Back to Journals » Infection and Drug Resistance » Volume 11
Clustering and recent transmission of Mycobacterium tuberculosis in a Chinese population
Authors Xu G, Mao X, Wang J ![]() , Pan H
, Pan H ![]()
Received 9 November 2017
Accepted for publication 30 December 2017
Published 6 March 2018 Volume 2018:11 Pages 323—330
DOI https://doi.org/10.2147/IDR.S156534
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Eric Nulens
Guisheng Xu,1,* Xuhua Mao,2,* Jianming Wang,1 Hongqiu Pan3
1Department of Epidemiology, Key Laboratory of Infectious Diseases, School of Public Health, Nanjing Medical University, Nanjing, China; 2Department of Clinical Laboratory, Yixing People’s Hospital, Wuxi, China; 3Department of Tuberculosis, The Third Hospital of Zhenjiang, Zhenjiang, China
*These authors contributed equally to this work
Purpose: The objectives of the present study were to characterize the clinical isolates prevailing in the northeast of Jiangsu and to investigate the mode of transmission. The study also aimed to explore the extent to which Mycobacterium tuberculosis strains contributed to drug resistance and the possible factors related to the recent transmission.
Patients and methods: We consecutively enrolled 912 culture-confirmed pulmonary tuberculosis (TB) cases from 1 January 2013 to 31 December 2014 in Lianyungang City, which is located in the center of China’s vast ocean area and the northeast of Jiangsu province. Isolates were genotyped using 15-locus mycobacterial interspersed repetitive unit-variable number tandem repeat (MIRU-VNTR) typing. The Hunter–Gaston discrimination index (HGDI) was used to estimate the discriminatory power and diversity of molecular markers.
Results: Among 741 successfully genotyped isolates, 144 (19.43%) strains formed 46 clusters, while 597 (80.57%) isolates had the unique MIRU pattern. The total HGDI for all 15 loci was 0.999. The average cluster size was 3 (2–13) patients. The estimated proportion of recent transmission was 13.34%. Patients with unfavorable treatment outcomes were infected with clustered strains at a higher proportion than were those with favorable treatment outcomes (adjusted OR: 1.78, 95% CI: 1.14–2.85, P=0.012).
Conclusion: The probability of recent TB transmission was relatively low in the study site, while the cases mainly arose from the activation of previous infection. Spatial analysis showed that strains forming larger clusters had the characteristics of regional aggregation.
Keywords: tuberculosis, molecular epidemiology, genotype, transmission, drug resistance
Introduction
Tuberculosis (TB) remains a global public health threat, especially in developing countries.1 Although TB control has been effective in some regions of the world, the emergence of multidrug-resistant (MDR) and extensively drug-resistant TB during the past decade threatens to undermine these advances.2,3
Molecular epidemiology is increasingly used as a complementary tool to conventional epidemiology.4 Genotyping of Mycobacterium tuberculosis (Mtb) has been used to enhance our understanding of the TB epidemic and determine the routes of transmission. Cases with indistinguishable molecular characteristics of strains more likely contribute to recent transmission, whereas cases caused by strains with unique fingerprints are typically due to the reactivation of latent infection.5 Restriction fragment length polymorphism (RFLP) targeting the insertion sequence 6110 (IS6110) transposable element has been widely used and considered as a gold standard tool. However, this method requires large amounts of high-quality DNA and has poor discriminatory power for isolates with >6 copies of IS6110.6 Other alternative molecular tools include mixed linker PCR (ML-PCR), double-repetitive-element PCR (DRE-PCR), fast ligation-mediated PCR (FliP), spacer oligonucleotide typing (spoligotyping), and mycobacterial interspersed repetitive unit-variable number tandem repeat (MIRU-VNTR) typing, among others.4,6
TB is highly prevalent in China, and studies on the mode of transmission have been carried out in some regions; however, few efforts in fingerprinting analysis have been made based on the mutation profiles of isolates from the east region.7–10 Jiangsu is located in the east part of China, absorbs a large number of migrations and has the highest population density in China.11 Exploring the pattern of Mtb transmission and drug resistance in this area could substantially facilitate local and countrywide TB control. The objectives of the present study were to characterize and classify the clinical isolates prevailing in the northeast of Jiangsu and to investigate the mode of transmission. The study also aimed to explore the extent to which Mtb strains contributed to drug resistance and the possible factors related to the recent spread of TB strains.
Patients and methods
Study setting and participants
This study was conducted in Lianyungang City, which is located in the center of China’s vast ocean area, the northeast of Jiangsu province and the extreme north of the Yangtze River Delta. We have consecutively enrolled 912 culture-confirmed TB cases from 1 January 2013 to 31 December 2014. The Institutional Review Board of Nanjing Medical University approved this study. Written informed consent was obtained from all participants included in the study.
