with EC. This could suggest an interaction of naringin with EC on kneading with the three-release media.
In ALG spectrum, there were bands at 1625 and 1420 cm−1 of asymmetric and symmetric stretching of carboxylate salt groups, respectively (Figure 1C). In addition, the bands around 1094 cm−1 (C–O stretching), 1031 cm−1 (C–O–C stretching) and 947 cm−1 (C–O stretching) were related to the saccharide structure. Similar spectral characteristics of ALG have been documented in the literature.57 The spectrum of naringin-ALG PM exhibited all characteristic bands of the drug. In comparison with PM, KMs with ALG showed some spectral changes. For example, new peaks at 1545 cm−1 and 1680 cm−1 were observed in the spectrum of KM at pH 7.4. In addition to the new peak at 1545 cm−1, the peak of C=O group appeared in PM spectrum at 1645 cm−1 was divided into two peaks at 1631 cm−1 and 1650 cm−1 in the spectrum of KM at pH 1.2. Such a divided peak was also observed in the spectrum of ALG KM at pH values 6.8 and 7.4. As well, the intensity of the bands ranged from 1000 to 1500 cm−1 was considerably reduced on kneading of naringin-ALG PM with the different release media. These spectral modifications may be explained on the basis that there was an interaction via hydrogen bonding between C=O and –OH groups in these compounds on kneading with the used release media.
In SCMC spectrum, a strong peak at 3448 cm−1 assigned to –OH stretching vibration was obtained (Figure 1D). The band due to ether bond (–O–) was recorded at the range of 1023–1112 cm−1. The peak related to the carboxylate was observed at 1622 cm−1. The peak of methylene at 2923 cm−1 was present. Reported spectral characteristics were similar to those obtained in this study.58 The characteristic peaks of the drug were noticed in its PM with SCMC. On the other hand, the band of carbonyl group in naringin was shifted from 1643 cm−1 in SCMC PM to 1631 cm−1 in the spectra of KMs with release media of pH 6.8 and 7.4. In the spectrum of KM at pH 1.2, a new peak at 1545 cm−1 was recorded. Moreover, there was a noticeable reduction in the peak’s intensity at the range of 1000–1500 cm−1 in all KMs with SCMC. These data may reflect the possibility of an interaction between the drug and this polymer possibly through hydrogen bonding following the exposure to the different release media.
Differential Scanning Calorimetry (DSC)
Figure 2A illustrates DSC thermograms of free naringin, EUD-L, their 1:1 PM and KMs. According to this figure, naringin showed two endothermic peaks at 95.51°C and 162.93°C corresponding to the successive loss of two water molecules.54 A sharp endothermic peak at 245.57°C is related to naringin melting.53,59 EUD-L exhibited an endotherm at 202.28°C corresponding to its glass transition temperature.55 Naringin-EUD-L PM showed the melting endothermic peak of the drug, yet the intensity was reduced possibly due to the dilution effect in the mixture. Also, it was shifted to a lower temperature since the drug may disperse in the molten mass of the polymer.60 Compared to PM with EUD-L, the absence of a drug endothermic peak in DSC curves of all KMs with EUD-L was recorded. In accordance with FT-IR data, these results may suggest an interaction between the drug and EUD-L following exposure to the release media.
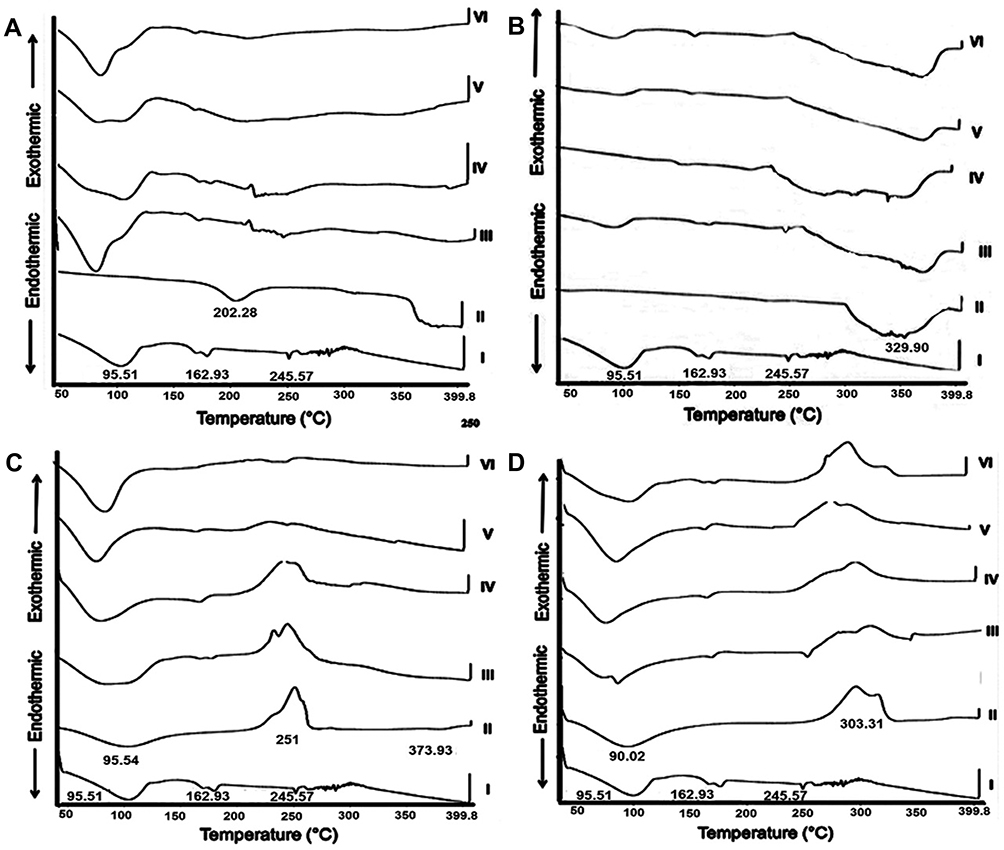 |
Figure 2 DSC thermograms. Notes: (A) EUD-L, (B) EC, (C) ALG, and (D) SCMC. (I) naringin, (II) polymer, (III) 1:1 PM, and (IV) 1:1 KM at pH 1.2, (V) 1:1 KM at pH 6.8, and (VI) 1:1 KM at pH 7.4. Abbreviations: DSC, differential scanning calorimetry; EUD-L, Eudragit L; EC, ethylcellulose; ALG, sodium alginate; SCMC, sodium carboxymethyl cellulose; PM, physical mixture; KM, kneaded mixture. |
DSC thermogram of EC showed no sharp peaks indicating its amorphous nature (Figure 2B). The peaks recorded above 300°C may indicate the decomposition of this polymer. Similar results have been reported.56 The drug endothermic peak was still seen in DSC curves describing naringin-EC PM. However, this peak was absent in the DSC curves of all KMs with EC. In agreement with FT-IR data, an interaction between the drug and EC at different release media can be proposed.
Loss of loosely bound water from ALG was indicated by an endothermic peak at 95.54°C (Figure 2C). The exothermic peak at 251°C can be explained by the polymer decomposition. The recorded thermal behavior was similar to that reported.61–63 Drug endotherm and ALG exotherm overlapped in their PM. Naringin endothermic peak was absent in DSC thermograms of its KMs with ALG at all media. Consequently, an interaction of the drug and ALG in the presence of release media as suggested by FT-IR may be still expected.
DSC curve of SCMC alone showed an endotherm at 90.02°C possibly due to its dehydration (Figure 2D). The exothermic peak at 303.31°C may be related to the transition temperature of SCMC.64 The drug endothermic peak appeared together with the polymer exotherm in DSC thermogram of their PM. On the other hand, the drug peak was absent in its KMs with SCMC. Accordingly, an interaction of naringin with SCMC may occur on the contact with the release media.
Swelling Study
welling of compression coatings was investigated to clarify the influences of release medium pH as well as natural release retardant and concentration on coats swelling, drug release and possibility of colon targeting. The average values of percentage swelling of different compression coatings based on 5% and 10% w/w of release retardants in combination with EUD-L using three successive media of pH 1.2 for 2 hrs, followed by PB pH 6.8 for 3 hrs and finally PB pH 7.4 up to 3 hrs are depicted in Figure 3A and B, respectively.
