")
Back to Journals » International Journal of Nanomedicine » Volume 17
Co-Delivery of Dexamethasone and Captopril by α8 Integrin Antibodies Modified Liposome-PLGA Nanoparticle Hybrids for Targeted Anti-Inflammatory/Anti-Fibrosis Therapy of Glomerulonephritis
Authors Zhou L, Ye Z , Zhang E, Chen L, Hou Y , Lin J, Huang F, Yuan Z
Received 1 November 2021
Accepted for publication 10 March 2022
Published 30 March 2022 Volume 2022:17 Pages 1531—1547
DOI https://doi.org/10.2147/IJN.S347164
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yan Shen
Liuting Zhou,1,2 Zhenyan Ye,3 E Zhang,4 Li Chen,1 Yitong Hou,1 JuChun Lin,2 Fenglan Huang,1 Zhixiang Yuan5
1Department of Osteoporosis, West China School of Public Health and West China Fourth Hospital, Sichuan University, Chengdu, Sichuan, People’s Republic of China; 2Department of Pharmacy, College of Veterinary Medicine, Sichuan Agricultural University, Chengdu, Sichuan, People’s Republic of China; 3School of Clinical Medical; Chengdu Medical College, Chengdu, People’s Republic of China; 4Officers college of PAP, Chengdu, Sichuan, People’s Republic of China; 5College of Pharmacy, Southwest Minzu University, Chengdu, Sichuan, People’s Republic of China
Correspondence: Fenglan Huang, Email [email protected]
Purpose: Mesangial cells-mediated glomerulonephritis refers to a category of immunologically mediated glomerular injuries characterized by infiltration of circulating inflammatory cells, proliferation of mesangial cells, and the common pathological manifestation to the later stage is renal fibrosis, accompanied by excessive accumulation of extracellular matrix (ECM). Treatment regimens include glucocorticoids and immunosuppressive agents, but their off-target distribution causes severe systemic toxicity. Hence, specific co-delivery of “anti-inflammatory/anti-fibrosis” drugs to the glomerular mesangial cell (MC) region is expected to produce better therapeutic effects.
Methods: A novel kidney-targeted nanocarrier drug delivery system targeting MCs was constructed using passive targeting resulting from the difference in pore size between the glomerular endothelial layer and the basement membrane, and active targeting based on the specific binding of antibodies and antigens. Specifically, a liposome-nanoparticle hybrid (PLGA-LNHy) was formed by coating the surface of PLGA nanoparticles (NPs) with a phospholipid bilayer, and then PLGA-LNHy was co-modified with PEG and α 8 integrin antibodies to obtain PLGA immunoliposomes (PLGA-ILs).
Results: The results showed that the obtained NPs had a core-shell structure, uniform and suitable particle size (119.1 ± 2.31 nm), low cytotoxicity, and good mesangial cell-entry ability, which can successfully accumulate in the glomerular MC region. Both dexamethasone (DXMS) and captopril (CAP) were loaded onto PLGA-ILs with a drug loading of 10.22 ± 1.00% for DXMS and 6.37 ± 0.25% for CAP (DXMS/CAP@PLGA-ILs). In vivo pharmacodynamics showed that DXMS/CAP@PLGA-ILs can effectively improve the pathological changes in the mesangial area and positive expression of proliferating cell nuclear antigen (PCNA) in glomeruli as well as reduce the expression of inflammatory factors, fibrotic factors and reactive oxygen species (ROS). Thus, renal inflammation and fibrosis were relieved.
Conclusion: We have provided a strategy to increase nanoparticle accumulation in MCs with the potential to implement regulatory effects of anti-inflammatory and anti-fibrosis in glomerulonephritis (GN).
Keywords: MCs, renal inflammatory, renal fibrosis, tem
Introduction
The prevalence of chronic kidney disease (CKD) is increasing worldwide, and the mortality rate continues to be unacceptably high. CKD is estimated to affect more than 100 million patients in China.1,2 Glomerulonephritis (GN) is the major component of chronic kidney disease, and it continues to be one of the leading causes of end-stage kidney disease (ESKD) in China and worldwide3 with a high prevalence and strong concealment.4 The main pathogenesis is immune inflammation injury; the initial phase of the disease is based on inflammation with the activation of immune cells. As the disease progresses, many immunoglobulins deposit in glomeruli leading to glomerular sclerosis associated with renal fibrosis and tubular atrophy, and the main clinical symptoms include proteinuria, hematuria, hypertension, and edema.5 Furthermore, oxidative stress is also one of the main causes of kidney injury. Wojcicka et al proposed the possible role of ROS in the pathogenesis of glomerulonephritis and proved that ROS was involved in glomerular injury.6 Hence, early detection and timely intervention can significantly reduce the complications of patients with GN and improve their survival rates. Unfortunately, there is no specific radical cure for GN, and therapy based on the use of glucocorticoids (GC) and non-specific immunosuppressive drugs commonly administered in GN remains the primary therapeutic weapon. Moreover, this long-term treatment able to moderate immune glomerular injury remains a nonspecific therapy with severe side effects and insufficient efficacy.7,8 In addition, anti-inflammatory therapy with these drugs alone does not reverse part of the pathological tissue where fibrosis has begun, making it difficult to obtain a good prognosis for patients with GN.9
Based on the above clinical challenge in the treatment of GN, we propose the following scientific hypotheses: simultaneous restraint of the stage of inflammatory response and prevention of the stage of fibrosis formation can effectively control the inflammatory response and slow down or even reverse the fibrosis process of GN using a renal targeting delivery system with high efficiency and low toxicity. First, glomerular mesangial cells (MCs) are one of the most important intrinsic cells in the glomeruli, accounting for 30% to 40% of the total number of glomerular cells, and maintaining glomerular hemodynamics and structural integrity of the microvascular bed. The proliferation and fibrosis of glomerular MCs are the basic pathological processes of GN.10 Hence, MCs are an ideal target for the treatment of GN. Research into the development of renal targeting delivery systems began in the 1990s,11 and the concurrent rapid development of nanotechnology, macromolecular carrier technology, and nanotechnology-based renal targeting delivery systems have been successfully developed.12 At present, novel renal delivery systems focusing on different lesions of nephropathy have also been designed to further improve specific targeting. Interestingly, an injectable hydrogel crosslinked via self-assembled triblock polymeric micelles is developed to help reduce renal interstitial fibrosis.13 Guo et al found that tripterin-loaded albumin NPs with a particle size of 95 nm easily accumulated in the MC region, which could improve the curative effect on MC proliferative nephritis.14 Virus-mimetic NPs have also been designed to specifically target renal MCs and therefore increase intracellular drug accumulation, providing a more refined therapy for diabetic nephropathy.15 Wang et al designed glomerular-targeted liposomes loaded with siRNA, which could reduce proteinuria, inflammation, and excessive ECM deposition in IgA nephropathy model mice.16 In our previous study, 3,5-dipentadecyloxybenzamidine hydrochloride (TRX-20) ligand-modified PEGylated liposomes were designed to explore the possibility of targeting glomerular MCs.17 Combined with the renal ultrastructure and previous studies, we found that NPs with tunable diameters can extravasate the effective filtration area of glomerular endothelial cells (10–70 nm), but cannot traverse the mesangial support area (70–130 nm, under pathological conditions: > 200 nm), thus remaining in MCs.
Apart from passive targeting via the enhanced permeability and retention effect, active targeting could be pursued with the use of specific ligands or antibodies in liposomes.18 According to the in vitro study by Scindia et al, the immune complex prepared using the MC antibody molecule α8 integrin as a targeted molecule had good MC targeting properties.19 Therefore, we believe that the most effective treatment strategy for GN is to realize high efficiency and low toxicity through the combination of passive and active MC-targeted drug delivery systems.
DXMS is currently the first-line drug used in the clinical treatment of GN,20 which can combine with the unique GC-reactive molecules on the DNA sequence to activate the anti-inflammatory gene transcription process and play an anti-inflammatory role. Additionally, several studies have shown that DXMS can inhibit the growth and reproduction of cells by regulating members of the MAPK family, such as ERK and JNK, which probably also accounts for certain anti-fibrosis effects of DXMS.21,22 However, many studies have shown that the use of glucocorticoids could not contribute to an adequate anti-fibrosis effect, which is not conducive to the prognosis of GN.9 Therefore, the introduction of anti-fibrosis drugs is necessary. CAP is a synthetic angiotensin converting enzyme (ACE) inhibitor with antioxidant effect and can also effectively inhibit the formation of renal fibrosis and scarring by inhibiting the production of transforming growth factor-β (TGF-β).23–25 In view of their action characteristics of, simultaneous targeted delivery of DXMS and CAP to the focal sites of GN is a meaningful strategy for achieving the regulation of ” inflammation/ fibrosis.”
In this study, both α8 integrin antibodies and PEG-modified liposome-PLGA nanoparticle hybrids termed PLGA-ILs were prepared and DXMS-and CAP-loaded PLGA-IL delivery systems termed DXMS/CAP@PLGA-ILs were constructed for anti-inflammatory/anti-fibrosis targeting therapy to GN (Figure 1). This co-delivery system was characterized, and the ability of PLGA-ILs to target MCs was evaluated by cellular uptake, section staining, and in vivo imaging. The establishment of an animal model of GN and pharmacodynamic evaluation was performed to evaluate the anti-inflammatory and anti-fibrosis profiles of DXMS/CAP@PLGA-ILs.