Data collection
Sputum samples from each TB suspect were collected for acid-fast bacilli (AFB) smear microscopy, which was performed at local hospitals. All AFB smear-positive samples were routinely transported to the Fourth People’s Hospital of Lianyungang for culture and drug susceptibility testing (DST). Drug resistance to rifampicin (RIF), isoniazid (INH), ofloxacin (OFLX), and kanamycin (KA) was assessed based on the proportion method as recommended by WHO/IUATLD (International Union against Tuberculosis and Lung Disease). Genomic DNA was obtained by suspending mycobacterial colonies in 100–200 μl distilled H2O and incubating at 85°C for 30 min. All isolates were genotyped using 15-locus MIRU-VNTR typing (Mtub04, ETRC, ETRD, MIRU40, MIRU10, MIRU16, Mtub21, Qub11b, ETRA, Mtub30, MIRU26, ETRE, Mtub39, Qub26, and Qub4156). The primers used to amplify specific loci are described in Table 1. PCR was performed on an amplifier (MJ Research, Inc., Watertown, MA, USA), and the amplification products were separated by agarose gel electrophoresis. The product sizes were estimated by comparison with the 50 bp and 100 bp ladder markers. The copy number per locus was calculated using the method described by Supply et al.12 We applied the “pheatmap” package of R software to perform the clustering analysis.13
 | Table 1 Primers and HGDI for the 15 MIRU-VNTR loci Abbreviations: HGDI: Hunter–Gaston discrimination index; MIRU, mycobacterial interspersed repetitive unit; VNTR, variable number tandem repeat. |
Data analysis
Data were entered and analyzed using SPSS 18.0 software (SPSS Inc., Chicago, IL, USA). Characteristics of cases with different strains were compared using the χ2 or Fisher’s exact test. A multivariate logistic regression model was applied to estimate the effect of factors related to drug resistance, clustering, and cluster size. The test level was set at 0.05. A cluster was defined as two or more isolates from different patients with identical MIRU patterns, whereas nonclustered patterns were referred to as unique. If the cluster contained five or more cases, it was defined as