Back to Journals » Blood and Lymphatic Cancer: Targets and Therapy » Volume 4
Coexistence of primary myelofibrosis and chronic lymphocytic leukemia
Authors Burgstaller S, Thaler J
Received 12 June 2014
Accepted for publication 22 July 2014
Published 3 October 2014 Volume 2014:4 Pages 101—105
DOI https://doi.org/10.2147/BLCTT.S29592
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Sonja Burgstaller, Josef Thaler
Klinikum Wels-Grieskirchen, Abteilung für Innere Medizin IV, Wels, Austria
Abstract: Coexistence of two hematologic malignancies in one patient is generally a rare phenomenon. The topic of this article is the coincidence of primary myelofibrosis and chronic lymphocytic leukemia, which has been reported up to now in only 16 patients. In summary, simultaneous detection of both diseases was notable in half of the patients at presentation. In the case of a subsequent diagnosis of both disorders, primary myelofibrosis preceded the lymphoproliferative disease in the majority of patients. The clinical course seems to be more benign than for each disorder itself. A substantial proportion of patients did not require any treatment at all. Knowledge about the pathogenetic mechanisms, treatment approaches, and prognosis of these patients is limited.
Keywords: primary myelofibrosis, chronic lymphocytic leukemia, coexistent
Introduction
Recently, a case report on a patient with coincidence of primary myelofibrosis (PMF) and chronic lymphocytic leukemia (CLL) was published by our group.1 At the beginning of this article, we present a brief summary and an update of this patient, followed by a review of the current literature.
A 61-year-old woman with transfusion-dependent normochromic, normocytic anemia (hemoglobin 6.0 g/dL) was admitted to our department. The white blood cell count and platelets were within the reference interval. Differential blood count and smear revealed leukoerythroblastosis, anisocytosis, and poikilocytosis, indicating the presence of a myelofibrosis. Additionally, lymphocytosis with typical lymphocytes and some Gumprecht shadows were present.
Immunophenotyping confirmed the presence of a clone positive for CD5, CD19, and CD45. Bone marrow histology showed a hypercellular marrow with atypical megakaryopoiesis and fibrosis classified as World Health Organization grade 2. In addition to that, a small lymphocytic infiltrate accounting for 5% of the marrow volume was described. All these findings together were consistent with the coexistence of a myeloproliferative neoplasm and a lymphoproliferative neoplasm. Cytogenetic analysis revealed a normal karyotype, and JAK2 V617F could not be detected on a molecular level. Splenomegaly with a diameter of 17 cm on ultrasound was present at diagnosis.
Anemia with a hemoglobin level of 6.0 g/dL at diagnosis was considered to be caused by the myelofibrosis and not to be associated with the incipient lymphoproliferative disorder. Due to transfusion dependency, treatment was indicated and lenalidomide was started. At 6 months of treatment, hemoglobin levels increased to 8.5 g/dL and the patient became transfusion-independent. Spleen size decreased to 15 cm after 3 months of therapy. Differential blood count was normal after 9 months. Lymphocytosis disappeared after 24 months of treatment. After a follow-up period of 6 years, the patient remained transfusion-free with a hemoglobin level of 11.8 g/dL. White blood cells and platelets were within the reference interval. Spleen size decreased further to 13 cm. Bone marrow examination still showed a hypercellular marrow, with persistent World Health Organization grade 2 fibrosis, while lymphocytic infiltration disappeared. The patient is currently completely asymptomatic, with an Eastern Cooperative Oncology Group performance status of 0.1
Chronic lymphocytic leukemia
CLL is the most common type of leukemia in developed countries. It is characterized by the presence of monoclonal lymphocytes revealing expression of B-cell associated antigens (CD19, CD20, and CD23), the T-cell associated antigen CD5, and low levels of surface antigens by flow cytometry. Additionally, an absolute B lymphocyte count ≥5,000/μL in the peripheral blood is required for establishing a diagnosis of CLL.2 The course of patients with CLL is highly variable, and survival times from initial diagnosis range from 2 to 20 years. Median survival is approximately 10 years. Diagnosis of CLL is often made at an early asymptomatic stage (RAI 0,3 Binet A4). These patients should be monitored without treatment until they show evidence of disease progression. Patients presenting with an intermediate or higher risk disease according