Back to Archived Journals » Advances in Genomics and Genetics » Volume 5
COL1A2 gene analysis in a Czech osteogenesis imperfecta patient: a candidate novel mutation in a patient affected by osteogenesis imperfecta type 3
Authors Hrušková L, Mařík I, Mazurová S, Martásek P, Mazura I
Received 16 June 2014
Accepted for publication 14 September 2014
Published 5 August 2015 Volume 2015:5 Pages 275—281
DOI https://doi.org/10.2147/AGG.S58766
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr John Martignetti
Lucie Hrušková,1 Ivo Mařík,2,3 Stella Mazurová,1 Pavel Martásek,1 Ivan Mazura1
1Department of Pediatrics and Adolescent Medicine, First Faculty of Medicine, Charles University in Prague, Prague, Czech Republic; 2Ambulant Centre for Defects of Locomotor Apparatus 1.1.c., Prague, Czech Republic; 3Faculty of Medical Studies, West Bohemia University, Pilsen, Czech Republic
Abstract: Osteogenesis imperfecta is a heritable bone fragility disease with a heterogenic genetic origin. Most cases result from mutations of either the COL1A1 gene or the COL1A2 gene. We identified a novel COL1A2 gene mutation in a Czech patient, born to unaffected parents, who was diagnosed according to clinical and anthropometric findings and radiographic features as having type 3 osteogenesis imperfecta, which is a severe form of this disease. The identified Gly814Trp mutation was predicted by a number of complementary bioinformatic programs to result in functional alteration of the protein. This case report provides both evidence of a novel COL1A2 mutation resulting in type 3 osteogenesis imperfecta and a genotype:phenotype correlation in this affected individual.
Keywords: osteogenesis imperfecta type 3, collagen, alpha-2 (I) chain, substitution, sequencing
Introduction
Osteogenesis imperfecta (OI) is a disorder of bone formation with a worldwide prevalence of 1:16–20,000. In addition to typical skeletal signs, such as low bone density, high risk for fractures, and bone deformities, there are many clinical signs observed in affected patients. These include the blue color of the sclera, dentinogenesis imperfecta, joint hypermobility, skin hyperlaxity, and hearing loss, among others. The clinical picture of OI is heterogenic, and the presence of individual signs differs from type to type and patient to patient.1 The most autosomal-dominant forms of OI result from mutations of two genes coding the collagen type 1 protein (COL1A1 and COL1A2). Other dominantly or recessively inherited OI types are caused by mutations in eleven different genes (wingless-type mouse mammary tumor virus [MMTV] integration site family, member 1 [WNT1]’ interferon-induced transmembrane protein 5 [IFITM5]; serpin peptidase inhibitor, clade F, member 1 [SERPINF1]; cartilage-associated protein [CRTAP]; leucine- and proline-enriched proteoglycan 1 [LEPRE1]; peptidyl-prolyl isomerase 1 [cyclophilin B; PPIB]; serpin peptidase inhibitor, clade H, member 1 [SERPINH1]; FK506-binding protein 10 [FKPB10]; osteoblast-specific transcription factor SP7 [SP7]; bone morphogenetic protein 1 [BMP1]; and transmembrane protein 38B [TMEM38B]).1–8
OI type 3 is the most severe autosomal dominant form of OI. First fractures are described as occurring in the perinatal period and at birth. Patients are of subnormal stature and have deformed short and long bones. Radiographic findings include low mineralization of the skull, with the presence of Wormian bones, and popcorn-like calcification of meta- and epiphyses, especially of the long bones, which, when compared with the diaphysis, are broad and flattened. In some severe sporadic cases, OI type 3 or 4 can present with a secondary basilar impression (BI) in the second to fourth decade of life. It was presumed that BI occurs equally in all types of OI.9 Other clinical signs observed in patients affected by OI type 3 include triangular face, dentinogenesis imperfecta, blue sclera (the color could turn to white with age), barrel-shaped chest, weak muscles, hyperlaxity of joints and ligaments, and severe scoliosis. Deformities of the chest are the most common reason for pulmonary deficiency.1,10
Materials and methods
In this study, we analyzed the DNA sample of a single Czech individual diagnosed with a severe type of OI originally diagnosed according to clinical and anthropometric findings and radiographic features as type 3 OI.11 This study was approved (project 83/14) by the Ethics committee of General University Hospital, Prague.