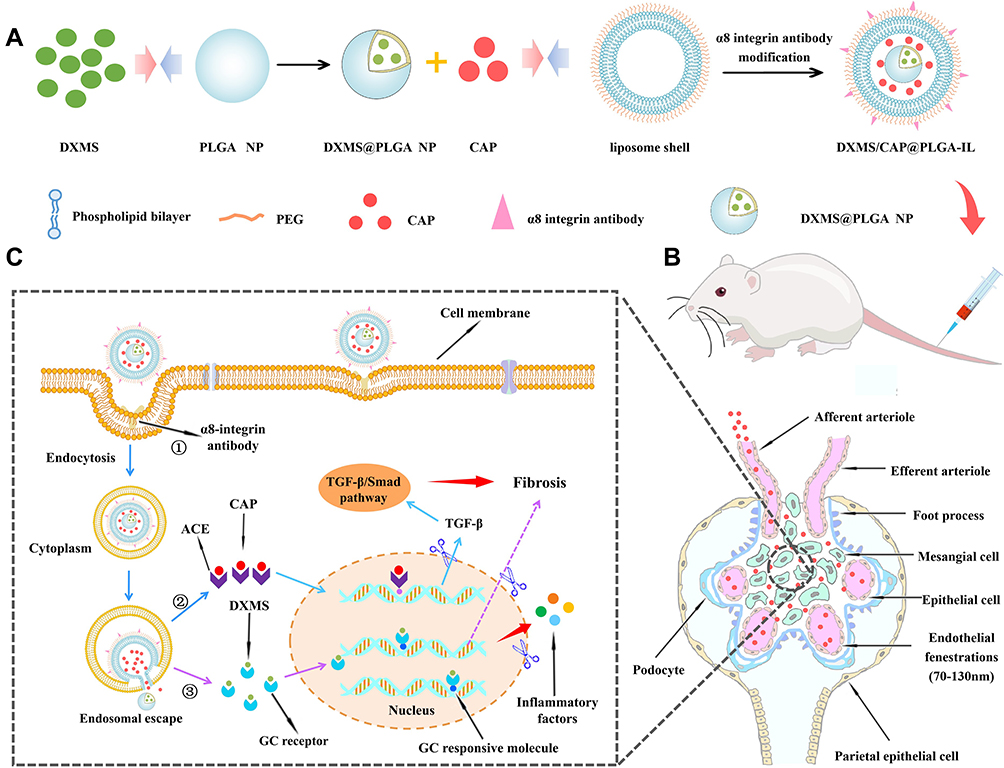 |
Figure 1 The overall strategy and mechanism of DXMS/CAP@PLGA-ILs targeting MCs. (A) The novel functionalized NPs (DXMS/CAP@PLGA-ILs) were constructed in this study. (B) DXMS/CAP@PLGA-ILs in blood circulation enter the glomerulus. The NPs between 70–130 nm can penetrate the glomerular endothelial space (70–130 nm) into the glomerular MCs area, but cannot penetrate the glomerular basement membrane (10–70 nm) and passively target to the region of glomerular MCs. (C) The mechanism of “anti-inflammation/anti-fibrosis”for GN after DXMS/CAP@PLGA-ILs MCs. “①” DXMS/CAP@PLGA-ILs enters cells through receptor-mediated endocytosis; “②” Lipid bilayer releases CAP through membrane fusion, Inhibits the production of TGF-β, and exerts anti-fibrosis effect; “③” DXMS molecules are released into the cytoplasm and enter the nucleus to exert anti-inflammatory effects. Abbreviations: DXMS, dexamethasone; CAP, captopril; MCs, mesangial cells; GN, glomerulonephritis; TGF-β, transforming growth factor-β. |
Materials and Methods
Materials
Poly (D,L-lactic-co-glycolide) (PLGA, Mw 4000−15,000, lactide:glycolide 75:25), 2-Iminothiolane hydrochloride (2-IT), coumarin-6 (C6), mouse anti-proliferating cell nuclear antigen (PCNA) polyclonal antibodies, mouse anti-the inflammatory chemokine CCL2 (CCL2) polyclonal antibodies and mouse anti-β-actin polyclonal antibodies were purchased from Sigma-Aldrich (USA); 1.2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide (polyethylene glycol) −2000] (Mal-DSPE-PEG2000) and 1, 2-distearoyl-sn-glycero-3-phosphoethanolamine -N-[methoxy (polyethylene glycol)-2000] (DSPE-PEG2000) were obtained from Laysan Bio (USA); vitamin E-TPGS (TPGS) was purchased from Macklin Biochemical Co., LTD; (Shanghai, China); TGF-β ELISA Kits were obtained from Ponstar Biotech Co., LTD (Chengdu, China), DXMS and CAP were supplied by Meilun Biotech Co., LTD (purity ≥ 99%; Dalian, China); α8 integrin antibodies was supplied from Santa Cruz (USA). The near-infrared dye cyanine 5.5 NS acid was purchased from AAT Bioquest (USA). Rhodamine (RHOD), 4’,6-diamidino-2-phenylindole (DAPI), BSA, and a dialysis membrane with a 25−7000 Da cut-off were provided by the Chinese Solarbio Company (Beijing, China). DMEM/F12 medium and FBS were purchased from Hyclone (USA), trypsin and fetal bovine serum (FBS) were obtained from Gibco (USA); Habu snake venom (HSV) was purchased from WAKO (Japan). The other chemicals used in this study were of analytical grade.
Male SPF Kunming mice (20 ± 2 g) were purchased from Chengdu Dossy Experimental Animals Co., Ltd. (Chengdu, China) and maintained in a pathogen-free environment under controlled humidity and temperature. The animal experiments were conducted in accordance with the protocol of the National Act on the Use of Experimental Animals (China) and were approved by the Sichuan Committee on Laboratory Animals (approval number SYXK2014-187). All experiments were performed according to the requirements of the People ’s Republic of China National Act on the use of experimental animals.
Preparation and Characterization of the NPs
A nanometer-scale precipitation approach was used to produce NPs with particle sizes ranging from 70 nm to 130 nm. Briefly, 10 mg PLGA, 3 mg DXMS, and 3 mg TPGS were dissolved in 3 mL of acetone as an organic phase solution, and then slowly added to 15 mL of 1% poloxam 188 aqueous solution under magnetic stirring at 40 ± 2°C.26 Subsequently, the formation of PLGA NPs was further driven by continuous stirring for 15 min, and the organic solvent was allowed to evaporate simultaneously. The solution was then applied to a filter to remove the oversized NPs, and then the solution was centrifuged for 15 min at 12,000 rpm, and the DXMS@PLGA NPs were harvested as the sediment. Finally, the mixture was resuspended in 10 mL of deionized water.
A PEG-modified liposome-PLGA nanoparticle hybrid termed PLGA-LNHy was produced by the thin film ultrasonic dispersion method with liposomes. First, a lipid mixture containing 16 mg soy lecithin, 4 mg cholesterol, 2 mg mPEG2000-DSPE, 0.5 mg Mal-PEG2000-DSPE and 7.9 mg TPGS27,28 was dissolved in 10 mL of chloroform, and dried under vacuum at 50°C in a rotary evaporator to remove the organic solvent and form a dry film. Subsequently, 10 mg CAP was added to the PLGA NPs colloidal solution, and then the colloidal solution was applied to a rotary evaporator to hydrate the dried film. The resulting suspension was ultrasonicated in an ice bath and successively extruded through an extrusion apparatus to obtain the DXMS/CAP@PLGA-LNHy colloidal solution. Finally, the DXMS/CAP@PLGA-LNHy was collected by centrifuging at 12,000 rpm and the free DXMS and CAP in the supernatant were removed. The drug loading rate of DXMS/CAP@PLGA-LNHy was determined by using HPLC method after demulsification by adding methanol.
The solutions of 0.2 mL α8 integrin antibodies (200 g/mL) and 0.8 mL 2-IT28,29 (3.75 μg/mL, oxygen removal) were incubated at 37°C in the dark for 1 h, then the activated 1 mL antibody solution was added to the dialysis bag (deionized water pretreatment). Dialysis bags were placed into 100 mL dialysate at 4°C for 4 h with magnetic stirring. The dialysate was changed every 1 h, and the dialysis bags were kept suspended all the time. Ultimately, DXMS/CAP@PLGA-LNHy colloidal solution (2 mL) and the purified antibody solution (1 mL) were added into a sterile centrifuge tube and incubated at 37°C overnight in the dark to obtain a DXMS/CAP@PLGA-IL colloidal solution.19 PLGA NPs, PLGA-LNHys, and PLGA-ILs without drug loading were also prepared.
C6-loaded PLGA NPs (C6@PLGA NPs) were prepared as described above except 0.5 mg C6 was dissolved in 3 mL of the organic phase solution at the beginning. RHOD-loaded PLGA-LNHys (RHOD@PLGA-LNHys) were prepared as described above, except that 1 mg RHOD was dissolved in 10 mL of chloroform at the beginning. C6 and RHOD-loaded PLGA-ILs (C6/RHOD@PLGA-ILs) were prepared as above except 0.5 mg C6 was dissolved in 3 mL organic phase solution and 1 mg RHOD was dissolved in 10 mL of chloroform at the beginning.
Free Cy5.5 was prepared using 0.1 mg Cy5.5NS dissolved in 10 mL deionized water. Cy5.5@PLGA NPs, Cy5.5@PLGA-LNHys, and Cy5.5@PLGA-ILs were prepared as above except 0.1 mg Cy5.5NS were dissolved in 3 mL organic phase solution at the beginning.
The particle size and zeta potential of PLGA NPs, PLGA-LNHys, and PLGA-ILs were determined by using a zeta potential/particle sizer (NanoZS ZEN3600, Malvern, UK). After negative staining with 2% sodium phosphotungstate solution, the morphology of PLGA NPs, PLGA-LNHys, and PLGA-ILs were examined by transmission electron microscopy (TEM, Libra200 FE, ZEISS, Germany).
In vitro Release of DXMS and CAP from NPs
The in vitro DXMS release profile of DXMS/CAP@PLGA-ILs was determined using the dialysis method. Briefly, 2 mL of nanoparticle colloidal solution and 3 mL PBS were added to the dialysis bag (7000 Da) suspended in 95 mL of release medium of PBS at 37 ± 0.5°C. The solution (1 mL) was removed from the outside medium for measurements at 0 h, 0.5 h, 1 h, 2 h, 4 h, 8 h, 12 h, and 24 h, and replaced with 1 mL of fresh medium at the same time. The sample solution was filtered through a 0.45 μm microporous filter membrane. The amount of DXMS and CAP released in the filtrate was determined by HPLC.
