")
Back to Journals » International Journal of Nanomedicine » Volume 13
Colon-targeted delivery of cyclosporine A using dual-functional Eudragit® FS30D/PLGA nanoparticles ameliorates murine experimental colitis
Authors Naeem M, Bae J, Oshi MA , Kim M, Moon HR, Lee BL, Im E, Jung Y, Yoo J
Received 20 November 2017
Accepted for publication 6 February 2018
Published 28 February 2018 Volume 2018:13 Pages 1225—1240
DOI https://doi.org/10.2147/IJN.S157566
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Muhammad Naeem,* Junhwan Bae,* Murtada A Oshi, Min-Soo Kim, Hyung Ryong Moon, Bok Luel Lee, Eunok Im, Yunjin Jung, Jin-Wook Yoo
College of Pharmacy, Pusan National University, Busan, Republic of Korea
*These authors contributed equally to this work
Background: Colon-targeted oral nanoparticles (NPs) have emerged as an ideal, safe, and effective therapy for ulcerative colitis (UC) owing to their ability to selectively accumulate in inflamed colonic mucosa. Cyclosporine A (CSA), an immunosuppressive agent, has long been used as rescue therapy in severe steroid-refractory UC. In this study, we developed CSA-loaded dual-functional polymeric NPs composed of Eudragit® FS30D as a pH-sensitive polymer for targeted delivery to the inflamed colon, and poly(lactic-co-glycolic acid) (PLGA) as a sustained-release polymer.
Methods: CSA-loaded Eudragit FS30D nanoparticles (ENPs), PLGA nanoparticles (PNPs), and Eudragit FS30D/PLGA nanoparticles (E/PNPs) were prepared using the oil-in-water emulsion method. Scanning electron microscope images and zeta size data showed successful preparation of CSA-loaded NPs.
Results: PNPs exhibited a burst drug release of >60% at pH 1.2 (stomach pH) in 0.5 h, which can lead to unwanted systemic absorption and side effects. ENPs effectively inhibited the burst drug release at pH 1.2 and 6.8 (proximal small intestine pH); however, nearly 100% of the CSA in ENPs was released rapidly at pH 7.4 (ileum–colon pH) owing to complete NP dissolution. In contrast to single-functional PNPs and ENPs, the dual-functional E/PNPs minimized burst drug release (only 18%) at pH 1.2 and 6.8, and generated a sustained release at pH 7.4 thereafter. Importantly, in distribution studies in the gastrointestinal tracts of mice, E/PNPs significantly improved CSA distribution to the colon compared with PNPs or ENPs. In a mouse model of colitis, E/PNP treatment improved weight loss and colon length, and decreased rectal bleeding, spleen weight, histological scoring, myeloperoxidase activity, macrophage infiltration, and expression of proinflammatory cytokines compared with PNPs or ENPs.
Conclusion: Overall, this work confirms the benefits of CSA-loaded E/PNPs for efficiently delivering CSA to the colon, suggesting their potential for UC therapy.
Keywords: cyclosporine A, ulcerative colitis, colon-targeted nanoparticles, sustained and pH-sensitive release
Introduction
Ulcerative colitis (UC) is a chronic inflammatory disorder of the rectum and colon, which is caused by unknown genetic, environmental, and bacterial factors.1 The symptoms of UC range from bloody diarrhea and weight loss to ulceration and complete obstruction of the gastrointestinal tract (GIT), which can negatively affect the daily life of a patient.2 Furthermore, one of the most important consequences of chronically active UC is the development of colorectal cancer.3 Since the etiology of UC is unknown and there is currently no cure, major therapeutic strategies are aimed at inducing remission, preventing inflammatory episodes, and ensuring colectomy-free survival. Drug therapies for UC include aminosalicylates, corticosteroids, immunosuppressants, and biologicals.4 Although intravenous corticosteroids are used as first-line therapy for severe UC, approximately 30%–40% of patients are resistant to steroid treatment.5 Cyclosporine A (CSA) is an immunosuppressant drug that has been used as a rescue therapy in clinical practice owing to its rapid onset of action in severe, steroid-refractory UC. Acute remission rates with CSA ranged between 63% and 82% in previous trials.6,7
CSA was originally marketed in soft-gel form (Sandimmune® oral) as a crude, oil-in-water-based delivery system, where the bioavailability of CSA from Sandimmune was rate limited by its solubility. Sandimmune Neoral® (hereafter referred to as Neoral), a stable microemulsion-based soft-gel formulation, enhanced solubility and absorption in the upper intestine, resulting in increased systemic bioavailability.8 However, systemic CSA therapy has been limited in clinical practice owing to significant side effects, such as nephrotoxicity, hypertension, seizures, renal dysfunction, and opportunistic infection, as well as the need for careful monitoring of drug plasma concentration during treatment to prevent toxicity.9,10 Moreover, Sandimmune and Neoral contain a high concentration of polyoxyethylated castor oil, Cremophor EL®, which can cause nephrotoxicity and anaphylactic reactions.11 The use of rectal enemas that provide localized colonic topical delivery of CSA has been reported as an alternative to intravenous/oral administration, with reduced systemic toxicity.12 However, a rectal enema of CSA is suitable only for treating a distal region of the colon.13 In addition, patient compliance is a significant limiting factor for the long-term use of enemas. Because of the limitations of the current commercial formulations of CSA in UC therapy, there is an unmet need to develop advanced delivery systems that reduce systemic CSA concentrations and efficiently deliver CSA to the target, inflamed regions of the colon.
Colonic inflammation-targeted oral nanoparticles (NPs) have gained much attention as an ideal drug delivery system for treating inflammatory bowel disease (IBD). Such an inflammation-targeted local drug delivery approach allows a lower dose than that needed with intravenous applications, and minimizes systemic drug absorption to avoid adverse effects. Several studies have demonstrated that NPs exhibit size-dependent adhesion to inflamed tissues of the colon, combined with an enhanced therapeutic effect in experimental colitis.14–17 Guada et al evaluated the effect of CSA-loaded lipid NPs in a mouse model of dextran sodium sulfate (DSS)-induced colitis, using a commercial formulation (Neoral) as a reference.18 However, the lipid NPs did not satisfactorily ameliorate the colitis and the authors suggested further studies. In another study in mice, superior alleviation of trinitrobenzene–sulfonic acid (TNBS)-induced colitis by intrarectal administration of CSA-loaded NPs was reported.19 However, rectal administration is not easily managed by patients and the most desired and accepted way to administer the drug in UC therapy is orally. CSA-loaded poly(lactic-co-glycolic acid) (PLGA) NPs and microparticles have been evaluated for the treatment of DSS-induced colitis in mice and their efficacy has been compared with that of Neoral. The study showed that orally administered NPs improved colitis parameters compared to microparticles or Neoral.20 However, PLGA NPs, which were designed for sustained drug release, have an intrinsic limitation during colon-targeted drug delivery; an initial burst release in the upper GIT (ie, stomach and small intestine) leads to less efficient colon-specific delivery and unwanted systemic absorption.15,21,22 Similarly, the single pH-dependent NP system is not recommended for UC therapy owing to rapid and complete drug release at ileum–colonic pH.15,22 These reports emphasize the potential use of sustained-release polymeric NPs for targeted drug delivery to inflamed intestinal mucosa, and that this method can be further improved for oral administration by implementing pH-dependent drug release. Thus, an ideal colon-specific delivery system for CSA in UC therapy should prevent premature drug release before reaching the colon and should deliver significant amounts of the drug to the inflamed colon tissue in a controlled manner.
In this study, we prepared dual-functional, CSA-loaded polymeric NPs using a combination of sustained-release PLGA copolymer and pH-responsive Eudragit® FS30D polymer (Eudragit® FS30D/PLGA nanoparticles [E/PNPs]). The objectives of using this polymeric combination were to minimize early CSA release in the stomach and small intestine, to target the inflamed colon tissue, and to allow a slow and sustained drug release, which is beneficial for oral UC therapy. For comparison, CSA-loaded PLGA nanoparticles (PNPs) and CSA-loaded Eudragit FS30D nanoparticles (ENPs) were also prepared. The size, shape, and drug-loading capability of the NPs were characterized. The CSA release of the NPs was assessed under different pH environments, resembling those of the GIT. In addition, the in vivo therapeutic efficacy and GIT distribution of the NPs were evaluated in a mouse model of DSS-induced colitis.
Materials and methods
Materials
PLGA (PLGA 5050 5E, inherent viscosity 0.41 dL/g) was purchased from Evonik (Darmstadt, Germany). CSA, polyvinyl alcohol (PVA, molecular weight=30,000–70,000 Da), and tin octanoate were purchased from Sigma-Aldrich (St Louis, MO, USA). A hydrophobic near-infrared dye, 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide (DiR), was purchased from Invitrogen (Carlsbad, CA, USA). Eudragit FS30D was generously donated by Evonik Korea (Seoul, Republic of Korea). DSS (molecular weight=36,000–50,000 Da) was obtained from MP Biomedicals (Irvine, CA, USA). All other reagents and solvents were the highest analytical grade commercially available.
Preparation of CSA-loaded NPs
The CSA-loaded NPs were prepared with PLGA, Eudragit FS30D, or mixtures thereof (1:1 w/w) using a previously reported oil-in-water emulsification solvent evaporation method, with slight modifications.23 In brief, PLGA and/or Eudragit FS30D (50 mg) was dissolved in 3 mL dichloromethane with CSA (5 mg). This organic mixture was then emulsified in 10 mL aqueous acidic PVA solution (1% w/v) by sonication (amplitude 30%, 2 min) using an ultrasound probe sonicator in an ice bath. This emulsion was then diluted with 30 mL of acidic PVA solution (0.2% w/v). The organic solvent was allowed to evaporate for 4 h at room temperature in a fume hood with a magnetic stirrer (500 rpm). After evaporating the residual solvent, the NPs were collected by centrifugation at 20,000 × g for 30 min and washed with deionized water 3 times. The obtained NPs were immediately used for the following experiments.
