")
Back to Journals » International Journal of Nanomedicine » Volume 13
Co-delivery of Aurora-A inhibitor XY-4 and Bcl-xl siRNA enhances antitumor efficacy for melanoma therapy
Authors Duan X, Mu M, Yan J, Bai L, Zhong L, Zhu Y, Pan H, Zhang M, Shi J
Received 30 July 2017
Accepted for publication 7 December 2017
Published 9 March 2018 Volume 2018:13 Pages 1443—1456
DOI https://doi.org/10.2147/IJN.S147759
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Xingmei Duan,1,2,* Minjie Mu,3,* Junfeng Yan,1 Lan Bai,1 Lei Zhong,1 Yuxuan Zhu,1 Haixia Pan,1 Mei Zhang,3 Jianyou Shi1,3
1Personalized Drug Therapy Key Laboratory of Sichuan Province, Department of Pharmacy, Sichuan Provincial People`s Hospital, Chengdu, 2State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, West China Medical School, Sichuan University, Chengdu, 3Key Laboratory Standardization of Chinese Herbal Medicines of Ministry of Education, State Key Laboratory Breeding Base of Systematic Research, Development and Utilization of Chinese Medicine Resources, School of Pharmacy, Chengdu University of Traditional Medicine (TCM), Chengdu, People’s Republic of China
*These authors contributed equally to this work
Background: The newly synthesized Aurora-A kinase inhibitor XY-4 is a potential anti-cancer agent, but its hydrophobicity and limited efficiency restrict further application. Nanotechnology based combined therapy provides an optimized strategy for solving these issues.
Methods: In this study, the newly synthesized Aurora-A kinase inhibitor XY-4 and Bcl-xl targeted siRNA were co-delivered by cationic liposomes, creating an injectable co-delivery formulation. The anti-cancer ability and mechanisms of XY-4/Bcl-xl siRNA co-loaded cationic liposomes were studied both in vitro and in vivo.
Results: The prepared liposomes had a mean particle size of 91.3±4.5 nm with a zeta potential of 38.5±0.5 mV and were monodispersed (Polydispersity index =0.183) in water solution, with high drug loading capacity and stability. Intriguingly, the positive charges of co-delivery liposomes not only facilitated gene delivery, but also obviously enhanced drug uptake. The XY-4/Bcl-xl siRNA co-loaded cationic liposomes demonstrated enhanced anti-cancer effects on B16 melanoma cells in vitro by activation mitochondrial apoptosis pathway. Moreover, intratumoral injection of this co-delivery formulation efficiently inhibited the growth of a B16 melanoma xenograft model in vivo.
Conclusion: By co-delivering Aurora-A kinase inhibitor XY-4 and Bcl-xl targeting siRNA in a nanoformulation, our study supplied a potential combination strategy for melanoma therapy.
Keywords: RNA interference, Aurora-A kinase inhibitor, liposome, co-delivery, melanoma, apoptosis
Introduction
Cancer is a leading cause of death worldwide, while melanoma accounts for most skin cancer deaths, with nearly 191,100 estimated new cases in 2012.1,2 Although significant efforts have been made to increase in-depth understanding and treatment of melanoma, there is still a great need for more efficient therapeutic agents and strategies.3
Cell cycle kinase inhibitors are a broad range of molecules with therapeutic potential.4–6 The cell cycle is driven by families of protein kinases that orchestrate the complex events, and their activity is frequently deregulated in cancer cells.7,8 Several key nuclear kinases are involved in cell cycle progression and cell division, including cyclin-dependent kinases (CDKs), checkpoint kinases (CHKs), Aurora kinases (AURKs) and polo-like kinases (PLKs).6,9,10 Inhibitors of these classes have received much attention, with some currently under clinical development.9,11 Among them, Aurora-A kinase localizes to the centrosomes and spindle poles and recruits the cyclin B1-CDK1 complex to drive cell mitosis.10,12 Amplification of Aurora-A kinase is oncogenic and has been shown to be associated with drug resistance.13–17 In a previous study, we successfully designed and synthesized a novel AURK inhibitor XY-4, which specifically targets Aurora subtype A (Figure 1).18 XY-4 belongs to a series of new compounds bearing a pyrazolo[3,4-b] pyridine scaffold, and interacts with Aurora-A kinases in a similar mode to Danusertib. This new inhibitor showed strong efficiency in G2/M cell cycle arrest, demonstrating good anti-proliferative activities in several cancers including lung cancer, colon cancer and ovarian cancer. However, the issue of water insolubility greatly limits its further application as an injectable formulation. Meanwhile, it is necessary to further improve its therapeutic efficacy in different types of cancer.
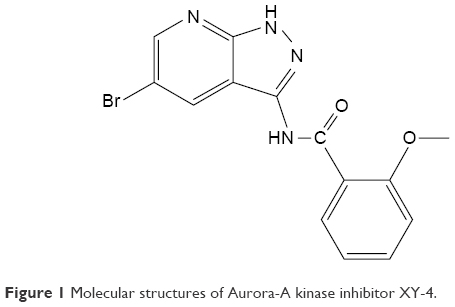 | Figure 1 Molecular structures of Aurora-A kinase inhibitor XY-4. |
The use of multiple therapeutic agents in combination has become one of the primary strategies in cancer therapy. It is generally acknowledged that proper combinational treatment can promote enhanced or synergistic actions, improved target selectivity, and decreased cancer drug resistance.19 In previous reports, cell cycle kinase inhibitors have been successfully applied in cancer therapy in combination with other chemotherapeutics.20 For example, Russell et al21 reported effective inhibition of neuroblastoma both in vivo and in vitro by simultaneous administration of CHK1 and Wee1 inhibitors, while other groups reported enhanced effects of combination therapy with Chk1 inhibitor in the treatment of acute myeloid leukemia and neuroblastoma.21,22 Meanwhile, Aurora kinase inhibitors including MK-5108, AT9283 and MLN8237 were also reported to exhibit enhanced antitumor activities when combined with docetaxel and lenalidomide.23–25 These strategies of combining cell cycle kinase inhibitors with other chemotherapy agents have provided optimized effects. However, adverse effects and issues of administration may prevent these strategies from being developed into a single formulation. Unlike chemical agents, inhibitory nucleotides such as siRNA and shRNA bind to targeted genes specifically, providing alternative choices for combination therapy with high safety and efficacy. However, delivery of these therapeutic nucleotides requires carriers which are largely different from those used in chemotherapeutics. Moreover, balancing of amounts of two cargoes in a single formulation is critical for the treatment outcome. Therefore, developing a suitable strategy for an Aurora kinase inhibitor-based combinational therapy essential.
Nanotechnology provides an optimized method to overcome the solubility issue of hydrophobic drugs.25 Encapsulation of hydrophobic drugs in nanoparticles could render them completely dispersible in aqueous solutions, and lead to prolonged drug release behavior and high stability.26,27 Nanocarriers with positive charge optimized surfaces could also be an ideal vector for therapeutic nucleotides through electronic binding. Moreover, we have previously indicated that cationic nanovectors could effectively enhance drug uptake by cancer cells, suggesting strong potential in drug delivery.28 Thus, their capacity for simultaneous delivery of multiple cargos facilitates combination therapy. Liposomal nanoparticles are widely recognized to be excellent drug delivery systems for both chemical and biological therapeutics, and some liposome based drugs have already been marketed.27,29–33 Therefore, to develop an injectable formulation with enhanced efficacy, we co-delivered the newly synthesized Aurora-A kinase inhibitor XY-4 with Bcl-xl targeting siRNA using cationic liposomes. We hypothesize that the treatment efficacy of XY-4 through cell cycle arrest could be improved by enhanced drug uptake and solubility. Meanwhile, simultaneous silencing of Bcl-xl might further strengthen the therapeutic effect by inducing apoptosis. In this work, the anti-cancer ability of prepared liposomes on melanoma was evaluated both in vitro and in vivo, and the relevant molecular mechanisms were studied.
Materials and methods
Materials
N-[1-(2,3-dioleoyloxy) propyl]-N,N,N-trimethylammonium methyl sulfate (DOTAP) and cholesterol were purchased from Avanti Polar Lipids (Alabaster, AL, USA). DMEM and MTT were obtained from Sigma-Aldrich Co., (St Louis, MO, USA) and used without further purification. Chloroform and other organic solvents were purchased from ChengDu KeLong Chemical Co., Ltd (ChengDu, People’s Republic of China) Hoechst 33258 was purchased from Beyotime, (Shanghai, People’s Republic of China). XY-4 was previously synthesized in our laboratory. Mouse B16 melanoma cell line was purchased from the American Type Culture Collection (Manassas, VA, USA). Cells were cultured in DMEM medium supplemented with 10% fetal calf serum, incubated at 37°C with 5% CO2 humidified air atmosphere. Antibodies against mouse cytochrome c, β-actin, Bcl-xl, caspase 3 and caspase 9 were purchased from Abcam (Cambridge, MA, USA). C57 mice were obtained from Beijing HFK Bio-technology Co., Ltd (Beijing, People’s Republic of China) and maintained under specific pathogen-free conditions. All animal procedures were approved and controlled by the Institutional Animal Care and Treatment Committee of Sichuan Provincial People’s Hospital and carried out according to the Animal Care and Use Guidelines of Sichuan Provincial People’s Hospital.