Cell Toxicity Assay
The cytotoxicity of the different formulations was evaluated using a CCK-8 assay. MCs in the logarithmic growth phase were incubated in 96-well plates at 1×105 cells per well and incubated for 24 h at 37°C. The medium was then removed, followed by treatment with PLGA NPs, PLGA-LNHys, and PLGA-ILs for 24 h (with an equivalent dose of PLGA: 0.125, 0.25, 0.5, 1, and 2 μg/mL). After the medium was discarded, fresh medium (90 μL) and CCK8 reagent (10 μL) were added to each well and incubated for another 3 h before measurement. Absorbance at 450 nm was measured using a microplate reader (Multiskan FC, Thermo Scientific, USA). Relative cell viability was calculated using the following equation:
Cell viability = (Asample-Ablank)/(Anegative control-Ablank) ×100%.
Immunofluorescence and Cellular Uptake
A sterile cover glass was placed in a 6-well culture plate and MCs in the logarithmic growth phase were seeded in 6-well plates at a density of 2×105 cells per well and incubated overnight. The cells were fixed and permeabilized with 0.1% Triton X-100 after incubation. Cells were blocked with 3% BSA for 30 min and incubated with primary antibodies (α8 integrin antibody) in 0.2% (v/v) BSA in PBS overnight at 4°C. The diluted secondary antibody (Cy3 labeled) solution was incubated at 37°C for 50 min. Images were acquired using a fluorescence microscope.
The cells were prepared as described above and incubated to a fusion degree of more than 90% at 37°C. After the medium were discard, 0.5 mg/mL (with an equivalent dose of PLGA) of C6/RHOD@PLGA-ILs, RHOD@PLGA-LNHys, and C6@PLGA NPs were added to the wells (n = 3) and placed in an incubator for 22 h. Then, the 6-well plates were washed three times with PBS after discarding the medium, and 1 mL of 4% paraformaldehyde solution was added and fixed for 15 min. Next, the cover glass was cleaned twice with PBS and anti-fluorescence quenching sealant containing DAPI was dropped on it for staining and sealing. The fluorescence distribution was captured with a fluorescence microscope (BX 53, Olympus, Tokyo, Japan), and the relative fluorescence intensity was analyzed using ImageJ software for semi-quantitative comparison.
Fluorescent in situ Hybridization
To further verify the targeting location of different types of nanocarriers in the kidney, fluorescent in situ hybridization was performed. Kunming mice were randomly divided into C6/RHOD@PLGA-IL, RHOD@PLGA-LNHy, C6@PLGA NP, and normal groups. Mice in the experimental group were injected with C6/RHOD@PLGA-ILs, RHOD@PLGA-LNHys, and C6@PLGA NPs at an equivalent dose of 10 mg/kg PLGA, while mice in the normal group were injected with the same volume of normal saline through the tail vein. All mice were sacrificed and dissected 4 h after injection, and then the kidneys were collected and rinsed with PBS, frozen in optimal cryotomy temperature (OCT) compound, and sectioned into 4−5 µm thick. Before staining, sections were fixed at 4°C with precooled acetone for 10−20 min, and then rinsed three times with PBS solution for 5 min each. Finally, the sections were stained and sealed with an anti-fluorescence quenching sealant containing DAPI. The fluorescent distribution was captured using a fluorescence microscope (BX 53, Olympus, Tokyo, Japan).
In vivo and ex vivo Imaging
To further verify the distribution of NPs in vivo, in vivo and ex vivo imaging was carried out. Kunming mice were randomly divided into saline, free Cy5.5, Cy5.5@PLGA NP, Cy5.5@PLGA-LNHy and Cy5.5@PLGA-IL groups (n = 3). The experimental group of 5-week-old Kunming male mice were injected into the tail vein at an equivalent dose of 10 mg/kg PLGA, while the control group was injected with the same amount of free Cy5.5, and the normal group was injected with the same volume of saline. At 0.5, 1, 2, 4, 12, and 24 h after injection, the mice were anesthetized and imaged using an in vivo imaging system (IVIS Spectrum, Maestro, USA). The excitation wavelength was measured at 683 nm, and the emission wavelength was set at 703 nm. The mice were sacrificed and dissected after live imaging. The anatomical heart, liver, spleen, lung, kidney, and brain were washed with PBS and subjected to ex vivo fluorescence imaging.
In vivo Efficacy Studies
Animal Model and Treatment
To demonstrate the anti-inflammatory and anti-fibrosis therapeutic efficacy, we evaluated the therapeutic effects of DXMS/CAP@PLGA-ILs in a mouse model of mesangial proliferative glomerulonephritis (MesPGN), the most common pathological form of GN, which is characterized by MC proliferation and mesangial matrix accumulation, and inflammation-related cytokines play an important role in its pathology.30,31 Habu snake venom (HSV) was used to induce the mouse model of MesPGN.32,33 To improve the success rate of modeling, HSV was injected twice through the caudal vein. Normal rats were injected with normal saline. HSV was injected through the tail vein (0.5 mg/kg) and repeatedly injected once again after 3 days. The HSV-induced mouse models were randomly divided into five groups: (1) normal group, (2) DXMS/CAP@PLGA-IL group, (3) a mixture of DXMS and CAP treatment group (DXMS/CAP group); (4) DXMS treatment group (DXMS group); and (5) CAP treatment group (CAP group). The treatment groups were administered DXMS/CAP@PLGA-ILs, DXMS, CAP, and DXMS/CAP at an equivalent dose of 1 mg/kg of DXMS or 2 mg/kg of CAP, respectively, and the normal group was administered normal saline instead. The injection was carried out once every two days, three times a week.
Periodic Acid-Schiff (PAS) Staining
After treatment for a week, the renal tissues of mice were collected. Kidney tissues were fixed in a 4% paraformaldehyde solution. The samples were then transferred to 70% ethanol. They were then processed using a graded ethanol series and embedded in paraffin. The paraffin sections were cut into 5 μm-thick slices using a microtome (Microm HM 315 microtome, Walldorf, Germany) and stained with PAS. Tissue sections were examined for glomerular histopathology and photographed under a light microscope (BX5, Olympus, Tokyo, Japan).
Immunohistochemical Analysis
To evaluate the in vivo efficacy, immunohistochemical staining of proliferating cell nuclear antigen (PCNA) were conducted on kidney tissue sections to observe the expression of PCNA in glomerulus of each group. Sections of kidney tissues were deparaffinized and neutralization with endogenous peroxidase using 3% H2O2 for 10 min at room temperature. After washing with 0.01 M PBS (pH=7.4), the sections were immersed in citrate buffer, followed by microwaving for antigen retrieval (30 min). Sections were preincubated with blocking serum for 20 min and then incubated with primary antibodies (anti-PCNA antibody was diluted to 1:500) overnight at room temperature. Following washing with PBS, the sections were incubated with HRP-labeled secondary antibodies at 37 °C for 40 min and then washed in PBS. Finally, the color was developed via a 15 min incubation with DAB solution and the sections were weakly counterstained with hematoxylin at room temperature for 3 min. The slides were observed under a light microscope (BX5, Olympus, Tokyo, Japan).
Quantitative RT-PCR
After treatment for a week, the renal tissues of mice were collected. Total RNA was extracted from isolated renal cortex using the RNAsimple Total RNA Kit (TianGen, China) following the manufacturer’s instructions. First-strand cDNA was obtained by reverse transcription of total RNA using the RevertAid First Strand cDNA Synthesis Kit (Thermo, USA). The resulting cDNA and corresponding primers were used for SYBR Green quantitative real-time PCR to assay the levels of CCL2. The primers were designed based on the mRNA sequences in GenBank and were synthesized by Shanghai Shenggong Biotechnology (Shanghai, China). The forward primer sequence was 5’TGTGCTGACCCCAAGAAGG3’. The reverse primer sequence was 5’CACTGTCACACTGGTCACTCCT3’. Mouse specific primers were used and β-actin was used as the endogenous control and the sequence of the forward primer was 5’CACGAAACTACCTTCAACTCC3’, while the sequence of the reverse primer was 5’CATACTCCTGCTTGCTGATC3’. The experiment PCR conditions followed the manufacturer’s instructions. The normalized fold expression of the tested gene relative to the normal control was calculated based on the 2−ΔΔCt method,34 where Ct is the mean threshold cycle difference.
Western Blot
Similarly, to demonstrate the anti-inflammatory therapeutic efficacy, the expression levels of CCL2 was measured using Western blot. After treatment for a week, the renal tissues of mice were collected. Protein lysates from the kidney tissues were prepared following standard protocols, and the protein content was determined using the BCA protein assay kit (ThermoFisher, USA). Then, proteins were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis under reducing conditions. After electrophoresis, samples were transferred to polyvinylidene fluoride (PVDF) membranes, blocked with 5% skimmed milk in TBS-T for 1 h, washed with TBS-T, and then incubated overnight at 4°C with primary antibodies (anti-CCL2-antibody was diluted to 1:200). The membranes were then washed and incubated with secondary antibodies for 1 h at room temperature. After washing with TBS-T, the blots were developed using the chemiluminescence method and observed under a ChemiDoc imaging system (ChemiDoc, BioRad, Singapore).
ELISA
After treatment for a week, the renal tissues of mice were collected. The levels of inflammatory factors including tumor necrosis factor α (TNF-α), Interleukin-6 (IL-6), Interleukin-1β (IL-1β), and fibrotic factors including TGF-β, alpha-smooth muscle actin (α-SMA), fibronectin (Fn) in kidney tissue homogenate were measured using ELISA kits according to the manufacturers’ protocols. In addition, ROS levels was also measured using ELISA kits.
Statistical Analysis
All the experiments were repeated at least three times. The results were presented as mean±SD. Statistical analysis was produced by SPSS 23.0 software and graphed by Graphpad Prism 8.0.2. Statistical significance was defined at *P < 0.05, **P < 0.01, ***P < 0.001. The threshold for statistical significance was P < 0.05.