Genetic analysis was focused on the coding sequence of the COL1A2 gene coding the alpha-2 (I) chain of the collagen type 1. The genomic DNA was isolated from the leucocytes of the peripheral blood. The quality of isolated samples was measured by gel electrophoresis, and the quantity was detected spectrophotometrically. DNA samples were amplified, using a polymerase chain reaction and specially designed 100% complementary primers (Table 1). Sequences of electrophoretically checked polymerase chain reaction products were obtained on the ABI PRISM 3730xl Genetic Analyzer automated sequencer (Applied Biosystems, Foster City, CA, USA), using the BigDye Terminator v3.1 chemistry.
 | Table 1 Sequences of used primers |
Obtained data were compared with the wild-type sequence, as submitted to Ensembl (accession no ENST00000297268 http://www.ensembl.org/Homo_sapiens/Transcript/Exons?db=core;g=ENSG00000164692;r=7:94394561-94431232; t=ENST00000297268). Nucleotides are numbered from the first base of the start codon (ATG) of the complementary DNA reference sequence, according to GenBank NM_000089.3. Amino acid residues were numbered from the start codon for methionine (ATG) of the alpha-2 chain of type 1 collagen (reference sequence GenBank NP_000080.2).
Novel mutation was identified by absence from the Type 1 Collagen Mutation Database (https://oi.gene.le.ac.uk/variants.php?action=search_unique&limit=100&order=Variant% 2FDNA%2CASC&page=1), the Human Genome Mutation Database (https://portal.biobase-international.com/hgmd/pro/allmut.php), and the Ensembl database. (http://www.ensembl.org/Homo_sapiens/Transcript/Exons?db=core;g=ENSG00000164692;r=7:94023873-94060544;t=ENST00000297268)
The case
A diagnosis of OI, type 3, of the female patient was established at the birth of the individual, and she has been followed up and treated by the same orthopedic surgeon (and pediatrician) to date.
The patient comes from the second gravidity (one miscarriage) of healthy parents. At her delivery, by C-section at term, with a weight of 2,450 g and a length of 45 cm, the patient was resuscitated, and oxygen therapy was applied in the incubator for 24 hours. A mild icterus neonatorum disappeared spontaneously. After birth, a triangular face; incomplete mineralization of the calvaria, with multiple Wormian bones in the occipital region; healed fractures of ribs and the left clavicle; and fresh fractures of both femurs were verified with X-rays. Before age 15 years, the patient suffered from 16 fractures (both femurs and the left tibia were repeatedly treated, as well as the right clavicle, right tibia, left humerus, right and left ulna or radius, right scapula, and ribs).
Starting when she was 2 years old, the patient has undergone a lot of surgeries, especially corrective and multiple osteotomies of both femurs and forearms (usually fixation with intramedullary nailing, according to Küntscher nail technique12). As a result of surgery and the orthotic fitting of her legs, she was able to walk from age 6 to 11 years, using support aids. Later, she was confined to a wheelchair. Her mental development was normal.
Medication was indicated with the aim of decreasing bone turnover. From age 7–11 years, calcitonin was administrated, and from age 14–16 years, bisphosphonates were administrated, together with vitamin D and calcium.
Physical examination at the age of 10 years revealed severe shortening of the trunk and neck, a barrel-shaped thorax, narrow shoulders, and a small umbilical hernia. There was dextroscoliosis of the lower thoracic part of the spine, as well as accentuation of thoracic kyphosis. Rhizomelic shortening of the legs and saber-shaped right humerus were noticeable, as was both tibia being saber-shaped. Scars on both thighs are sheer (Figure 1A). Permanent teeth were healthy, without dentinogenesis imperfecta. A triangular face with a prominent nose and protuberant eyes with bluish sclera are presented (Figure 1A and B). The shape of the hands and feet was normal. Examination of the range of joint motion revealed bilateral flexion contractures of the hips (approximately 40°). Other big and small joints were hypermovable. The patient was able to crawl on all fours. She did not suffer from hearing loss, and her mental development was normal.
 | Figure 1 The patient aged 10 years. |
Anthropological examination at age 15 years
Body length according to the right leg was 90 cm (−11.7 standard deviation [SD]), body weight was 21 kg (−8.6 SD), and body weight to height was above norm, but skinfold thickness was normal, between the 50th and 75th percentiles. Both the trunk and the extremities were severely shortened: sitting height was 52 cm (−11.6 SD), subischial length of the right leg was −9.9 SD, and the left leg was shortened by 10 cm (−12.5 SD).