Preparation of liposomes
XY-4 loaded cationic liposomes were prepared as previously described.34 Briefly, DOTAP, cholesterol and XY-4 were co-dissolved at 15:5:1 mass ratio in chloroform, and the lipid film was formed by evaporation using a rotary evaporation for 2 hours. To remove the residual organic solvent, the flask was placed on a vacuum pump for 20 minutes. The dried film was subsequently rehydrated in distilled water to a final drug concentration of 0.5 mg/mL, which was finally filtered through a 0.45 μm nylon filter to remove unloaded XY-4.
XY-4/Bcl-xl siRNA co-loaded cationic liposomes (XY-4/siBcl-xl-CLP) were prepared by further incubating Bcl-xl targeting siRNAs with prepared XY-4 loaded cationic liposomes in fresh serum-free medium before administration.
Due to its specific fluorescence property and similar hydrophobic nature to XY-4, coumarin-6 (Sigma-Aldrich Co.) was used as a model drug to evaluate the cell uptake efficiency. The preparation method for coumarin-6 loaded cationic liposomes was the same as that of XY-4 loaded liposomes. The coumarin-6 loaded neutral liposomes were also prepared in a similar way by substituting DOTAP with soybean phospholipid.
Characterization of XY-4 loaded cationic liposomes
Size distribution and zeta potential of the XY-4 loaded cationic liposomes were measured using a Zetasizer Nano ZS (Malvern Instruments, Malvern, UK). Measurements were performed at 25°C after equilibration for 2 minutes. All results were the mean of three test runs.
The concentration of XY-4 was determined by high performance liquid chromatography (HPLC; Waters Alliance 2695; Waters, Milford, MA, USA). The solvent delivery system was equipped with a column heater and a plus autosampler. Detection was carried out on a Waters 2996 detector. Chromatographic separations were performed on a reversed phase C18 column (4.6×150 mm-5 mm, Sunfire Analysis column; Waters) with the column temperature kept at 28°C. Methanol/water (75/25, v/v) was used as eluent at a flow rate of 1 mL/min.
Drug loading (DL) and encapsulation efficiency (EE) of prepared liposomes were determined as follows. Briefly, 10 mg of lyophilized XY-4 loaded cationic liposomes were dissolved in 0.1 mL dichloromethane (DCM) and diluted with methanol. The amount of XY-4 in the solution was determined by HPLC. Finally, the DL and EE of XY-4 loaded cationic liposomes were calculated according to Equations 1 and 2:
|
|
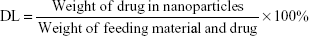
|
|
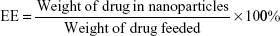
In vitro release study
In vitro release behavior of XY-4 loaded liposomes was studied using a dialysis method. XY-4 cationic liposomes and equivalent free XY-4 were loaded into dialysis tubes (molecular weight cutoff 3 kDa) which had been soaked in PBS, and the ends were tightened. The dialysis tubes were incubated in 30 mL PBS (pH =7.4) containing 0.5% of Tween-80 at 37°C with gentle shaking. Release media were collected at a fixed time point and replenished with the same amount of fresh dialysis solution. The samples were determined by HPLC to analyze the concentration of XY-4. All results were the mean of the three samples.
In vitro gene transfection
B16 cells were seeded into 6-well plates at a density of 5×104 cells per well in 2 mL DMEM medium containing 10% FBS, before transfection. The 5′-carboxyfluorescein labeled siRNA (FAM siRNA) labeled negative control siRNA was used as a reporter gene. After replacing the medium with 1 mL of fresh serum-free medium, gene transfer complexes including 100 pmol of siRNA in serum-free medium were pipetted into each well, and the mass ratio of liposome/siRNA and polyetherimide 25K (PEI25K)/siRNA were 5:1 and 2:1, respectively. After incubation for 4 h, the media were replaced by complete medium. Twenty-four hours later, pictures of each well were taken under a microscope and luciferase activity was measured by flow cytometry (Epics Elite ESP; Coulter Corp., Miami, FL, USA).
In vitro gene silencing study
siRNA targeting mouse Bcl-xl (target sequence: 5′-AAG GAU ACA GCU GGA GUC AGU-3′), scrambled siRNA (target sequence: 5′-AAU UCU CCG AAC GUG UCA CGU-3′) and FAM-labeled negative control siRNA were purchased from GenePharma Co., Ltd (Shanghai, People’s Republic of China) in unprotected, desalted, annealed form.
To determine the level of Bcl-xl mRNA, total RNA was extracted from B16 cells using TRIzol™ Reagent (Thermo Fisher Scientific, Waltham, MA, USA), and individual cDNAs were synthesized with a SuperScript II reverse transcriptase assay (Thermo Fisher Scientific). Real-time quantitative polymerase chain reaction (PCR) was performed with an SYBR GreenER quantitative PCR SuperMix Universal kit (Thermo Fisher Scientific). Reactions were run with a standard cycling program: 50°C for 2 minutes, 95°C for 10 minutes, 40 cycles of 95°C for 15 seconds, and 60°C for 1 minute, on an AB7500 real-time PCR system (Applied Biosystems, Foster City, CA, USA). The PCR primers to detect mouse Bcl-xl (forward; 5′-TCG GGA TGG AGT AAA CTG GG-3′, reverse; 5′-CCA CGC ACA GTG CCC C-3′) and GAPDH (forward; 5′-ATG GGG AAG GTG AAG GTC G-3′, reverse; 5′-TAA AAG CAG CCC TGG TGA CC-3′) were synthesized and purified by TSINGKE Biological Technology (Chengdu, People’s Republic of China).
Cellular uptake in vitro
B16 mice melanoma cancer cells were cultured on 6-well plates for 24 hours to achieve approximately 70% confluence. Cationic liposomes or neutral liposomes encapsulating coumarin-6 were added to the designated wells. After incubation for 6 hours, the cells were collected for measurement of coumarin-6-derived green fluorescence. The fluorescence was observed using a Zeiss M2Bio fluorescence microscope (Carl Zeiss Meditec AG, Jena, Germany). To identify each cell, we simultaneously stained cell nucleus with blue fluorescent dye Hoechst 33258.
Cell proliferation study
The anti-proliferation abilities of free XY-4, cationic liposome delivered Bcl-xl siRNA, and drug loaded liposomes to B16 cell lines were evaluated by the MTT method. Briefly, B16 cells were plated at a density of 5×103 cells per well in 100 μL of DMEM containing 10% FBS in 96-well plates and grown for 24 hours. Cells were then exposed to a series of free XY-4 or cationic liposome delivered Bcl-xl siRNA or drug loaded liposomes at different concentrations for 72 hours. The viability of cells was measured using the MTT method. Results were the mean of six test runs.
Western blotting analysis
Cellular proteins were extracted by using RIPA cell lysate containing 1% (v/v) phenylmethane sulfonyl fluoride (PMSF) and collected by centrifugation at 13,000 rpm for 15 minutes at 4°C. The total protein concentration was determined using the BCA Protein Assay Kit (Vazyme Biotech Co., Ltd, Nanjing, People’s Republic of China). Proteins were separated using 10% sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis and transferred to a polyvinylidene fluoride membrane. Target proteins were then detected with primary antibodies and horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse secondary antibodies. All these bands are detected on the same gel, and after each exposure we stripped the membrane and incubated it with another antibody. Protein blots were visualized with SuperLumia ECL Plus (Thermo Fisher Scientific).
Flow cytometry assay
The cellular apoptosis and cell cycle status were tested via propidium iodide (PI) staining. In brief, B16 cells were seeded into 6-well plates and exposed to a series of free XY-4, cationic liposome delivered Bcl-xl siRNA, and drug loaded liposomes, separately. The treated cells were collected, washed, and stained with PI at room temperature for 15 minutes. The percentage of apoptotic cells and the property of cell cycle arrest were analyzed by flow cytometry (Becton Dickinson, Franklin Lakes, NJ, USA).