Results
Preparation and Characterization of NPs
We prepared stabilized PLGA NPs with a size of around 112.5 ± 2.52 nm (Table 1), and then encapsulated them with liposomes and modified with PEG2000 and α8 integrin antibodies to prepare PLGA-LNHys and PLGA-ILs. The prepared PLGA NPs, PLGA-LNHys and PLGA-ILs all showed blue opalescence and the “Dundall effect” was produced by laser irradiation. The size, potential, and aggregation state of nanocarriers are key factors for their application. The sizes of PLGA-LNHy and PLGA-IL were controlled by a miniature liposome extruder to ensure that the prepared NPs had an ideal particle size. The particle size and zeta potential were examined; results displayed that the average particle size of PLGA-LNHy was 115.1 ± 3.51 nm, and the zeta potential was −15.2 ± 0.4 mV with PDI of 0.259 ± 0.026. With the gradual assembly of PLGA-IL, the particle size increased to 119.1 ± 2.31 nm, and the zeta potential decreased to −10.1 ± 0.2 mV with PDI of 0.211 ± 0.07 (Table 1). TEMconfirmed that the particle sizes corresponded with the mean particle size determined by the Malvern Nano-ZS instrument, PLGA NPs were spherical in structure, and the morphological structure of PLGA-LNHys were consistent with that of PLGA-ILs (Figure 2A–C). In brief, the particles were generally spheroids, with a liposome layer as the outer shell and a PLGA NPs as the core. Furthermore, the drug loading of DXMS/CAP@PLGA-ILs was directly evaluated using HPLC, which revealed that DXMS and CAP were successfully encapsulated with 10.22 ± 1.00% and 6.37 ± 0.25% of drug loading, respectively. Moreover, the in vitro release of DXMS and CAP from DXMS/CAP@PLGA-ILs was examined in PBS (pH 7.4). HPLC method determined that the cumulative release of DXMS in DXMS/CAP@PLGA-ILs for 24 h was 77.23 ± 2.96% and the cumulative release of CAP in DXMS/CAP@PLGA-ILs for 24 h was 83.18 ± 3.77% (Figure 2D).
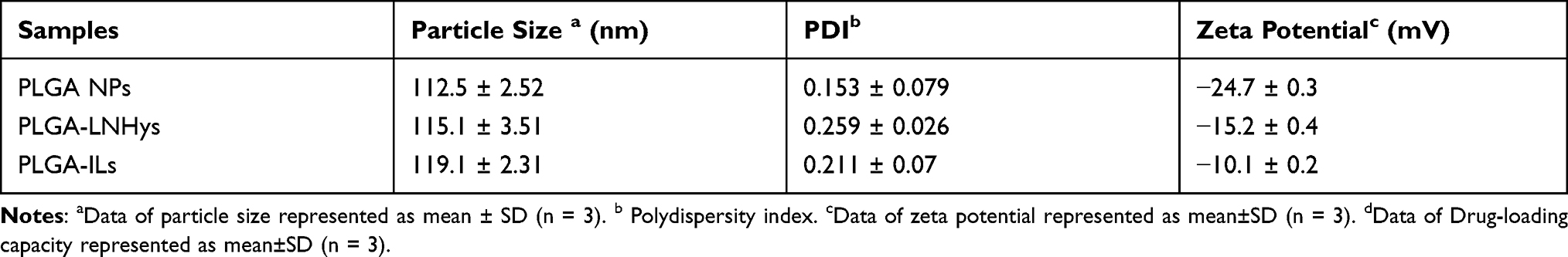 |
Table 1 Physicochemical Properties of the Prepared NPs |
 |
Figure 2 The characterization of nanocarriers and in vitro release of DXMS/CAP@PLGA-ILs. (A) TEM of PLGA NPs (Scale bars: 100 nm). (B) TEM of PLGA-LNHys (Scale bars: 100 nm). (C) TEM of PLGA-ILs (Scale bars: 100 nm). (D) In vitro DXMS and CAP cumulative release profiles under PBS. Data were expressed as mean ± SD (n = 3). Abbreviations: DXMS, dexamethasone; CAP, captopril; TEM, transmission electron microscopy; PBS, phosphate-buffered saline. |
Cytotoxicity
The toxicity of nanoparticle carriers is usually related to their particle size, surface charge, and concentration properties, and the effect varies among different types of cells. Cell viability indicated that the cell survival rate of MCs cultured with PLGA NPs, PLGA-LNHys, or PLGA-ILs was above 83.6% at PLGA concentrations ranging from 0.125 to 2 mg/mL (Figure 3A). According to ISO10993-5, the survival rate of cells above 83.6% was considered to be level 0 or 1 toxicity,35 which implies that all groups were considered safe for MCs.
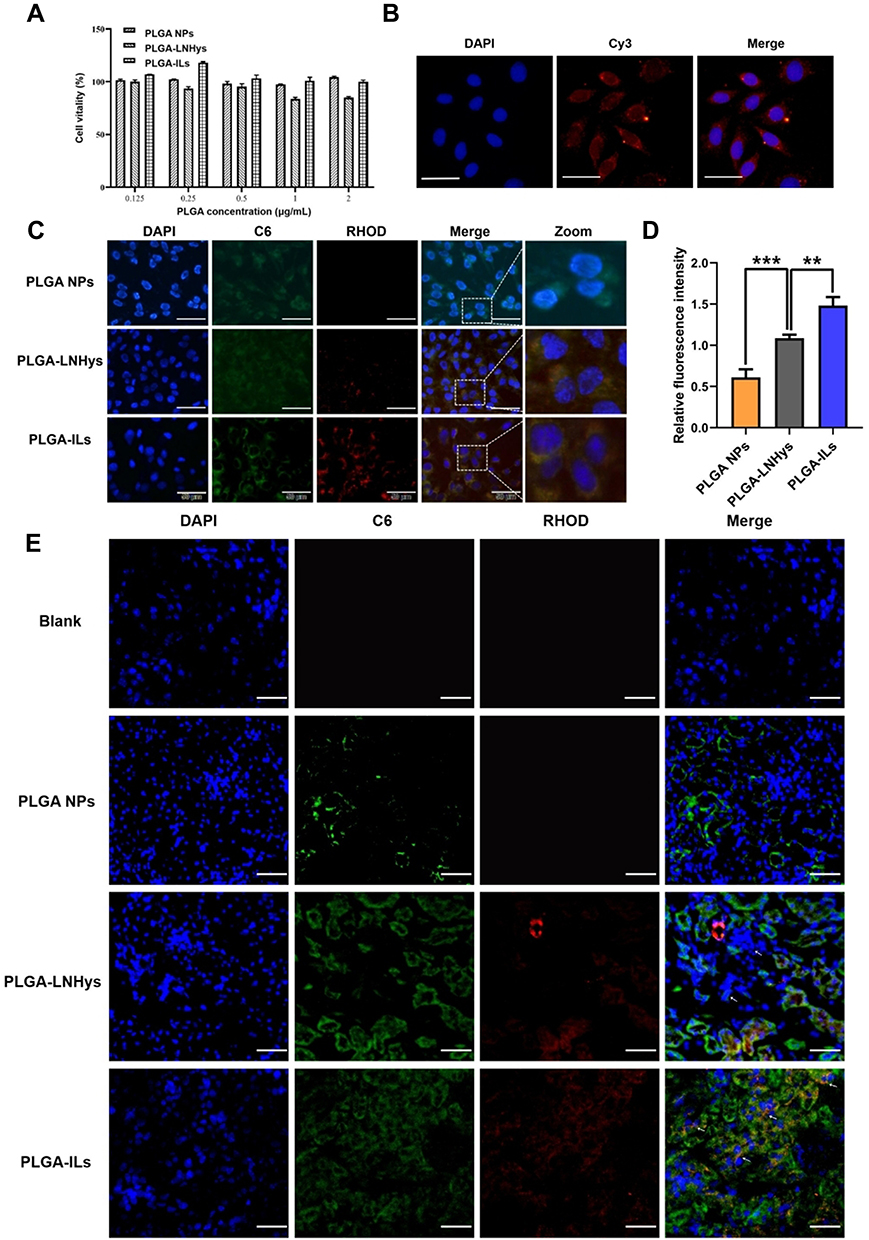 |
Figure 3 Cell studies of PLGA-ILs in vitro. (A) Cytotoxicity results of PLGA NPs, PLGA-LNHys and PLGA-ILs at PLGA concentrations ranging from 0.125 to 2 mg/mL. (B) Immunofluorescence detection of MCs surface protein α8-integrin (×400). (C) Cellular uptake of PLGA NPs, PLGA-LNHys and PLGA-ILs in vitro. Blue, green and red represent DAPI, C6 and RHOD fluorescence, respectively. Scale bars: 50 μm. (D) Relative fluorescence density of PLGA NPs, PLGA-LNHys and PLGA-ILs uptake by MCs. (E) Distribution of NPs in the kidney, “→” represents mesangial region. Blue, green and red represent DAPI, C6 and RHOD fluorescence, respectively. Scale bars: 25 μm. All data were measured as mean ± S.D, ** = P < 0.01, *** = P < 0.001, n=3. Statistical significance was determined by one-way ANOVA with Tukey post hoc test. Abbreviations: MCs, mesangial cells; C6, coumarin-6; RHOD, Rhodamine. |
Immunofluorescence and Cellular Uptake
Under the fluorescence microscope, the nucleus (DAPI staining) is blue when excited by the corresponding UV excitation wavelength, and the red on the cell surface is the binding of antigen and antibody (the second antibody is marked red by Cy3) (Figure 3B). Red fluorescence was visible on the cell surface, indicating that the surface of MCs showed the expression of α8 integrin antigen and confirming that the MC characteristics were not altered in the cultured cells.