The patient had a barrel-shaped thorax with a thoracic index of 104. Her head circumference was 56 cm (0.8 SD; 75th centile), and her skull was hyperbrachycephalic (cephalic index, 88.1), with a very wide forehead, wide nasal bridge, and triangular face.
At 25 years of age, her body weight was 31.7 kg; her body length according to the right leg was 87 cm; her arm span, measured by segments, was 118 cm; and her sitting height was 57.5 cm.
Radiographic examination at different ages shows generalized skeletal changes, such as typical deformities of long bones, spine, thorax, and bone structure that are consistent with a severe OI type (Figure 2A–J).
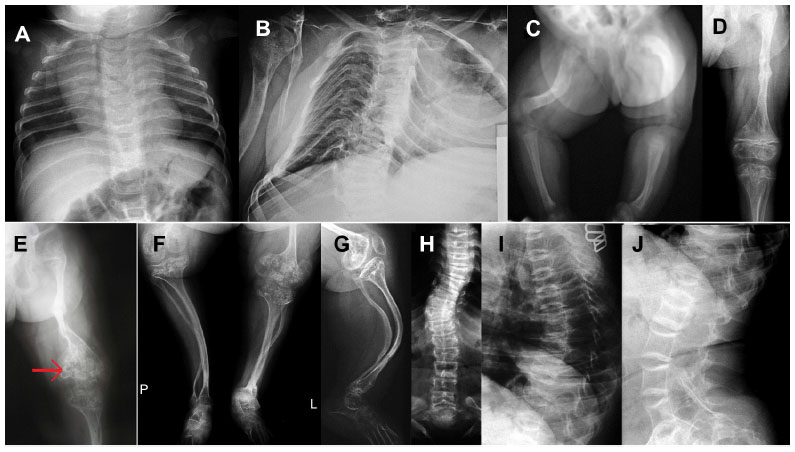 | Figure 2 X-rays of the presenting osteogenesis imperfecta patient. |
The routine blood and urine examinations during the patient’s growth period were all normal. Markers of bone formation and resorption repeatedly proved higher bone turnover. Vitamin D and parathormone in serum were in the reference values.
Genetic examination determined that the family history was negative, and the affected girl represents an isolated case in the family. Karyotype 46, XX was ascertained. We supposed an autosomal dominant and/or recessive inheritance.
Densitometric examination, Hologic, Discovery A (S/N 85046), at 22 years of age proved the patient has significant low bone density in the lumbar region, as well as the low whole-body density (T-score, −3.2 SD, and T-score, −5.0 SD, respectively). Her fat tissue percentage was 38.4%, or in the 65th percentile (according to a reference database of the American population).13
At the whole-body scan (Figure 3A and B), the severe skeletal deformities, disproportionate stature, and shortening of the left lower extremity and the right upper extremity are well-documented (Figure 3A). The patient’s fat tissue distribution and muscle hypotrophy are visible in Figure 3B.
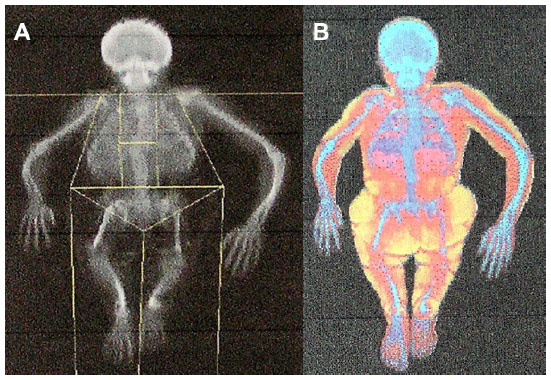 | Figure 3 Whole-body scans. The scans approximately document both the skeleton deformities and disproportionate stature of the patient at the age of 25 years (A) and her fat tissue distribution (B). |
After puberty, only a few fractures were diagnosed, and they were conservatively treated. At 25 years of age, the woman did not suffer from hearing loss, dentinogenesis imperfecta, BI or pulmonary insufficiency. She is in a wheelchair and is self-sufficient. She lives alone in a barrier-free flat. She draws a disability pension, but she successfully studied at a technical high school. At present, she works half-time as a clerk.