In vitro characterization of NPs
Morphology of NPs
The external morphology of the CSA-loaded NPs was analyzed by scanning electron microscopy (SEM). NPs suspended in water were dropped on a carbon tape and air dried at room temperature in a fume hood or desiccator. Samples were then coated with platinum for 2 min in a vacuum and viewed by field-emission SEM (FE-SEM, S4800; Hitachi, Tokyo, Japan) at an acceleration voltage of 1–5 kV.
Size and size distribution analysis of NPs
The hydrodynamic diameter and polydispersity index (PDI) of CSA-loaded NPs were measured by dynamic light scattering (Zetasizer Nano ZS; Malvern Instruments, Malvern, UK) in double-distilled water at 25°C and a fixed angle of 173°. The measurements were performed for each batch in triplicate, and the mean and SD were calculated.
Determination of yield (%), loading capacity (%), and encapsulation efficiency (%)
The freeze-dried NPs were weighed to calculate the yield per batch. The percentage yield was calculated using Equation (1). The drug content in the NPs was determined directly by measuring the encapsulated CSA amount in the NPs using high-performance liquid chromatography (HPLC), according to an established method.23 The HPLC system used was an LC-20AT (Shimadzu, Tokyo, Japan) equipped with an autosampler processor, an SPD-20A ultraviolet (UV) detector, and a Luna C18 column (5 μm, 150 × 4.6 mm; Phenomenex, Torrance, CA, USA). The UV detector wavelength was set at 210 nm. The mobile phase was a mixture of acetonitrile and water (75:25) at a flow rate of 1.5 mL/min, and a column temperature of 65°C was used. A calibration curve using standard CSA solution was generated. To determine CSA content, a known weight of CSA-loaded NPs was dissolved in acetonitrile:water (7:3) and placed in an ultrasonic bath for 1 h. After centrifugation for 30 min at 14,000 rpm, the CSA content of the samples was determined by HPLC, using the calibration curve. Samples were prepared in triplicate, and the percentage yield, drug loading (DL), and encapsulation efficiency (EE) were calculated using Equations (1), (2), and (3), respectively:
|
|
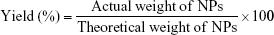
|
|
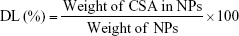
|
|

In vitro CSA release profile from NPs at different pH values
The in vitro drug release of CSA from NPs was evaluated in release medium with gradually changing pH values (ie, 1.2, 6.8, and 7.4).24,25 This gradually changing medium was selected based on the normal variations in pH and general transit time along the GIT from the stomach (pH 1.2, 0–2 h) and small intestine (pH 6.8, 2–6 h) to the colon region (pH 7.4, 6–24 h).26 CSA-loaded NPs (10 mg) were added to 50 mL of the release medium in triplicate and incubated in a shaking water bath (60 rpm, 37°C). Polysorbate 80 (0.2% w/v) was added to the release medium to facilitate the solubilization of CSA. At predetermined time intervals, 150 μL aliquots were sampled, and the release medium was replenished with an equal volume of fresh buffer. The aliquots were centrifuged at 17,000 × g for 30 min, and supernatants containing CSA released from the NPs were analyzed by HPLC, as described previously.23
NP size measurements at different pH values
CSA-loaded PNPs, ENPs, and E/PNPs were suspended in buffer solutions with different pH values (ie, 1.2, 6.8, or 7.4) and incubated in a shaking water bath (60 rpm, 37°C) for specific times. NPs were centrifuged, redispersed in distilled water, and washed twice. Water was used as the dispersant for the particles to measure their size. The NP sizes were measured using a Zetasizer Nano-ZS (Malvern Instruments), as described above.
Animal studies
All in vivo experiments were approved and performed in accordance with the regulations of Pusan National University and Korean legislation on animal studies. Male imprinting control region (ICR) mice (7 weeks of age; body weight 30–32 g) were purchased from Samtako Bio Korea (Osan, Republic of Korea) and acclimatized in the university animal facility at 25°C±3°C under a 12 h light/dark cycle for 1 week before experimentation.
DSS-induced colitis in mice
Colitis was induced in ICR male mice by oral administration of 3% (w/v) DSS in tap water ad libitum for 7 days. Age-matched male ICR mice receiving normal tap water served as controls. Mean DSS water consumption, body weight, and development of clinical symptoms of colitis, such as diarrhea and rectal bleeding, were assessed daily during the colitis induction period.
Ex vivo imaging
To track the colon-specific drug delivery potential of PNPs, ENPs, and E/PNPs after oral administration, an in vivo imaging system (IVIS) (FOBI; Neoscience, Suwon, Republic of Korea) was used. The near-infrared dye, DiR, was loaded as a fluorescent probe into the NPs. Colitis mice, fasted for 24 h, were orally administered the DiR-loaded NPs at an equivalent DiR concentration (0.5 mg DiR/kg). Blank NPs (without DiR) were also tested. After oral administration for 6 or 12 h, the mice were euthanized to obtain the GIT. The fluorescence signals from the DiR-loaded NPs were imaged in the GIT at an excitation wavelength of 720 nm and an emission wavelength of 790 nm using the IVIS spectrum.
Quantification of CSA-loaded NPs in the GIT
The in vivo quantitative distribution of the CSA-loaded PNPs, ENPs and E/PNPs was evaluated in the GIT of the colitis mice. Food was withheld from all mice for 24 h before the administration of the NPs. The NPs were administered by oral gavage under isoflurane anesthesia, and the mice were euthanized 6 and 12 h post-administration. Then, GIT segments, including luminal contents, were collected and divided into stomach, small intestine, and colon (including cecum). The tissue samples were homogenized in PBS and then CSA was extracted with acetonitrile by shaking overnight. Specimens excised from the mice administered blank NPs were homogenized as mentioned earlier and used as controls. Samples were centrifuged and supernatants were collected and analyzed by HPLC for CSA.
Drug treatment of DSS-induced colitis mice
Mice were divided into 5 groups (8 mice per group): the healthy control, colitis control, PNP-treated, ENP-treated, and E/PNP-treated groups. First, experimental colitis was induced in mice by administering 3% (w/v) DSS in drinking water for 7 days. Drug-treated groups received an equal dose of CSA (15 mg/kg) in the form of suspended NPs administered orally by gavage for 7 days. The healthy control and the colitis control groups received normal saline by gavage.
Evaluation of colitis severity by body weight changes and disease activity index (DAI)
The clinical course of colitis was monitored daily using a DAI consisting of 3 parameters: weight loss, stool consistency, and anal bleeding, as previously described.27 In brief, inflammation was scored on a scale of 0 to 4. No weight loss was scored as 0 points, 1%–5% weight loss as 1 point, 5%–10% as 2 points, 10%–20% as 3 points, and >20% as 4 points. For stool consistency, 0 points were given for well-formed pellets, 2 points for pasty and semi-formed stools that did not stick to the anus, and 4 points for liquid stools that stuck to the anus. Bleeding was scored as 0 points for no blood, 2 points for positive findings, and 4 points for gross bleeding. The mean of these scores formed the clinical score, ranging from 0 (healthy) to 4 (maximal colitis).
Colon length, colon weight/length ratio, and spleen weight
All mice were euthanized 24 h after the last drug treatment and their entire colons were resected, from the cecum to the anus. Colon length was measured before dividing the colon for histology and evaluation of immune cell infiltration by immunostaining. Colon tissue samples (5 cm) were resected, opened longitudinally, and rinsed with ice-cold buffer to remove the luminal content. The ratio of the colon sample wet weight to colon length was determined as an index of colonic inflammation.28 In addition to the colon, for all mice, the spleen was separated, and the weight increase was recorded as an indication of inflammation.29
Histological assessment of colitis
For histological examinations, small segments of the colon from the healthy control, colitis control, and CSA-loaded NP-treated mice were fixed in phosphate-buffered 10% formalin and embedded in paraffin. Sections (5 μm thick) were cut with a microtome (Reichert, Munich, Germany) and stained with H&E, followed by light microscopy analysis (Zeiss, Axioskop, Germany). The degree of inflammation and epithelial injury on microscopic cross-sections of the colon was graded semi-quantitatively from 0 to 5.30 Grading was done in a blinded fashion on coded slides.
Infiltration of macrophages in colitis tissue
For the determination of macrophage infiltration in the inflamed colonic tissues, fluorescence immunostaining was conducted. Paraffin-embedded colon sections (5 μm thick) were prepared as described earlier. After deparaffinization and rehydration, sections were incubated at 4°C overnight with primary antibody (anti-F4/80; 1:200, Abcam) for the detection of macrophages. Sections were then incubated with secondary antibody, anti-rabbit immunoglobulin G labeled with Alexa Fluor® 568 (Thermo Fisher Scientific, Waltham, MA, USA), for 2 h at room temperature and then incubated with DAPI solution (5 μg/mL) at room temperature for 20 min. Images were acquired using an FV10i FLUOVIEW Confocal Microscope (Olympus, Tokyo, Japan).
Myeloperoxidase (MPO) activity assay
MPO activity was measured according to an established method.31 In brief, distal colon specimens were minced with sharp scissors in 1 mL of hexadecyltrimethylammonium bromide buffer (0.5% in 50 mM phosphate buffer, pH 6.0) on ice and then homogenized with a tissue homogenizer for 4 min at 30 Hz. The mixture was then sonicated for 10 s, subjected to 3 freeze–thaw cycles, and centrifuged at 14,000 rpm at 4°C for 3 min. The supernatant was collected and stored at –70°C until use. Then, 7 μL of supernatant was added in triplicate into a 96-well plate, and 200 μL of o-dianisidine solution containing H2O2 (16.7 mg o-dianisidine dihydrochloride, 90 mL distilled water, 10 mL potassium phosphate buffer, and 50 μL 1.2% H2O2 in distilled water) was added to the each of the wells. The absorbance was measured at 450 nm by a spectrophotometer. MPO activity was expressed in units per milligram of wet tissue. One unit of MPO activity is defined as the amount of enzyme degrading 1 μmol of peroxide per minute at 25°C.