a large cluster. The index case in each large cluster was temporally defined as the first case diagnosed within the cluster. The rate of recent transmission was calculated by the formula: [Tc–Nc]/Ta*100%, where Tc was the total number of clustered isolates, Nc was the number of clusters, and Ta was the total number of isolates. The “pheatmap” package of R software was used to plot the MIRU–VNTR results. The dendrograms based on 15 VNTR locus data were conducted using the unweighted pair group method with arithmetic mean protocol. According to the first-class clustering results, the samples were divided into MIRU-type I and II. The Hunter–Gaston discrimination index (HGDI) for the loci in MIRU-15 sets was used to estimate the discriminatory power of the genotyping method and the diversity of molecular markers.14,15  , where N is the total number of strains, nj is the number of strains of the j genotype, and s is the MIRU-VNTR locus genotype number.
, where N is the total number of strains, nj is the number of strains of the j genotype, and s is the MIRU-VNTR locus genotype number.
Results
We genotyped 912 clinical samples, and 741 (81.25%) isolates provided complete data for the entire MIRU-15 set. Among the 741 TB patients, 135 (18.22%) were previously treated, 274 (36.98%) were aged over 60 years, and 587 (79.22%) were male. DST showed that the drug resistance to RIF, INH, OFLX, and KA was 8.37%, 11.74%, 9.04% and 0.68%, respectively. The proportion of MDR-TB was 5.94%, which was higher among patients aged <60 years (Table 2).
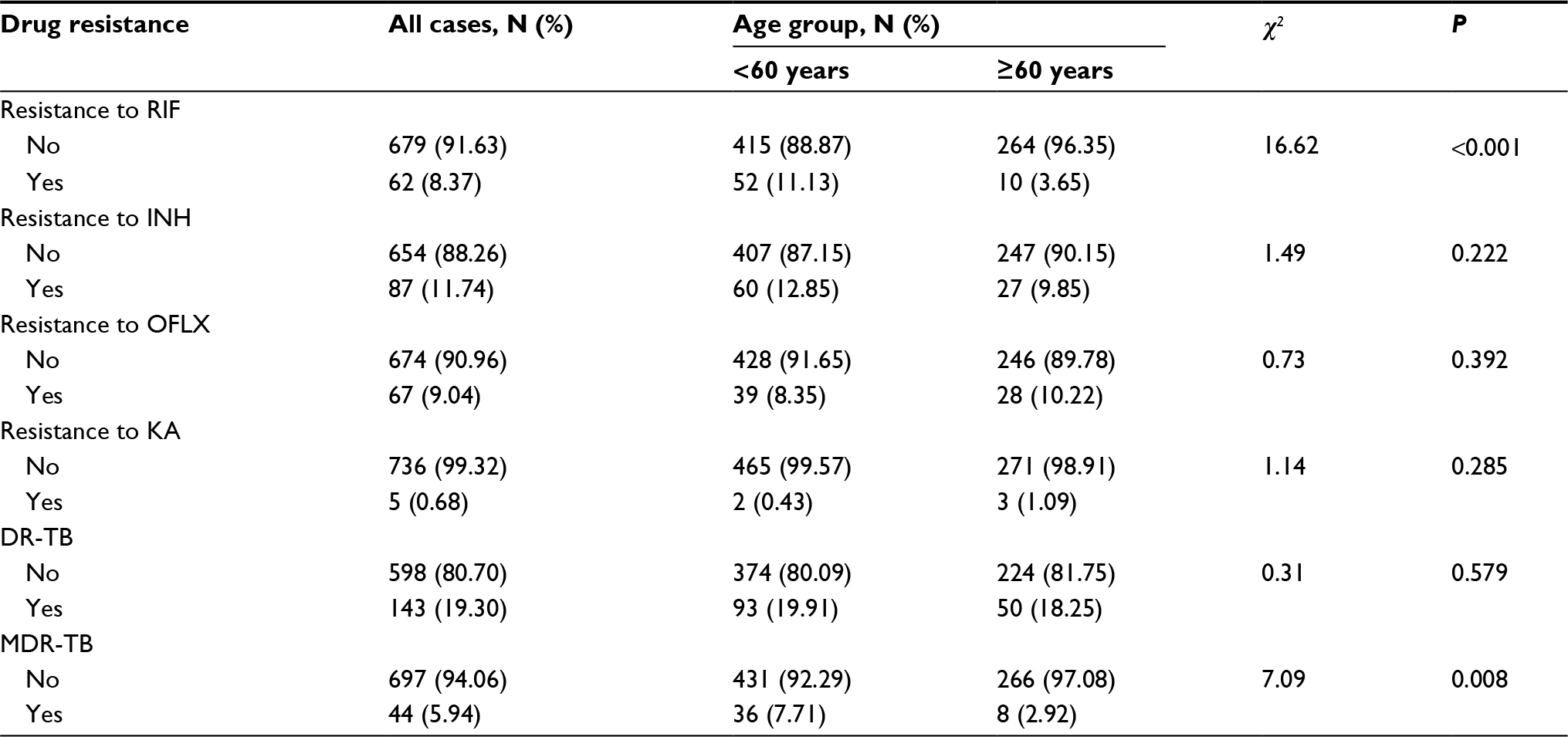 | Table 2 Resistance to antituberculosis drugs stratified by age groups Notes: DR-TB: drug resistance to RIF, INH, OFLX or KA; MDR-TB: drug resistance to RIF and INH. Abbreviations: RIF, rifampicin; INH, isoniazid; OFLX, ofloxacin; KA, kanamycin; DR, drug-resistance; TB, tuberculosis; MDR, multidrug-resistant. |