In vivo tumor xenograft study
For the abdominal cavity metastatic model, 1×105 B16 cells were injected subcutaneously into the right flanks of female C57 mice (6–8 weeks old) on day 0. On day 3, the mice were randomized into four groups (five mice per group) and numbered. The four groups were intratumorally injected with five groups of normal saline (control), XY-4 loaded cationic liposome (XY-4: 10 mg/kg), cationic liposome delivered Bcl-xl siRNA (siRNA: 12 μg), or XY-4/Bcl-xl-CLP (siRNA: 12 μg; XY-4: 10 mg/kg) every other day. The weight of mice was recorded every day. Tumor burden was measured every 2 days with a caliper (calculated volume [mm3] =π/6× length × width × width). On day 20, when the mice in the control group were very weak, all mice were euthanized by cervical vertebra dislocation, and their tumors were immediately harvested, weighed, and analyzed.
Histological analysis
The excised tissues were fixed in 4% neutral-buffered formalin solution for more than 24 hours and embedded in paraffin. Sections of the tissues (3–5 μm) were stained with hematoxylin and eosin. This analysis was performed following the manufacturer’s protocol, and the samples were examined with a fluorescence microscope (400×).
A commercially available TdT-mediated dUTP nick-end labeling (TUNEL) kit (Promega, Madison, WI, USA) was used to analyze the apoptotic effects in B16 mouse melanoma xenograft tumor tissues. This analysis was performed following the manufacturer’s protocol.
Statistical analysis
Data are expressed as the mean value ± SD. Statistical analysis was performed with one-way analysis of variance (ANOVA) using SPSS software (IBM Corporation, Armonk, NY, USA). P-values less than 0.05 were considered to be statistically significant.
Results
Preparation and characterization of XY-4/Bcl-xl siRNA co-loaded cationic liposomes
To combine hydrophobic Aurora-A kinase inhibitor XY-4 with Bcl-xl targeting siRNA, we developed a drug and gene co-delivery system based on cationic liposomes. A general view of XY-4/siBcl-xl-CLP is presented in Figure 2. In this system, the hydrophobic XY-4 was encapsulated in the core area of liposomes while positive charges provided by DOTAP were utilized to bind and transfer Bcl-xl targeting siRNA. The prepared liposomes were characterized in detail. As presented in Figure 3A, the prepared liposomes were monodispersed (PDI =0.183) with a mean particle size of 91.3±4.5 nm, indicating that they had a very narrow particle size distribution. The spectrum of co-delivery liposomes is presented in Figure 3B with a zeta potential of 38.5±0.5 mV. The DNA-binding ability of prepared cationic liposomes was evaluated by gel retardation assay, and is shown in Figure 3C. When the N/P ratio was 8, complete retardation of DNA was achieved. It was also found that XY-4 could be efficiently encapsulated into cationic liposomes, creating XY-4 loaded cationic liposomes with encapsulation efficiency of 84.6% and drug loading rate of 4.76%.
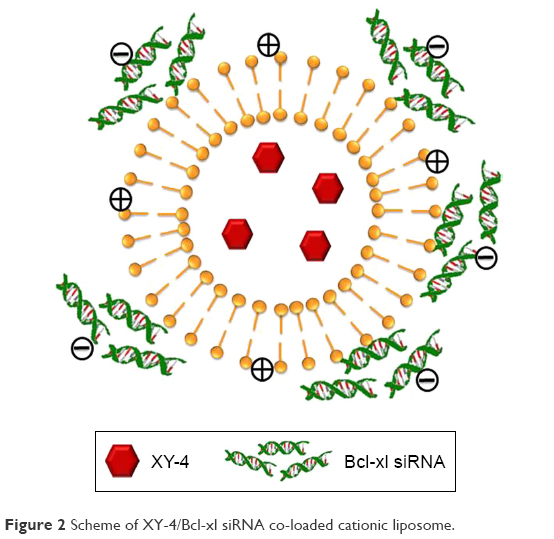 | Figure 2 Scheme of XY-4/Bcl-xl siRNA co-loaded cationic liposome. |
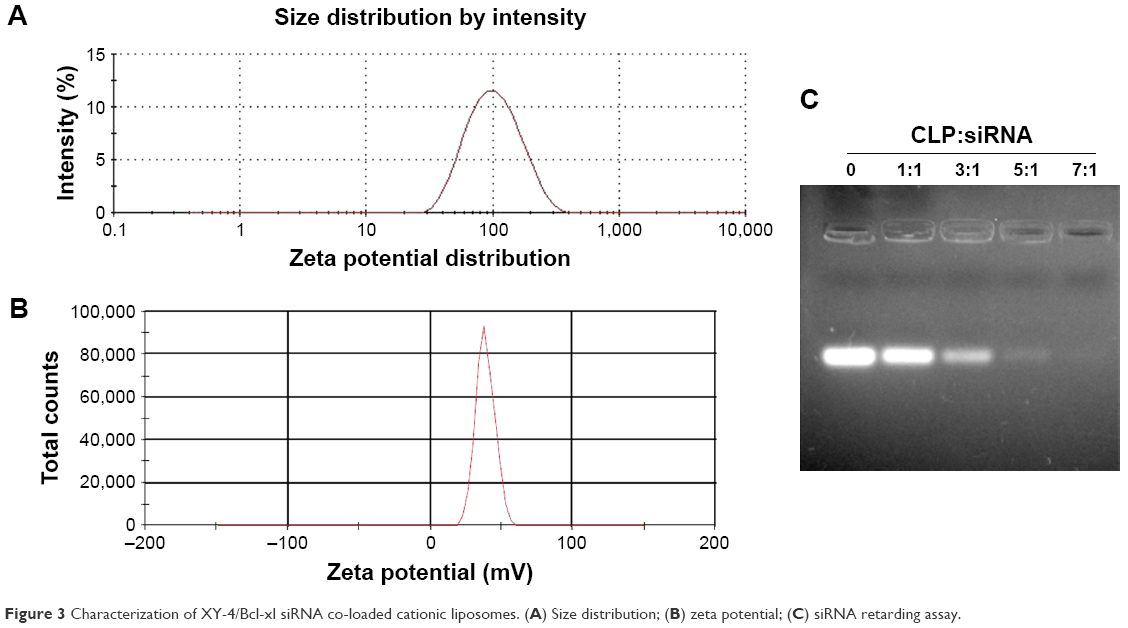 | Figure 3 Characterization of XY-4/Bcl-xl siRNA co-loaded cationic liposomes. (A) Size distribution; (B) zeta potential; (C) siRNA retarding assay. |
To confirm whether cationic liposome-encapsulated XY-4 could be released, the release profile of XY-4 loaded liposomes was studied using a dialysis method in vitro. Our results showed a slow-releasing behavior of XY-4 loaded liposomes in vitro with about 64.9% of total XY-4 released in 4 days. Compared to the free XY-4 group, the release of liposomal XY-4 was much slower (Figure 4).
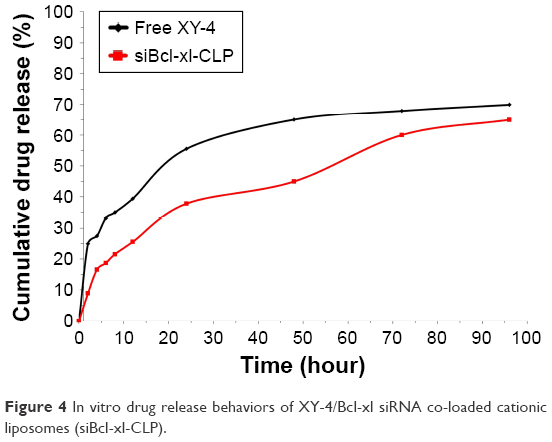 | Figure 4 In vitro drug release behaviors of XY-4/Bcl-xl siRNA co-loaded cationic liposomes (siBcl-xl-CLP). |
Further to biophysical characterization, we compared the transfection efficiency of prepared cationic liposomes with PEI25K (the “gold standard” transfection agent) in vitro. The FAM labelled negative control siRNA was used as a reporter nucleotide. As can be seen from Figure 5A, B16 cells could be efficiently transfected by cationic liposome/FAM negative control (FAM-NC) siRNA complexes. The transfection efficiency on B16 cells was 32%±3.8% compared to that of 40%±2.6% for PEI25K. In addition, Figure 5B and C demonstrate the transfection ability of cationic liposomes with an FAM siRNA-based reporter gene. The high siRNA transfection performance suggested that cationic liposomes may be efficient in siRNA delivery.