Quantification of proinflammatory cytokines by enzyme-linked immunosorbent assay (ELISA)
The concentrations of proinflammatory cytokines, tumor necrosis factor-α (TNF-α) and IL-6, in the colons of mice from all groups were determined by a sandwich-type ELISA using commercially available kits (Biolegend, San Diego, CA, USA, and R&D Systems, Minneapolis, MN, USA, respectively) according to the manufacturers’ instructions. In brief, the frozen distal colon specimens were homogenized with potassium phosphate buffer (pH 6.0) and centrifuged at 2,500 × g and 4°C for 5 min. The supernatants were centrifuged at 10,000 × g and 4°C for 10 min, and were assayed by ELISA for TNF-α and IL-6 levels. Total protein concentration was determined by the Bradford protein assay using the BCA kit (Fisher Scientific, Waltham, MA, USA).
Statistical analysis
The results are expressed as the mean±SD. Statistical comparisons of the in vitro and in vivo data between two groups were performed using the Student’s t-test in GraphPad Prism 5.0 (GraphPad Software, La Jolla, CA, USA); one-way analysis of variance (ANOVA) followed by the Tukey–Kramer test was used to compare multiple groups. A p-value <0.05 was accepted as a statistically significant difference.
Results
Physicochemical characterization of CSA-loaded NPs
Particle size is an important factor in the development of colon-specific drug delivery strategies for UC treatment because it affects drug accumulation in the inflamed colon tissue.14,32,33 In the current study, CSA was loaded in PNPs, ENPs, and E/PNPs using the oil-in-water emulsion/solvent evaporation method.22 The morphology of CSA-loaded NPs was evaluated by SEM and a representative photograph of each formulation is shown in Figure 1A. All NPs exhibited a well-defined spherical shape with a smooth surface and homogeneous nanosize distribution. Further, size distribution and PDI values were determined by the Zetasizer and the data are shown in Figure 1B and Table 1. Most of the NPs fell in the size range of 200–260 nm with PDI values of less than 0.3, which indicates a monodisperse size distribution. The % yield, % DL, and % EE are shown in Table 1. The high CSA loading (~6.5%) into NPs can be attributed to the partition phenomenon in the organic phase of the initial emulsion and the poor water solubility of CSA, resulting in little loss to the aqueous phase.11 Overall, the CSA-loaded NPs were spherical and of uniform size, with a high yield and high drug loading.
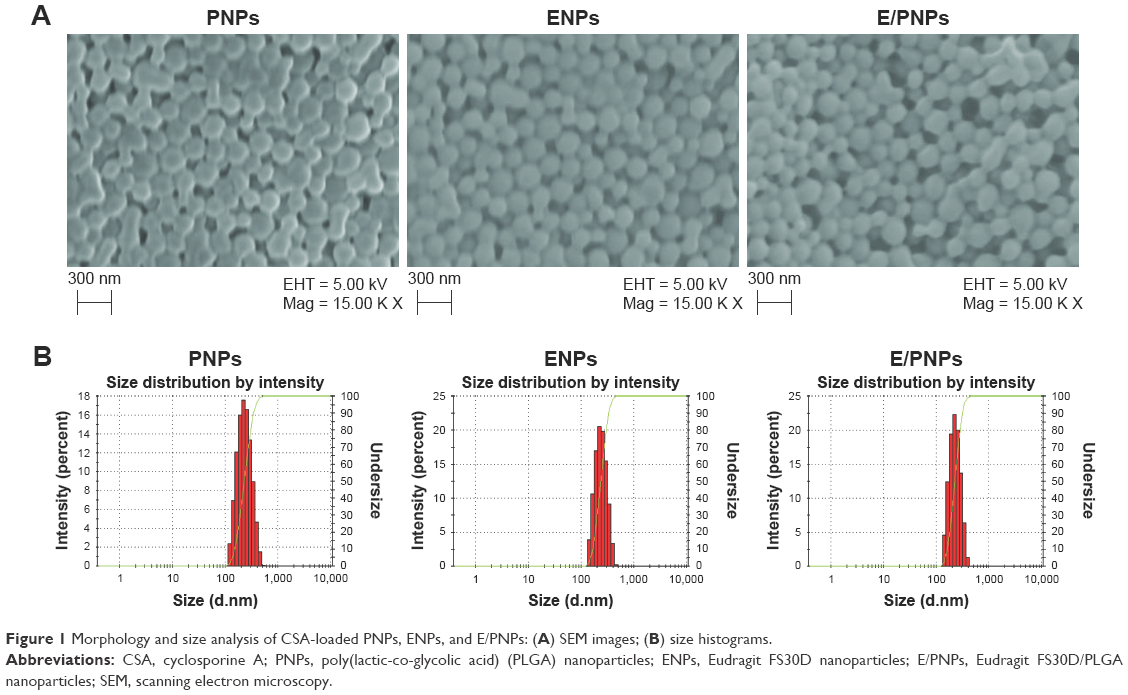 | Figure 1 Morphology and size analysis of CSA-loaded PNPs, ENPs, and E/PNPs: (A) SEM images; (B) size histograms. |
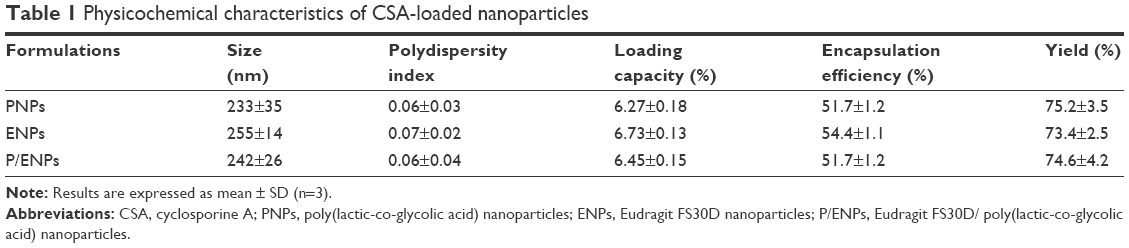 | Table 1 Physicochemical characteristics of CSA-loaded nanoparticles |
pH-dependent in vitro CSA release from NPs and size analysis
The release profiles of CSA from NPs were determined in gradually pH-changing medium (pH 1.2, 6.8, and 7.4) for 24 h (Figure 2A). PNPs exhibited a burst drug-release profile, which was pH independent. Approximately 70% of the CSA was released in the first 6 h at pH 1.2 and 6.8, which represents the pH of the stomach and the upper part of the small intestine, respectively. There was no significant difference in drug-release profile between the E/PNPs or ENPs at pH 1.2 and 6.8, with less than 20% of the CSA released during the first 6 h. However, at the ileum pH (pH 7.4), E/PNPs and ENPs showed markedly different release profiles. ENPs showed a sudden burst release (nearly 100%) of the drug, owing to their complete dissolution at pH >7. In contrast, E/PNPs avoided complete drug release at pH 7.4 and released CSA in a sustained manner. The pH-dependent size measurement data of the NPs agree with the release behavior of the respective NPs (Figure 2B). All NPs exhibited no size changes at pH 1.2 and 6.8. However, ENP particles were not detected at pH 7.4, indicating complete dissolution and CSA release. However, PNPs and E/PNPs maintained their nanoparticulate morphology at pH 7.4 and no significant change in size was observed. Taken together, it was demonstrated that E/PNPs possessed pH-dependent and sustained drug-release characteristics, while maintaining nanoparticle morphology; these are desirable characteristics for colon-targeted UC therapy.
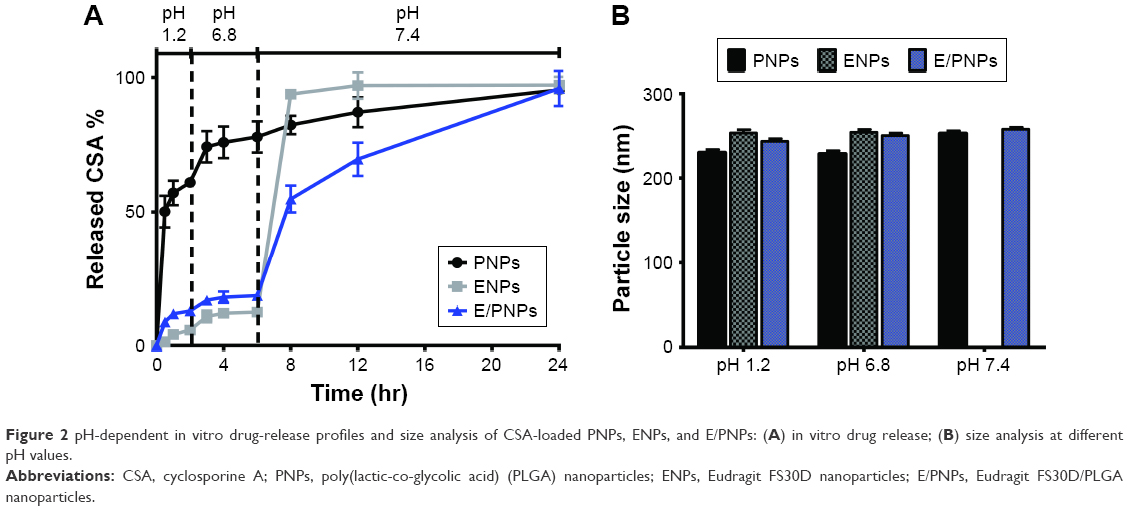 | Figure 2 pH-dependent in vitro drug-release profiles and size analysis of CSA-loaded PNPs, ENPs, and E/PNPs: (A) in vitro drug release; (B) size analysis at different pH values. |
Ex vivo imaging of GIT after oral administration of DiR-loaded NPs in colitis mice
The biodistribution of DiR-loaded PNPs, ENPs, and E/PNPs along the GIT following oral administration in mice was analyzed using an IVIS to investigate the specificity of the NPs to target the colon. For accurate measurement of particle biodistribution, the GIT was removed at 6 and 12 h post-administration of NPs and imaged as shown in Figure 3. Mice administered blank NPs were used to discriminate the effect of carrier material, and no fluorescence signals were observed. In contrast, DiR-loaded NPs gave rise to fluorescence signals at all time points. As can be seen in the representative ex vivo images of the GIT (Figure 3), the PNPs showed weak fluorescence signals in all major sections of the GIT (ie, stomach, small intestine, and colon) at all time points. This indicates that maximum dye was released and absorbed systemically before reaching the colon. In contrast, at 6 h, ENPs exhibited strong fluorescence signals in the distal small intestine, indicating the complete dissolution and release of the dye before reaching the colon. However, weak signals were found in the colon at all time points, indicating that the dye was absorbed or cleared quickly from the GIT after the dissolution of the ENPs. Mice administered dual-functional E/PNPs exhibited dominant fluorescence signals in the colon compared to the signals in the other regions of the GIT at all time points, indicating enhanced drug delivery and accumulation in inflamed colon tissue. This observation is significant because it confirms in vivo that dual-functional E/PNPs overcome the limitations of a single delivery system (ie, the bursting and premature drug release in early small intestine of PNPs, and the complete dissolution and release in the distal small intestine of ENPs), delivering maximum dye to the colon.