to the modified RAI classification or with a Binet B or C stage usually have a benefit from initiating therapeutic interventions.4,5 First-line treatment for fit patients with normal renal function and no relevant comorbidities consists of rituximab, fludarabine, and cyclophosphamide.6 Alternatively, a combination of rituximab and bendamustine can be used, especially for patients with uncontrolled autoimmune hemolysis or impaired renal function.7 Patients with impaired renal function and/or relevant comorbidities can be treated with bendamustine or chlorambucil, although chlorambucil should be reserved for patients with comorbidities. Combination of either agent with rituximab produces higher response rates.8 Phase III studies with older patients and comorbidities are ongoing. Patients exhibiting a deletion 17p13 or a p53 mutation show lower response rates, a shorter duration of response, and a shorter overall survival after treatment with chemotherapy. Selected patients in this group (younger age, no comorbidities) should receive alternative treatment options, such as alemtuzumab and allogeneic stem cell transplantation.8,9 These patients should be preferably treated within clinical trials. Lenalidomide alone or in combination with mainly rituximab showed efficacy in patients with CLL. Toxic side effects such as tumor lysis syndrome and tumor flare reaction make the management of treatment with lenalidomide in CLL different from that in other hematologic malignancies.10 The incidence of secondary malignancies in patients with CLL is higher compared with the general population, but the exact reason for that remains unknown.11,12 Immunodeficiency caused by multiple abnormalities within the immune system is regarded as a factor contributing to the emergence of secondary hematologic or solid neoplasms in patients with CLL.13
Primary myelofibrosis
PMF is a myeloproliferative neoplasm characterized by bone marrow fibrosis, splenomegaly, and progressive anemia.14,15 Patients are compromised by symptoms such as fatigue, dyspnea, early satiety, abdominal pain, and constitutional symptoms. Myelofibrosis is also a very heterogeneous disease, with a median survival time ranging from 16 to 185 months.16 Prognostic risk factors are age >65 years, hemoglobin <10 g/dL, white blood cell count >25 G/L, and circulating blasts >1%. Beside these risk factors, the prognosis of patients with PMF is strongly dependent on the presence of cytogenetic abnormalities, transfusion dependency, platelets <100 G/L, and the presence of other comorbidities. These risk factors were used to establish prognostic scoring systems such as the International Prognostic Scoring System (IPSS),17 the Dynamic IPSS (DIPPS),18 and the DIPPS plus.16 The only curative treatment option available for patients with PMF is allogeneic stem cell transplantation. However, as the majority of patients are not in the transplant age group at the time of diagnosis, allogeneic stem cell transplantation is only an option for a very limited number of PMF patients defined by an expected survival time <5 years, transfusion dependency, increased risk of leukemic transformation, and the availability of a suitable donor.19 For symptomatic splenomegaly, radiation and splenectomy can be tried. Cytoreductive treatment with hydroxyurea and management of anemia with red blood cell transfusions, erythropoiesis-stimulating agents, androgens, danazol, corticosteroids, and immunomodulatory agents are additional treatment options with limited activity.20 Immunomodulatory agents show response rates for anemia of approximately 20% throughout all clinical trials.21–24
Reduction of bone marrow fibrosis could not be demonstrated. Until recently, apart from allogeneic stem cell transplantation, no other therapy was able to influence survival of patients with PMF. With the discovery of the JAK2 V617F mutation, found in approximately 50%–60% of patients with PMF, our understanding of myeloproliferative neoplasms and the molecular mechanisms of the diseases has been expanded substantially.25–28 This new knowledge led to the development of small-molecule JAK inhibitors, with ruxolitinib to be the first that was approved in the USA, Canada, and Europe. Ruxolitinib showed profound effects on splenomegaly, but also improved patient symptoms and quality of life in two Phase III trials (COMFORT-I and COMFORT-II; ClinicalTrials.gov identifiers NCT00952289 and NCT00934544, respectively).29,30 Additionally, a survival advantage for patients treated with ruxolitinib could be demonstrated in both COMFORT trials.29,30 Several other JAK inhibitors are in various stages of development.