According to the genotypes of the MIRU-15 loci, strains were categorized into different groups. The total HGDI for all loci was 0.999 (Table 1). The estimated proportion of recent transmission was 13.34%. As shown in Figure 1, two main groups were categorized, and we defined them as MIRU-type I (n=214, 28.88%) and MIRU-type II (n=527, 71.12%). The ETRE, MIRU10, MTUB04, MIRU40, Mtub21, Mtub30, Mtub39, and MIRU26 loci had more repeated sequences in the MIRU-type II group than in the MIRU-type I group. The MIRU-type I and II classification was significantly associated with gender (χ2=6.21, P=0.013), age (χ2=4.67, P=0.031), and clustering (χ2=53.15, P<0.001) (Table 3).
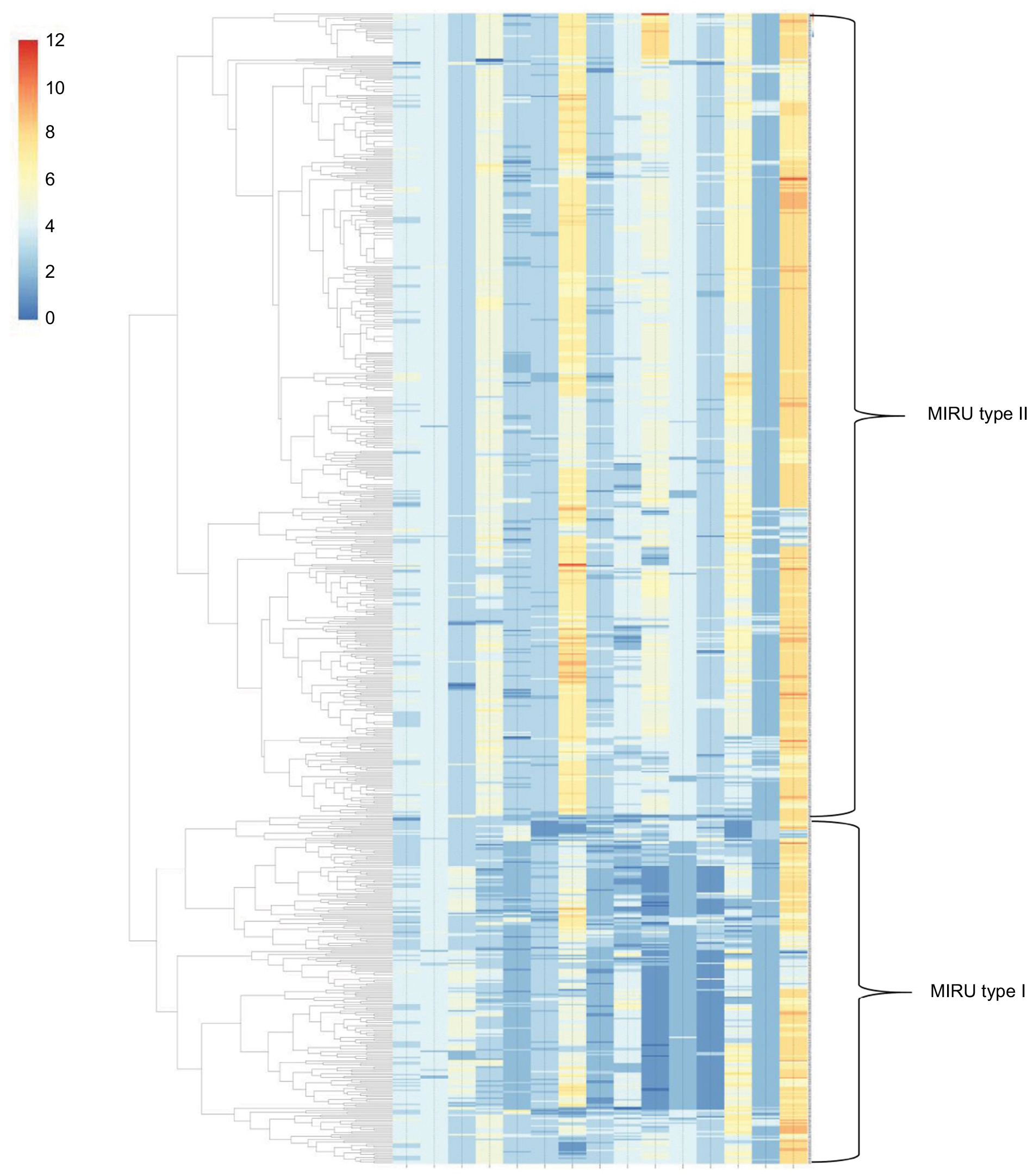 | Figure 1 Dendrogram of 741 Mtb isolates from Lianyungang, China. Note: The phylogenetic tree was generated from the 15-locus MIRU-VNTR profile: ETRA – ETRC – ETRD – ETRE – MIRU10 – MIRU16 – MIRU26 – MIRU40 – MTUB04 – MTUB21 – MTUB30 – MTUB39 – Qub11b – Qub4156 – Qub26. Changes in strip color from dark blue to orange correspond to increasing numbers of repeats, from 0 to 12. Abbreviations: MIRU, mycobacterial interspersed repetitive unit; VNTR, variable number tandem repeat. |
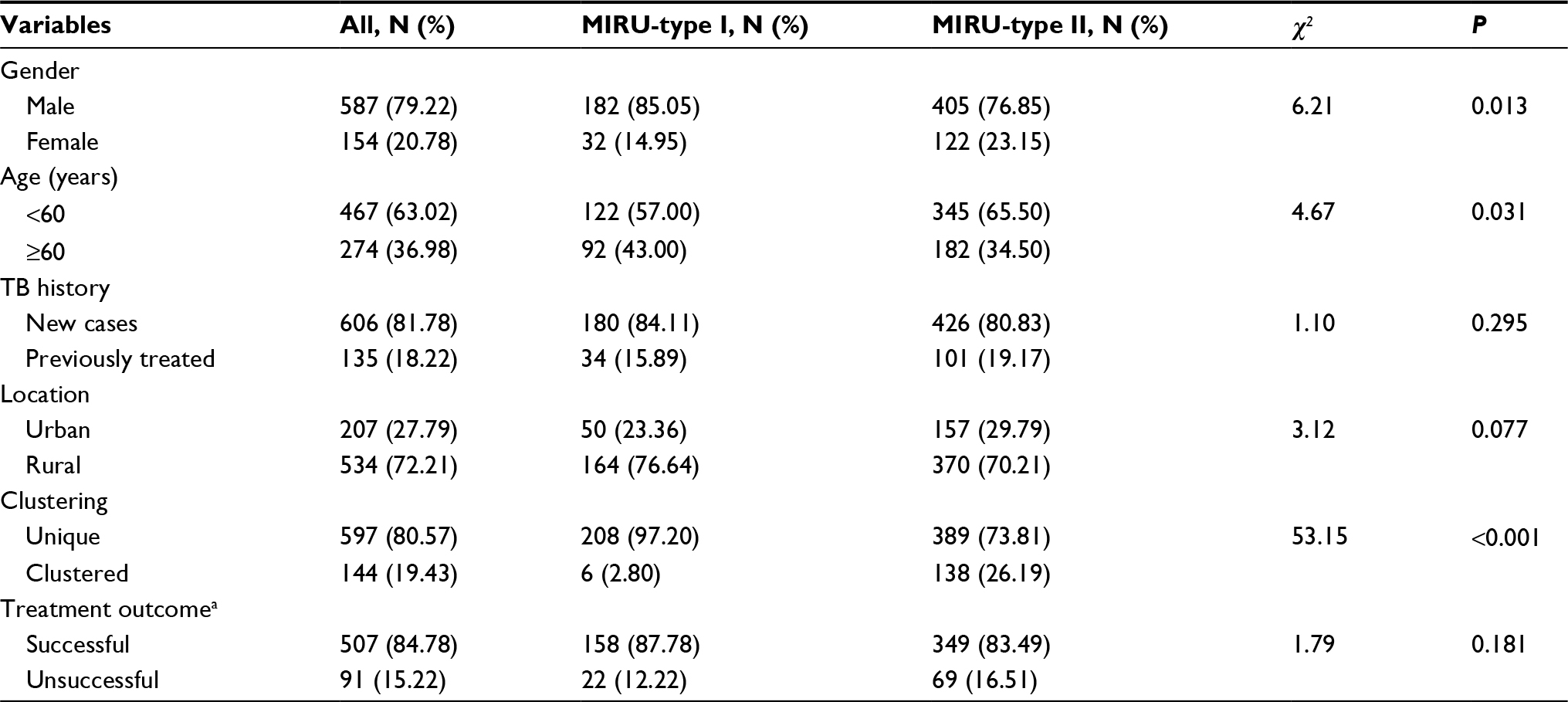 | Table 3 Characteristics of strains genotyped in Lianyungang Note: aWith missing values. Abbreviations: MIRU, mycobacterial interspersed repetitive unit; TB, tuberculosis. |
One hundred forty-four (19.43%) strains formed 46 clusters, while 597 (80.57%) isolates had a unique MIRU pattern (Figure 1). The average cluster size was 3 (2–13) cases. Over 38.19% of clusters contained two cases, and the largest cluster included 13 cases. We have plotted patients in a geographic information system map. There were 9 patients in cluster 1, which was mainly distributed in the north part of Lianyungang. Cluster 2 included 5 patients, all living in the south part of the study area. There were 5 patients in cluster 3, and they were distributed throughout the whole city. Cluster 4 included 12 patients, mainly distributed in the east or south area (Figure 2). We have further categorized 144 clustered strains into two groups: small clusters (including 2–4 cases) and large clusters (including 5 or more cases). No significant factors were observed to be related with cluster size (Table 4).