A diagnosis of OI type 3 was established at birth and confirmed at 15 years of age on the basis of typical clinical and anthropological findings and radiographic features that presented. This case was one of a cohort 55 Czech OI patients who were evaluated in the frame of a PhD thesis.14 All collected patient data were gathered into the complex database and statistically evaluated. The patients were reclassified on the basis of clinical and radiological signs of OI types defined by Sillence.15,16
Anthropological characteristics within OI types were specified. Types of OI differed from each other remarkably. Type 3 (3/2B) was the most severe type and was diagnosed in nine cases.11
Results
Molecular genetic analysis identified a mutation of the coding sequence the COL1A2 gene in the presented case of OI type 3. This mutation results in the substitution of glycine by tryptophan at position 814 of the alpha-2 (I) chain (p.Gly814Trp, c.2440G>T, g.7:940522305) (Figure 4). This mutation was using the program Alamut (used algorithms: SpliceSiteFinder, MaxEntScan, NNSPLICE, GeneSplicer, Human Splicing Finder) predicted as a disease-causing (P=1.0). The substituted nucleotide is situated in the highly conserved region (phylo-P=5.94) (Figure 5).
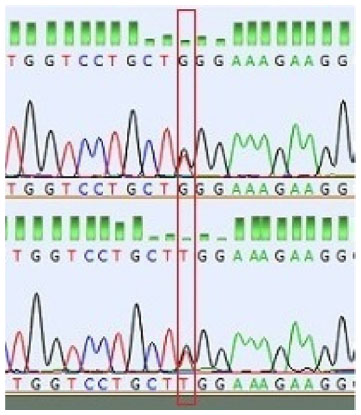 | Figure 4 The p.Gly814Trp substitution. |
 | Figure 5 Alamut mutation analysis. |
Discussion
Identified mutation p.Gly814Trp (c.2440G>T, g.7:940522305) was found in an individual of a family with a negative OI family history. The mutation is presented in the heterozygote state and is localized in exon 40 of the COL1A2. From the molecular point of view, glycine is a crucial amino acid that occurs in 338 Gly-X-Y repetitive sequences and, in this way, ensures the tight approach of collagen type 1 alpha chains. This increases the stability of the protein molecule. Furthermore, the mutation of Gly814 is situated in the α1β1 integrin binding region of the alpha chains. Integrins are a family of proteins that mediate cell–extracellular matrix interaction. Another function of these molecules is participation in tissue morphogenesis, matrix assembly, and cell signaling. Integrins are also a crucial part of collagen type 1 fibrillogenesis and induce conformation changes in collagen, and thereby accelerate the fibrillogenesis. Integrins are situated on the plasma membrane and support the close proximity of collagen molecules.17,18
Glycine 814 is situated in multiligand binding region 2 (MLBR2), which is defined by codons 656–824. This area includes a part of coding sequence of exons 38 and 42 and the whole coding sequences of exons 39–41 of the COL1A2 gene. The clinical outcome of mutations localized in this part of the gene is in most of cases a severe form of OI (compared with this, mutations of MLBR2 of the alpha-1[I] chain especially result in type 2 OI, the lethal form of the disease). Identified substitution of glycine at position 814 corresponds with the phenotype picture of other mutations localized in the MLBR2 region.
The Gly814Trp mutation affects the structure of the final protein, and therefore the bone formation. Affected morphogenesis could also result from interrupted integrin α1β1 binding.
Comparing databases, we concluded that the p.Gly814Trp substitution is a novel mutation identified in the cohort of Czech OI affected individuals.
Conclusion
In conclusion, a novel candidate COL1A2 Gly814Trp mutation was identified in a female Czech patient diagnosed with OI type 3. The possibility always exists that mutations in other genes related to an autosomal recessive form of OI may have caused this individual’s disease and necessitates consideration of this possibility in genetic counseling of the family. We believe, however, that the in silico analysis suggesting a pathogenic role for the Gly814Trp mutation we identified in this individual supports an autosomal dominant mode of transmission. Now that this candidate mutation has been described in this individual, it should provide useful information for helping diagnose others with the same mutation and for establishing transmission patterns.