 | Figure 3 Representative ex vivo images of the GIT after oral administration of blank and DiR-loaded NPs, showing colon-targeted drug delivery and accumulation potential of PNPs, ENPs, and E/PNPs. |
In vivo quantification of CSA-loaded NPs in the GIT
Next, we quantified CSA-loaded NPs in the GIT of colitis mice (Figure 4). The percentage dose of CSA in major GIT segments (ie, stomach, small intestine, and colon including cecum) was assayed 6 and 12 h post-administration of NPs. Distribution results in the colon showed that CSA levels were significantly higher in mice administered E/PNPs than in mice administered PNPs or ENPs. At 6 h post-administration of NPs, the percentage dose of CSA in the colons of E/PNP-administered mice was 5-fold higher than that of the PNP- and ENP-administered mice. A similar trend was observed after 12 h. Indeed, the results obtained from the in vitro drug-release study (Figure 2), biodistribution of NPs in the GIT of the colitis mouse model using ex vivo imaging (Figure 3), and quantitative determination of CSA (Figure 4) collectively imply that, compared with CSA-loaded PNPs and ENPs, the dual-functional E/PNP system has a greater therapeutic potential for colitis therapy.
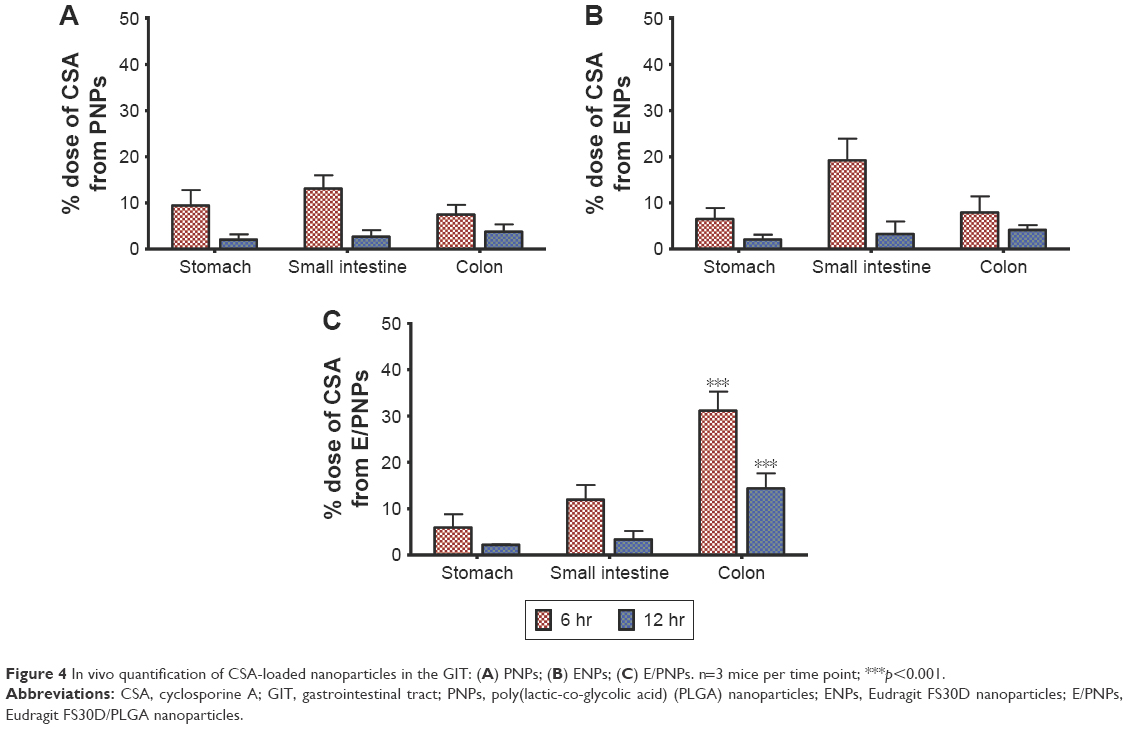 | Figure 4 In vivo quantification of CSA-loaded nanoparticles in the GIT: (A) PNPs; (B) ENPs; (C) E/PNPs. n=3 mice per time point; ***p<0.001. |
In vivo therapeutic efficacy
Body weight changes and DAI of colitis
The body weight changes of mice in all groups are shown in Figure 5A. Before DSS administration, there were no intergroup differences in body weight. However, the administration of 3% DSS in the drinking water to induce colitis significantly decreased body weights in the colitis groups compared to the body weights in the healthy control group. Improvement in body weight was observed in the CSA-loaded NP-treated groups compared to that in the colitis control group. However, the recovery of body weight was faster in the E/PNP-treated group than in the PNP- or ENP-treated group. We also evaluated DAI (Figure 5B), which considers weight loss, stool consistency, and rectal bleeding. In the colitis control group, the DAI increased in response to intestinal inflammation. All CSA-treated groups showed reduced DAI compared to the colitis control group. The E/PNP-treated group showed the most prominent reduction in DAI, with a 2-fold and 3-fold reduction compared to the single-function NPs (PNPs and ENPs) and colitis control group, respectively.