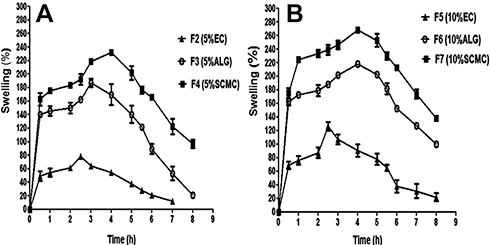 |
Figure 3 Swelling of naringin compression-coated tablets containing different release retardants in combination with EUD-L at pH 1.2 (2 hrs), 6.8 (3 hrs) and 7.4 (up to 3 hrs). Notes: (A) 5% w/w and (B) 10% w/w of each release retardant.Abbreviations: EUD-L, Eudragit L; EC, ethylcellulose; ALG, sodium alginate; SCMC, sodium carboxymethyl cellulose. |
Generally, release medium pH as well as natural release retardant and concentration affected the swelling behavior of the coats. Regarding release medium pH, all release retardants at the two concentrations (5% and 10% w/w) showed high percentage swelling of the coats at pH 1.2 particularly throughout the first half hour probably due to the rapid hydration and the high affinity to the test medium. At PB pH 6.8, the increase in the percentage swelling continued reaching a maximum at 2.5 hrs for EC-coated tablets (F2 and F5), 3 hrs for 5% w/w ALG coating (F3) and 4 hrs for both 10% w/w ALG (F6) as well as SCMC-coated tablets at both ratios (F4 and F7), followed by a slight decline. On the other hand, a decline in the swelling of all coats was observed at PB pH 7.4 beyond the fifth hour that could suggest erosion of swollen coats. Based on the comparison of the maximum percentage swelling (MPS), polymer nature significantly (P<0.05) influenced the coat swelling in the order of SCMC > ALG > EC at both concentrations of 5% w/w (Figure 3A) and 10% w/w (Figure 3B). As well, the increase in the polymer concentration from 5% to 10% w/w significantly (P<0.05) enhanced coats swelling as reflected by significantly higher (P<0.05) MPS of coats containing 10% w/w EC, ALG or SCMC (126.10 ± 6.38, 218.50 ± 3.85 and 267.90 ± 3.57, respectively) when compared to those based on 5% w/w of EC, ALG or SCMC (78.31± 2.34, 186.90 ± 6.26 and 231.90 ± 2.56, respectively).
Scanning Electron Microscope (SEM) Examination
As clarified later based on an in vitro release study, the optimized tablets that provided minimal drug release at pH values 1.2 and 6.8 were those coated with 10% w/w EC (F5) and 5% w/w of each of ALG (F3) and SCMC (F4), all together with EUD-L. SEM microphotographs of the optimized tablets before and after in vitro release at different media (pH 1.2, 6.8 and 7.4) are illustrated in Figure 4. Before release, 10% w/w EC-coated tablets (F5) showed a nonporous, compact and homogenous structure (Figure 4A). Following in vitro release at pH 1.2, small pores were observed possibly because EC coat started to swell at this pH. Both the number and size of the pores in this coat increased after release at PB pH 6.8. This could be related to higher swelling rates of the coat at this pH as clarified by swelling study. The increase in the pore size of this coat continued following in vitro release at PB pH 7.4 since the swollen coat could be exposed to erosion by the diffused release medium as suggested by swelling study. Similar behavior was noticed in the case of optimized tablets coated with 5% w/w of each of ALG (Figure 4B) and SCMC (Figure 4C) before and after release. Larger pores were noticed in case of EC coats followed by ALG coats and finally those of SCMC. These results could be related to the faster erosion of the swollen EC coats by diffused release medium than those of ALG and SCMC as suggested by respective times of MSP of 2.5, 3 and 4 hrs (Figure 3).
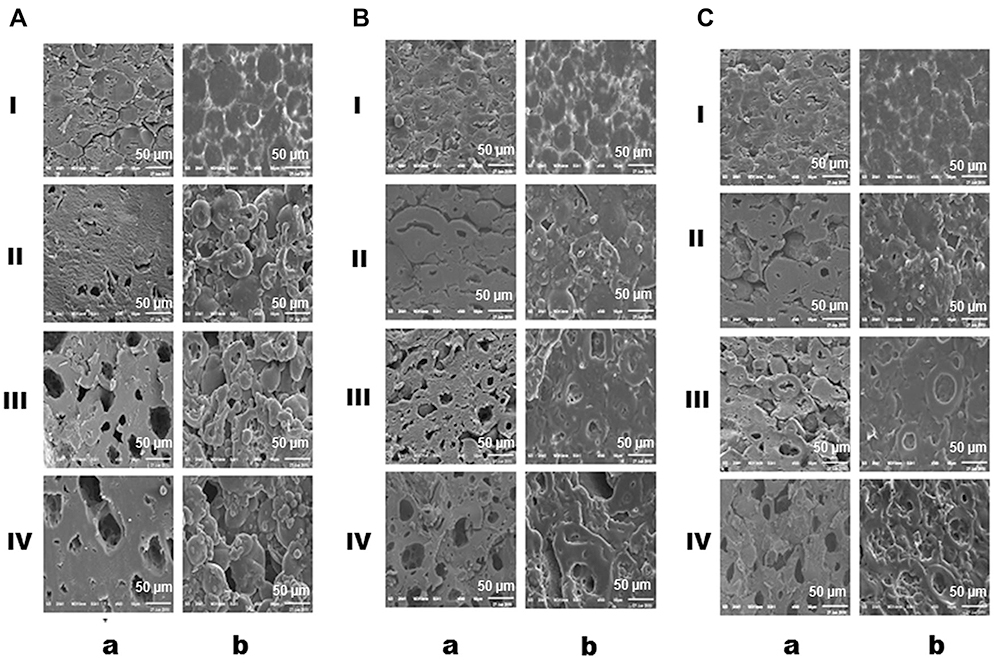 |
Figure 4 SEM microphotographs of cut-coated tablets. Notes: (A) 10% w/w EC, (B) 5% w/w ALG, and (C) 5% w/w SCMC, all in combination with EUD-L. (a) side view of the coat, (b) surface view of the coat, (I) before in vitro release, as well as after in vitro release at pH (II) 1.2, (III) 6.8, (IV) 7.4.Abbreviations: SEM, scanning electron microscopy; EUD-L, Eudragit L; EC, ethylcellulose; ALG, sodium alginate; SCMC, sodium carboxymethyl cellulose. |
In vitro Release Study
In vitro release of naringin from core tablets coated with EUD-L in combination with each of EC, ALG and SCMC at 5% w/w (F2, F3 and F4, respectively) as well as at 10% w/w (F5, F6 and F7, respectively) compared to both core tablets and those coated with EUD-L alone (F1) is illustrated in Figure 5A and B, respectively. The results revealed that core tablets showed almost a complete release after 1.5 hrs in 0.1N HCl. On the other hand, significantly (P<0.05) slower release in 0.1N HCl was observed on the coating of core tablets with EUD-L alone (F1), yet a complete release (98.14% ± 1.86) after 5 hrs in PB pH 6.8 was seen negating the possibility of colon targeting. Consequently, incorporation of different release retardants in combination with EUD-L in the coats was attempted for drug release retardation in PB pH 6.8. The effects of natural release retardant and concentration as well as release medium pH on naringin in vitro release from the coated tablets were studied.