Acknowledgments
This study was supported by grants from Charles University in Prague, Czech republic (SVV-2013-266517, SVV-2014-260034, SVV-2015-260158, PRVOUK P24/1LF/3 and UNCE 204011). The authors’ hearty thanks belong to the patient for her permission to publish unmasked photos from her childhood in the scientific journal.
Disclosure
The authors report no conflicts of interest in this work.
References
Endotext [homepage on the Internet]. MDText.com, Inc. Available from: http://www.endotext.org/chapter/osteogenesis-imperfecta/7/. Accessed April 15, 2015. Accessed June 16, 2014. | |
Fahiminiya S, Majewski J, Mort J, Moffatt P, Glorieux FH, Rauch F. Mutations in WNT1 are a cause of osteogenesis imperfecta. J Med Genet. 2013;50(5):345–348. | |
Cho TJ, Lee KE, Lee SK, et al. A single recurrent mutation in the 5′-UTR of IFITM5 causes osteogenesis imperfecta type V. Am J Hum Genet. 2012;91(2):343–348. | |
Cundy T. Recent advances in osteogenesis imperfecta. Calcif Tissue Int. 2012;90(6):439–449. | |
Lapunzina P, Aglan M, Temtamy S, et al. Identification of a frameshift mutation in Osterix in a patient with recessive osteogenesis imperfecta. Am J Hum Genet. 2010;87(1):110–114. | |
Martínez-Glez V, Valencia M, Caparrós-Martín JA, et al. Identification of a mutation causing deficient BMP1/mTLD proteolytic activity in autosomal recessive osteogenesis imperfecta. Hum Mutat. 2012;33(2):343–350. | |
Shaheen R, Alazami AM, Alshammari MJ, et al. Study of autosomal recessive osteogenesis imperfecta in Arabia reveals a novel locus defined by TMEM38B mutation. J Med Genet. 2012;49(10):630–635. | |
Zhang ZL, Zhang H, Ke YH, et al. The identification of novel mutations in COL1A1, COL1A2, and LEPRE1 genes in Chinese patients with osteogenesis imperfecta. J Bone Miner Metab. 2012;30(1):69–77. | |
Hayes M, Parker G, Ell J, Sillence D. Basilar impression complicating osteogenesis imperfecta type IV: the clinical and neuroradiological findings in four cases. J Neurol Neurosurg Psychiatry. 1999;66:357–364. | |
Primorac D, Rowe DW, Mottes M, et al. Osteogenesis imperfecta at the beginning of bone and joint decade. Croat Med J. 2001;42(4):393–415. | |
Hudaková-Maříková O, Mařík I, Zemková D, Šedová M, Mazura I, Kuklík M. Osteogenesis imperfecta from the point of view clinical and anthropological characteristics of the disease and differential diagnostics of its types. Pohybové ústrojí. 2007;14(3–4):325–329. Czech. | |
Maruenda-Paulino JI. Kuntscher nailing of femoral shaft fractures in children and adolescents. Int Orthop. 1993;17(3):158–161. | |
Laurson KR, Eisenmann JC, Welk GJ. Body Fat Percentile Curves for US Children and Adolescents. Am J Prev Med. 2011;41(4S2):S87–S92. | |
Hudáková O. Skeletal defomities in bone dysplasias with decreased bone density: osteogenesis imperfecta [PhD thesis]. Prague: Faculty of Science, Charles University. 2007;p187. | |
Sillence D. Osteogenesis imperfecta: An expanding panorama of variants. Clin Orthop. 1981;159:11–25. | |
Rowe DW, Shappiro JR. Osteogenesis imperfecta. In: Avioli LV, Krane SM, editors. Metabolic Bone Disease and Clinically Related Disorders. 3rd ed. San Diego: Academic Press; 1998:651–695. | |
Sweeney SM, Orgel JP, Fertala A, et al. Candidate cell and matrix interaction domains on the collagen fibril, the predominant protein of vertebrates. J Biol Chem. 2008;283(30):21187–21197. | |
Kadler KE, Hill A, Canty-Laird EG. Collagen fibrillogenesis: fibronectin, integrins, and minor collagens as organizers and nucleators. Curr Opin Cell Biol. 2008;20(5):495–501. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.