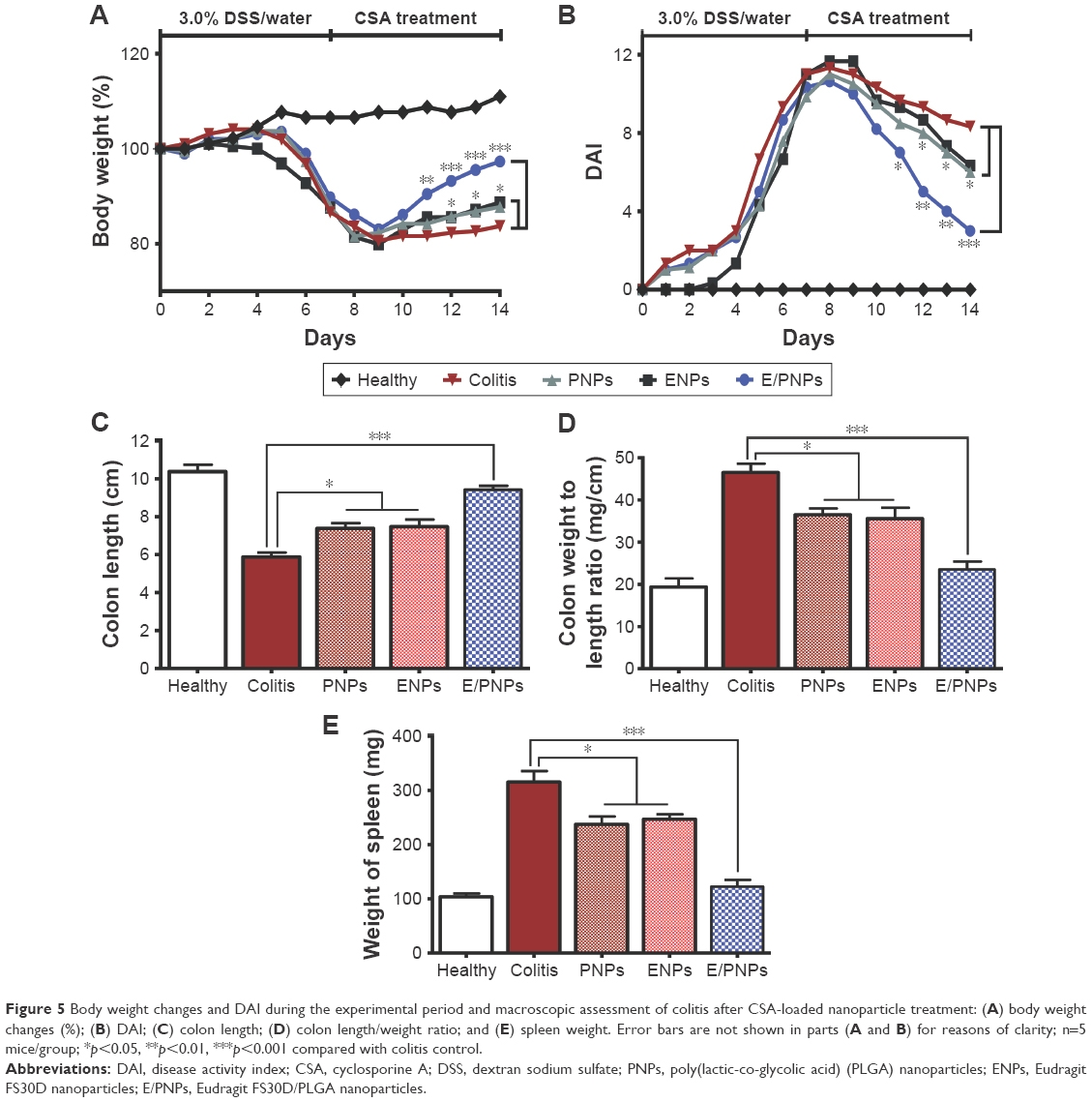 | Figure 5 Body weight changes and DAI during the experimental period and macroscopic assessment of colitis after CSA-loaded nanoparticle treatment: (A) body weight changes (%); (B) DAI; (C) colon length; (D) colon length/weight ratio; and (E) spleen weight. Error bars are not shown in parts (A and B) for reasons of clarity; n=5 mice/group; *p<0.05, **p<0.01, ***p<0.001 compared with colitis control. |
Macroscopic assessment of colitis
All mice were euthanized 24 h after the last drug treatment. The abdominal cavity was surgically opened, and the colon (with cecum) and spleen were resected. DSS administration reduced the size of the colon by several centimeters, which is the main indicator of DSS-induced colitis. Therefore, an expected therapeutic index of treatment efficacy would be the recovery of the drug-treated colon length to initial values. The results of the colon length of healthy and DSS-treated mice are shown in Figure 5C. The average colon length of the colitis control mice decreased to 5.8±0.4 cm, compared to 10.5±0.6 cm in the healthy control mice. As the administration of CSA-loaded NPs alleviated inflammation, the colon length also increased relative to that of the colitis control mice. However, PNPs and ENPs only showed a slight increase in colon length (7.3±0.7 cm and 7.4±0.9 cm, respectively). In contrast, E/PNPs showed the greatest increase in colon length to 9.6±0.3 cm, which is much closer to the colon length of the healthy mice (10.8±1.3 cm). The results for the colon weight/length ratio are shown in Figure 5D. Similar to the observation made regarding colon length, the colon weight/length ratio after E/PNP treatment was found to be improved (23.5±3.2 mg/cm) in comparison to that of the PNP- or ENP-treated group (36.5±2.6 mg/cm and 35.1±4.3 mg/cm, respectively) or colitis control group (46.5±3.6 mg/cm). The results of the spleen weights are shown in Figure 5E. The spleen is an important organ of the body’s immune system and an increase in spleen weight is used as a marker of inflammation severity.29 The spleen weight was significantly higher (312±52.8 mg) in the colitis control mice than in the healthy control mice (103.4±11.1 mg). When the colitis was alleviated, the spleen weight also decreased in the CSA-treated mice. However, the E/PNP-treated mice exhibited a lower average spleen weight (125.2±17.9 mg) than the PNP- or ENP-treated mice (237.4±20.8 mg and 243.5±24.9 mg, respectively), indicating that the E/PNPs ameliorate colitis to a greater extent.
Microscopic assessment of colitis
Histological evaluation of the colon tissue from all groups was performed to determine the colon-targeted delivery and therapeutic potential of E/PNPs for UC treatment. As shown in Figure 6A, the mucosa from the healthy control group showed no signs of disrupted morphology. In contrast, tissue sections from the colitis control group exhibited disruption, irregular morphology, edema, and infiltration of inflammatory cells. Colitis subsided substantially in the groups treated with CSA-loaded NPs. However, histological findings from E/PNP-treated mice demonstrated morphological tissue structures resembling healthy mouse tissue, indicating epithelial restoration and alleviation of inflammation. Histological scoring of the colon tissue sections confirmed the above observational findings, indicating that inflammation was most significantly alleviated (p<0.001) in the E/PNP-treated mice, compared to that in the PNP- or ENP-treated mice (p<0.05) (Figure 6B). Overall, these findings indicate that dual-functional E/PNPs deliver sufficient CSA to the inflamed colon, compared with CSA delivery by PNPs or ENPs, thereby significantly alleviating inflammation.
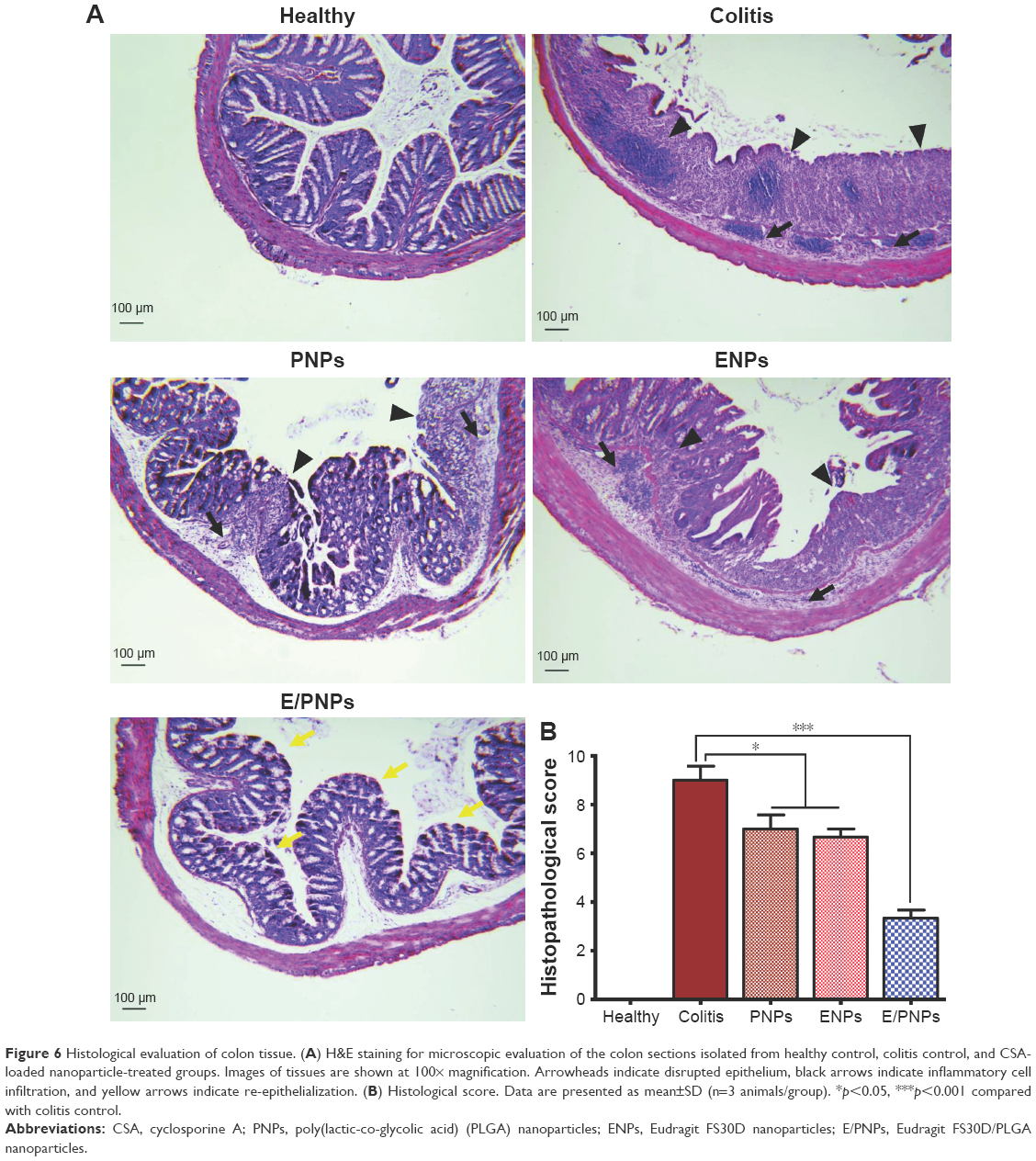 | Figure 6 Histological evaluation of colon tissue. (A) H&E staining for microscopic evaluation of the colon sections isolated from healthy control, colitis control, and CSA-loaded nanoparticle-treated groups. Images of tissues are shown at 100× magnification. Arrowheads indicate disrupted epithelium, black arrows indicate inflammatory cell infiltration, and yellow arrows indicate re-epithelialization. (B) Histological score. Data are presented as mean±SD (n=3 animals/group). *p<0.05, ***p<0.001 compared with colitis control. |
Immunofluorescence staining of macrophages
An immunofluorescence study of F4/80-positive cells in the colonic mucosa was undertaken to investigate the relationship between macrophage infiltration and colitis severity. Macrophages produce proinflammatory cytokines, such as IL-6 and TNF-α, at intestinal inflammation sites and are believed to be involved in the pathogenesis of colitis.34 Increased macrophage infiltration into the epithelium was induced by DSS intake in colitis control mice, confirming inflammation severity (Figure 7). Compared to colitis control mice, CSA-loaded PNP- or ENP-treated mice showed reduced macrophage counts. More importantly, E/PNP treatment suppressed the increased macrophage infiltration in the colonic mucosa. These results further confirm more efficient CSA delivery to the colon with dual-functional E/PNPs than with PNPs or ENPs for UC therapy.