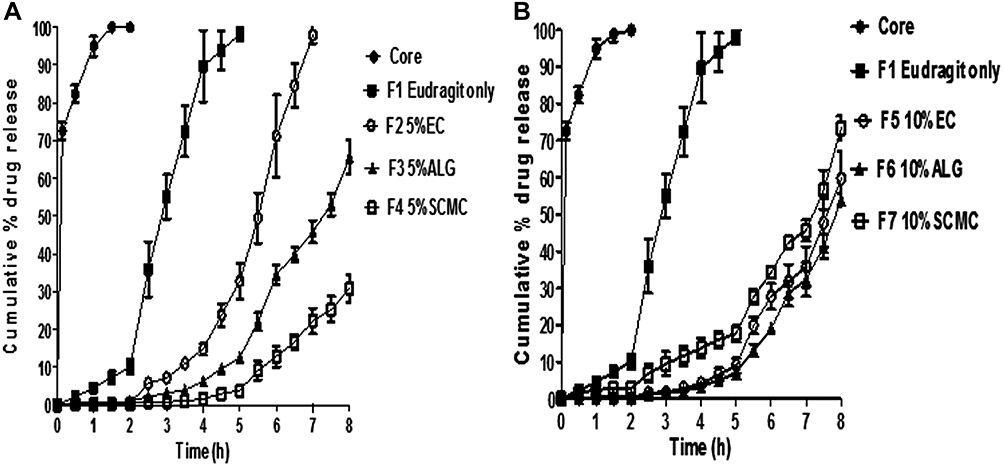 |
Figure 5 In vitro release of naringin at pH 1.2 (2 hrs), 6.8 (3 hrs) and 7.4 (up to 3 hrs) from compression-coated tablets containing different release retardants in combination with EUD-L in comparison with core tablets and cores coated with 100% EUD-L (F1). Notes: (A) 5% w/w and (B) 10% w/w of each release retardant.Abbreviations: EUD-L, Eudragit L; EC, ethylcellulose; ALG, sodium alginate; SCMC, sodium carboxymethyl cellulose. |
Regarding release medium pH, drug release from coated tablets in 0.1N HCl was significantly (P<0.05) lower than that seen at PB pH 6.8 and 7.4. These results could be attributed to the polymers swelling at pH 1.2 (Figure 3) that would increase the diffusional path length of the release medium, and hence retard the drug release. The low porosity of the swollen coat at this pH as recorded by SEM may account for the minimal drug release at the acidic medium (Figure 4). The enhanced swelling (Figure 3) as well as the more porous structure of the coats as illustrated by SEM microphotographs (Figure 4) could explain the higher drug release at PB pH 6.8 relative to pH 1.2. The noticeable erosion of all swollen polymeric matrices suggested by a decline in percentage swelling beyond the fifth hour at the two concentrations used (5% and 10% w/w) may also explain the significantly (P<0.05) higher drug release at PB pH 7.4 compared to PB pH 6.8 (Figure 3). Such erosion can be also suggested by the increase in pores size at PB pH 7.4 as clarified by SEM examination (Figure 4).
Drug release was significantly (P<0.05) affected by the natural release retardant at both concentrations of 5% and 10% w/w. The retardation of drug release can be arranged in the following order: SCMC > ALG > EC resembling that of swelling percentage. This indicates that the retarded release of naringin in PB pH 6.8 on mixing these polymers with EUD-L in the coats can be attributed to the remarkable polymer swelling at this pH (Figure 3). SEM examination revealed that the pore size increased in the reverse order: EC coat > ALG coat > SCMC coat (Figure 4). The increase in the pore size and number would facilitate the diffusion of the release medium into and out the swollen coats enhancing the drug release.65
Generally, more retarded drug release was noticed on the increase in release retardant concentration from 5% w/w (Figure 5A) to 10% w/w (Figure 5B) possibly due to the enhanced coat swelling (Figure 3). As the amount of swellable polymer increases in the coat, the gel layer formed surrounding the tablet thickens increasing the diffusion path length of the release medium and slowing the drug release.
It has been reported that the mean time for a formulation to reach colon can be estimated as 5 hrs since the average residence time in stomach is 2 hrs, and that in small intestine is 3 hrs.30,31 Accordingly, several studies have reported that cumulative percentage release after 5 hrs (CR5h) less than 20% can refer to gastric and intestinal protection against considerable drug release, and hence colon targeting can be expected.30,66,67 Therefore, CR5h was used to compare the coated tablets with respect to their ability to retard naringin release particularly at PB 6.8 and possibly target it to colon. Lag time taken for <10% drug to be released was also used to evaluate the ability of compression coats to minimize the drug release in the upper gut.
Significant (P<0.05) retardation of drug release in PB pH 6.8 was obtained on incorporating 5% w/w of EC, ALG or SCMC with 95% w/w EUD-L (F2, F3 and F4, respectively) as clarified by significantly (P<0.05) lower respective CR5h (32.81% ± 4.70, 12.57% ± 1.44 and 3.91% ± 1.24) than that of cores coated with EUD-L alone (98.14% ± 1.86). In accordance, respective lag times were 3.5, 4.5 and 5.5 hrs versus 2 hrs for tablets coated with EUD-L alone. Only compression coatings of EUD-L in combination with each of ALG and SCMC at a respective weight ratio of 95:5 can be expected to target naringin to colon since they exhibited CR5h < 20%. Lowering in drug release as a result of increasing the release retardant concentration in the coat was only significant (P<0.05) in case of 10% w/w EC (F5, CR5h = 8.96 ± 2.09) when compared to tablets coated with 5% w/w EC (F2, CR5h = 32.81% ± 4.70). More prolonged lag time of 5 hrs was observed for tablets coated with 10% w/w EC (F5) or ALG (F6). Surprisingly, an increase of drug release in PB pH 6.8 as a result of increased SCMC concentration in the coating from 5% w/w (F4) to 10% w/w (F7) was recorded possibly due to the hydrophilic nature of this polymer that may have allowed the penetration of the release medium.25 However, minimal drug release was still found in pH 1.2 and 6.8 (CR5h = 17.65% ± 2.21). Therefore, it can be said that all tablets coated with a mixture of EUD-L and each of the investigated release retardants at a concentration of 10% w/w can be described as possible colon targeted systems of naringin.
Considering the lower release retardant concentration required to obtain colon targeting to comply with the economical point of view for large-scale production, tablets coated with 5% w/w of each of ALG and SCMC (F3 and F4, respectively) and those coated with 10% w/w EC (F5) were selected to investigate the cytoprotective activity of naringin against indomethacin-induced colitis in rabbits.
Release Kinetics
Table 3 illustrates the results of kinetic analysis of release data of naringin from the prepared core and compression-coated tablets. According to this table, the release mechanism varied at pH 1.2 while it obeyed zero order for the majority of studied tablets at PB pH 6.8 and 7.4. Korsmeyer–Peppas model was used to verify the release mechanism. According to this model, n ≤ 0.5 may indicate Fickian diffusion and values of (n) between 0.5 and 1 correspond to non-Fickian release.37 At pH 1.2, core tablets showed predominant erosion suggested by n > 1. Fickian release mechanism described the drug release from the examined coated tablets indicating diffusion-controlled release of the drug that may account for its slow release at pH 1.2. The high swelling rate of the release retardants used at this pH at both concentrations of 5% and 10% w/w may explain these results (Figure 3). And, non-Fickian release can be suggested for the tested coated tablets at pHs 6.8 and 7.4 indicating drug release controlled by both diffusion and erosion possibly due to the increased swelling percentages till reaching a maximum value followed by a decline (Figure 3) as well as more porous coats at the buffers relative to the acidic medium as suggested by SEM examination (Figure 4).