 | Figure 2 Distribution of large clustered strains. Notes: Circle with date indicates the index case. The red boxes indicate that cluster 1 strains are located in the north part while cluster 3 strains are located in the south part. Abbreviation: GIS, geographic information system. |
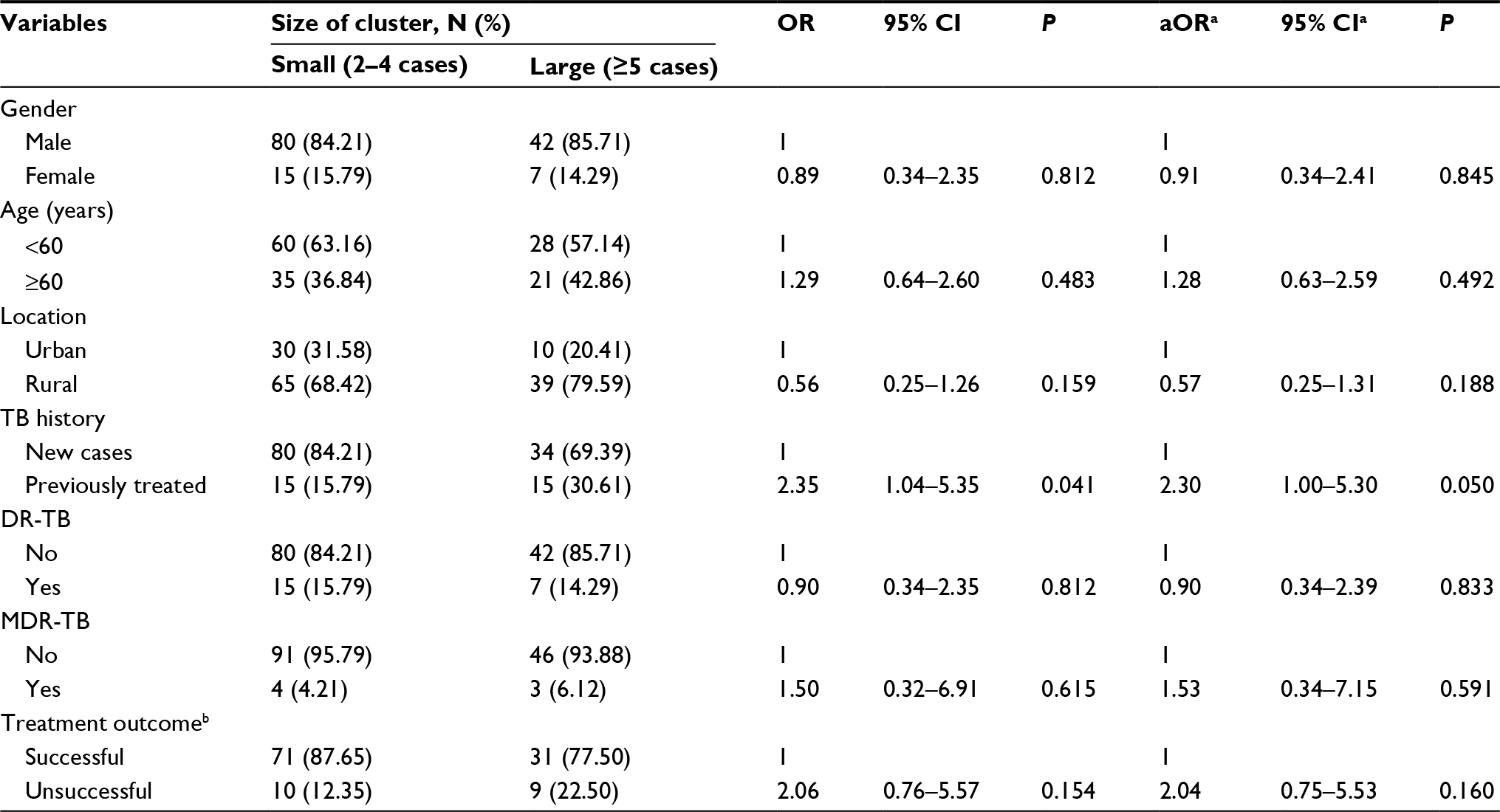 | Table 4 Factors related to cluster size Notes: aAdjusted for age and gender; bwith missing values; DR-TB: drug resistance to RIF, INH, OFLX or KA; MDR-TB: drug resistance to RIF and INH. Abbreviations: TB, tuberculosis; DR, drug-resistance; MDR, multidrug-resistant; MIRU, mycobacterial interspersed repetitive unit; RIF, rifampicin; INH, isoniazid; OFLX, ofloxacin; KA, kanamycin. |
As shown in Table 5, strains in the MIRU-type II group were more likely to form a cluster (adjusted OR: 13.25, 95% CI: 5.73–30.62, P<0.001). Patients with unfavorable treatment outcomes were infected with clustered strains at a higher proportion than were those with favorable treatment outcomes (adjusted OR:1.78, 95% CI: 1.14–2.85, P=0.012).