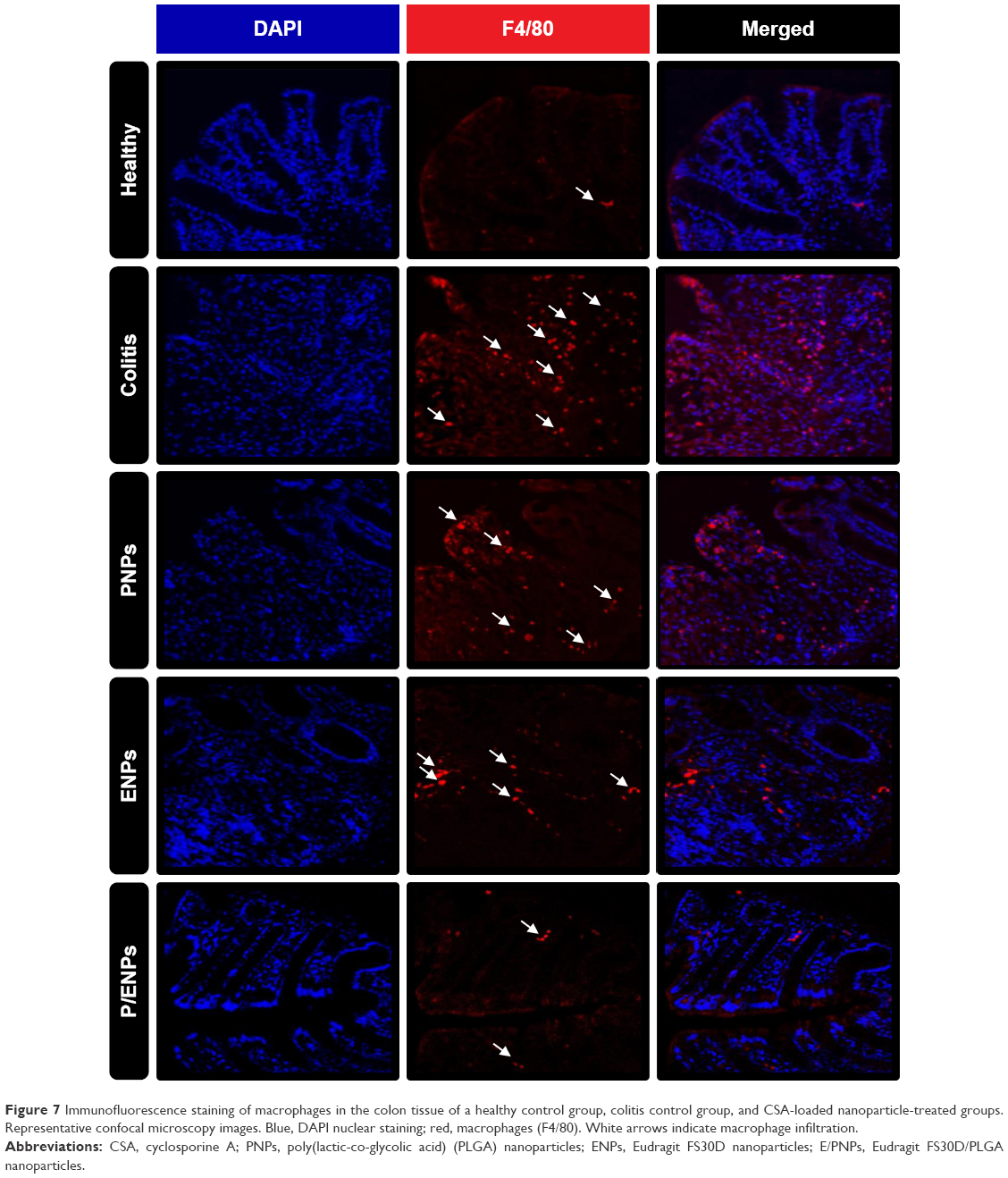 | Figure 7 Immunofluorescence staining of macrophages in the colon tissue of a healthy control group, colitis control group, and CSA-loaded nanoparticle-treated groups. Representative confocal microscopy images. Blue, DAPI nuclear staining; red, macrophages (F4/80). White arrows indicate macrophage infiltration. |
Proinflammatory cytokine levels and MPO activity in the colon
The expression profiles of the proinflammatory cytokines, TNF-α and IL-6, in the treated and control groups are shown in Figure 8A and B, respectively. All animals with colitis in this study exhibited elevated proinflammatory tissue cytokine expression, compared to healthy control mice. However, proinflammatory cytokine expression substantially decreased in the CSA-loaded NP-treated groups, compared to the expression in the untreated colitis group. It is noteworthy that cytokine concentrations decreased more significantly in the E/PNP-treated group (p<0.01) than in the PNP- or ENP-treated (p<0.05) group. An improvement in inflammation based on MPO activity was also recorded after administration of CSA-loaded NPs to colitis mice. The activity of the MPO enzyme is usually associated with the presence of neutrophils in the mucosa and submucosa of the inflamed tissues. Therefore, measurement of MPO activity can be used as a reliable index to quantify the degree of inflammatory colitis. The MPO activity in the colitis group markedly increased compared with that in the healthy control group (Figure 8C). However, the CSA-loaded NP-treated groups showed reduced MPO activity. Importantly, the E/PNP-treated group showed significantly lower MPO activity than the PNP- and ENP-treated groups, indicating efficient delivery of CSA to the inflamed colon and superior therapeutic activity.
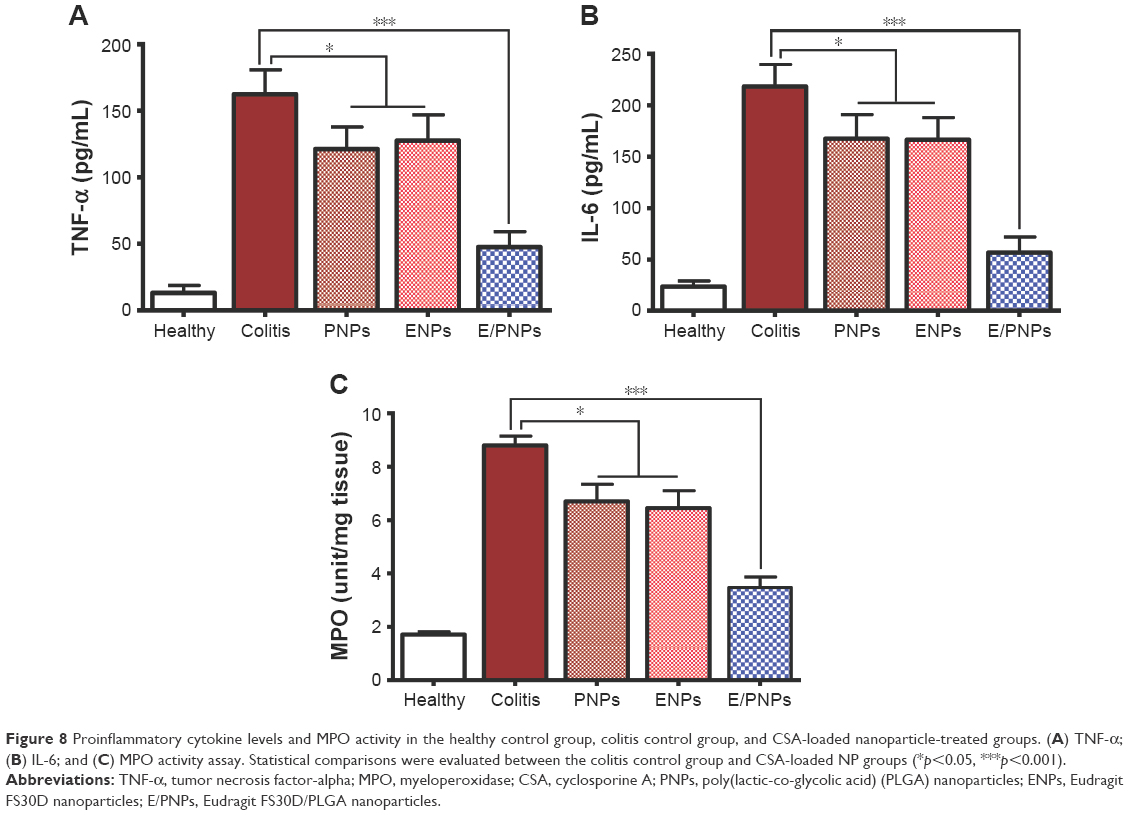 | Figure 8 Proinflammatory cytokine levels and MPO activity in the healthy control group, colitis control group, and CSA-loaded nanoparticle-treated groups. (A) TNF-α; (B) IL-6; and (C) MPO activity assay. Statistical comparisons were evaluated between the colitis control group and CSA-loaded NP groups (*p<0.05, ***p<0.001). |
Discussion
Approximately 10%–15% of UC patients have a severe attack requiring hospitalization at some time during the course of the disease.35 Intravenous corticosteroid use is a standard treatment for severe UC flare-ups; however, it fails to induce remission in up to 40% of patients.36 Treatment options for UC patients resistant to intravenous corticosteroids are limited to either CSA therapy or emergency colectomy.37 CSA is an immunosuppressant drug that was first shown to be effective in corticosteroid-resistant UC in the late 1990s, with a 64%–82% response rate.35,38 However, while currently available oral and intravenous CSA formulations are efficacious, the potentially serious side effects, such as nephrotoxicity, neurotoxicity, and opportunistic infections, can cause death, are highly limiting, and their use is generally restricted to short-term treatment regimens of no longer than 3 months.35 Focusing on the colon from a UC disease perspective, there is an unmet clinical need to improve CSA targeted and sustained delivery to the inflamed colon mucosa. Such targeted CSA delivery would allow a dose reduction compared to that required for systemic applications, avoiding high systemic concentrations and minimizing side effects.