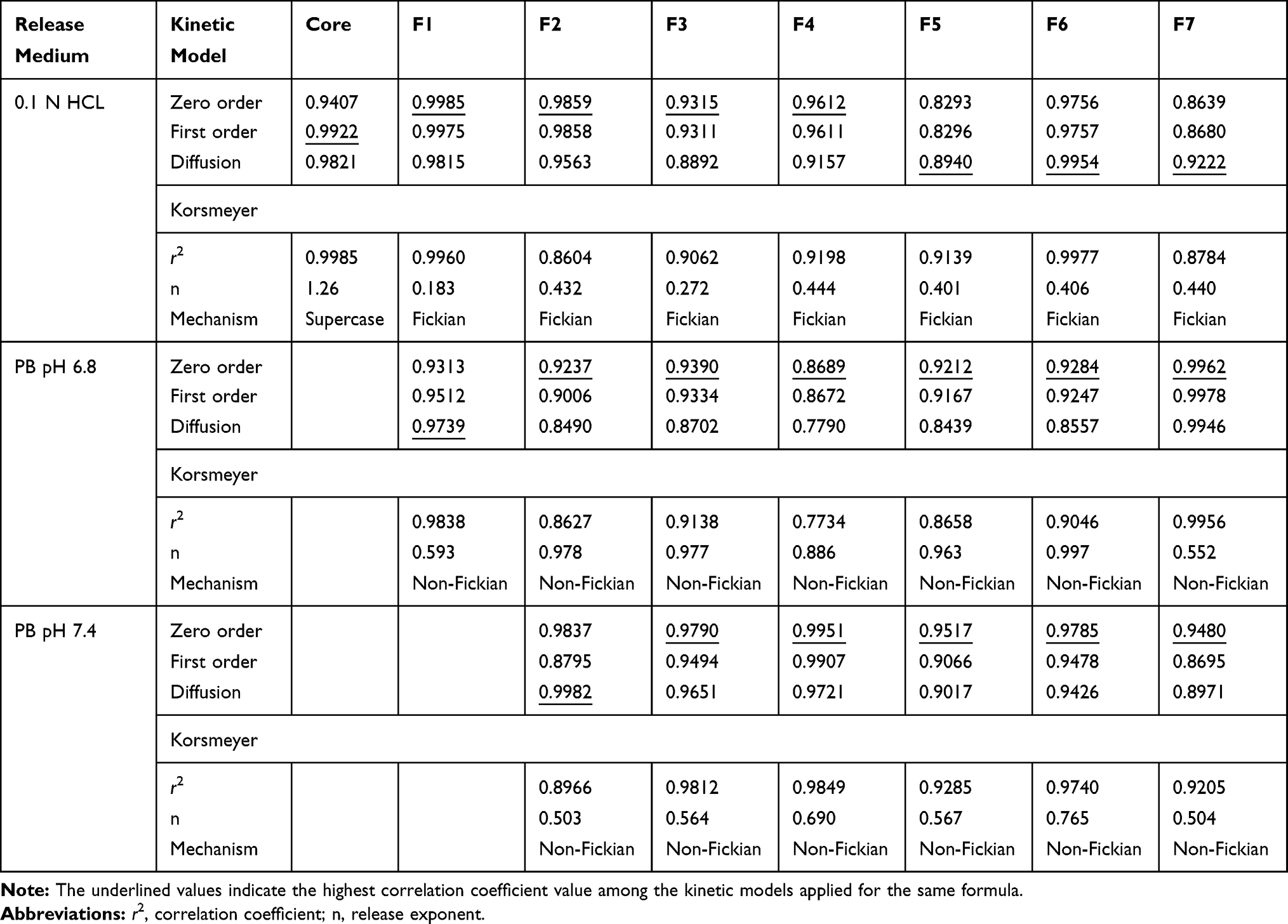 |
Table 3 Kinetic Analysis of Release Data |
Histopathological Examination of Rabbit’s Colon
Colon of the normal control group (I) showed normal mucosa with normal lining enterocytes and normal submucosa (Figure 6A). Administration of indomethacin to group II without naringin pretreatment resulted in ulceration of entire thickness of mucosa and intense neutrophilic infiltration confirming that indomethacin at an oral dose of 8 mg/kg induced a detectable ulceration in rabbit’s colon (Figure 6B). Similar findings have been obtained in the colon of rabbits received naringin cores (group III) 5 days before indomethacin administration (Figure 6C). These results can be explained on the basis that naringin cores did not provide gastric and intestinal protection against drug release as reflected by complete in vitro drug release at 0.1N HCl during the first 1.5 hrs (Figure 5), and hence no colon targeting can be expected. In addition, low naringin absorption (about 8%) following oral administration of its cores can be proposed due to cleavage in lumen or cell gut at harsh pH (1–2.5) and enzymatic conditions of upper GIT.8–10
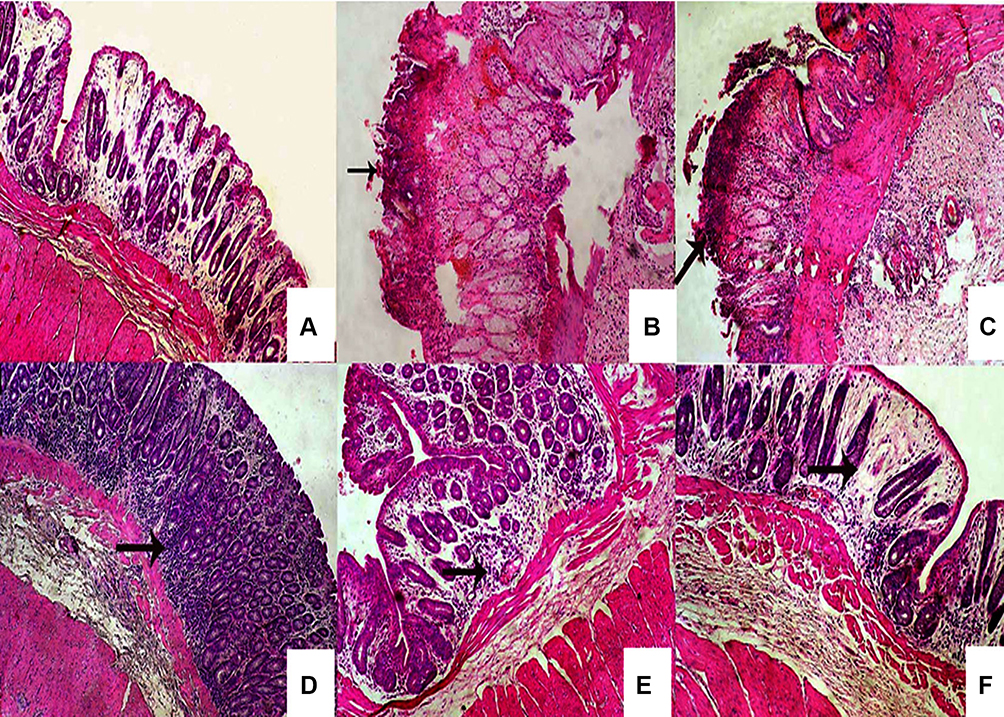 |
Figure 6 Histopatholological examination of rabbit colonic tissues. Notes: (A) Normal mucosa and submucosa of normal control animals, ulceration of entire thickness of mucosa and intense neutrophilic infiltration (arrow) of (B) untreated colitis group (indomethacin, 8 mg/kg), and (C) those pretreated with core tablets, (D) normal epithelial lining of colonic mucosa and lymphohistiocytic infiltration in the lamina propria (arrow) in rabbits pretreated with cores coated with 10% w/w EC, (E) normal epithelial layer and mild edema and histiocytic recruitment in the lamina propria of the colonic mucosa (arrow) of rabbits pretreated with 5% w/w ALG-coated tablets, (F) mild edema in the lamina propria and normal epithelial lining of the mucosa in rabbits pretreated with 5% w/w SCMC coated tablets. (H&E, 100×).Abbreviations: EC, ethylcellulose; ALG, sodium alginate; SCMC, sodium carboxymethyl cellulose; H&E, hematoxylin and eosin. |
On the other hand, group IV that received naringin tablets coated with 10% w/w EC exhibited normal epithelial lining of colonic mucosa and lymphohistiocytic infiltration in the lamina propria (Figure 6D). Oral administration of tablets coated with 5% w/w ALG (group V) resulted in normal epithelial layer and mild edema and histiocytic recruitment in the lamina propria of the colonic mucosa (Figure 6E). Rabbits pretreated with 5% w/w SCMC coated tablets (group VI) showed mild edema in the lamina propria and normal epithelial lining of the mucosa (Figure 6F). Colon displayed normal mucosa with normal supporting stromal cells in rabbits of normal control (Figure 7A). Predominant neutrophilic infiltration was observed in the colonic mucosa of rabbits of untreated colitis group (II) and those pretreated with core tablets (Figure 7B and C, respectively). Lymphocytic and histiocytic infiltration in the colonic mucosa was recorded following pretreatment with tablets coated with 10% w/w EC (Figure 7D). There was a histiocytic recruitment in the colonic mucosa of animals that received 5% w/w ALG-coated tablets (Figure 7E). Rabbits administered tablets coated with 5% w/w SCMC showed mild edema in the colonic mucosa (Figure 7F).