Coincidence of PMF and CLL
Coincidence of PMF and CLL in one patient is a rare phenomenon. Up to now only 16 cases (summarized in Table 1) have been described in the literature, with the first one published in 1957.1,31–39 Due to low numbers of patients, a potential bias in the following interpretation of data cannot be ruled out. Half of these patients developed both entities simultaneously, whereas in six cases the myeloproliferative neoplasm preceded the lymphoproliferative disorder. CLL as the underlying disease was described only in two cases. A male predominance is remarkable, with 13 male patients out of these 16 cases, and only three being female. The median age of this population was 66 (range 45–77) years. The median time from diagnosis of the first entity until emergence of the second disease was 37.5 (range 24–156) months. Five of eight patients with a sequential evolution of both entities did not have any prior cytostatic therapy. Therefore, a chemotherapy-induced genesis can be excluded in these patients. Eight of 16 patients had no need for treatment at all, which might indicate a more indolent clinical disease course in these cases. A follow-up of 16 years for the patient reported by Böhm et al showed stable disease under cytostatic chemotherapy.31 The patient from our group has been asymptomatic and transfusion-independent on a low dose of lenalidomide for the last 6 years. This observation of a more indolent course than one would expect for each disorder itself was also found by an Italian group reporting on 46 patients with coexisting CLL and various myeloproliferative neoplasms, such as chronic myeloid leukemia, polycythemia vera, essential thrombocythemia, PMF, or myeloproliferative/myelodysplastic syndromes. Only nine of these 46 patients experienced a progression of CLL, and only six patients required treatment at all. Twenty patients were treated with chemotherapy for their myeloproliferative neoplasms without influencing the course of CLL. A clinical indolent course, especially of the lymphoproliferative disorder was observed.39
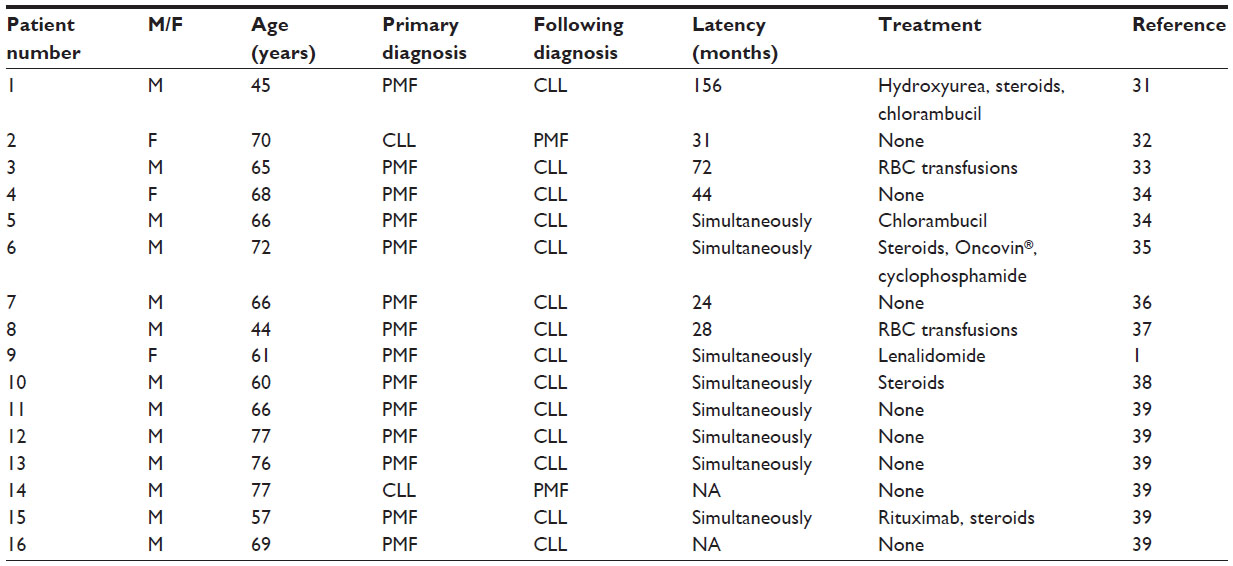 | Table 1 Cases of PMF and CLL in the literature |
The pathogenetic mechanisms underlying the association between PMF and CLL are unknown. The rare cases with development of myelofibrosis subsequent to previously treated CLL might have been induced by a leukemogenic effect of the administered chemotherapy (usually containing alkylating agents). In the majority of cases, PMF was the primary disease as mentioned above. The incidence of malignancies secondary to myeloproliferative neoplasms is not known. As the majority of patients did not need any treatment at all, the influence of chemotherapy on development of a secondary lymphoproliferative disorder seems to be unlikely. Both PMF and CLL are diseases with the highest prevalence in elderly people, regardless of occurrence alone or in combination. So a true coincidence of two distinct and independent diseases might be the case. The estimated low probability of a coincidental appearance would explain the very low number of cases reported in the literature.40 Proliferation of two unrelated cell lines driven by a common stimulus is another potential explanation for the coexistence of PMF and CLL. Impaired T-cell immunity commonly observed in CLL patients might predispose to the emergence of a second malignant clone.13 Proliferation of a lymphocytic and a myelogenous cell line from one common pluripotent stem cell is the hypothesis favored by the majority of authors. The presence of a cytogenetic or a molecular marker in both the lymphocytic and myelogenous cell line would support this hypothesis. Carulli et al described a chromosomal aberration (18p+) in a patient with CLL following a diagnosis of polycythemia vera. This cytogenetic abnormality (18p+) was demonstrated in bone marrow cells as well as in peripheral lymphocytes.41 Until now, no common marker has been identified for PMF and CLL. Some similarities in pathogenetic mechanisms might be seen with mixed-phenotype acute leukemia. In approximately 2%–5% of patients with acute leukemia, the lineage origin remains ambiguous. Myeloid, B-cell, and T-cell lineages are involved in triphenotypic leukemia. These types of leukemia are also thought to derive from transformation of one hematopoietic stem cell with the capability of differentiating along both myeloid and lymphoid lineages. In contrast with patients having a combination of a chronic myeloproliferative and lymphoproliferative neoplasm, those with mixed-phenotype acute leukemia have a poor prognosis and probably need more intensive treatment.42
As indicated by the retrospective analysis from the Italian group neither the myeloproliferative neoplasm nor the lymphoproliferative disorder is associated with a more aggressive course when occurring together, which seems also to be valid for the cohort of 16 patients with coexisting PMF and CLL summarized in this article.39 Recommendations for treatment cannot be given due to the heterogeneity and limited number of patients.