One of the major limitations of currently marketed dosage forms (tablets and capsules) in UC therapy is the lack of targeted delivery of drugs specifically to the inflamed colon tissue.39 In addition, unreliable colonic release of a majority of pH-based marketed dosage forms, which is caused by inter- and intra-individual gut pH variability, makes doubtful their suitability for UC therapy.15,40 Furthermore, dosage forms with a size of over 200 μm are subjected to diarrhea, which results in short gastrointestinal transit time followed by decreased therapeutic outcomes.32 To maintain an adequate concentration of drug in the inflamed colon tissues, those dosage forms may require large doses and multiple administrations, which reduce patient compliance and increase side effects. Thus, a targeted delivery of drugs to the inflamed tissues with low doses is an ideal strategy to treat UC. In this regard, particle size has emerged as a crucial parameter in colon-specific drug delivery strategies for the treatment of UC, because carrier size impacts accumulation in the inflamed colon.32,41 According to previous reports, there are several pathophysiological changes due to mucosal inflammation in UC that are involved in preferential NP accumulation, including elevated mucus production, disrupted intestinal barriers, and infiltration of immune-related cells.42 A subsequent increase in residence time is postulated for NPs, compared to that of conventional dosage forms (eg, tablets, capsules, and beads), allowing for dose reduction and their frequency of administration, thereby improving adherence to treatment. In this regard, we loaded CSA in colon-targeted dual-functional NPs composed of sustained-release (PLGA) and pH-dependent (Eudragit FS30D) polymers. The objectives of such a combination are to minimize the early release of CSA in the stomach and small intestine by using Eudragit FS30D and, at the same time, to prevent complete drug release prior to reaching the inflamed colon mucosa and achieve a sustained release thereafter by using PLGA.43,44 Results from the in vitro drug-release study in the medium with a gradually changing pH that mimics the environment of the GIT demonstrated that dual-functional E/PNPs minimize the burst CSA release under stomach conditions (pH 1.2) and the complete release under ileum conditions (pH 7.4), showing sustained release thereafter (Figure 2A). Furthermore, size analysis at different pH values revealed that, unlike pH-dependent ENPs, dual-functional E/PNPs maintained their nanoparticulate size at pH 7.4, which demonstrated the presence of PLGA. Accordingly, the CSA-loaded dual-functional E/PNPs combined the advantages of both pH-sensitive and sustained-release properties, as well as nanoparticulate properties, which indicates a promising strategy for CSA delivery to the inflamed colon.
The goal of colon-targeted delivery in UC therapy is to deliver the maximum amount of drug to the inflamed colon, while minimizing systemic drug absorption and unwanted side effects in the upper regions of the GIT. In this regard, we attempted to measure plasma CSA concentration in mice by means of HPLC after oral administration of respective nanoparticles. However, plasma samples did not provide detectable CSA peaks. Alternatively, to achieve a greater understanding of the colon-targeted drug delivery potential of dual-functional E/PNPs, the fluorescence signals of DiR-loaded E/PNPs in colitis GIT were compared to those of single sustained-release PNPs and pH-dependent ENPs. At 6 h post-administration of PNPs and ENPs, weak fluorescence signals in the colon were observed and comparatively high fluorescence signals in the small intestine were noted, indicating unwanted premature release from both formulations. Similarly, at 12 h, PNPs and ENPs gave rise to negligible fluorescence signals in the colon, which reflected that encapsulated dye was prematurely released and absorbed systemically in the small intestine before reaching the target sites of the inflamed colon. Dual-functional E/PNPs showed strong fluorescence signals in the colon at 6 and 12 h, demonstrating accumulation in the inflamed colon tissues and enhanced drug delivery potential. This behavior of dual-functional E/PNPs is expected to facilitate localized release of CSA in the inflamed colon tissue and to enhance the therapeutic and safety profile in UC therapy. Therefore, in vivo distribution of CSA in the colitis mice GIT was further evaluated to assess the colon-targeting properties of dual-functional E/PNPs compared to those of single-functional NPs that have been most recently reported for CSA delivery in experimental colitis.20 At 6 and 12 h post administration, the dual-functional E/PNPs achieved considerably higher levels of CSA in the colon in comparison with the CSA levels from single-functional NPs. These results indicate that in the case of dual-functional E/PNPs, attenuation of the burst drug release in the upper small intestine, slow and incomplete release of the CSA in the terminal ileum, and accumulation in the colon resulted in higher levels of CSA in the colon.
After assessing the in vitro and in vivo colon-targeted drug delivery potential, the therapeutic efficacy of dual-functional E/PNPs was evaluated in DSS-induced colitis mice in comparison with the therapeutic efficacy of PNPs and ENPs. We used CSA at a dose of 15 mg/kg because higher doses are needed in mice than those used for humans to reach the therapeutic level of CSA by the oral route.45,46 The DSS-induced colitis model was chosen because it exhibits certain characteristics present in human UC.47 UC clinical parameters, such as colon length and body weight, were markedly reduced and DAI, and spleen weights increased in the colitis control mice compared to those in the NP-treated mice. However, treatment with the dual-functional E/PNPs ameliorated DSS-induced colitis better than did single-functional NP treatment. A significant recovery in body weight, colon length, spleen weight, and DAI was observed for dual-functional E/PNP-treated mice compared to that for PNP- or ENP-treated mice. In all cases, the dual-functional E/PNPs exhibited significantly better therapeutic efficacy than did the PNPs or ENPs. The colitis-ameliorating effect of CSA-loaded NPs on DSS-induced colitis was also demonstrated by histological examination of mouse colonic tissue. The healthy control group showed normal colon histology that showed no evidence of inflammation or disruption of healthy tissue morphology. However, colon tissues from the colitis control group exhibited clear signs of inflammation, such as cell infiltration, goblet cell depletion, and an irregular mucosal structure. Tissues from the PNP- or ENP-treated group showed slight decreases in the level of inflammation; however, tissues from the dual-functional E/PNP-treated group had no obvious inflammation, and the tissue morphology was much closer to that of the healthy baseline tissue, with no severe symptoms of colitis.
The grade of colitis and recovery was also evaluated by assessing MPO and proinflammatory cytokine levels. MPO, which is the most abundant protein in neutrophils, is widely used as a standard test in IBD patients and IBD animal models. This is especially true in the DSS-induced colitis model, where significantly high neutrophil infiltration is observed at all stages of DSS administration, and even after DSS withdrawal.48 In the colitis control group, the MPO level was significantly elevated, whereas MPO was downregulated in the colonic tissues of the CSA-loaded NP-treated groups. However, the dual-functional E/PNP-treated mice displayed significantly lower MPO levels than did the PNP- or ENP-treated mice, suggesting that the efficient delivery of CSA to the colon by the dual-functional E/PNPs induced a reduction in inflammation in the DSS-induced colitis mice. Along with the neutrophil infiltration in the inflamed colon area, the activation of macrophages leads to proinflammatory cytokine secretion, including TNF-α and IL-6.21 Thus, quantification of TNF-α and IL-6 by ELISA was essential in the colonic tissues. The expression of both TNF-α and IL-6 decreased in the colon tissues of the CSA-treated mice compared to the expression in the untreated colitis mice. However, treatment with dual-functional E/PNPs suppressed TNF-α and IL-6 expression more prominently than did treatment with PNPs or ENPs, indicating efficient delivery of CSA to the inflamed colon to enhance therapeutic efficacy in UC. These cytokine reductions can be correlated with the results of the macrophage infiltration in the colon, which displayed the same trend of decreased inflammation with the CSA-loaded dual-functional E/PNP-treated groups. Overall, the findings in this study justify the use of dual-functional E/PNPs composed of PLGA and Eudragit FS30D for efficient delivery of CSA to the inflamed colon in UC treatment.
Conclusion
CSA-loaded dual-functional E/PNPs composed of sustained-release and pH-dependent polymers for colon targeting and sustained delivery during UC therapy have been presented in this study. Based on our findings, the dual-functional E/PNPs have the ability to avoid burst release in the stomach; they exhibit incomplete and slow release at the pH of the ileum and colon, followed by a sustained release, thus delivering a sufficient amount of CSA specifically to the inflamed colon. The dual-functional E/PNPs loaded with CSA showed a significant effect in alleviating colitis in DSS-induced mice compared with that of CSA-loaded sustained-release PNPs or pH-dependent ENPs. The CSA-loaded dual-functional E/PNPs presented in this study appear to be a promising safe and effective drug delivery system for UC therapy.
Abbreviations
UC, ulcerative colitis; GIT, gastrointestinal tract; CSA, cyclosporine A; NPs, nanoparticles; IBD, inflammatory bowel disease; DSS, dextran sodium sulfate; TNBS, trinitrobenzene–sulfonic acid; PLGA, poly(lactic-co-glycolic acid); PNPs, PLGA NPs; ENPs, Eudragit® FS30D NPs; E/PNPs, Eudragit FS30D/PLGA NPs; PVA, polyvinyl alcohol; DiR, 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide; SEM, scanning electron microscopy; PDI, polydispersity index; HPLC, high-performance liquid chromatography; UV, ultraviolet; DL, drug loading; EE, encapsulation efficiency; ICR, imprinting control region; IVIS, in vivo imaging system; DAI, disease activity index; MPO, myeloperoxidase; ELISA, enzyme-linked immunosorbent assay; ANOVA, analysis of variance; TNF-α, tumor necrosis factor-α.
Acknowledgment
This study was financially supported by the 2017 Post-Doc. Development Program of Pusan National University.
Disclosure
The authors report no conflicts of interest in this work.