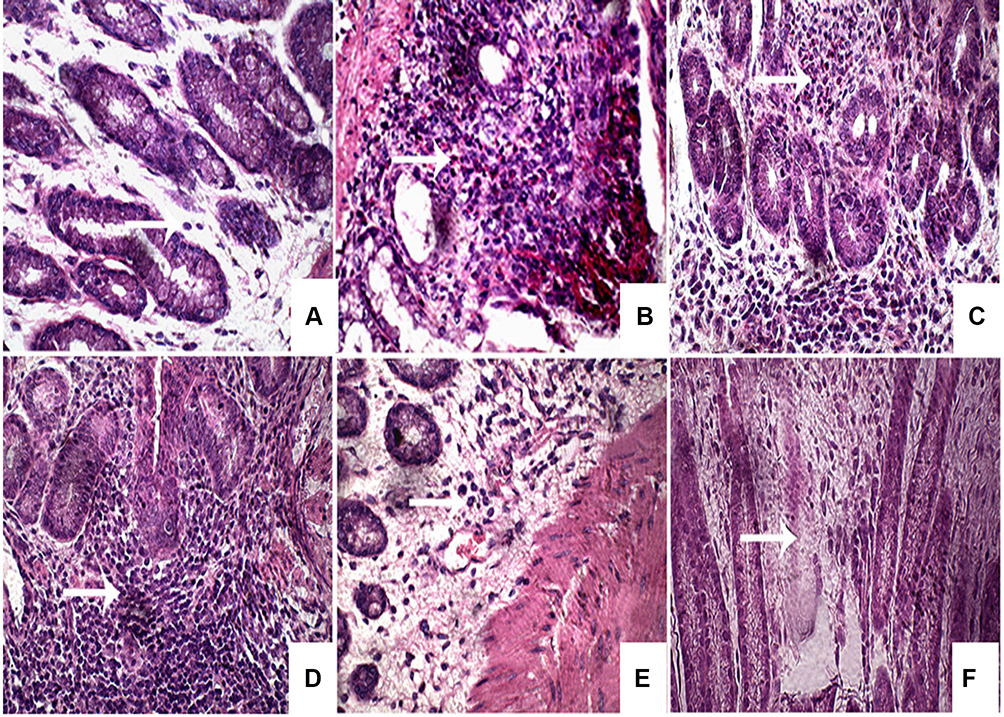 |
Figure 7 Microphotographs illustrating the type of leukocytes infiltration in rabbit colonic tissues. Notes: (A) Normal mucosa with normal supporting stromal cells in normal control, predominant neutrophilic infiltration in the colonic mucosa of untreated colitis group (B) and those pretreated with core tablets (C), (D) lymphocytic and histiocytic infiltration in the colonic mucosa of rabbits pretreated with 10% w/w EC-coated tablets, (E) histiocytic recruitment in the colonic mucosa of animals that received 5% w/w ALG-coated tablets, (F) mild edema in the colonic mucosa of rabbits that administered tablets coated with 5% w/w SCMC. (H&E, 400×).Abbreviations: EC, ethylcellulose; ALG, sodium alginate; SCMC, sodium carboxymethyl cellulose; H&E, hematoxylin and eosin. |
Figure 8 depicts results describing the mucosal damage scores of the different studied groups. In agreement with the histopathological examination, the core tablet-pretreated and untreated colitis groups (II and III, respectively) showed significantly (P<0.05) higher mucosal damage scores than normal control (group I), yet there was insignificant difference between them. The coated tablets (groups IV–VI) produced significantly (P<0.05) lower mucosal damage scores than that of untreated colitis group (II). In comparison with rabbits received core tablets (group III), the coating with 5% w/w of each of ALG and SCMC resulted in a significant reduction in the mucosal damage.
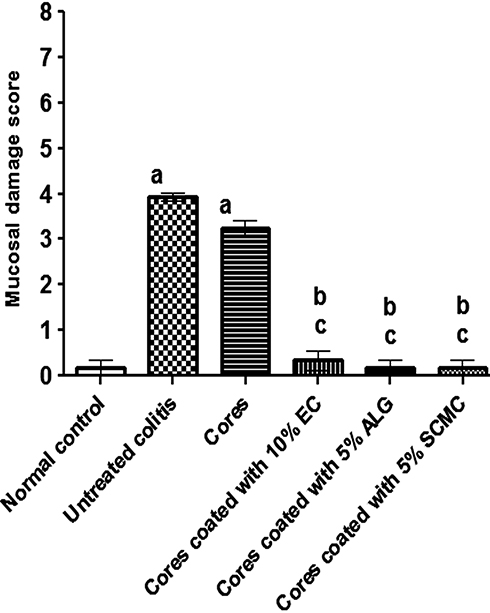 |
Figure 8 Mucosal damage scoring of (H&E) stained sections of rabbits colon in all studied groups. Notes: Data are expressed as mean ± SEM. a means significantly (P<0.05) different from normal control animals. b indicates significantly (P<0.05) different from untreated colitis group. c means significantly (P<0.05) different from animals pretreated with core tablets.Abbreviations: H&E, hematoxylin and eosin; SEM, standard error of mean; EC, ethylcellulose; ALG, sodium alginate; SCMC, sodium carboxymethyl cellulose. |
Consequently, it can be said that compression coating of naringin cores with these polymers, particularly ALG and SCMC in combination with EUD-L, potentially improved its protective action against indomethacin-induced colitis in rabbits as suggested by insignificant difference in mucosal damage scores from that of the normal control group (Figure 8). These results could be explained on the basis that the swelling of these coated tablets (Figure 3) and the drug release retardation (Figure 5) may have resulted in colon targeting. At lower concentration (5% w/w), tablets coated with ALG and SCMC showed insignificantly different mucosal damage scores in comparison with 10% w/w EC-coated tablets. Hence, these two release retardants can be considered more effective than EC to potentiate the cytoprotective activity of naringin against indomethacin-induced colitis in rabbits. These results may be attributed to the significantly higher (P<0.05) MPS for coats containing 5% w/w ALG or SCMC (186.90 ± 6.26 and 231.90 ± 2.56, respectively) when compared to those based on 10% w/w of EC (126.10 ± 6.38). Inspite of the significantly higher MPS of coated tablets based on 5% w/w SCMC, their mucosal damage score did not significantly differ from that imparted by 5% w/w ALG-coated tablets (Figure 8). ALG inhibitory effects on cytokine such as TNF-α and eicosanoid production as well as matrix metalloproteinase-2 (MMP-2) activity may explain these findings.68 The successful use of compression-coated tablets of budesonide in the treatment of ulcerative colitis has been documented.32
Serum Levels of pANCA
Within the group, statistical analysis using Kolmogorov–Smirnov normality test revealed that pANCA levels were normally distributed (data not shown). The serum levels of pANCA, as an important quantitative index of colonic inflammation, were demonstrated in Figure 9. Surprisingly, the high basal levels of pANCA antibodies in the normal control group were recorded. This can be explained on the basis that pANCA identifies an antigen(s) expressed by bacteria resident in the colonic mucosa including Bacteroides caccae and Escherichia coli.69 However, the pANCA serum levels of untreated colitis group (II) were significantly (P<0.05) higher than the normal control group (I) confirming the induction of a detectable colitis using indomethacin at a dose of 8 mg/kg orally. Significantly (P<0.05) increased serum levels of pANCA in rabbits received naringin core tablets (group III) were obtained in comparison with those of normal control group (I), yet it did not significantly differ from that of untreated colitis group (II). These results may be due to the inability of core tablets to protect the drug against excessive release and degradation at upper GIT. In comparison with untreated colitis group (II) and that pretreated with core tablets (group III), the serum levels of pANCA were significantly (P<0.05) lower in rabbits given naringin tablets coated with 10% w/w EC (F5, group IV), 5% w/w ALG (F3, group V) and 5% w/w SCMC (F4, group VI). These results may be explained on the basis that the swelling and the delayed drug release obtained on coating of naringin cores with these release retardants in combination with EUD-L may have allowed colon targeting and potentiated cytoprotection of this drug. There was an insignificant difference in pANCA serum levels among these groups (IV, V and VI) when compared to the normal group (I). In accordance with histopathological examination, these results may indicate the potentiated cytoprotective activity of these coated tablets of naringin against indomethacin-induced colitis in rabbits.