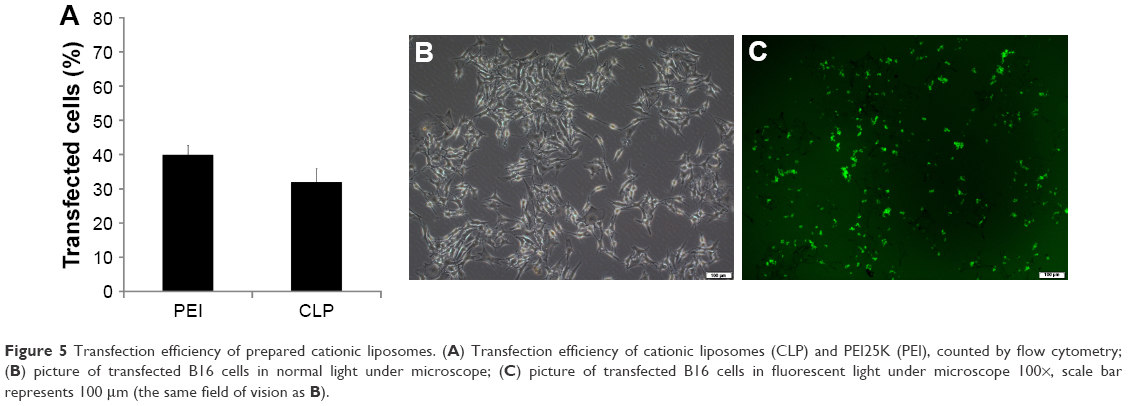 | Figure 5 Transfection efficiency of prepared cationic liposomes. (A) Transfection efficiency of cationic liposomes (CLP) and PEI25K (PEI), counted by flow cytometry; (B) picture of transfected B16 cells in normal light under microscope; (C) picture of transfected B16 cells in fluorescent light under microscope 100×, scale bar represents 100 μm (the same field of vision as B). |
Cellular uptake of cationic liposome delivered cargo by B16 cells
The capacity of cationic liposomes to deliver XY-4 into B16 melanoma cancer cells was determined. The cellular uptake ability of cationic liposomes was compared with that of neutral liposomes in vitro. As a fluorescence probe, coumarin-6 was encapsulated into cationic liposomes or neutral liposomes. After B16 melanoma cancer cells were exposed to coumarin-6 containing cationic liposomes or neutral liposomes for 2 hours, cells were collected for analysis of coumarin-6-derived fluorescence. As shown in Figure 6, B16 cells took up cationic liposomes in preference to neutral liposomes. After incubation for 2 hours, more cells with stronger green fluorescence were observed in the former group. These results suggested that cationic liposomes were more efficient than neutral liposomes for delivering their cargo to melanoma cells. This result is consistent with our previous reports in which bladder cancer cells exhibited higher drug cell uptake when treated with cationic polymeric nanoparticles rather than neutral nanoparticles. In this work, our results also indicated that the cationic surface of cationic liposomes could improve the cellular uptake of cargos, suggesting that the delivery of hydrophobic XY-4 into cancer cells was facilitated by the cationic carrier.
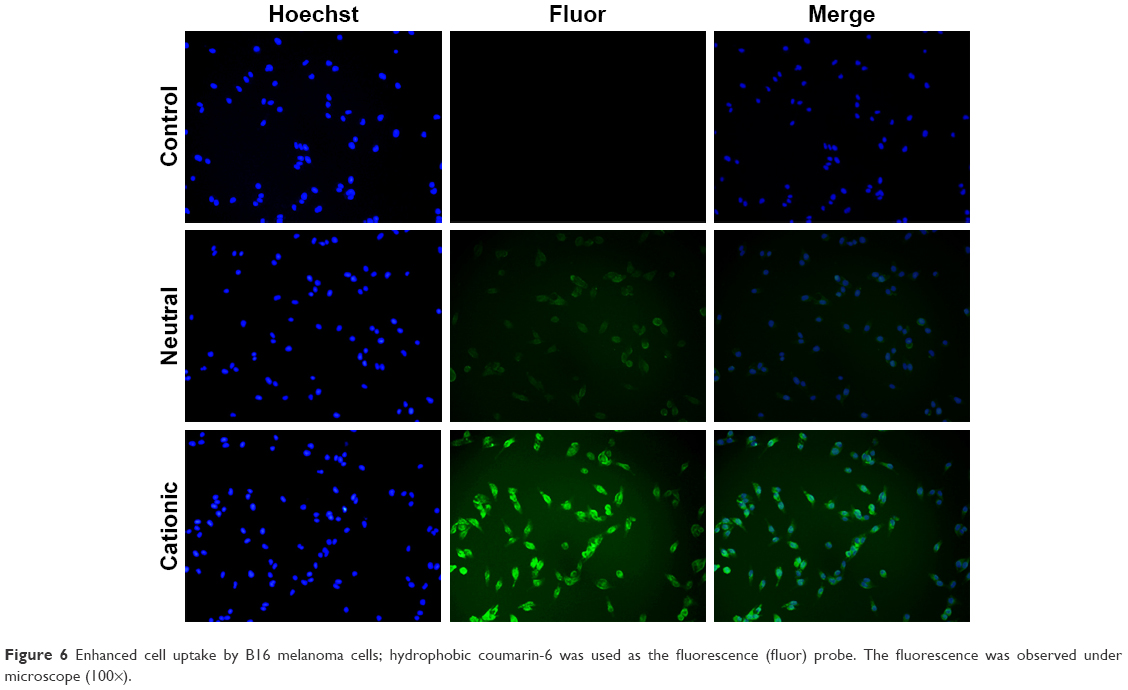 | Figure 6 Enhanced cell uptake by B16 melanoma cells; hydrophobic coumarin-6 was used as the fluorescence (fluor) probe. The fluorescence was observed under microscope (100×). |
Anticancer activity in vitro
To further enhance the therapeutic ability of XY-4, the combination of XY-4 with Bcl-xl targeting siRNA was analyzed. As an Aurora-A kinase inhibitor, XY-4 is capable of inducing cell cycle arrest in cancer cells. Thus, we first evaluated this property on B16 melanoma cells by flow cytometry. As shown in Figure 7A, after 24 hours’ exposure to XY-4, a significant inhibition of cell cycle progression could be observed.Compared with the control group, the percentages of G2/M fraction in the XY-4 treated group were increased from 11.8% to 79% (p<0.05). Meanwhile, the cell cycle distribution of XY-4 treated cell in G1 and S phase were obviously decreased (p<0.05), demonstrating significant G2/M phase arrest (Figure 7B). We studied the potential anti-cancer ability of XY-4 on B16 melanoma cells in vitro. As presented in Figure 8A, according to our MTT results, XY-4 could inhibit the proliferation of B16 melanoma cells with a IC50 of 5.5 μM. On the other hand, in this study, a Bcl-xl targeting siRNA was designed to silence the expression of Bcl-xl gene. As a member of the Bcl-2 family of proteins, Bcl-xl acts as an anti-apoptotic protein by preventing the release of mitochondrial contents such as cytochrome c, which leads to caspase activation.35 According to our real-time quantitative PCR (qPCR) results, the designed siRNA (50 nM and 100 nM) delivered by cationic liposomes efficiently reduced Bcl-xl mRNA level (p<0.05, Figure 8B). As expected, after 72 hours exposure, the cationic liposome delivered Bcl-xl siRNA (50 nM) showed obvious anti-proliferation ability (Figure 9). We then evaluated the possible enhanced anti-cancer effect of free XY-4 and Bcl-xl siRNA combination on B16 cell lines. In our study, cationic liposome delivered Bcl-xl siRNA (50 nM) was combined with XY-4 at different molar ratios (1:25, 1:50, 1:100, 1:200, 1:400 and 1:800). By evaluating cell proliferation status, the combination treatment showed obvious anti-proliferation effects on B16 cells at the ratio of 1:400 and 1:800 (Figure 9). In these two groups, cell survival rates were much lower than either free XY-4 or the Bcl-xl siRNA group. These results suggested that possible enhanced therapeutic effect might exist by the combination of XY-4 and Bcl-xl siRNA.