References
Travis SP, Danese S, Kupcinskas L, et al. Once-daily budesonide MMX in active, mild-to-moderate ulcerative colitis: results from the randomised CORE II study. Gut. 2014;63(3):433–441. | ||
Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A. 2007;104(34):13780–13785. | ||
Rogler G. Chronic ulcerative colitis and colorectal cancer. Cancer Lett. 2014;345(2):235–241. | ||
Burger D, Travis S. Conventional medical management of inflammatory bowel disease. Gastroenterology. 2011;140(6):1827–1837.e2. | ||
Park SC, Jeen YT. Current and emerging biologics for ulcerative colitis. Gut Liver. 2015;9(1):18–27. | ||
Actis GC, Fadda M, David E, Sapino A. Colectomy rate in steroid-refractory colitis initially responsive to cyclosporin: a long-term retrospective cohort study. BMC Gastroenterol. 2007;7(1):13. | ||
Cohen RD, Stein R, Hanauer SB. Intravenous cyclosporin in ulcerative colitis: a five-year experience. Am J Gastroenterol. 1999;94(6):1587–1592. | ||
Beauchesne PR, Chung NS, Wasan KM. Cyclosporine A: a review of current oral and intravenous delivery systems. Drug Dev Ind Pharm. 2007;33(3):211–220. | ||
Eun CS, Han DS. Does the cyclosporine still have a potential role in the treatment of acute severe steroid-refractory ulcerative colitis? Gut Liver. 2015;9(5):567–568. | ||
Aberra FN, Lichtenstein GR. Review article: monitoring of immunomodulators in inflammatory bowel disease. Aliment Pharmacol Ther. 2005;21(4):307–319. | ||
Dai J, Nagai T, Wang X, Zhang T, Meng M, Zhang Q. pH-sensitive nanoparticles for improving the oral bioavailability of cyclosporine A. Int J Pharm. 2004;280(1):229–240. | ||
Mahesh VN, Campbell R, Ahluwalia NK. W1330 cyclosporine enema in conventional ± immunomodulator refractory distal ulcerative colitis: ready for prime time or hype? Gastroenterology. 2010;138(5):S-700. | ||
Sandborn WJ, Strong RM, Forland SC, Chase RE, Cutler RE. The pharmacokinetics and colonic tissue concentrations of cyclosporine after i.v., oral, and enema administration. J Clin Pharmacol. 1991;31(1):76–80. | ||
Youshia J, Lamprecht A. Size-dependent nanoparticulate drug delivery in inflammatory bowel diseases. Expert Opin Drug Deliv. 2016;13(2):281–294. | ||
Naeem M, Cao J, Choi M, et al. Enhanced therapeutic efficacy of budesonide in experimental colitis with enzyme/pH dual-sensitive polymeric nanoparticles. Int J Nanomedicine. 2015;10:4565–4580. | ||
Vong LB, Mo J, Abrahamsson B, Nagasaki Y. Specific accumulation of orally administered redox nanotherapeutics in the inflamed colon reducing inflammation with dose–response efficacy. J Control Release. 2015;210:19–25. | ||
Hua S, Marks E, Schneider JJ, Keely S. Advances in oral nano-delivery systems for colon targeted drug delivery in inflammatory bowel disease: selective targeting to diseased versus healthy tissue. Nanomedicine. 2015;11(5):1117–1132. | ||
Guada M, Beloqui A, Alhouayek M, et al. Cyclosporine A-loaded lipid nanoparticles in inflammatory bowel disease. Int J Pharm. 2016;503(1):196–198. | ||
Courthion H, Mugnier T, Rousseaux C, Möller M, Gurny R, Gabriel D. Self-assembling polymeric nanocarriers to target inflammatory lesions in ulcerative colitis. J Control Release. 2017;275:32–39. | ||
Melero A, Draheim C, Hansen S, et al. Targeted delivery of Cyclosporine A by polymeric nanocarriers improves the therapy of inflammatory bowel disease in a relevant mouse model. Eur J Pharm Biopharm. 2017;119:361–371. | ||
Ali H, Weigmann B, Neurath M, Collnot E, Windbergs M, Lehr C-M. Budesonide loaded nanoparticles with pH-sensitive coating for improved mucosal targeting in mouse models of inflammatory bowel diseases. J Control Release. 2014;183:167–177. | ||
Naeem M, Choi M, Cao J, et al. Colon-targeted delivery of budesonide using dual pH-and time-dependent polymeric nanoparticles for colitis therapy. Drug Des Devel Ther. 2015;9:3789–3799. | ||
Aksungur P, Demirbilek M, Denkbaş EB, Vandervoort J, Ludwig A, Ünlü N. Development and characterization of Cyclosporine A loaded nanoparticles for ocular drug delivery: cellular toxicity, uptake, and kinetic studies. J Control Release. 2011;151(3):286–294. | ||
Sun S, Liang N, Yamamoto H, Kawashima Y, Cui F, Yan P. pH-sensitive poly(lactide-co-glycolide) nanoparticle composite microcapsules for oral delivery of insulin. Int J Nanomedicine. 2015;10:3489–3498. | ||
Beloqui A, Coco R, Memvanga PB, Ucakar B, des Rieux A, Préat V. pH-sensitive nanoparticles for colonic delivery of curcumin in inflammatory bowel disease. Int J Pharm. 2014;473(1–2):203–212. | ||
Vandamme TF, Lenourry A, Charrueau C, Chaumeil J-C. The use of polysaccharides to target drugs to the colon. Carbohydr Polym. 2002;48(3):219–231. | ||
Hartmann G, Bidlingmaier C, Siegmund B, et al. Specific type IV phosphodiesterase inhibitor rolipram mitigates experimental colitis in mice. J Pharmacol Exp Ther. 2000;292(1):22–30. | ||
Makhlof A, Tozuka Y, Takeuchi H. pH-Sensitive nanospheres for colon-specific drug delivery in experimentally induced colitis rat model. Eur J Pharm Biopharm. 2009;72(1):1–8. | ||
Viennois E, Xiao B, Ayyadurai S, et al. Micheliolide, a new sesquiterpene lactone that inhibits intestinal inflammation and colitis-associated cancer. Lab Invest. 2014;94(9):950–965. | ||
Weigmann B, Lehr HA, Yancopoulos G, et al. The transcription factor NFATc2 controls IL-6-dependent T cell activation in experimental colitis. J Exp Med. 2008;205(9):2099–2110. | ||
Kim JJ, Shajib MS, Manocha MM, Khan WI. Investigating intestinal inflammation in DSS-induced model of IBD. J Vis Exp. 2012(60):e3678. | ||
Lamprecht A, Schäfer U, Lehr C-M. Size-dependent bioadhesion of micro- and nanoparticulate carriers to the inflamed colonic mucosa. Pharm Res. 2001;18(6):788–793. | ||
Watanabe A, Tanaka H, Sakurai Y, et al. Effect of particle size on their accumulation in an inflammatory lesion in a dextran sulfate sodium (DSS)-induced colitis model. Int J Pharm. 2016;509(1–2):118–122. | ||
Reinecker HC, Steffen M, Witthoeft T, et al. Enhanced secretion of tumour necrosis factor-alpha, IL-6, and IL-1 beta by isolated lamina propria mononuclear cells from patients with ulcerative colitis and Crohn’s disease. Clin Exp Immunol. 1993;94(1):174–181. | ||
Arts J, D’Haens G, Zeegers M, et al. Long-term outcome of treatment with intravenous cyclosporin in patients with severe ulcerative colitis. Inflamm Bowel Dis. 2004;10(2):73–78. | ||
Järnerot G, Rolny P, Sandberg-Gertzén H. Intensive intravenous treatment of ulcerative colitis. Gastroenterology. 1985;89(5):1005–1013. | ||
Lichtiger S, Present DH, Kornbluth A, et al. Cyclosporine in severe ulcerative colitis refractory to steroid therapy. N Engl J Med. 1994;330(26):1841–1845. | ||
D’Haens G, Lemmens L, Nevens F, et al. Intravenous cyclosporine versus intravenous corticosteroids as single therapy for severe attacks of ulcerative colitis. Gastroenterology. 2001;120(6):1323–1329. | ||
Lamprecht A. IBD: selective nanoparticle adhesion can enhance colitis therapy. Nat Rev Gastroenterol Hepatol. 2010;7(6):311–312. | ||
Nugent S, Kumar D, Rampton DS, Evans DF. Intestinal luminal pH in inflammatory bowel disease: possible determinants and implications for therapy with aminosalicylates and other drugs. Gut. 2001;48(4):571–577. | ||
Viscido A, Capannolo A, Latella G, Caprilli R, Frieri G. Nanotechnology in the treatment of inflammatory bowel diseases. J Crohns Colitis. 2014;8(9):903–918. | ||
Lamprecht A, Yamamoto H, Takeuchi H, Kawashima Y. Nanoparticles enhance therapeutic efficiency by selectively increased local drug dose in experimental colitis in rats. J Pharmacol Exp Ther. 2005;315(1):196–202. | ||
Ma X, Williams RO. Polymeric nanomedicines for poorly soluble drugs in oral delivery systems: an update. J Pharm Invest. 2018;48(1):61–75. | ||
Pawar PK, Gautam C. Design, optimization and evaluation of mesalamine matrix tablet for colon drug delivery system. J Pharm Invest. 2016;46(1):67–78. | ||
Guada M, Lana H, Gil AG, del Carmen Dios-Viéitez M, Blanco-Prieto MJ. Cyclosporine A lipid nanoparticles for oral administration: pharmacodynamics and safety evaluation. Eur J Pharm Biopharm. 2016;101:112–118. | ||
Sann H, von Erichsen J, Hessmann M, Pahl A, Hoffmeyer A. Efficacy of drugs used in the treatment of IBD and combinations thereof in acute DSS-induced colitis in mice. Life Sci. 2013;92(12):708–718. | ||
Wirtz S, Neurath MF. Mouse models of inflammatory bowel disease. Adv Drug Deliv Rev. 2007;59(11):1073–1083. | ||
Mendoza J, Abreu M. Biological markers in inflammatory bowel disease: practical consideration for clinicians. Gastroenterol Clin Biol. 2009;33:S158–S173. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.