To function as a drug delivery vehicle, PLGA-ILs must enter the cells effectively and release their payload into the cytoplasm.36 For this reason, cellular uptake was performed on MCs to elucidate the uptake ability of various nanocarriers, including PLGA-ILs, PLGA-LNHys, and PLGA NPs labeled with fluorescent probes. Under the same exposure, DAPI binding to DNA in the nucleus exhibited dark blue fluorescence for labeling the nucleus, C6 encapsulated in PLGA NPs showed green fluorescence at a certain excitation wavelength, while RHOD loaded into the liposome bilayer turned out red; after excitation green and red fluorescent signals overlapped into bright yellow fluorescent signals after combination. As shown in Figure 3C, as expected, the bright yellow fluorescence intensity of C6/RHOD@PLGA-ILs is stronger than that of C6@PLGA NPs and RHOD@PLGA-LNHys, indicating that α8 integrin antibody was crucial for the intracellular aggregation of PLGA-ILs. Semi-quantitative fluorescence analysis was performed by dividing the optical densities of C6 and DAPI (Figure 3D). The amount of PLGA-ILs taken up by MCs was 2.43 times higher than that of PLGA NPs, while PLGA-LNHys uptake was only 1.79 times higher than that of PLGA NPs. The semi-quantitative fluorescence results were consistent with the fluorescence observations.
Subcellular Localization of NPs
MCs are the major target in the progression of GN because of their ability to clear circulating immune complexes, produce pro-inflammatory mediators, and deposit immunoglobulin and immune complexes in the mesangial matrix.37 To determine the exact location of PLGA-ILs in the kidney, frozen kidney sections were collected and stained with DAPI. According to the characteristics of the glomerular physiological structure, all three NPs should theoretically pass freely through the endothelial fenestrations and enter the mesangium. C6@PLGA NPs had a small distribution of particles in the MC region, even in the kidney, suggesting that PLGA NPs could not quickly escape the capture of the reticuloendothelial system to target MCs. In contrast to C6@PLGA NPs, RHOD@PLGA-LNHys slightly distributed in the MC region because of PEG modification in phospholipid bilayers. However, it was found that C6/RHOD@PLGA-ILs showed higher intensity and good co-localization with MCs than the other two NPs (Figure 3E), suggesting that α8 integrin antibodies of PLGA-ILs specifically combined with the α8 integrin antigen on the MC surface plays a role in active targeting. These results further demonstrate that MCs have a strong retention force on PLGA-ILs.
In vivo Imaging and Tissue Distribution
The renal targeting efficiency of Cy5.5@PLGA NPs, Cy5.5@PLGA-LNHys, and Cy5.5@PLGA-ILs after intravenous injection in Kunming mice were further evaluated using an in vivo imaging system. During 0.5 h−4 h after injection, the distribution of each nanocarrier mostly accumulated in the liver of mice, and the near-infrared fluorescence signal was stronger than that in the kidney, which may be attributed to both NPs and long-circulating liposomes being mainly captured by the liver system.38 Surprisingly, the fluorescence intensity in the liver significantly decreased and even vanished during 12 h−24 h. At the same time, the fluorescence intensity in the kidneys became stronger (Figure 4A). In general, the fluorescence intensity of Cy5.5@PLGA-ILs in the kidney was stronger than that of free Cy5.5, Cy5.5@PLGA NPs, and Cy5.5@PLGA-LNHys, which confirmed that this particular size and antibodies-mediated active targeting maximized nanoparticle content and glomerular targeting and could accumulate more in the kidney. The treated mice were sacrificed, and the main organs were removed and analyzed at different times. In vitro images of the organs (Figure 4B) and the semi-quantitative fluorescence both confirmed this result (Figure 4C) that due to combining the effect of a phospholipid bilayer and α8 integrin antibodies, the distribution of Cy5.5@PLGA-ILs in the kidney was significantly higher than that of free Cy5.5, PLGA NPs and Cy5.5@PLGA-LNHys fluorescence.
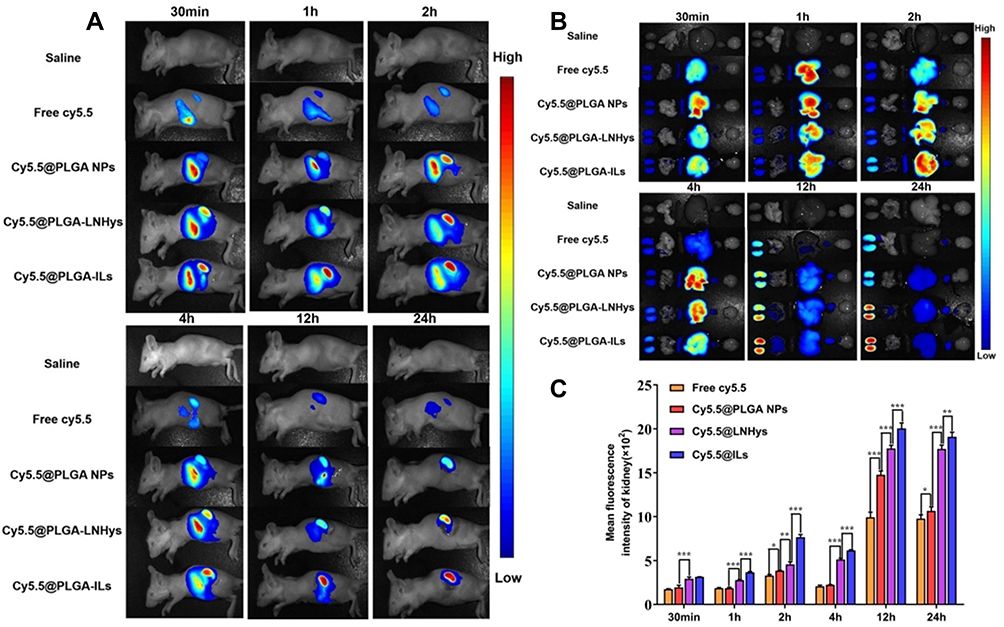 |
Figure 4 (A) In vivo bioluminescence images of mice receiving saline, free Cy5.5, Cy5.5@PLGA NPs, Cy5.5@PLGA-LNHys, or Cy5.5@PLGA-ILs at different time point. (B) Ex vivo imaging of organs in mice at different time point. (C) Mean fluorescence intensity of kidney at different time points ex vivo imaging. All data were measured as mean ± S.D, * = P < 0.05, ** = P < 0.01, *** = P < 0.001, n=3. Statistical significance was determined by one-way ANOVA with Tukey post hoc test. |
In vivo Anti-Inflammatory/Anti-Fibrosis Therapy
The in vitro and in vivo biodistribution results showed excellent glomerulus accumulation of PLGA-ILs; therefore, we investigated the therapeutic efficacy of DXMS/CAP@PLGA-ILs against GN. In view of this simple, feasible, and short-time consuming method, the HSV-induced MesPGN model was used in this study. The lesions stimulate the production of numerous cytokines, such as TGF-β, accompanied by the infiltration of neutrophils and macrophages, and the proliferation of MCs, leading to an increase in the number of nucleated cells in the glomeruli.39 The PAS technique is perhaps the most versatile and widely used technique for the demonstration of glycoproteins, carbohydrates, and mucins. Therefore, we stained the sections with PAS and compared the therapeutic effects of the drugs based on the pathological changes observed. PAS stained kidney sections from the normal group displayed prominent glomerular complete structure and a clearly visible profile, while the disordered glomeruli began to show significant MC cluster-like proliferation and various special proteins were deposited in the region of MCs from the HSV model group, resulting in the proliferation of the mesangial region severely compressing the glomerular capillary loop and narrowing the lumen, and even glomerular sclerosis in some cases. From PAS-stained sections, the pathological manifestations of the mesangial region in the mouse MesPGN model were largely attenuated by DXMS/CAP@PLGA-ILs treatment, and rescued glomerular lesions significantly more efficiently than other treatments (Figure 5A), indicating that all types of cell infiltration and proliferation were effectively controlled after treatment with DXMS/CAP@PLGA-ILs.