Summary
The coexistence of two different hematologic malignancies in one patient is generally a rare phenomenon. Concurrent diagnosis of PMF and CLL has so far been described in only 16 patients. Little is known about the pathogenetic mechanisms, treatment approaches, and prognosis of these patients. Taking all published reports together, PMF is the initially present disorder in the majority of cases. Treatment for either disease was needed less often than expected in the patients of this cohort. A clinically indolent course is remarkable in a substantial proportion of patients. The reason for that is unknown.
Disclosure
The authors report no conflicts of interest in this work.
References
Burgstaller S, Wimmer S, Mayrbaeurl B, et al. Coexistence of primary myelofibrosis and chronic lymphocytic leukaemia: treatment of two different diseases with one agent. Blood Cancer J. 2011;1:e20. | |
Hallek M, Cheson BD, Catovsky D, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute – Working Group 1996 guidelines. Blood. 2008;111:5446–5456. | |
Rai KR, Sawitsky A, Cronkite EP, et al. Clinical staging of chronic lymphocytic leukemia. Blood. 1975;46:219–234. | |
Binet JL, Auquier A, Dighiero G, et al. A new prognostic classification of chronic lymphocytic leukemia derived from a multivariate analysis. Cancer. 1981;48:198–204. | |
Rai KR. A critical analysis of staging in CLL. In: Gale RP, Rai KR, editors. Chronic Lymphocytic Leukemia: Recent Progress and Future Directions. New York, NY, USA: Liss; 1987. | |
Hallek M, Fingerle-Rowson G, Fink A-M, et al. First-line treatment with fludarabine (F), cyclophosphamide (C), and rituximab (R) (FCR) improves overall survival (OS) in previously untreated patients (pts) with advanced chronic lymphocytic leukemia (CLL):results of a randomized Phase III trial on behalf of an international group of investigators and the German CLL Study Group. Lancet. 2010;376:1164–1174. | |
Knauf WU, Lissichkov, Aldaoud A, et al. Phase III randomized study of bendamustine compared with chlorambucil in previously untreated patients with chronic lymphocytic leukemia. J Clin Oncol. 2009;27: 4378–4384. | |
Gribben JG, O’Brien S. Update on therapy of chronic lymphocytic leukemia. J Clin Oncol. 2011;29:544–550. | |
Schetelig J, van Biezen A, Brand R, et al. Allogeneic hematopoietic stem-cell transplantation for chronic lymphocytic leukemia with 17p deletion: a retrospective European Group for Blood and Marrow Transplantation analysis. J Clin Oncol. 2008;26:5094–5100. | |
Gonzales-Rodriguez AP, Payer AR, Acebes-Huerto A, et al. Lenalidomide and chronic lymphocytic leukemia. BioMed Res Int. 2013;932010. | |
Berg JW. The incidence of multiple primary cancers. I. Development of further cancers in patients with lymphomas, leukemias, and myeloma. J Natl Cancer Inst. 1967;38:741–752. | |
Hyman GA. Increased incidence of neoplasia in association with chronic lymphocytic leukaemia. Scand J Haematol. 1969;6:99–104. | |
Bartik MM, Welker D, Kay NE. Impairments in immune cell function in B cell chronic lymphocytic leukemia. Semin Oncol. 1998;25:27–33. | |
Tefferi A. Essential thrombocythemia, polycythemia vera, and myelofibrosis: current management and the prospect of targeted therapy. Am J Hematol. 2008;83:491–497. | |
Vanucchi AM. Management of myelofibrosis. Hematology Am Soc Hematol Educ Program. 2011;2011:222–230. | |
Gangat N, Caramazza D, Vaidya R, et al. DIPSS plus: a refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol. 2011;29:392–397. | |
Cervantes F, Dupriez B, Pereira A, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood. 2009;113:2895–2901. | |