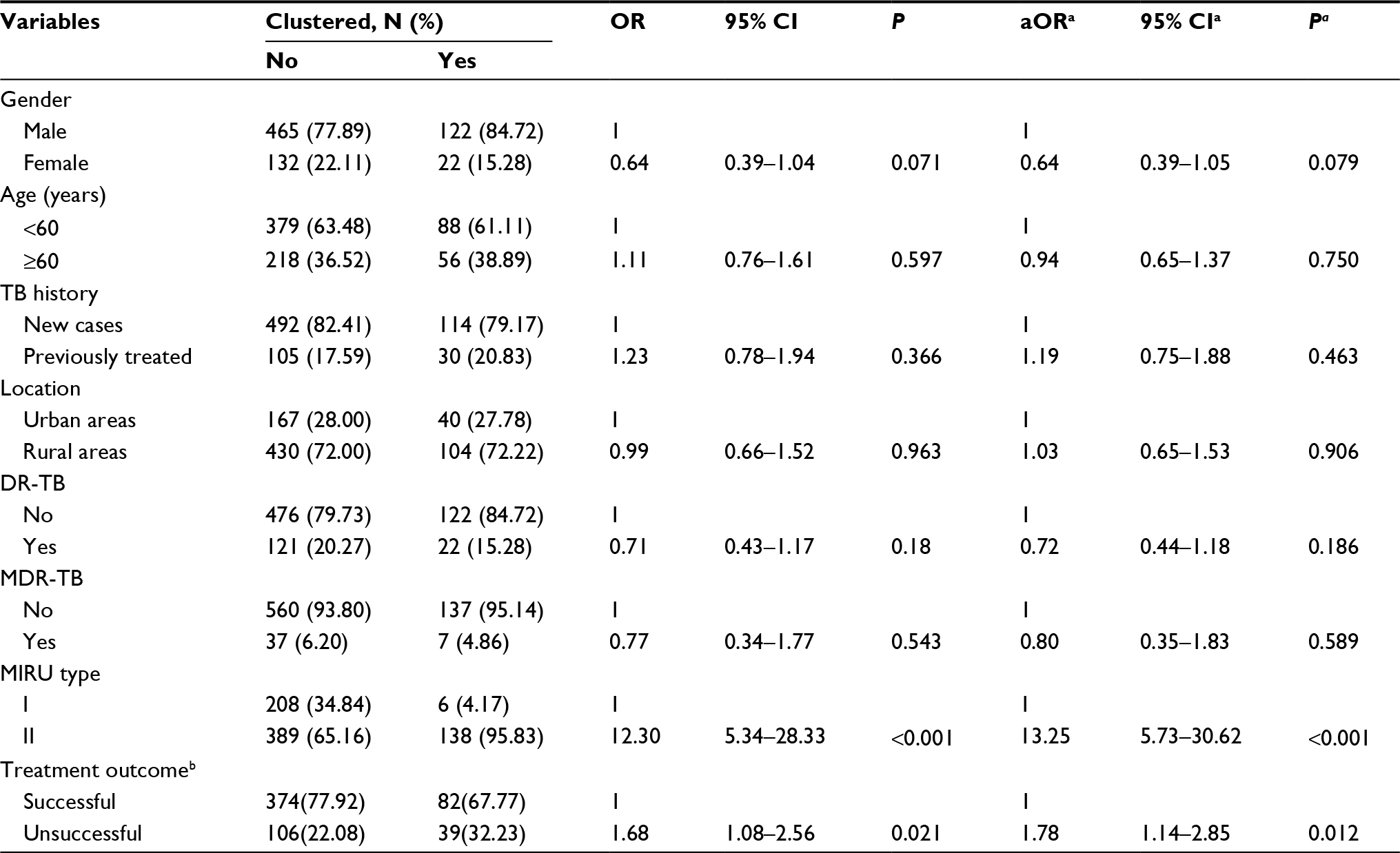 | Table 5 Factors related to the clustering of strains Notes: aAdjusted for age and gender; bwith missing values; DR-TB: drug resistance to RIF, INH, OFLX or KA; MDR-TB: drug resistance to RIF and INH. Abbreviations: TB, tuberculosis; DR, drug-resistance; MDR, multidrug-resistant, RIF, rifampicin; INH, isoniazid; OFLX, ofloxacin; KA, kanamycin. |
Discussion
We have consecutively enrolled TB cases from one region of China and genotyped them using the 15-locus MIRU-VNTR. The proportion of clustering in the study area was 19.43%, and the recent transmission rate was 13.34%, which was lower than that reported in some developed countries. For example, a study conducted in London reported that 46% were clustered and the estimated proportion attributable to recent transmission was 34%.16 Another study performed in the UK reported that the proportion of cases attributable to recent transmission was 15%.17 In the Netherlands, 46% of strains were found in clusters with identical RFLPs, and 35% were attributed to active transmission.18 In the United States, recent transmission was estimated to be 48% from 1996 to 2000 but decreased to 23% in 2005–2009.19,20 The proportion of recent transmission was relatively low in our study, indicating the higher possibility of recrudescence in the study area. This may partly contribute to the higher prevalence of latent infection in the Chinese population.21 In 2013, a population-based study conducted in rural China reported that the age-standardized and sex-standardized rates of skin-test positivity (≥10 mm) ranged from 15% to 42%, and the QuantiFERON positivity rates ranged from 13% to 20%.22 Another cross-sectional study that was undertaken in eastern of China reported that the positive rate of the interferon-gamma release assay was 19.98%.
According to the genotypes of MIRU-15 loci, two main groups were categorized (MIRU-type I and II). The classification was significantly associated with gender and age. This finding might be explained as different recent transmission rates among specific populations. Previous studies have reported that recent TB infection is associated with a variety of factors, including youth, male sex, belonging to an ethnic minority, homelessness, drug use, excessive alcohol consumption, homelessness, and previous incarceration.23 Factors related to clustering varied greatly between areas. In a UK study conducted by Hamblion et al, the clustered patients were more likely to be born in the UK, to have been diagnosed with pulmonary TB, to have a previous TB history, to have issues with substance abuse or alcohol abuse, and to have been imprisoned.16 Another UK study conducted by Anderson et al found that being born in the UK and using illicit drugs were significantly associated with clustering.17 Using the 15-locus MIRU method, a study in Jiangxi province, China, reported that patients who failed treatment or patients with MDR isolates were more likely to be in clusters.24 In our study, we observed that clustered cases were more likely to have a unfavorable treatment outcome. With the extension of sputum bacteriological conversion, the risk of ongoing transmission among the community also increased. Compared with that in other studies, the size of clustered cases was small in the current study, which may result in a relatively small sample size over a short time period.