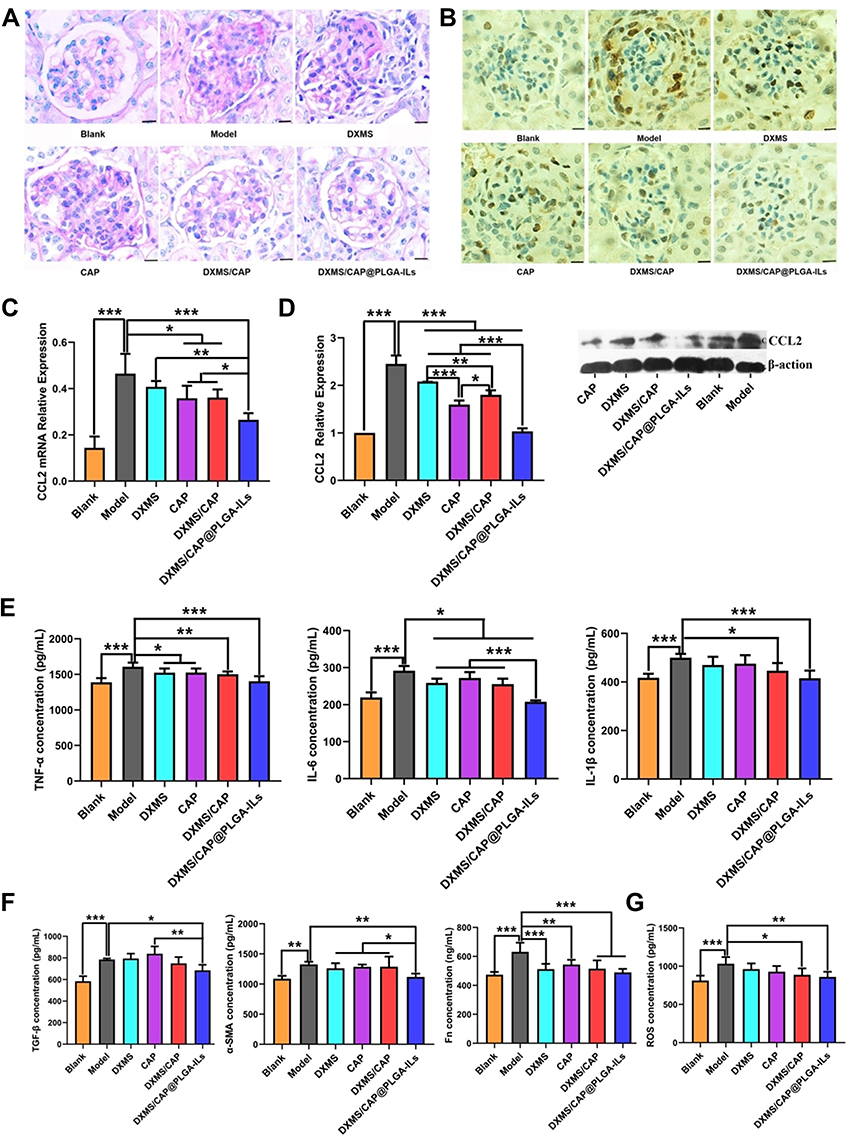 |
Figure 5 DXMS/CAP@PLGA-ILs supressed glomerular inflammation and fibrosis. (A) Glomerular histology revealed by PAS staining of kidney tissue sections. The pathological change was rescued by various degrees with different treatments. Scale bars, 25 μm. (B) Immunohistochemical staining of PCNA in kidney tissue sections. DXMS/CAP@PLGA-ILs displayed strong therapeutic effects. Scale bars, 25 μm. (C) Real-time PCR kidney inflammatory chemokines CCL2 test results. (D) Western blot inflammatory chemokines CCL2 protein expression test results. (E) Inflammatory factor (TNF-α, IL-6, and IL-1β) levels in renal cortical tissues. (F) Fibrosis factor (TGF-β, α-SMA and Fn) levels in renal cortical tissues. (G) The expression levels of ROS in renal cortical tissues. All data were measured as mean ± S.D, * = P < 0.05, ** = P < 0.01, *** = P < 0.001. Statistical significance was determined by one-way ANOVA with Tukey post hoc test. Abbreviations: DXMS, dexamethasone; CAP, captopril; PAS, periodic acid-Schiff; PCNA, proliferating cell nuclear antigen; CCL2, the inflammatory chemokine CCL2; TNF-α, tumor necrosis factor α; IL-6, Interleukin-6; IL-1β, Interleukin-1β; TGF-β, transforming growth factor-β; α-SMA, alpha-smooth muscle actin; Fn, fibronectin; ROS, reactive oxygen species. |
PCNA is a well-known scaffold for many DNA replication and repair proteins and plays an important role in the regulation of DNA replication.14 The expression of PCNA began to rise in the late G1 cell proliferation cycle, which indicated that the cells were in a proliferating state and is commonly used as a cell proliferation marker.40 Immunohistochemical staining was used to observe the expression of PCNA in the glomeruli of each group. The immunopositive reaction products were brown particles in the nucleus, and positive expression was also observed in some cytoplasm (Figure 5B). According to the immunostaining results, moderate positive expression of PCNA was detected in glomeruli on day 3 after disease induction compared to the normal group, which was prominently reduced following treatment with DXMS/CAP@PLGA-ILs. Taken together, kidney sections from mice treated with DXMS or CAP showed a mild positive reaction for PCNA expression as compared to the faint positive reaction in the DXMS/CAP and DXMS/CAP@PLGA-IL groups.
CCL2 is formed under the stimulation of pathological conditions and is actively involved in the inflammatory response, attracting immune cells to the inflammatory site.41,42 Therefore, in order to investigate the anti-inflammatory effect of DXMS/CAP@PLGA-ILs, the expression levels of CCL2 were determined by real-time PCR and Western blotting. We studied the mRNA expression levels of CCL2 in the renal cortex of DXMS/CAP@PLGA-ILs-treated MesPGN mice, and found that the increase in inflammatory chemokines was significantly reduced (Figure 5C). Unsurprisingly, the results of Western blotting were consistent with the results of real-time PCR testing (Figure 5D). These results show that DXMS/CAP@PLGA-ILs could dramatically inhibit the protein expression of CCL2 in the renal cortex by targeting and releasing drugs, thus reducing stimulation to MCs and inhibiting the proliferation of MCs.
To further evaluate the anti-inflammatory and anti-fibrosis effect of DXMS/CAP@PLGA-ILs in the mouse MesPGN model of this study. Hence, we measured the expression levels of inflammatory factors and fibrosis factors, including TNF-α, IL-6, IL-β1, TGF-β, α-SMA and Fn in the renal cortices of mice in the different groups (Figure 5E and F). Compared with the blank group, the MesPGN model group showed the higher expression levels of TNF-α, IL-6, IL-β1, TGF-β, α-SMA and Fn. However, these increased indexes were seldom improved after treated with CAP or DXMS or DXMS/ CAP combination, respectively. Fortunately, DXMS/CAP@PLGA-ILs group demonstrated positive therapeutic efficacy with reduced the expression levels of TNF-α, IL-6, IL-β1, TGF-β, α-SMA and Fn, indicating that they can be used for the targeted treatment of inflammation and fibrosis in MesPGN. Moreover, in order to evaluate the antioxidant stress effect of DXMS/CAP@PLGA-ILs, the expression levels of ROS was determined by ELISA kits (Figure 5G). We found that the level of ROS was significantly increased in the MesPGN model group. Similar trend of positive therapeutic efficacy was observed in the DXMS/CAP@PLGA-ILs group compared with other groups, indicating that DXMS/CAP@PLGA-ILs could play an effective role in the inhibition of oxidant stress. Overall, the results demonstrated that DXMS/CAP@PLGA-ILs could inhibit the expression of inflammatory factors, fibrosis factors and ROS of MCs in MesPGN model.
Discussion
GN continues to be one of the leading causes of ESKD in China and worldwide, and renal damage is already in an irreversible stage when some patients are first diagnosed.43 Currently, treatment remains controversial because none of the current treatment regimens produce a convincing benefit. As introduced, in the existing targeted therapy studies for MCs lesions, most researchers think that suppressing inflammation responses of GN is a key to prevent pathological damage. But the truth is that GN is often accompanied by inflammation and fibrotic lesions, and inflammation runs through the entire process of renal fibrosis. Therefore, we believe that the most effective treatment strategy for GN is to realize inhibition of inflammation and fibrosis through the MC-targeted dual drug delivery system.
In this study, we prepared PLGA NPs, PLGA-LNHys and PLGA-ILs, we selected PLGA NPs-based core-shell liposome polymer NPs as the drug delivery system mainly due to following reasons: (i) PLGA nanoparticle are prepared by a facile and highly reproducible method; (ii) the nano-cores of PLGA-ILs can be used as internal support skeletons to provide mechanical stability to the shell. Liposomes as shells are advantageous in that they have good biocompatibility and can be easily modified; (iii) core-shell nanocarriers could have a synergistic therapeutic effect as they allow for the loading of multiple drug molecules. Our experimental data also proved the advantages mentioned above. All NPs with PDI values less than 0.3 are considered to be uniformly dispersed,44 and a negative zeta potential value indicates that there is a charge difference between PLGA-ILs and the liquid, and that the large repulsive force could maintain a stable nanoparticle system.45 Previous study results indicated that the drug loading of nanoparticle-coated DXMS usually ranges from 1% to 5%.46,47 However, as a semicrystalline polymer, PLGA can not only incorporate DXMS into its structure and have a high drug load of approximately 8%,48,49 but also release them over time by a combination of diffusion and degradation. As expected, the drug load of DXMS and CAP was consistent with expectations. More importantly, the drug loading of DXMS was more than 8%, which could be related to the use of TPGS as a matrix material of NPs blended with PLGA polymer, enabling the efficient encapsulation of DXMS.50 It has been reported that DXMS released from PLGA NPs could quickly reach saturation during the first 4 h of incubation51 with an average cumulative release of approximately 90%. Surprisingly, the release profile of DXMS/CAP@PLGA-ILs showed a fast release such that the average cumulative release of DXMS and CAP were only approximately 25% and 29%, respectively, in the first 0.5 h, afterward DXMS and CAP sustained release reached about 77.23 ± 2.96% and 83.18 ± 3.77%, respectively, and remained stable over time. This proved that DXMS/CAP@PLGA-ILs could partly attenuate the effect of burst release and lengthen the effect of sustained release, which may be due to the PLGA NPs as nano-cores that can be used as internal support skeletons to provide mechanical stability to the shell. In addition, differences in release between the two drugs may be attributed to CAP in the empty liposome, and the permeable liposome layer would cause the faster release of CAP, accompanied by the accumulation of CAP being higher than that of DXMS. DXMS was wrapped in PLGA NPs and drug release from PLGA microspheres was controlled by polymer degradation,52 which is a result of the polymer backbone hydrolyzing into oligomers and monomers. Theoretically, the release rate of DXMS should be significantly slower than the release rate of CAP. While this result had no significant difference, DXMS would most probably be in the vicinity of the nanoparticle surface, leading to an increase in the release rate.