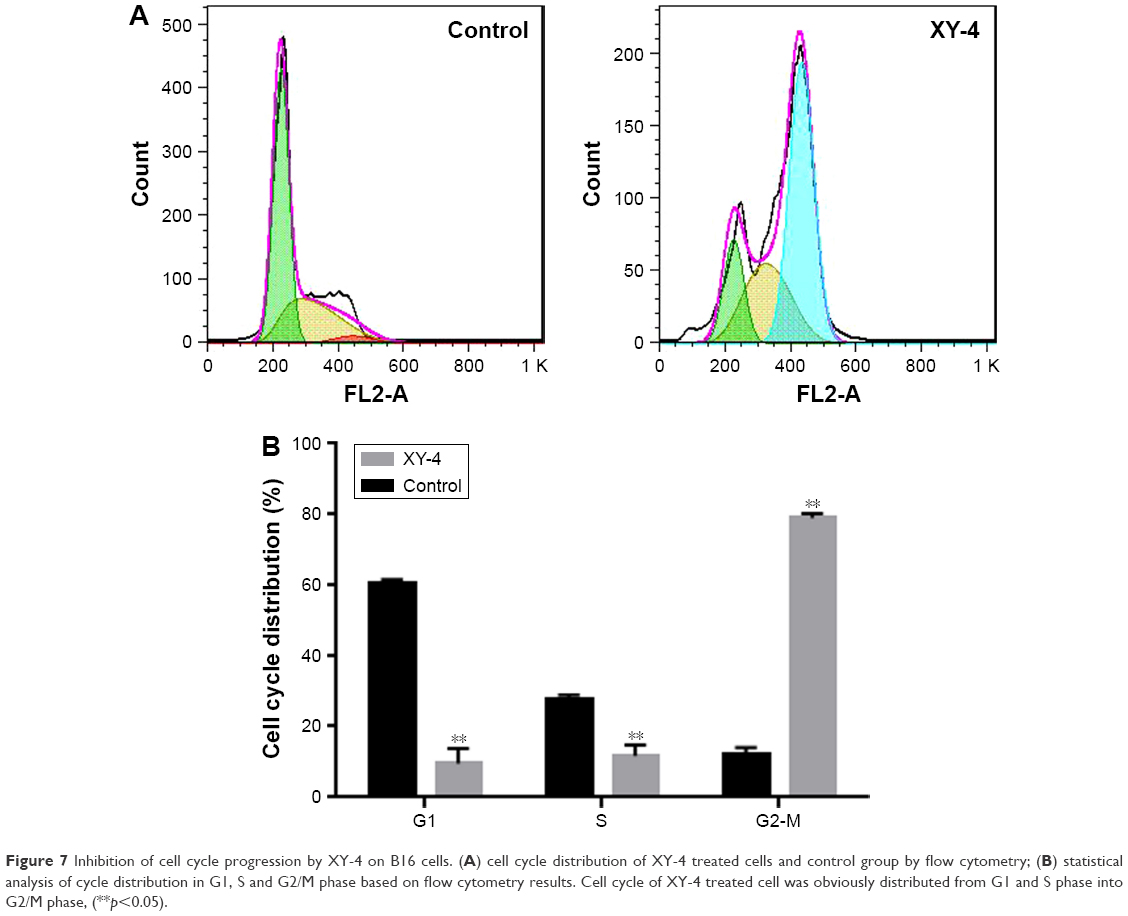 | Figure 7 Inhibition of cell cycle progression by XY-4 on B16 cells. (A) cell cycle distribution of XY-4 treated cells and control group by flow cytometry; (B) statistical analysis of cycle distribution in G1, S and G2/M phase based on flow cytometry results. Cell cycle of XY-4 treated cell was obviously distributed from G1 and S phase into G2/M phase, (**p<0.05). |
 | Figure 8 (A) Anti-cancer abilities of free XY-4 on B16 cells; (B) mRNA level of Bcl-xl was obviously reduced by both two concentration of cationic liposome delivered Bcl-xl siRNA (**p<0.05). |
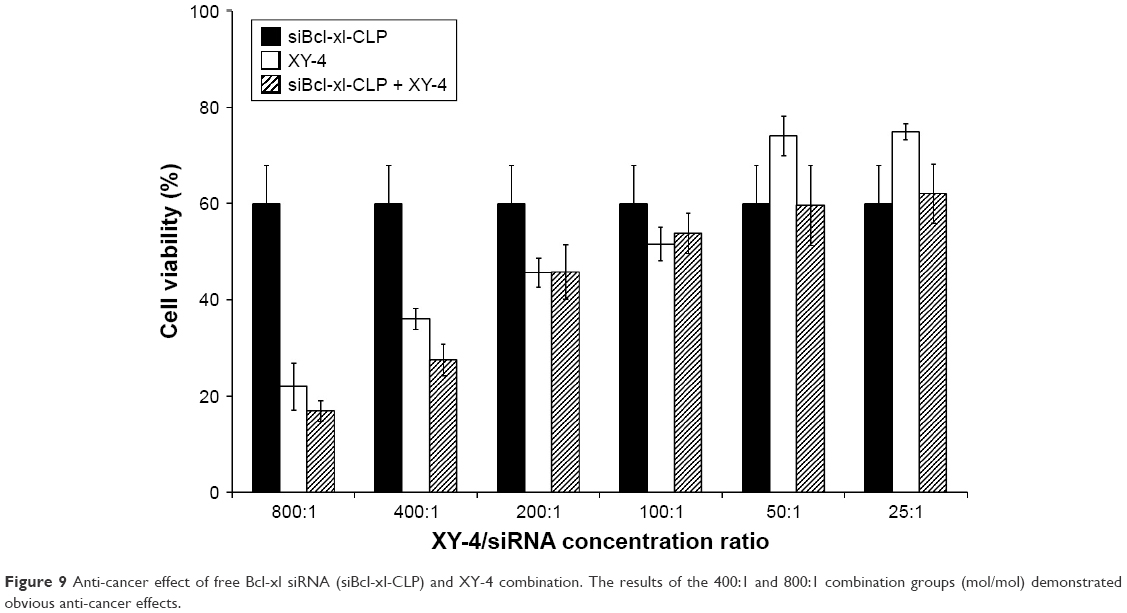 | Figure 9 Anti-cancer effect of free Bcl-xl siRNA (siBcl-xl-CLP) and XY-4 combination. The results of the 400:1 and 800:1 combination groups (mol/mol) demonstrated obvious anti-cancer effects. |
Despite their anti-cancer ability, the poor water solubility of XY-4 still limited the further application of this combination. In order to address this issue, we utilized cationic liposomes to co-deliver XY-4 and Bcl-xl siRNA, forming XY-4/siBcl-xl-CLP. In this formulation, the hydrophobic XY-4 was encapsulated in the core area while Bcl-xl siRNA was bound to the surface by electronic interaction. The anti-cancer abilities of XY-4/siBcl-xl-CLP (Bcl-xl siRNA/XY-4 mass ratio: 1/400; XY-4 concentration: 2.5–80 μM) on the B16 cell line were then studied. According to our results, XY-4/siBcl-xl-CLP showed strong anti-cancer effects in an obvious concentration dependent manner (Figure 10). It showed better anti-cancer effects than free XY-4 or cationic liposome delivered Bcl-xl siRNA (siBcl-xl-CLP) alone on B16 cells. In addition, the XY-4/siBcl-xl-CLP had similar anti-cancer ability to free XY-4 and siBcl-xl-CLP in combination. Our results indicated that co-delivery of the two agents by cationic liposomes not only retained their cancer controlling ability, but also demonstrated enhanced efficacy when administrated together.
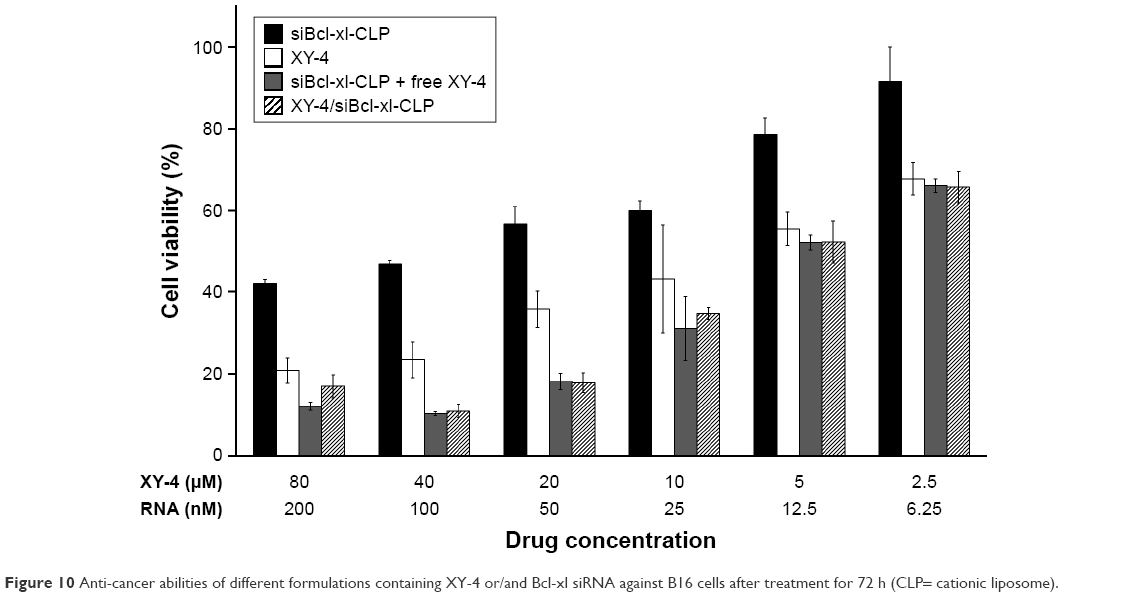 | Figure 10 Anti-cancer abilities of different formulations containing XY-4 or/and Bcl-xl siRNA against B16 cells after treatment for 72 h (CLP= cationic liposome). |
XY-4/Bcl-xl siRNA co-loaded cationic liposomes induced apoptosis in B16 melanoma cells
To determine whether the XY-4/siBcl-xl-CLP-induced loss of proliferation capacity and cell viability of melanoma cancer cells was associated with the induction of apoptosis, B16 cells were treated with free XY-4 (10 μM), siBcl-xl-CLP (Bcl-xl siRNA concentration: 50 nM) and XY-4/siBcl-xl-CLP (10 μM for XY-4 and 50 nM for Bcl-xl siRNA). The numbers of apoptotic cells were assessed using flow cytometry. As shown in Figure 11A and B, after 48 hours of treatment, the apoptosis rates of free XY-4 and siBcl-xl-CLP treated cells were 7.7% and 14.6%, respectively, while that of the XY-4/siBcl-xl-CLP group was 30.4% (vesus 1.5% of control group, p<0.05). The apoptosis assay results were consistent with the MTT results above, demonstrating enhanced effects between XY-4 and Bcl-xl siRNA, and suggesting inducing of apoptosis as a possible mechanism.