Strain genotyping data, when combined with epidemiological data, enabled the identification of TB patients involved in the same chain of transmission. We assume that cases sharing the same strain (in the same cluster) were recently infected in Lianyungang. We have plotted cases forming large clusters on the map. Cases in cluster 1 or 2 mainly lived together in a small area. We hypothesized that the strains were probably transmitted through the same source of infection.25 The MIRU-VNTR loci and drug resistance have been explored in previous studies. For example, the ETRB locus was associated with INH resistance,26 and the MIRU20 locus was associated with EMB resistance.27,28
In 1997, Supply et al identified a novel minisatellite-like structure in the Mtb genome and named such structures MIRUs.29 These MIRUs are located mainly in intergenic regions and are dispersed throughout the Mtb genome. In 2001, Supply et al proved the usefulness of MIRUs for epidemiologic study by conducting PCR analysis of 12 variable tandem repeat loci with specific primers followed by gel electrophoresis.12 Supply et al proposed a 15-locus system as a new standard for routine epidemiological discrimination of Mtb isolates and a 24-locus system as a high-resolution tool for phylogenetic studies.30 Although several optimal loci have been suggested for genotyping homogenous Mtb,16,31,32 15 loci have been widely used in China.33–35 In the current study, the total HGDI for all 15 loci was 0.999. The locus Qub11b had the highest HGDI (0.762), while the HGDI of ETRC was very low (0.036). Identical MIRU-VNTR patterns are considered to be in a cluster. It was reported that the proportion of clustering decreased with a greater number of MIRU-VNTR loci typed, with increasing TB incidence and with increasing maximum cluster size.36
Limitations
Our study has some limitations. First, samples were collected over a relatively short time period. The probability of clustering might be underestimated. A long-term cohort study can solve this problem. Second, China has a high proportion of the Beijing lineage of Mtb. In this study, we did not analyze the lineage of the Beijing family. Third, epidemiological data are essential for identifying the route of transmission. In this study, we lack necessary information such as the history of contact with the patients or the frequency of visiting areas with high population density. Thus, we could not clearly explain the chain of transmission for patients in the same cluster.
Conclusion
The probability of recent TB transmission was relatively low in the study site, and the cases mainly came from the activation of previous infection. Spatial analysis showed that some strains in the larger clusters had the characteristics of regional aggregation.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81473027), Qing Lan Project (2014), Six Talent Peaks Project in Jiangsu province (2014-YY-023), Jiangsu province Science and Technology Funds (BL2014067), Key Research and Development Plan of Zhenjiang (SHW2016008, SH2017022), and Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author contributions
GX and JW conceived, initiated and led the study. GX, XM, JW and HP analyzed the data with input from all the authors. GX and JW prepared the manuscript. All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work. All authors reviewed and approved the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Raviglione M, Sulis G. Tuberculosis 2015: Burden, challenges and strategy for control and elimination. Infect Dis Rep. 2016;8(2):6570. | ||
Gandhi NR, Nunn P, Dheda K, et al. Multidrug-resistant and extensively drug-resistant tuberculosis: a threat to global control of tuberculosis. Lancet. 2010;375(9728):1830–1843. | ||
Dheda K, Gumbo T, Maartens G, et al. The epidemiology, pathogenesis, transmission, diagnosis, and management of multidrug-resistant, extensively drug-resistant, and incurable tuberculosis. Lancet Respir Med. 2017;5(4):291–360. | ||
Bonura C, Gomgnimbou MK, Refregier G, et al. Molecular epidemiology of tuberculosis in Sicily, Italy: what has changed after a decade? BMC Infect Dis. 2014;14:602. | ||
Banu S, Uddin MK, Islam MR, et al. Molecular epidemiology of tuberculosis in rural Matlab, Bangladesh. Int J Tuberc Lung Dis. 2012;16(3):319–326. | ||
Ei PW, Aung WW, Lee JS, Choi GE, Chang CL. Molecular strain typing of mycobacterium tuberculosis: a review of frequently used methods. J Korean Med Sci. 2016;31(11):1673–1683. | ||
Chen H, He L, Huang H, et al. Mycobacterium tuberculosis lineage distribution in Xinjiang and Gansu Provinces, China. Sci Rep. 2017;7(1):1068. | ||
Liu HC, Deng JP, Dong HY, et al. Molecular typing characteristic and drug susceptibility analysis of mycobacterium tuberculosis isolates from Zigong, China. Biomed Res Int. 2016;2016:6790985. | ||
Hu Y, Mathema B, Zhao Q, et al. Acquisition of second-line drug resistance and extensive drug resistance during recent transmission of Mycobacterium tuberculosis in rural China. Clin Microbiol Infect. 2015;21(12):1093 e1099–e1093 e1018. | ||
Luo T, Yang C, Peng Y, et al. Whole-genome sequencing to detect recent transmission of Mycobacterium tuberculosis in settings with a high burden of tuberculosis. Tuberculosis (Edinb). 2014;94(4):434–440. | ||