According to the characteristics of glomerular physiological structure, all NPs with with diameters between 70 and 130 nm in the blood circulation to penetrate the glomerular endothelial space into the glomerular MCs region, rather than further penetrate the glomerular basement membrane, so as to passively deposit and retain in the area of MCs. Unlike PLGA NP, PLGA-LNHy and PLGA-IL with a phospholipid bilayer that forms the basic scaffolds of cell membranes leads to higher uptake capacity by MCs.53 Moreover, PEG modification of PLGA-LNHy and PLGA-IL resulted in longer circulation time in the body,54,55 making PLGA-LNHy and PLGA-IL repeatedly cross the glomeruli to the MC area. In contrast with PLGA-LNHy, PLGA-IL by virtue of α8 integrin antibodies modification appear to be superior in specificity towards MCs. This may be because α8 integrin antigen is only expressed by MCs in the glomerulus, which mediates cell adhesion and cytoskeleton organization in addition to acting as a signal receptor,56 thus leading to quick binding to the surface of MCs by PLGA-IL and mediating their internalization. Thus, it is concluded that dual modification with different functional ligands is superior to single modification. This might also be the reason that an increasing number of researchers have dedicated their efforts to multifunctional kidney-targeting drug delivery systems.57,58
Traditional anti-Thy-1 nephritis is a classic reversible model of MesPGN.59,60 However, this method cannot be used to prepare a mouse MesPGN model because of the lack of Thy1 antigen on the surface of mouse MCs. HSV-induced acute MesPGN model can produce pathological manifestations similar to GN. The injection of HSV through the tail vein can cause lesions in the mesangial area of the glomerulus, including mesangial dissolution, MC apoptosis, proliferation, and accumulation of extracellular matrix.61 As verified by a number of studies, kidney inflammation and subsequent fibrosis are key processes that lead to ESKD. On the one hand, of the many pro-inflammatory cytokines/chemokines, CCL2, TNF-α, IL-6 and IL-β1 have emerged as the key mediator of inflammation following kidney injury,62,63 and aberrantly increased inflammatory mediator expression is responsible for sustaining and exacerbating cell recruitment and the resultant inflammation. During chronic inflammation, due to repetitive or prolonged injury from the inflammatory process, which evolves into a progressive and irreversible fibrotic process.64,65 On the other hand, TGF-β is a cytokine essential for the induction of the renal fibrotic response, and the notion that TGF-β mediate tissue fibrosis is supported by cell biological studies, animal model experiments, and clinical evidence.66 α-SMA and Fn are universally known and characterized protein used for assessment of fibrosis in several tissues and organs including the kidney and the relative incorporation of the fibrotic markers α-SMA and Fn into the matrix increases in response to TGF-β.67,68 Considering that inflammation-related cytokines and fibrosis-related factors play an important role in the pathological process of MesPGN, we investigated the expression levels of inflammatory chemokine CCL2, TNF-α, IL-6, IL-β1, TGF-β, α-SMA and Fn. We found that the expression levels of the above factors were significantly increased after HSV modeling, indicating that HSV can effectively promote the expression of inflammation and fibrosis factors in nephritis mice, and the MesPGN model was successfully induced by injected HSV twice through tail vein for evaluating the therapeutic effects of DXMS/CAP@PLGA-ILs in MesPGN. As expected, the kidney of MesPGN model mice showed increased expression of inflammatory factors and fibrosis factors compared with blank group, but the induction of these factors was significantly inhibited after administration. The comprehensive situation of cytokine measurement indexes indicated that the antibodies modified nanoparticle DXMS/CAP@PLGA-ILs had stronger “anti-inflammatory/anti-fibrosis” effect on the kidney of MesPGN model. Furthermore, ROS serve as cell signaling molecules generated in oxidative metabolism and excessive oxidative stress derived from ROS accumulation deregulates the antioxidative defense system, which is closely associated with various diseases.69 In glomeruli, ROS are generated by both infiltrating cells and resident glomerular cells and excessive ROS levels lead to cell death by damaging cellular components, including proteins, lipid bilayers, and chromosomes. The participation of ROS in glomerular damage was proved in many experimental studies.6 CAP could exerts treatment effects by increasing tissue antioxidant activity, scavenging different types of ROS.70 Through experiment, we verified optimistic therapeutic effect of DXMS/CAP@PLGA-ILs in vivo MesPGN model, thereby more powerfully proving the excellence of the treatment effect of DXMS/CAP@PLGA-ILs.
Conclusion
In this paper, we prepared PLGA-ILs modified with both α8 integrin antibodies and PEG using a facile nanometer precipitation methodology and film dispersion method, which exhibited obvious core-shell structure with a uniform particle size of 119.1 ± 2.31 nm and had remarkable MC targeting ability. In vitro and in vivo experiments revealed that the mesangial targeting profile of PLGA-ILs was significantly improved compared to PLGA-LNHys and PLGA NPs. Besides the characteristic of co-delivering anti-inflammatory and anti-fibrosis loaded drugs, PLGA-ILs are not only a promising delivery platform for MC related diseases, but also have the potential to be more widely investigated for other applications in the pharmaceutical field. Ultimately, DXMS/CAP@PLGA-ILs showed an outstanding advantage in anti-inflammation and anti-fibrosis efficiency in MesPGN model mice. Co-delivery of the “anti-inflammatory/anti-fibrosis” drugs targeting MCs is a novel concept for GN treatment.
Acknowledgments
The study was funded by the National Natural Science Foundation of China (81973265).
Disclosure
The authors declare no conflict of interest, financial or otherwise.
References
1. Yuan J, Zou XR, Han SP, et al. Prevalence and risk factors for cardiovascular disease among chronic kidney disease patients: results from the Chinese cohort study of chronic kidney disease (C-STRIDE). BMC Nephrol. 2017;18(1):23.
2. Lousa I, Reis F, Beirão I, Rui A, Santos-SilVa A. New Potential Biomarkers for Chronic Kidney Disease Management—A Review of the Literature. Int J Mol Sci. 2020;22(1):43.
3. Gao JR, Jiang NN, Jiang H, et al. Effects of Qi Teng Xiao Zhuo granules on circRNA expression profiles in rats with chronic glomerulonephritis. Drug Des Devel Ther. 2019;13(1):1901–1913.
4. Infante BRM, Leo S, Troise D, et al. Recurrent Glomerulonephritis after Renal Transplantation: the Clinical Problem. Int J Mol Sci. 2020;21(17):5954.
5. Li W, Qu W, Zhou H. Assessment of Bailing Capsule in the Treatment of Chronic Glomerulonephritis: a Meta-analysis. Indian J Pharm Sci. 2020;82(1):59–63.
6. Wójcicka G, Bełtowski J. [Oxidative stress in glomerulonephritis]. Postpy Higieny I Medycyny Dowiadczalnej. 2001;55(6):855–869. Spanish.
7. Curran CS, Kopp JB. PD-1 immunobiology in glomerulonephritis and renal cell carcinoma. BMC Nephrol. 2021;22(1):1–19.
8. Zwiech R. Macrophage migration inhibitory factor urinary excretion revisited–MIF a potent predictor of the immunosuppressive treatment outcomes in patients with proliferative primary glomerulonephritis. BMC Immunol. 2015;16(1):1–9.
9. Klinkhammer BM, Goldschmeding R, Floege J, Boor P. Treatment of renal fibrosis—turning challenges into opportunities. Adv Chronic Kidney Dis. 2017;24(2):117–129.
10. Tumlin JA, Madaio MP, Hennigar R. Idiopathic IgA nephropathy: pathogenesis, histopathology, and therapeutic options. Clin J Am Soc Nephrol. 2007;2(5):1054–1061.
11. Liu CP, Hu Y, Lin JC, Fu HL, Lim LY, YZ X. Targeting strategies for drug delivery to the kidney: from renal glomeruli to tubules. Med Res Rev. 2019;39(2):561–578.
12. Yuan ZX, Shang ZH, Gu J, He LL. Renal targeting delivery systems. Future Med Chem. 2019;11(17):2237–2240.
13. Qin X, Xu Y, Zhou X, et al. An injectable micelle-hydrogel hybrid for localized and prolonged drug delivery in the management of renal fibrosis. Acta Pharmaceutica Sinica B. 2021;11(3):835–847.
14. Guo L, Luo S, Du Z, et al. Targeted delivery of celastrol to mesangial cells is effective against mesangioproliferative glomerulonephritis. Nat Commun. 2017;8(1):878.
15. Fleischmann DHM, Maslanka Figueroa S, Schlossmann J, Goepferich A. Targeted Delivery of Soluble Guanylate Cyclase (sGC) Activator Cinaciguat to Renal Mesangial Cells via Virus-Mimetic Nanoparticles Potentiates Anti-Fibrotic Effects by cGMP-Mediated Suppression of the TGF-β Pathway. Int J Mol Sci. 2021;22(5):2557.
16. Yw A, Qw A, Jw B, et al. Co-delivery of p38α MAPK and p65 siRNA by novel liposomal glomerulus-targeting nano carriers for effective immunoglobulin a nephropathy treatment - ScienceDirect. J Controlled Release. 2020;320:457–468.
17. Yuan Z-X, Jia L, Lim LY, et al. Renal-targeted delivery of triptolide by entrapment in pegylated TRX-20-modified liposomes. Int J Nanomedicine. 2017;12(1):5673–5686.
18. Fatemeh SA. Active targeting drug delivery nanocarriers: ligands. Nano Stru Nano-Objects. 2019;23:100370.
19. Yogesh SUDPR, Harini T. Anti-alpha8 integrin immunoliposomes in glomeruli of lupus-susceptible mice: a novel system for delivery of therapeutic agents to the renal glomerulus in systemic lupus erythematosus. Arthritis Rheum. 2008;58(12):3884–3891.
20. Yuan F, Nelson RK, Tabor DE, Zhang Y, Wang D. Dexamethasone prodrug treatment prevents nephritis in lupus-prone (NZB × NZW)F1 mice without causing systemic side effects. Arthritis Rheumatol. 2014;64(12):4029–4039.
21. Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids–new mechanisms for old drugs. N Engl J Med. 2005;353(16):1711–1723.
22. Ayroldi E, Cannarile L, Migliorati G, Nocentini G, Delfino DV, Riccardi C. Mechanisms of the anti-inflammatory effects of glucocorticoids: genomic and nongenomic interference with MAPK signaling pathways. FASEB J. 2012;26(12):4805–4820.
23. Mizar S, Kozman MR, Abo-Saif AA, et al. Combination of Captopril with Gliclazide Decreases Vascular and Renal Complications and Improves Glycemic Control in Rats with Streptozotocin-induced Diabetes Mellitus. Endocrine Metab Immune Disorders Drug Targets. 2021;21(6):1096–1106.
24. Corey SJ, Jha J, Mccart EA, Rittase WB, Day RM. Captopril mitigates splenomegaly and myelofibrosis in the Gata1low murine model of myelofibrosis. J Cell Mol Med. 2018;22(9):4274–4282.
25. Shirazi M, Noorafshan A, Bahri MA, Tanideh N. Captopril Reduces Interstitial Renal Fibrosis and Preserves More Normal Renal Tubules in Neonatal Dogs with Partial Urethral Obstruction: a Preliminary Study. Urol Int. 2007;78(2):173–177.