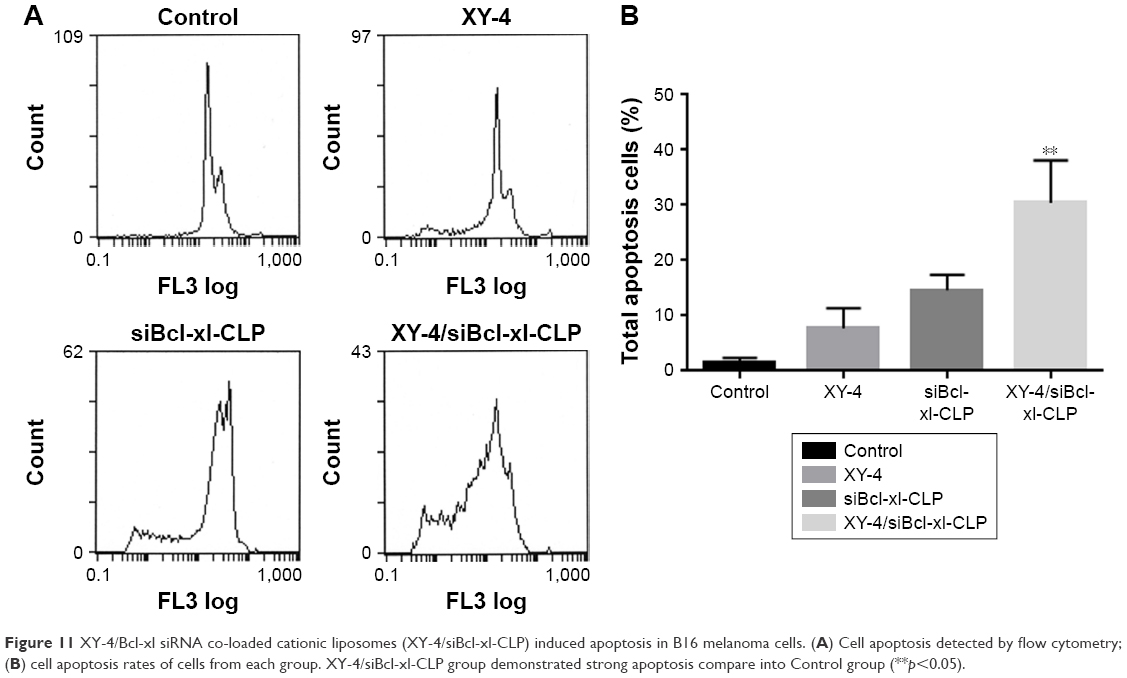 | Figure 11 XY-4/Bcl-xl siRNA co-loaded cationic liposomes (XY-4/siBcl-xl-CLP) induced apoptosis in B16 melanoma cells. (A) Cell apoptosis detected by flow cytometry; (B) cell apoptosis rates of cells from each group. XY-4/siBcl-xl-CLP group demonstrated strong apoptosis compare into Control group (**p<0.05). |
We further studied the possible anti-cancer mechanism by Western blotting. The protein levels of several related proteins suggested that the observed apoptosis might be induced through triggering a mitochondria-dependent apoptotic signaling pathway. As showed in Figure 12, compared to free XY-4 or the siBcl-xl-CLP treated group, XY-4/siBcl-xl-CLP obviously increased the cytochrome c levels in B16 cells, which is a critical step in the mitochondrial-dependent signaling pathways of apoptosis. Treatment of cells with the XY-4/siBcl-xl-CLP group also resulted in an obvious reduction in the levels of the anti-apoptotic proteins Bcl-xl. After the treatment with the combination group, increased protein levels of cleaved caspase-9 and cleaved caspase-3 were observed, while the expression of caspase-9 was decreased. This indicated that the XY-4/siBcl-xl-CLP had stronger effect on activating the mitochondrial-based apoptosis pathway than the single drug formulation. Our results suggested that apoptosis activation might be a possible explanation for the enhanced anti-cancer effects of the co-loaded liposome.
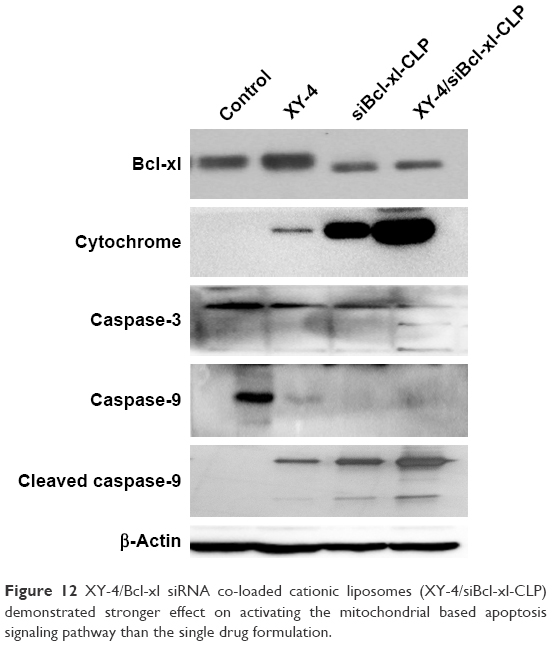 | Figure 12 XY-4/Bcl-xl siRNA co-loaded cationic liposomes (XY-4/siBcl-xl-CLP) demonstrated stronger effect on activating the mitochondrial based apoptosis signaling pathway than the single drug formulation. |
Anticancer effect of XY-4/Bcl-xl siRNA co-loaded cationic liposomes in vivo
We then examined the in vivo antitumor effect of the co-delivering liposomes in C57 mice. The anticancer activity through intratumoral injection on the xenograft model of B16 mouse melanoma is illustrated in Figure 13. According to our results, the mean tumor volume for the control group increased from 100.43±15.44 to 610.6±54.6 mm3, XY-4 loaded cationic liposome group increased from 100.5±14.68 to 358.1±54.92 mm3 and Bcl-xl siRNA group increased from 90.45±15.24 to 451.4±52.92 mm3, whereas the XY-4/siBcl-xl-CLP group only increased from 100±13.52 to 201.83±54.2 mm3, demonstrating higher anticancer efficacy (Figure 13A). At the end of the experiment, the mean tumor weight of the control group, the XY-4 loaded cationic liposome group and the Bcl-xl siRNA group were 0.82±0.09 g, 0.59±0.061 g and 0.54±0.06 g, respectively, while that of XY-4/siBcl-xl-CLP group was 0.25±0.12 g (p<0.01 versus control group, Figure 13B). Compared with other treatment groups, the co-delivery formulation group achieved a statistically significant reduction in tumor weight (P<0.001). In addition, gross changes such as weight loss, ruffling of fur, and changes in behavior were not seen in XY-4/siBcl-xl-CLP treated mice (data not shown). No obvious pathological changes of heart, liver, kidney, lung and spleen were found by histological analysis of tissue sections (Figure 13D). The in vivo gene silencing efficiency of Bcl-xl in each group was also evaluated (p<0.1 versus control group, Figure 13C).
 | Figure 13 Anticancer effects and safety of XY-4/Bcl-xl siRNA co-loaded cationic liposomes on B16 mouse melanoma xenograft model. (A) Tumor development curve, calculated by tumor volume (CLP= cation liposome); (B) average weight of tumors in each group. Compared with other treatment groups, the co-delivery formulation group achieved a statistically significant reduction in tumor weight (***P<0.001); (C) Bcl-xl mRNA levels in tumors from each group. The co-delivery formulation group achieved a statistically significant reduction in mRNA levels (*P<0.1); (D) histological analysis of mouse tissues; (E) TdT-mediated dUTP nick-end labeling (TUNEL) staining of B16 mouse melanoma xenograft. |
To study the mechanism associated with the anticancer activity of XY-4/siBcl-xl-CLP in vivo, a TUNEL assay was carried out. As shown in Figure 13E, numerous positive nucleus identified as apoptosis could be observed in the XY-4/siBcl-xl-CLP treated tumor tissue, whereas such nuclei were rare in other groups. This again implies that apoptosis induction may be an important mechanism of inhibiting melanoma tumor growth. This co-delivery liposomal formulation demonstrated much stronger apoptosis inducing capacity in vivo, which is consistent with our in vitro results above.