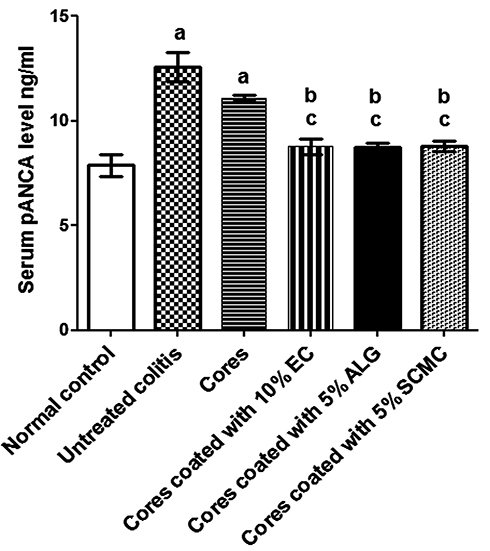 |
Figure 9 Serum levels of pANCA in rabbits pretreated with selected naringin-coated tablets compared to normal control and untreated colitis groups. Notes: Data are expressed as mean ± SEM. a means significantly (P<0.05) different from normal control animals. b indicates significantly (P<0.05) different from untreated colitis group. c means significantly (P<0.05) different from animals pretreated with core tablets.Abbreviations: pANCA, perinuclear antineutrophil cytoplasmic antibodies; SEM, standard error of mean; EC, ethylcellulose; ALG, sodium alginate; SCMC, sodium carboxymethyl cellulose. |
TNF-α Immunohistochemical Localization
Among the most relevant factors participating in the inflammatory process of patients with inflammatory bowel disease is TNF-α.2,70 TNF-α is a proinflammatory mediator that plays an evident role in the pathogenesis of inflammatory bowel disease, and a genetic association between TNF-α and ulcerative colitis has been proven.71 Studies involved patients with ulcerative colitis have indicated increased TNFα levels, and hence the biologic anti-TNF-α therapy has been found to be effective in ulcerative colitis treatment.71
The normal control group (I) exhibited almost negative immunostain for TNF-α (Figure 10A). The untreated colitis group (II) and the group that received naringin core tablets (group III) showed positive immunoreactivity for epithelial lining mucosa, inflammatory cells in the submucosa and endothelial lining blood vessels (Figure 10B and C, respectively). Colon of rabbits pretreated with naringin tablets coated with 10% w/w EC (F5, group IV) showed positive immunoreactivity for inflammatory cells in the lamina propria and the submucosa (Figure 10D). Groups V and VI that were given tablets coated with 5% w/w of each of ALG (F3) and SCMC (F4), respectively, experienced mild immunoreactivity limited to inflammatory cells in the submucosa (Figure 10E and F, respectively). The normal distribution of TNF-α colonic expression within the group was indicated by the statistical analysis applying Kolmogorov–Smirnov normality test (data not shown). The expression of colonic TNF-α was significantly (P<0.05) higher in untreated colitis group (II) and in the group that received naringin cores (group III) compared to the normal control group (I) (Figure 11). On the other hand, rabbits given naringin-coated tablets (groups IV, V and VI) experienced significantly (P<0.05) reduced TNF-α expression than that of untreated colitis group (II) and core tablets group (III) (Figure 11). In contrast to core tablets, swelling, subsequent release retardation and possible colon targetting of the selected coated tablets may still account for their ability to promote a significant reduction in TNF-α colonic expression. Furthermore, naringin is hydrolyzed into its aglycone naringenin by microflora in the colon.12 It has been reported that naringenin can significantly attenuate colitis through down-regulation of the mRNA expression of several proinflammatory mediators including TNF-α.13 There was insignificant difference between the normal control group (I) and the group that pretreated with tablets coated with 5% w/w ALG (group V). In accordance, it has been reported that ALG suppressed acetic acid-induced colitis in rats due to the inhibitory effects on cytokine and eicosanoid production as well as MMP-2 activity.68
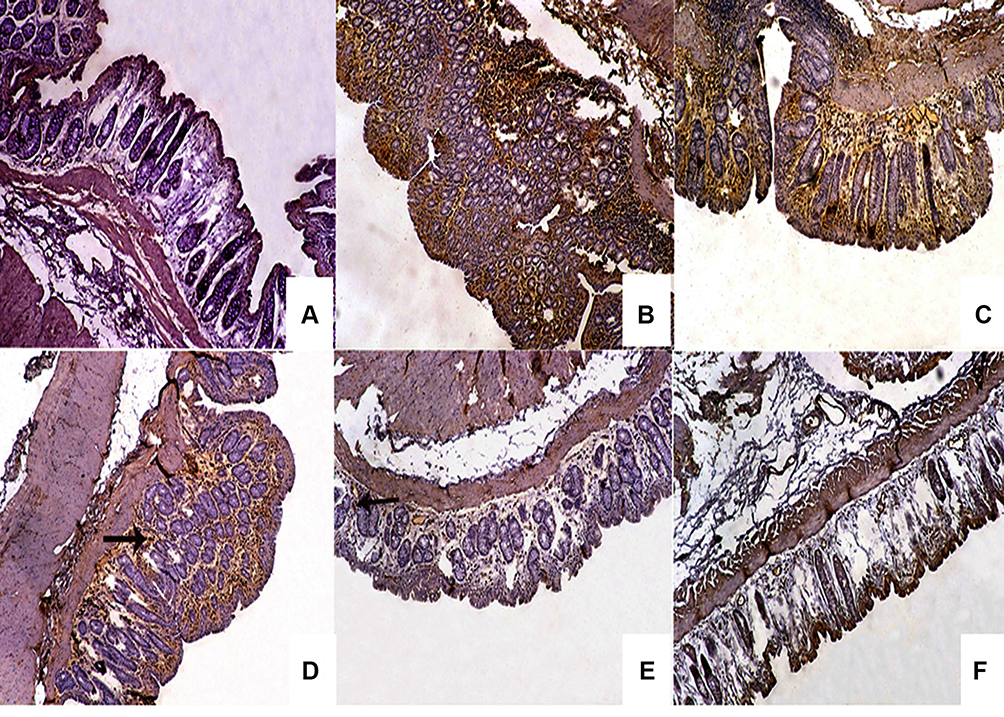 |
Figure 10 Microphotographs of rabbit’s colon sections using immunohistochemical staining for TNF-α localization. Notes: (A) negative immunostain for TNF-α in normal control group, positive immunoreactivity for epithelial lining mucosa, inflammatory cells in submucosa and endothelial lining blood vessels in rabbits of (B) untreated colitis group and (C) those pretreated with core tablets, (D) positive immunoreactivity for inflammatory cells in the lamina propria and submucosa (arrow) in rabbits pretreated with 10% w/w EC-coated tablets, and mild immunoreactivity limited to inflammatory cells in the submucosa in rabbits pretreated with (E) 5% w/w ALG or (F) SCMC coated tablets, respectively. (IHC, DAB immunostain, hematoxylin as counterstain, 100×).Abbreviations: TNF-α, tumor necrosis factor-alpha; EC, ethylcellulose; ALG, sodium alginate; SCMC, sodium carboxymethyl cellulose; IHC, immunohistochemical; DAB, 1,4-dideoxy-1,4-imino-d-arabinitol. |
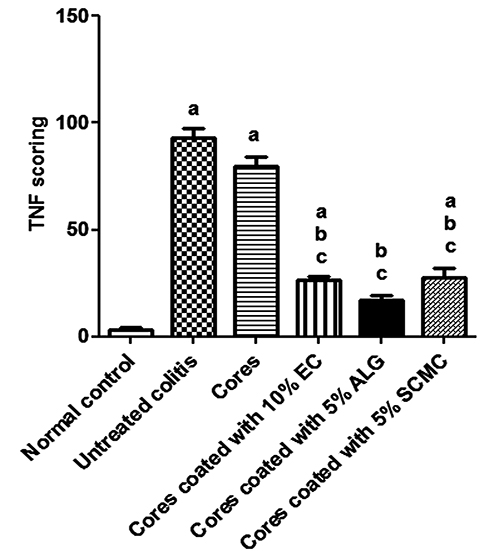 |
Figure 11 TNF-α scoring of rabbit’s colonic sections of all studied groups using ImageJ analysis following immunohistochemical staining for TNF-α localization. Notes: Data are expressed as mean ± SEM. a means significantly (P<0.05) different from normal control animals. b indicates significantly (P<0.05) different from untreated colitis group. c means significantly (P<0.05) different from animals pretreated with core tablets. (IHC, DAB immunostain, hematoxylin as counterstain, 100×).Abbreviations: TNF-α, tumor necrosis factor-alpha; SEM, standard error of mean; EC, ethylcellulose; ALG, sodium alginate; SCMC, sodium carboxymethyl cellulose; IHC, immunohistochemical; DAB, 1,4-dideoxy-1,4-imino-d-arabinitol. |
In agreement with histopathological examination and results describing pANCA serum levels, colonic TNF-α expression also represents the selected coated tablets as potential colon targeted delivery systems of naringin for effective cytoprotective action against indomethacin-induced colitis in rabbits.