26. Zhang MY, He JH, Zhang WL, Liu JP. Fabrication of TPGS-Stabilized Liposome-PLGA Hybrid Nanoparticle Via a New Modified Nanoprecipitation Approach: in Vitro and In Vivo Evaluation. Pharm Res. 2018;35(11):1–13.
27. Shin GH, Chung SK, Kim JT, Joung HJ, Park HJ. Preparation of Chitosan-Coated Nanoliposomes for Improving the Mucoadhesive Property of Curcumin Using the Ethanol Injection Method. J Agric Food Chem. 2013;61(46):11119–11126.
28. Petrilli R, Eloy JO, Lee RJ, Lopez RFV. Preparation of Immunoliposomes by Direct Coupling of Antibodies Based on a Thioether Bond. In: Picanço-Castro V, Swiech K, editors. Methods in Molecular Biology. New York: Humana Press; 2018:229–237.
29. Wang Y, Liu F, Wang Q, et al. A novel immunoliposome mediated by CD123 antibody targeting to acute myeloid leukemia cells. Int J Pharm. 2017;529(1):531–542.
30. Gao J, Wu L, Wang S, Chen X. Role of Chemokine (C–X–C Motif) Ligand 10 (CXCL10) in Renal Diseases. Mediators Inflamm. 2020;2020(1):1–16.
31. Li Y, Wang J, Zhu X, Feng Q, Li X, Feng X. Urinary protein markers predict the severity of renal histological lesions in children with mesangial proliferative glomerulonephritis. BMC Nephrol. 2012;13(1):1–10.
32. Neto OV, Russo EM, Costa RS, Coimbra TM, Dantas M. Effect of the absence of interleukin-12 on mesangial proliferative glomerulonephritis induced by habu snake venom. Ren Fail. 2009;31(10):964–970.
33. Wu L, Chen X, Mei Y, et al. CXCL10 expression induced by Mxi1 inactivation induces mesangial cell apoptosis in mouse Habu nephritis. Cell Signal. 2015;27(5):943–950.
34. Kenneth JL, Thomas DS. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2002;25:402–408.
35. PETRACHI T, GANZERLI F, CUOGHI A, et al. Assessing Biocompatibility of Face Mask Materials during COVID-19 Pandemic by a Rapid Multi-Assays Strategy. Int J Environ Res Public Health. 2021;18(10):5387.
36. Sun Y, Davis E. Nanoplatforms for Targeted Stimuli-Responsive Drug Delivery: a Review of Platform Materials and Stimuli-Responsive Release and Targeting Mechanisms. Nanomaterials. 2021;11(3):746.
37. Jan N, Jonathan B, Julian BA, Renfrow MB. Aberrant Glycosylation of the IgA1 Molecule in IgA Nephropathy. Semin Nephrol. 2018;38(5):461–476.
38. Zhang M, Gao S, Yang D, Fang Y, Shi K. Influencing factors and strategies of enhancing nanoparticles into tumors in vivo. Acta Pharmaceutica Sinica B. 2021;11(8):2265–2285.
39. Nikolay T, Chames K, Philippe C, Siem VDL, Dana H, Domenico M. PIP degron proteins, substrates of CRL4Cdt2, and not PIP boxes, interfere with DNA polymerase η and κ focus formation on UV damage. Nucleic Acids Res. 2014;42(6):3692–3706.
40. Strzalka W, Ziemienowicz A. Proliferating cell nuclear antigen (PCNA): a key factor in DNA replication and cell cycle regulation. Ann Bot. 2011;107(7):1127–1140.
41. Kanemitsu N, Kiyonaga F, Mizukami K, Maeno K, Ito H. Chronic treatment with the (iso-)glutaminyl cyclase inhibitor PQ529 is a novel and effective approach for glomerulonephritis in chronic kidney disease. Naunyn Schmiedeberg Arch Pharmacol. 2020;394(5):751–761.
42. Sonja D, Peter B. Cellular and molecular mechanisms of kidney fibrosis. Mol Aspects Med. 2018;65:16–36.
43. Ihm CG. Hypertension in Chronic Glomerulonephritis. Electrolyte Blood Pressure E Bp. 2015;13(2):41–45.
44. Anton N, Benoit JP, Saulnier P. Design and production of nanoparticles formulated from nano-emulsion templates-a review. J Controlled Release. 2008;128(3):185–199.
45. Guerra-Rosas MI, Morales-Castro J, Cubero-Márquez M, Salvia-Trujillo L, Martín-Belloso O. Antimicrobial activity of nanoemulsions containing essential oils and high methoxyl pectin during long-term storage. Food Control. 2017;77:131–138.
46. Ban J, Yan Z, Xin H, Deng G, Lu Z. dexamethasone. Int J Nanomedicine. 2017;12(1):1329.
47. Krishnan V, Xu X, Barwe SP, et al. Dexamethasone-Loaded Block Copolymer Nanoparticles Induce Leukemia Cell Death and Enhance Therapeutic Efficacy: a Novel Application in Pediatric Nanomedicine. Mol Pharm. 2013;10(6):2199–2210.
48. Bing G, Yan W, Burgess DJ. In vitro and in vivo performance of dexamethasone loaded PLGA microspheres prepared using polymer blends. Int J Pharm. 2015;496(2):534–540.
49. Yu D, Hao W, Sun C, Shi F, Wang X. A single dose of dexamethasone encapsulated in polyethylene glycol-coated polylactic acid nanoparticles attenuates cisplatin-induced hearing loss following round window membrane administration. Int J Nanomedicine. 2015;10:3567–3579.
50. Zhang Z, Tan S, Feng -S-S. Vitamin E TPGS as a molecular biomaterial for drug delivery. Biomaterials. 2012;33(19):4889–4906.
51. Gómez-Gaete C, Tsapis N, Besnard M, Bochot A, Fattal E. nanoparticles. Int J Pharm. 2007;331(2):153–159.
52. Dawes G, Fratila-Apachitei LE, Necula BS, Apachitei I, Witkamp GJ, Duszczyk J. Release of PLGA–encapsulated dexamethasone from microsphere loaded porous surfaces. J Mater Sci Mater Med. 2010;21(1):215–221.
53. Guo Y, Terazzi E, Seemann R, Fleury JB, Baulin VA. Direct proof of spontaneous translocation of lipid-covered hydrophobic nanoparticles through a phospholipid bilayer. Sci Adv. 2016;2(11):e1600261.
54. Patsula V, Horák D, Kuka J, Macková H, Efc L. Synthesis and modification of uniform PEG-neridronate-modified magnetic nanoparticles determines prolonged blood circulation and biodistribution in a mouse preclinical model. Sci Rep. 2019;9(1):10765.
55. Zhou H, Fan Z, Li PY, Deng J, Cheng H. Dense and Dynamic Polyethylene Glycol Shells Cloak Nanoparticles from Uptake by Liver Endothelial Cells for Long Blood Circulation. ACS Nano. 2018;12(10):10130–10141.
56. Bieritz B, Spessotto P, Colombatti A, Jahn A, Hartner A. Role of alpha8 integrin in mesangial cell adhesion, migration, and proliferation. Kidney Int. 2003;64(1):119–127.
57. Ray S, Li Z, Hsu C-H, et al. Dendrimer- and copolymer-based nanoparticles for magnetic resonance cancer theranostics. Theranostics. 2018;8(22):6322–6349.
58. Ma Y, Cai F, Li Y, Chen J, Lin W. A review of the application of nanoparticles in the diagnosis and treatment of chronic kidney disease. Bioactive Materials. 2020;5(3):732–743.
59. Wan Y, Gu L, Suzuki K, et al. Multi-Glycoside of Tripterygium wilfordii Hook f. Ameliorates Proteinuria and Acute Mesangial Injury Induced by Anti-Thy1.1 Monoclonal Antibody. Nephron Exp Nephrol. 2005;99(4):e121–e129.
60. Geng W, Wei R, Liu S, et al. Shenhua Tablet inhibits mesangial cell proliferation in rats with chronic anti-Thy-1 nephritis. Biol Res. 2016;49(1):17.
61. Cattell V, Bradfield JWB. Focal mesangial proliferative glomerulonephritis in the rat caused by habu snake venom. Am J Pathol. 1977;87(3):511.
62. Kashyap S, Osman M, Ferguson CM, et al. Ccl2 deficiency protects against chronic renal injury in murine renovascular hypertension. Sci Rep. 2018;8(1):8598.
63. Alvarez AM, Deocesano-Pereira C, Teixeira C, et al. IL-1β and TNF-α Modulation of Proliferated and Committed Myoblasts: IL-6 and COX-2-Derived Prostaglandins as Key Actors in the Mechanisms Involved. Cells. 2020;9(2005):1.
64. Gillespie SR, Tedesco LJ, Wang L, Bernstein AM. The deubiquitylase USP10 regulates integrin β1 and β5 and fibrotic wound healing. J Cell Sci. 2017;130(20):3481–3495.
65. Xu Y, Niu Y, Wu B, et al. Extended-release of therapeutic microRNA via a host-guest supramolecular hydrogel to locally alleviate renal interstitial fibrosis. Biomaterials. 2021;275:120902.
66. Frangogiannis NG. Transforming growth factor–β in tissue fibrosis. J Exp Med. 2020;217(3):e20190103.
67. Tang MK, Zhang YY, Xiao J, et al. Neural transcription factor Pou4f1 promotes renal fibrosis via macrophage–myofibroblast transition. Proce National Acad Sci. 2020;117(34):201917663.
68. Alcalde-Estévez E, Asenjo-Bueno A, Sosa P, et al. Endothelin-1 induces cellular senescence and fibrosis in cultured myoblasts. A potential mechanism of aging-related sarcopenia. Aging. 2020;12(12):11200–11223.
69. Wang Y, Qi H, Liu Y, et al. The double-edged roles of ROS in cancer prevention and therapy. Theranostics. 2021;11(10):4839–4857.
70. Tian Y, Li HB, Liu PY, et al. Captopril Pretreatment Produces an Additive Cardioprotection to Isoflurane Preconditioning in Attenuating Myocardial Ischemia Reperfusion Injury in Rabbits and in Humans. Mediators Inflamm. 2015;2015:819232.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.