Shao Y, Yang D, Xu W, et al. Epidemiology of anti-tuberculosis drug resistance in a Chinese population: current situation and challenges ahead. BMC Public Health. 2011;11:110. | ||
Supply P, Mazars E, Lesjean S, Vincent V, Gicquel B, Locht C. Variable human minisatellite-like regions in the Mycobacterium tuberculosis genome. Mol Microbiol. 2000;36(3):762–771. | ||
Gingeras TR, Ghandour G, Wang E, et al. Simultaneous genotyping and species identification using hybridization pattern recognition analysis of generic Mycobacterium DNA arrays. Genome Res. 1998;8(5):435–448. | ||
Chen J, Tsolaki AG, Shen X, Jiang X, Mei J, Gao Q. Deletion-targeted multiplex PCR (DTM-PCR) for identification of Beijing/W genotypes of Mycobacterium tuberculosis. Tuberculosis (Edinb). 2007;87(5):446–449. | ||
Imperiale BR, Moyano RD, AB DIG, et al. Genetic diversity of Mycobacterium avium complex strains isolated in Argentina by MIRU-VNTR. Epidemiol Infect. 2017;145(7):1382–1391. | ||
Hamblion EL, Le Menach A, Anderson LF, et al. Recent TB transmission, clustering and predictors of large clusters in London, 2010-2012: results from first 3 years of universal MIRU-VNTR strain typing. Thorax. 2016;71(8):749–756. | ||
Anderson LF, Tamne S, Brown T, et al. Transmission of multidrug-resistant tuberculosis in the UK: a cross-sectional molecular and epidemiological study of clustering and contact tracing. Lancet Infect Dis. 2014;14(5):406–415. | ||
van Soolingen D, Borgdorff MW, de Haas PE, et al. Molecular epidemiology of tuberculosis in the Netherlands: a nationwide study from 1993 through 1997. J Infect Dis. 1999;180(3):726–736. | ||
Ellis BA, Crawford JT, Braden CR, et al. Molecular epidemiology of tuberculosis in a sentinel surveillance population. Emerg Infect Dis. 2002;8(11):1197–1209. | ||
Moonan PK, Ghosh S, Oeltmann JE, Kammerer JS, Cowan LS, Navin TR. Using genotyping and geospatial scanning to estimate recent mycobacterium tuberculosis transmission, United States. Emerg Infect Dis. 2012;18(3):458–465. | ||
Liu Y, Huang S, Jiang H, et al. The prevalence of latent tuberculosis infection in rural Jiangsu, China. Public Health. 2017;146:39–45. | ||
Gao L, Lu W, Bai L, et al. Latent tuberculosis infection in rural China: baseline results of a population-based, multicentre, prospective cohort study. Lancet Infect Dis. 2015;15(3):310–319. | ||
Nava-Aguilera E, Andersson N, Harris E, et al. Risk factors associated with recent transmission of tuberculosis: systematic review and meta-analysis. Int J Tuberc Lung Dis. 2009;13(1):17–26. | ||
Chen KS, Liu T, Lin RR, Peng YP, Xiong GC. Tuberculosis transmission and risk factors in a Chinese antimony mining community. Int J Tuberc Lung Dis. 2016;20(1):57–62. | ||
Surie D, Fane O, Finlay A, et al. Molecular, spatial, and field epidemiology suggesting TB transmission in community, not hospital, Gaborone, Botswana. Emerg Infect Dis. 2017;23(3):487–490. | ||
Abrahams KA, Cox JA, Spivey VL, et al. Identification of novel imidazo[1,2-a]pyridine inhibitors targeting M. tuberculosis QcrB. PLoS One. 2012;7(12):e52951. | ||
Camacho LR, Ensergueix D, Perez E, Gicquel B, Guilhot C. Identification of a virulence gene cluster of Mycobacterium tuberculosis by signature-tagged transposon mutagenesis. Mol Microbiol. 1999;34(2):257–267. | ||
Zhang H, Li D, Zhao L, et al. Genome sequencing of 161 Mycobacterium tuberculosis isolates from China identifies genes and intergenic regions associated with drug resistance. Nat Genet. 2013;45(10):1255–1260. | ||
Supply P, Magdalena J, Himpens S, Locht C. Identification of novel intergenic repetitive units in a mycobacterial two-component system operon. Mol Microbiol. 1997;26(5):991–1003. | ||
Ravansalar H, Tadayon K, Ghazvini K. Molecular typing methods used in studies of Mycobacterium tuberculosis in Iran: a systematic review. Iran J Microbiol. 2016;8(5):338–346. | ||
Pan XL, Zhang CL, Nakajima C, et al. A quantitative and efficient approach to select MIRU-VNTR loci based on accumulation of the percentage differences of strains for discriminating divergent Mycobacterium tuberculosis sublineages. Emerg Microbes Infect. 2017;6(7):e68. | ||
Nabyonga L, Kateete DP, Katabazi FA, et al. Determination of circulating Mycobacterium tuberculosis strains and transmission patterns among pulmonary TB patients in Kawempe municipality, Uganda, using MIRU-VNTR. BMC Res Notes. 2011;4:280. | ||
Yuan X, Zhang T, Kawakami K, et al. Molecular characterization of multidrug- and extensively drug-resistant Mycobacterium tuberculosis strains in Jiangxi, China. J Clin Microbiol. 2012;50(7):2404–2413. | ||
Liu J, Li J, Liu J, et al. Genotypic diversity of mycobacterium tuberculosis clinical isolates in the multiethnic area of the Xinjiang Uygur autonomous region in China. Biomed Res Int. 2017;2017:3179535. | ||
Cheng XF, Jiang C, Zhang M, et al. Mycobacterial interspersed repetitive unit can predict drug resistance of Mycobacterium tuberculosis in China. Front Microbiol. 2016;7:378. | ||
Mears J, Abubakar I, Cohen T, McHugh TD, Sonnenberg P. Effect of study design and setting on tuberculosis clustering estimates using Mycobacterial Interspersed Repetitive Units-Variable Number Tandem Repeats (MIRU-VNTR): a systematic review. BMJ Open. 2015;5(1):e005636. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.