Conclusion
Compression coating of naringin cores with release retardants as EC, ALG and SCMC at concentrations of 10%, 5% and 5% w/w, respectively, together with EUD-L100-55 was successful to minimize in vitro drug release to <20% at media resembling gastric and intestinal environments; hence, colon targeting was expected. Following physical mixing, no detectable interaction of naringin with any of the polymers used was recorded. Drug–polymers interaction during in vitro release was suggested based on FT-IR and DSC results of the kneaded mixtures of the drug with each polymer. The low porosity of the swollen coats as indicated by SEM examination may explain the retarded drug release at the acidic medium relative to the buffers which resulted in more porous coats. The effective colon targeting of naringin in the optimized coated tablets and cytoprotection against indomethacin-induced colitis in rabbits was proven by histopathological evaluation of colon and significant (P<0.05) lowering in serum pANCA and colonic TNF-α compared to core tablets.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Strober W, Lúdvíksson BR, Fuss IJ. The pathogenesis of mucosal inflammation in murine models of inflammatory bowel disease and Crohn’s disease. Ann Intern Med. 1998;128:848–856. doi:10.7326/0003-4819-128-10-199805150-00009
2. Hagar HH, El-Medany A, El-Eter E, Arafa M. Ameliorative effect of pyrrolidinedithiocarbamate on acetic acid-induced colitis in rats. Eur J Pharmacol. 2007;554:69–77. doi:10.1016/j.ejphar.2006.09.066
3. Inoue S, Matsumoto T, Iida M, et al. Characterization of cytokine expression in the rectal mucosa of ulcerative colitis: correlation with disease activity. Am J Gastroenterol. 1999;94:2441–2446. doi:10.1111/j.1572-0241.1999.01372.x
4. Shanahan F. Inflammatory bowel disease: immunodiagnostics, immunotherapeutics, and ecotherapeutics. Gastroenterology. 2001;120:622–635. doi:10.1053/gast.2001.22122
5. Ogata H, Hibi T. Cytokine and anti-cytokine therapies for inflammatory bowel disease. Curr Pharm Des. 2003;9:1107–1113. doi:10.2174/1381612033455035
6. Vanamala J, Leonardi T, Patil BS, et al. Suppression of colon carcinogenesis by bioactive compounds in grapefruit. Carcinogenesis. 2006;27:1257–1265. doi:10.1093/carcin/bgi318
7. Amaro MI, Rocha J, Vila-Real H, et al. Anti-inflammatory activity of naringin and the biosynthesised naringenin by naringinase immobilized in microstructured materials in a model of DSS-induced colitis in mice. Food Res Int. 2009;42:1010–1017. doi:10.1016/j.foodres.2009.04.016
8. Choudhury R, Chowrimootoo G, Srai K, Debnam E, Rice-Evans CA. Interactions of the flavonoid naringenin in the gastrointestinal tract and the influence of glycosylation. Biochem Biophys Res Commun. 1999;265:410–415. doi:10.1006/bbrc.1999.1695
9. Hsiu SL, Huang TY, Han YC, Chin DH, Chao PD. Comparison of metabolic pharmacokinetics of naringin and naringenin in rabbits. Life Sci. 2002;70:1481–1489. doi:10.1016/S0024-3205(01)01491-6
10. Lauro MR, De Simone F, Sansone F, Iannelli P, Aquino RP. Preparations and release characteristics of naringin and naringenin gastro-resistant microparticles by spray-drying. J Drug Del Sci Tech. 2007;17:119–124. doi:10.1016/S1773-2247(07)50018-3
11. Rechner AR, Smith MA, Kuhnle G, et al. Colonic metabolism of dietary polyphenols: influence of structure on microbial fermentation products. Free Radic Biol Med. 2004;36:212–225. doi:10.1016/j.freeradbiomed.2003.09.022
12. Salaritabar A, Darvishi B, Hadjiakhoondi F, et al. Therapeutic potential of flavonoids in inflammatory bowel disease: a comprehensive review. World J Gastroenterol. 2017;23(28):5097–5114. doi:10.3748/wjg.v23.i28.5097
13. Kumar VS, Rajmane AR, Adil M, Kandhare AD, Ghosh P, Bodhankar SL. Naringin ameliorates acetic acid induced colitis through modulation of endogenous oxido-nitrosative balance and DNA damage in rats. J Biomed Res. 2014;28:132–145. doi:10.7555/JBR.27.20120082
14. Akala EO, Elekwachi O, Chase V, Johnson H, Lazarre M, Scott K. Organic redox initiated polymerization process for the fabrication of hydrogel for colon specific drug delivery. Drug Dev Ind Pharm. 2003;29:375–386. doi:10.1081/DDC-120018373
15. Orlu M, Cevher E, Araman A. Design and evaluation of colon specific drug delivery system containing flurbiprofen microsponges. Int J Pharm. 2006;318:103–117. doi:10.1016/j.ijpharm.2006.03.025
16. Karrout Y, Neut C, Wils D, et al. Novel polymeric film coatings for colon targeting: how to adjust desired membrane properties. Int J Pharm. 2009;371:64–70. doi:10.1016/j.ijpharm.2008.12.014
17. Yang L, Chu JS, Fix JA. Colon-specific drug delivery: new approaches and in vitro/in vivo evaluation. Int J Pharm. 2002;235:1–15. doi:10.1016/S0378-5173(02)00004-2
18. Singh BN. Modified-release solid formulations for colonic delivery. Recent Pat Drug Deliv Formul. 2007;1:53–63. doi:10.2174/187221107779814122
19. Ashford M, Fell J, Attwood D, Sharma H, Woodhead P. An evaluation of pectin as a carrier for drug targeting to the colon. J Control Release. 1993;26:213–220. doi:10.1016/0168-3659(93)90188-B
20. Kenyon CJ, Cole ET, Wilding IR. The effect of food on the in vivo behaviour of enteric coated starch capsules. Int J Pharm. 1997;112:207–213.
21. Gupta VK, Beckert TE, Price JC. A novel pH- and time-based multi-unit potential colonic drug delivery system. I. Development. Int J Pharm. 2001;213:83–91. doi:10.1016/S0378-5173(00)00649-9
22. Rujivipat S, Bodmeier R. Improved drug delivery to the lower intestinal tract with tablets compression-coated with enteric/nonenteric polymer powder blends. Eur J Pharm Biopharm. 2010;76:486–492. doi:10.1016/j.ejpb.2010.09.004
23. Vemula SK, Veerareddy PR, Devadasu VR. Pharmacokinetics of colon-specific pH and time-dependent flurbiprofen tablets. Eur J Drug Metab Pharmacokinet. 2015;40:301–3011. doi:10.1007/s13318-014-0210-0
24. Emeje M, Nwabunike P, Isimi Y, Kunle O, Ofoefule S. Preparation and evaluation of colon targeted drug delivery systems for albendazole using kneading, extrusion and compaction technology. Yao Xue Xue Bao. 2009;44:1152–1158.
25. Vemula SK, Veerareddy PR. Development, evaluation and pharmacokinetics of time-dependent ketorolac tromethamine tablets. Exp Opin Drug Deliv. 2013;10:33–45. doi:10.1517/17425247.2013.743528
26. Borg TM, Mohamed EA, El Naggar EE, El-Sheakh AR, Hamed MF. Colon targeting of naringin for cytoprotection against ulcerative colitis: in vitro-in vivo study. IOSR-JPBS. 2017;12(2):23–29.
27. Paget GE, Barnes JM. Evaluation of drug activities. In: Laurence DR, Bacharach AL, editors. Pharmacometrics. London: Academic Press; 1964:1–50.
28. The United States Pharmacopoeial Convention. The United States Pharmacopeia 28th, the National Formulary 23rd. Washington: The United States Pharmacopoeial Convention Inc.; 2005.
29. Late S, Banga A. Thermal and non-thermal methods to evaluate compatibility of granisetron hydrochloride with tablet excipients. Pharmazie. 2008;63:453–458.
30. Tadros MI. Controlled-release effervescent floating matrix tablets of ciprofloxacin hydrochloride: development, optimization and in vitro-in vivo evaluation in healthy human volunteers. Eur J Pharm Biopharm. 2010;74:332–339. doi:10.1016/j.ejpb.2009.11.010
31. Yusif RM, Abu Hashim II, Mohamed EA, Badria FA. Gastroretentive matrix tablets of Boswellia Oleogum resin: preparation, optimization, in vitro evaluation, and cytoprotective effect on indomethacin-induced gastric ulcer in rabbits. AAPS PharmSciTech. 2016;17:328–338. doi:10.2147/IJN.S154325
32. Yehia SA, Elshafeey AH, Sayed AH. Optimization of budesonide compression-coated tablets for colonic delivery. AAPS PharmSciTech. 2009;10:147–157. doi:10.1208/s12249-009-9188-3
33. Krishnaiah YS, Bhaskar Reddy PR, Satyanarayana V, Karthikeyan RS. Studies on the development of oral colon targeted drug delivery systems for metronidazole in the treatment of amoebiasis. Int J Pharm. 2002;236:43–55. doi:10.1016/S0378-5173(02)00006-6
34. Akhgari A, Sadeghi F, Afrasiabi Garekani H. Combination of time-dependent and pH-dependent polymethacrylates as a single coating formulation for colonic delivery of indomethacin pellets. Int J Pharm. 2006;320:137–142. doi:10.1016/j.ijpharm.2006.05.011