Passamonti F, Cervantes F, Vannucchi AM, et al. A dynamic prognostic scoring model to predict survival in primary myelofibrosis – a study by the IWG-MRT (International Working Group for Myelofibrosis Research and Treatment). Blood. 2010;116:1703–1708. | |
McLornan DP, Mead AJ, Jackson G, Harrison CN. Allogeneic stem cell transplantation for myelofibrosis in 2012. Br J Haematol. 2012;157: 413–425. | |
Cervantes F, Mesa R, Barosi G. New and old treatment modalities in primary myelofibrosis. Cancer J. 2007;13:377–383. | |
Tefferi A, Cortes J, Verstovsek S, et al. Lenalidomide therapy in myelofibrosis with myeloid metaplasia. Blood. 2006;108:1158–1164. | |
Quintas-Cardama A, Kantarjian HM, Manshouri T, et al. Lenalidomide plus prednisone results in durable clinical, histopathologic, and molecular responses in patients with myelofibrosis. J Clin Oncol. 2009;27: 4760–4766. | |
Mesa RA, Yao X, Cripe LD, et al. Lenalidomide and prednisone for myelofibrosis: Eastern Cooperative Oncology Group (ECOG) phase-2 trial E4903. Blood. 2010;116:4436–4438. | |
Daver N, Shastri A, Kadia T, et al. Modest activity of pomalidomide in patients with myelofibrosis and significant anemia. Leuk Res. 2013;37: 1140–1444. | |
James C, Ugo V, Le Couédic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythemia vera. Nature. 2005;434:1144–1148. | |
Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352: 1779–1790. | |
Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–1061. | |
Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7: 387–397. | |
Harrison C, Kiladjian JJ, Al-Ali HK, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366:787–798. | |
Verstovsek S, Mesa RA, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366: 799–807. | |
Böhm J, Schaefer HE, Fisch P. [Coincidence of chronic idiopathic myelofibrosis and chronic lymphocytic leukemia. A rare phenomenon]. Pathologe. 2002;23:480–485. German. | |
Nieto LH, Sanchez JMR, Arguelles HA, et al. A case of chronic lymphocytic leukemia overwhelmed by rapidly progressing idiopathic myelofibrosis. Haematologica. 2000;85:973–977. | |
Kaufman S, Iuclea S, Reif R. Idiopathic myelofibrosis complicated by chronic lymphocytic leukemia. Clin Lab Haematol. 1987;9:81–84. | |
Zylberait D, Audebert AA, Grolleau Y, et al. Association de leucémie lymphoide chronique et de splénomégalie myéloide. A propos de deux observations [Two cases of agnogenic myeloid metaplasia associated with chronic lymphocytic leukemia]. Nouv Rev Fr Hematol. 1981;23:111–113. French. | |
Guerci O, Floquet J, Thibaut G, et al. Splénomégalie myéloide avec lymphopathie maligne. Ann Med Nancy. 1978;12:187–189. French. | |
Louwagie AC, Desmet VJ, van den Berghe H. Coexistence of a myelo- and lymphoproliferative disorder. Scand J Haematol. 1973;11: 350–355. | |
Videbaek A, Poulsen H. Myelofibrosis complicated by lymphogenous leukaemia. Acta Haematol. 1957;17:250–256. | |
Palta A, Garg S, Chauhan S, et al. Simultaneous presence of two haematological malignancies: chronic lymphocytic leukemia and myelofibrosis in a patient. Indian J Hematol Blood Transfus. 2011;27:33–34. | |
Laurenti L, Tarnani M, Nichele I, et al. The coexistence of chronic lymphocytic leukemia and myeloproliferative neoplasms: a retrospective multicentric GIMEMA experience. Am J Hematol. 2011;86: 1007–1012. | |
Papayannis AG, Nikiforakis E, Aanagnostou-Keramida D. Devolpment of chronic lymphocytic leukemia in a patient with polycythemia vera. Scand J Haematol. 1982;29:65–69. | |
Carulli G, Marini A, Baicchi U, et al. Chronic lymphocytic leukemia (B-cell) in the course of polycythemia vera. Description of a case with an unusual chromosomic anomaly. Tumori. 1987;73:639–643. | |
Mautes E, Morilla R, Farahat N, et al. Definition of acute biphenotypic leukemia. Haematologica. 1989;82:64–66. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.