These results suggested that intratumoral injection of XY-4/siBcl-xl-CLP could efficiently inhibit the growth of B16 melanoma tumor in vivo through inducing apoptosis. Meanwhile, this co-delivery formulation demonstrated obviously higher therapeutic efficacy with high safety.
Discussion
In our previous work, a novel Aurora-A kinase inhibitor XY-4 with a potential anti-cancer agent was synthesized and characterized, but its hydrophobicity and limited efficiency restricts further development and application. In this study, co-delivered of XY-4 and Bcl-xl targeted siRNA by cationic liposomes created an injectable formulation for combinational therapy. The prepared liposomes successfully rendered XY-4 completely soluble in water solutions with controlled released behavior in vitro and efficient siRNA delivery capacity. By inducing apoptosis and cell cycle arrest, this co-delivery formulation demonstrated enhanced anti-melanoma ability both in vitro and in vivo.
Cell cycle kinase inhibitors are widely recognized as efficient anti-cancer agents, and the newly synthesized Aurora-A kinase inhibitor XY-4 has exhibited its potency in our previous work. However, XY-4 is water insoluble, which greatly limits its further administration. Meanwhile, its therapeutic potency should be further improved. Thus, there is a great need for developing better aqueous formulations and administration strategies for XY-4. A liposomal drug delivery system, which has potential application in drug delivery, can optimize strategies for solving the issue of poor water solubility of hydrophobic drugs. Liposome encapsulated drugs demonstrate several advantages such as high stability and carrier capacity, prolonged systemic circulation lifetime, controllable drug release as well as multi-drug encapsulation potential.30–33 In addition, cationic liposomes with positive charges are capable of co-delivering chemotherapy agents and genes including plasmid DNA and RNA, demonstrating strong potential in chemotherapy and gene therapy.31,36,37 On the other hand, combined therapy has been proven to be an alternative strategy for enhancing therapeutic ability. In addition to delivering single therapeutics, simultaneously incorporating and delivering multiple types of therapeutic payloads for combined therapy might potentially create enhanced or synergistic effects to eventually improve overall treatment outcomes.38,39 In reported pre-clinical and clinical studies, several cell cycle kinase inhibitors have been co-administrated with other chemotherapeutics, achieving enhanced actions including improved anticancer effects and safety.10,20–25 Compared to chemical drugs, inhibitory nucleotides such as siRNA and shRNA could bind to targeted genes with high specificity, offering higher safety and efficacy for combination strategy. Cationic liposomes with gene delivery properties are therefore a promising platform for this strategy. In previous reports, several studies have indicated the advantages of drug and gene combination in cancer therapy, especially with siRNA.40 For example, Yang et al41 developed a folate receptor targeted co-delivery system, folate-doxorubicin/Bmi1 siRNA liposome, and studied its combinational anti-cancer ability. They reported that the prepared liposomal combination system inhibited tumor growth by a combined role of Bmi1 siRNA and DOX both in vitro and in vivo, showing significantly higher efficacy than sole delivery. Meanwhile, higher accumulation of the siRNA and DOX in tumor cells was also achieved by liposomal co-delivery. Their results suggested that chemotherapy based cancer treatment can be further improved by co-delivery of gene drugs through liposomes. In our study, we used DOTAP modified liposomes to co-deliver Aurora-A kinase inhibitor XY-4 and Bcl-xl targeting siRNA through direct encapsulation and electronic interaction. In this process, XY-4 was expected to be encapsulated in the core area of liposomes while siRNA was bound to the surface. The obtained XY-4/siBcl-xl-CLP were monodispersed with small particle size, high encapsulation efficiency and drug loading rate, suggesting optimized solubility. The prepared liposomes could deliver Bcl-xl targeting siRNA into melanoma cells with high transfection efficiency. It is worth noting that we have previously indicated that cell uptake of encapsulated drugs would be enhanced by positive charges of certain cationic carriers. In this study, this phenomenon is also verified by cationic liposomes. According to our results, obvious enhancement of cell uptake was successfully observed. Combined with other in vivo and in vitro anti-cancer results, our study suggested that the positive property of XY-4/siBcl-xl-CLP might contribute to efficient cargo uptake and thus improved therapeutic ability. On the other hand, the in vitro release behavior of co-delivery liposomes demonstrated sustained drug releasing properties, which might further contribute to their safety and drug efficacy. Thus, our results showed that XY-4/siBcl-xl-CLP was an ideal aqueous formulation for combination therapy.
Most chemotherapeutic agents including cell cycle kinase inhibitors act on specific molecular targets of cancer cells. However, regulation of cell cycle and proliferation involve a complex network of signaling pathways and are facilitated by multiple genetic abnormalities in these pathways. Therefore, it is less likely that inhibition by one specific therapeutic agent such as Aurora kinase will be fully effective in curing a patient who has cancer. For this reason, in previous studies, combinational therapeutic approaches using different cell cycle kinase inhibitors have been tested. For example, Qi et al25 reported combining Aurora-A inhibitor MLN8237 with docetaxel in a leukemia lymphoma therapy study, and observed statistically enhanced cell apoptosis, significant tumor growth inhibition and enhanced survival compared to single agent therapy. Santo et al24 combined Aurora kinase inhibitor AT9283 with lenalidomide in multiple myeloma treatment. Their strategy showed significant synergistic cytotoxicity with increased inhibition of phosphorylated STAT3 and phosphorylated extracellular signal-regulated kinase. Shimomura et al23 also observed enhanced antitumor activities in multiple tumor cell lines by combining Aurora-A inhibitor MK-5108 with docetaxel, offering a new therapeutic approach to existing taxane therapies. In the above studies, Aurora kinase inhibitors were coupled with chemotherapeutics to acquire additional actions including mitosis inhibition, angiogenesis inhibition or immunomodulation. Although successful enhanced effects have been observed in these efforts, potential side effects still existed. For example, paclitaxel is known for its side effects of nausea, vomiting, thinned or brittle hair, and pain;42 while lenalidomide can cause teratogenesis, thrombosis and pulmonary embolus.43,44 These side effects cannot be ignored during clinical application. Compared to chemotherapeutics, small interfering RNAs silence target genes specifically, with the resulting mRNA degrading after transcription. In our study, in order to induce an additional apoptosis effect, Aurora-A kinase inhibitor XY-4 was combined with Bcl-xl targeting siRNA, which is commonly recognized to silence the anti-apoptotic protein. As expected, both apoptosis induction and cell cycle arrest of B16 melanoma cells have been indicated by our results. The combination of XY-4 and Bcl-xl siRNA demonstrated enhanced anti-cancer effect in vitro with enhanced anti-proliferation ability. Moreover, our preliminary mechanism studies revealed that XY-4/siBcl-xl-CLP had stronger effect on activating the mitochondrial-based apoptosis pathway than the single drug formulation. Intratumoral injection of this co-delivery formulation efficiently inhibited the growth of B16 melanoma xenograft model in vivo, which also indicated the obviously enhanced anti-cancer effect.
Conclusion
In this study, Aurora-A kinase inhibitor XY-4 and Bcl-xl targeting siRNA were co-delivered into cationic liposomes. Encapsulation of XY-4 makes it monodispersed in water solution, forming an injectable formulation. The prepared XY-4/siBcl-xl-CLP showed stability, slow-releasing behavior, high drug loading capacity and siRNA transfection efficiency as well as enhanced cell uptake. Obviously enhanced anti-cancer effect on B16 melanoma was obtained both in vitro and in vivo through gene and drug co-delivery. It is further suggested that the combination of XY-4 and Bcl-xl siRNA induced cell apoptosis through a mitochondrial based signaling pathway. Our results demonstrated that this XY-4/Bcl-xl siRNA co-delivery strategy and its formulation could have potential advantages for the treatment of melanoma.