35. Martin A, Bustamante P, Chun AHC. Kinetics. In: Physical Pharmacy.
36. Higuchi T. Mechanism of rate of sustained-action medication: the theoretical analysis of rate of release of solid drug dispersed in solid matrices. J Pharm Sci. 1963;52:1145–1149. doi:10.1002/jps.2600521210
37. Ritger PL, Peppas NA. A simple equation for description of solute release I. Fickian and non-Fickian release from non-swellable devices in the form of slabs, spheres, cylinders or discs. J Control Release. 1987;5:23–36. doi:10.1016/0168-3659(87)90034-4
38. Percy WH, Burton MB, Fallick F, Burakoff RA. comparison in vitro of human and rabbit distal colonic muscle responses to inflammatory mediators. Gastroenterology. 1990;99:1324–1332. doi:10.1016/0016-5085(90)91157-2
39. Wanga W, Liu Q, Wanga C, Menga Q, Kaku T, Liu K. Effects of JBP485 on the expression and function of PEPT1 in indomethacin-induced intestinal injury in rats and damage in Caco-2 cells. Peptides. 2011;32:946–955. doi:10.1016/j.peptides.2011.01.031
40. Reagn-shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2007;22:659–661. doi:10.1096/fj.07-9574LSF
41. Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharma. 2016;7:27–31. doi:10.4103/0976-0105.177703
42. Sharma PR, Lewis SA. Design and in vitro/in vivo evaluation of extended release matrix tablets of nateglinide. J Young Pharm. 2013;5:167–172. doi:10.1016/j.jyp.2013.11.003
43. Shehatou GS, Suddek GM. Sulforaphane attenuates the development of atherosclerosis and improves endothelial dysfunction in hypercholesterolemic rabbits. Exp Biol Med (Maywood). 2016;241(4):426–436. doi:10.1177/1535370215609695
44. Oda T. Role of mast cells in dextran sulfate sodium-induced experimental colitis in rats. J Kyoto Pref Univ Med. 1995;104:1069–1082.
45. Araki Y, Andoh A, Fujiyama Y, Bamba T. Development of dextran sulphate sodium-induced experimental colitis is suppressed in genetically mast cell-deficient Ws/Ws rats. Clin Exp Immunol. 2000;119:264–269. doi:10.1046/j.1365-2249.2000.01094.x
46. Austin GL, Herfarth HH, Sandler RS. A critical evaluation of serologic markers for inflammatory bowel disease. Clin Gastroenterol Hepatol. 2007;5:545–547. doi:10.1016/j.cgh.2007.03.006
47. Ruemmele FM, Targan SR, Levy G, Dubinsky M, Braun J, Seidman EG. Diagnostic accuracy of serological assays in pediatric inflammatory bowel disease. Gastroenterology. 1998;115:822–829. doi:10.1016/s0016-5085(98)70252-5
48. Lewis JD. The utility of biomarkers in the diagnosis and therapy of inflammatory bowel disease. Gastroenterology. 2011;140:1817–1826. doi:10.1053/j.gastro.2010.11.058
49. El-Sheakh AR, Suddek GM. Ammar el-SM. Antioxidant and anti-inflammatory effects of flavocoxid in high-cholesterol-fed rabbits. Naunyn Schmiedebergs Arch Pharmacol. 2015;388:1333–1344.
50. Mohamed EA, Abu Hashim II, Yusif RM, et al. Polymeric micelles for potentiated antiulcer and anticancer activities of naringin. Int J Nanomedicine. 2018;13:1009–1027.
51. Abdelaziz HA, Shaker ME, Hamed MF, Gameil NM. Repression of acetaminophen-induced hepatotoxicity by a combination of celastrol and brilliant blue G. Toxicol Lett. 2017;275:6–18. doi:10.1016/j.toxlet.2017.04.012
52. Sahu N, Soni D, Chandrashekhar B. Synthesis of silver nanoparticles using flavonoids: hesperidin, naringin and diosmin, and their antibacterial effects and cytotoxicity. Int Nano Lett. 2016;6:173–181.
53. Ficarra R, Tommasini S, Raneri D, et al. Study of flavonoids/β-cyclodextrins inclusion complexes by NMR, FT-IR, DSC, X-ray investigation. J Pharm Biomed Anal. 2002;29:1005–1014.
54. Cordenonsi LM, Sponchiado RM, Campanharo SC, et al. Study of flavonoids present in Pomelo (Citrus máxima) by DSC, UV-VIS, IR, 1H AND 13C NMR AND MS. Drug Anal. Res. 2017;1:31–37.
55. Shen X, Yu D, Zhu L, Branford-White C, White K, Chatterton NP. Electrospun diclofenac sodium loaded Eudragit® L 100-55 nanofibers for colon-targeted drug delivery. Int J Pharm. 2011;408:200–207. doi:10.1016/j.ijpharm.2011.01.058
56. Desai J, Alexander K, Riga A. Characterization of polymeric dispersions of dimenhydrinate in ethyl cellulose for controlled release. Int J Pharm. 2006;308:115–123. doi:10.1016/j.ijpharm.2005.10.034
57. Smitha B, Sridhar S, Khan AA. Chitosan–sodium alginate polyion complexes as fuel cell membranes. J Eur Polym. 2005;41:1859–1866. doi:10.1016/j.eurpolymj.2005.02.018
58. Bao Y, Ma J, Li N. Synthesis and swelling behaviors of sodium carboxymethyl cellulose-g-poly(AA-co-AM-co-AMPS)/MMT superabsorbent hydrogel. Carbohydr Polym. 2011;84:76–82.
59. Cui L, Zhang ZH, Sun E, Jia XB. Effect of β-cyclodextrin complexation on solubility and enzymatic conversion of naringin. Int J Mol Sci. 2012;13:14251–14261. doi:10.3390/ijms131114251
60. Liu J, Cao F, Zhang C, Ping Q. Use of polymer combinations in the preparation of solid dispersions of a thermally unstable drug by hot-melt extrusion. Acta Pharm Sin B. 2013;3:263–272. doi:10.1016/j.apsb.2013.06.007
61. González-Rodrı́guez M, Holgado M, Sánchez-Lafuente C, Rabasco A, Fini A. Alginate/chitosan particulate systems for sodium diclofenac release. Int J Pharm. 2002;232:225–234. doi:10.1016/S0378-5173(01)00915-2
62. Soares JP, Santos JE, Chierice GO, Cavalheiro ETG. Thermal behavior of alginic acid and its sodium salt. Eclética Química. 2004;29:57–64. doi:10.1590/S0100-46702004000200009
63. Jana S, Kumar Trivedi M, Tallapragada RM. Characterization of physicochemical and thermal properties of chitosan and sodium alginate after biofield treatment. Pharm Anal Acta. 2015;6:1–9.
64. El-Sayed S, Mahmoud KH, Fatah AA, DSC HA. TGA and dielectric properties of carboxymethyl cellulose/polyvinyl alcohol blends. Physica B Condens Matter. 2011;406:4068–4076. doi:10.1016/j.physb.2011.07.050
65. Abu Hashim II, Higashi T, Anno T, et al. Potential use of gamma-cyclodextrin polypseudorotaxane hydrogels as an injectable sustained release system for insulin. Int J Pharm. 2010;392:83–91. doi:10.1016/j.ijpharm.2010.03.026
66. Prasad YV, Krishnaiah YS, Satyanarayana S. In vitro evaluation of guar gum as a carrier for colon-specific drug delivery. J Control Release. 1998;51:281–287. doi:10.1016/S0168-3659(97)00181-8
67. Asghar LF, Chure CB, Chandran S. Colon specific delivery of indomethacin: effect of incorporating pH sensitive polymers in xanthan gum matrix bases. AAPS PharmSciTech. 2009;10:418–429. doi:10.1208/s12249-009-9223-4
68. Mirshafiey A, Khodadadi A, Rehm BH, et al. Sodium alginate as a novel therapeutic option in experimental colitis. Scand J Immunol. 2005;61:316–321. doi:10.1111/j.1365-3083.2005.01571.x
69. Cohavy O, Bruckner D, Gordon LK, et al. Colonic bacteria express an ulcerative colitis pANCA-related protein epitope. Infect Immun. 2000;68(3):1542–1548. doi:10.1128/IAI.68.3.1542-1548.2000
70. Rogler G, Andus T. Cytokines in inflammatory bowel disease. World J Surg. 1998;22:382–389. doi:10.1007/s002689900401
71. Sands BE, Kaplan GG. The role of TNF alpha in ulcerative colitis. J Clin Pharmacol. 2007;47(8):930–941. doi:10.1177/0091270007301623
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.