Acknowledgments
This work is financially supported by the National Natural Science Foundation of China (No 81302643), the National Key Specialty Construction Project of Clinical Pharmacy (No 30305030698), Youth Fund Projects of Sichuan Province (No 2017JQ0038), Construction of the Second Batch of National Chinese Medicine Clinical Research Base Research Subject (No JDZX2015210) and the Health and Family Planning Research Program of Sichuan Province (No 16PJ488).
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. | ||
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007;445(7130):851–857. | ||
Lapenna S, Giordano A. Cell cycle kinases as therapeutic targets for cancer. Nature Rev Drug Discov. 2009;8(7):547–566. | ||
Vermeulen K, Van Bockstaele DR, Berneman ZN. The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 2003;36(3):131–149. | ||
Bertoli C, Skotheim JM, de Bruin RA. Control of cell cycle transcription during G1 and S phases. Nature Rev Mol Cell Biol. 2013;14(8):518–528. | ||
Sherr CJ, Bartek J. Cell cycle–targeted cancer therapies. Ann Rev Cancer Biol. 2016;1:41–47. | ||
Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9(3):153–166. | ||
Pitts TM, Davis SL, Eckhardt SG, Bradshaw-Pierce EL. Targeting nuclear kinases in cancer: development of cell cycle kinase inhibitors. Pharmacol Ther. 2014;142(2):258–269. | ||
Peyressatre M, Prével C, Pellerano M, Morris MC. Targeting cyclin-dependent kinases in human cancers: from small molecules to Peptide inhibitors. Cancers. 2015;7(1):179–237. | ||
Bonifácio BV, da Silva PB, dos Santos Ramos MA, Negri KMS, Bauab TM, Chorilli M. Nanotechnology-based drug delivery systems and herbal medicines: a review. Int J Nanomedicine. 2014;9:1–15. | ||
Nikonova AS, Astsaturov I, Serebriiskii IG, Dunbrack RL, Golemis EA. Aurora A kinase (AURKA) in normal and pathological cell division. Cell Mol Life Sci. 2013;70(4):661–687. | ||
Hirota T, Kunitoku N, Sasayama T, et al. Aurora-A and an interacting activator, the LIM protein Ajuba, are required for mitotic commitment in human cells. Cell. 2003;114(5):585–598. | ||
Gu J, Gong Y, Huang M, Lu C, Spitz MR, Wu X. Polymorphisms of STK15 (Aurora-A) gene and lung cancer risk in Caucasians. Carcinogenesis. 2007;28(2):350–355. | ||
Sakakura C, Hagiwara A, Yasuoka R, et al. Tumour-amplified kinase BTAK is amplified and overexpressed in gastric cancers with possible involvement in aneuploid formation. Br J Cancer. 2001;84(6):824–831. | ||
Dar AA, Goff LW, Majid S, Berlin J, El-Rifai W. Aurora kinase inhibitors-rising stars in cancer therapeutics? Mol Cancer Ther. 2010;9(2):268–278. | ||
Anand S, Penrhyn-Lowe S, Venkitaraman AR. AURORA-A amplification overrides the mitotic spindle assembly checkpoint, inducing resistance to Taxol. Cancer Cell. 2003;3(1):51–62. | ||
Shi J, Xu G, Zhu W, et al. Design and synthesis of 1, 4, 5, 6-tetrahydropyrrolo[3,4-c] pyrazoles and pyrazolo[3,4-b] pyridines for Aurora-A kinase inhibitors. Bioorg Med Chem Lett. 2010;20(14):4273–4278. | ||
Hu C-MJ. Functionalization of nanocarriers for efficient combination drug delivery [dissertation]. San Diego: University of California; 2011. | ||
Dent P, Tang Y, Yacoub A, Dai Y, Fisher PB, Grant S. CHK1 inhibitors in combination chemotherapy. Mol Interv. 2011;11(2):133–140. | ||
Russell MR, Levin K, Rader J, et al. Combination therapy targeting the Chk1 and Wee1 kinases shows therapeutic efficacy in neuroblastoma. Cancer Res. 2013;73(2):776–784. | ||
Chaudhuri L, Vincelette ND, Koh BD, et al. CHK1 and WEE1 inhibition combine synergistically to enhance therapeutic efficacy in acute myeloid leukemia ex vivo. Haematologica. 2014;99(4):688–696. | ||
Shimomura T, Hasako S, Nakatsuru Y, et al. MK-5108, a highly selective Aurora-A kinase inhibitor, shows antitumor activity alone and in combination with docetaxel. Mol Cancer Ther. 2010;9(1):157–166. | ||
Santo L, Hideshima T, Cirstea D, et al. Antimyeloma activity of a multitargeted kinase inhibitor, AT9283, via potent Aurora kinase and STAT3 inhibition either alone or in combination with lenalidomide. Clin Cancer Res. 2011;17(10):3259–3271. | ||
Qi W, Cooke LS, Liu X, et al. Aurora inhibitor MLN8237 in combination with docetaxel enhances apoptosis and anti-tumor activity in mantle cell lymphoma. Biochem Pharmacol. 2011;81(7):881–890. | ||
Lee JJ, Yazan LS, Abdullah CAC. A review on current nanomaterials and their drug conjugate for targeted breast cancer treatment. Int J Nanomedicine. 2017;12:2373. | ||
Kneidl B, Peller M, Winter G, Lindner LH, Hossann M. Thermosensitive liposomal drug delivery systems: state of the art review. Int J Nanomedicine. 2014;9:4387–4398. | ||
Men K, Liu W, Li L, et al. Delivering instilled hydrophobic drug to the bladder by a cationic nanoparticle and thermo-sensitive hydrogel composite system. Nanoscale. 2012;4(20):6425–6433. | ||
Allen TM, Cullis PR. Drug delivery systems: entering the mainstream. Science. 2004;303(5665):1818–1822. | ||
Samad A, Sultana Y, Aqil M. Liposomal drug delivery systems: an update review. Curr Drug Deliv. 2007;4(4):297–305. | ||
Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Deliv Rev. 2013;65(1):36–48. | ||
Pattni BS, Chupin VV, Torchilin VP. New developments in liposomal drug delivery. Chem Rev. 2015;115(19):10938–10966. | ||
Patel N, Panda S. Liposome drug delivery system: a critic review. J Pharm Sci Biosci Res. 2012;2(4):162–194. | ||
Chen X, Wang X, Wang Y, et al. Improved tumor-targeting drug delivery and therapeutic efficacy by cationic liposome modified with truncated bFGF peptide. J Control Release. 2010;145(1):17–25. | ||
Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15(1):49–63. | ||
Balazs DA, Godbey W. Liposomes for use in gene delivery. J Drug Deliv. 2011;2011:326497. | ||
Kesharwani P, Gajbhiye V, Jain NK. A review of nanocarriers for the delivery of small interfering RNA. Biomaterials. 2012;33(29):7138–7150. | ||
He C, Tang Z, Tian H, Chen X. Co-delivery of chemotherapeutics and proteins for synergistic therapy. Adv Drug Deliv Rev. 2016;98:64–76. | ||
Kim J, Kim J, Jeong C, Kim WJ. Synergistic nanomedicine by combined gene and photothermal therapy. Adv Drug Deliv Rev. 2016;98:99–112. | ||
Wei T-YW, Wu P-Y, Wu T-J, et al. Aurora A and NF-κB survival pathway drive chemoresistance in acute myeloid leukemia via the TRAF-interacting protein TIFA. Cancer Res. 2017;77(2):494–508. | ||
Yang T, Li B, Qi S, et al. Co-delivery of doxorubicin and Bmi1 siRNA by folate receptor targeted liposomes exhibits enhanced anti-tumor effects in vitro and in vivo. Theranostics. 2014;4(11):1096–1111. | ||
Cella D, Peterman A, Hudgens S, Webster K, Socinski MA. Measuring the side effects of taxane therapy in oncology. Cancer. 2003;98(4):822–831. | ||
Kumar S, Rajkumar SV. Thalidomide and lenalidomide in the treatment of multiple myeloma. Eur J Cancer. 2006;42(11):1612–1622. | ||
Bwire R, Freeman J, Houn F. Managing the teratogenic risk of thalidomide and lenalidomide: an industry perspective. Expert Opin Drug Saf. 2011;10(1):3–8. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.