Back to Journals » Journal of Inflammation Research » Volume 13
Clusterin Deficiency Predisposes C57BL/6j Mice to Cationic Bovine Serum Albumin-Induced Glomerular Inflammation
Authors Sun P, Feng S, Guan Q, Adomat H, Barbour S, Gleave ME, Nguan CYC, Xu W, Du C ![]()
Received 9 October 2020
Accepted for publication 18 November 2020
Published 24 November 2020 Volume 2020:13 Pages 969—983
DOI https://doi.org/10.2147/JIR.S285985
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Pengcheng Sun,1,2 Shijian Feng,1,3 Qiunong Guan,1 Hans Adomat,1 Sean Barbour,4 Martin E Gleave,1 Christopher YC Nguan,1 Wanhai Xu,5 Caigan Du1
1Department of Urologic Sciences, University of British Columbia, Vancouver, BC V6H 3Z6, Canada; 2Department of Gynecology and Obstetrics, The Fourth Affiliated Hospital of Harbin Medical University, Harbin, Heilongjiang 150001, People’s Republic of China; 3Department of Urology, Institute of Reconstructive Urology, West China Hospital of Sichuan University, Chengdu, Sichuan 610041, People’s Republic of China; 4Division of Nephrology, Department of Medicine, University of British Columbia, Vancouver, BC V5T 3A5, Canada; 5Heilongjiang Key Laboratory of Scientific Research in Urology, Department of Urology, The Fourth Hospital of Harbin Medical University, Harbin, Heilongjiang 150001, People’s Republic of China
Correspondence: Caigan Du
Department of Urologic Sciences, University of British Columbia, Jack Bell Research Centre, 2660 Oak Street, Vancouver, British Columbia V6H 3Z6, Canada
Tel +1 604-875-4111 Ext 63793
Fax + 1 604-875-5654
Email [email protected]
Background: Membranous nephropathy (MN) is a specific entity of glomerulonephritis, and its glomerular inflammation is characterized by the deposition of immune complexes in the glomerular basement membrane and proteinuria. However, the molecular mechanisms underlying the glomerular inflammation of MN are not fully understood. This study was designed to investigate the role of clusterin (CLU) in the development of MN using a mouse model of cationic bovine serum albumin (cBSA)-induced MN.
Methods: Both wild-type C57BL/6j (WT) and CLU-knockout C57BL/6j (CLU-KO) mice were immunized with cBSA. The kidney function was determined by the levels of serum creatinine (SCr), blood urea nitrogen (BUN) and urinary protein. MN and glomerular deposits of CLU, complement C3 and immunoglobulins (Igs) were determined by histological analyses. Serum proteins were analyzed by the enzyme-linked immunosorbent assay, Western blot and liquid chromatography-mass spectrometry.
Results: Here, we showed that after cBSA immunization, SCr and proteinuria were increased in CLU-KO mice but not in WT mice. Similarly, severe glomerular atrophy and mesangial expansion along with C3 deposit were only found in the kidneys of CLU-KO mice but not in WT mice. However, there were no differences of serum IgG and complement 3 levels between CLU-KO and WT mice. In the serum of WT mice, CLU bound to anti-cBSA IgG, complements (eg, C8), proteinase/protease inhibitors and antioxidative proteins to form a complex, and incubation with WT serum reduced the complement-dependent lysis of podocytes in cultures.
Conclusion: Our data suggest that a CLU deficiency induces cBSA-initiated glomerular inflammation of MN in a disease-resistant strain of mice, suggesting an anti-glomerular inflammatory function of CLU in the resistance to MN development. This function may be at least in part due to the formation of CLU-anti-cBSA Igs complex that prevents glomerular inflammation or injury in the disease-resistant mice.
Keywords: inflammatory glomerular disease, membranous nephropathy, immune complex, glomerulonephritis, mouse model
Introduction
Glomerulonephritis is a term for various patterns of glomerular inflammation or injury and is typically presented by one or both of the nephrotic or nephritic syndromes.1 Membranous nephropathy (MN) is one entity of the glomerulonephritis, and it is the most common cause (20–40%) of the nephrotic syndrome in nondiabetic adults,2–4 and approximately 30% of MN cases eventually lead to end-stage kidney disease.4–8 Except of similar clinical features such as proteinuria, MN is classified into primary and secondary MN based on the different profiles of histopathology.9 The primary MN is typically caused by the binding of circulating autoantibodies to podocyte-expressing autoantigens such as M-type phospholipase A2 receptor (PLA2R) to form immune-complex deposit in the subepithelial zone between the basement membrane and the podocyte, and subsequent thickening of glomerular basement membrane (GBM), a typical lesion of MN.7,9–11 The secondary MN is associated with circulating antigens or pre-formed circulating immune complexes (CIC) that may be generated by the systemic diseases (eg, systemic lupus erythematosus, SLE), malignancy, medications, infections or exposure to foreign antigens.9,10 In the secondary MN, the antigens bound with antibodies or CIC accumulate not only in the subepithelial space but also likely in the subendothelial space and the mesangium, resulting in atypical lesions including mesangial expansion.7,10 However, the molecular mechanisms by which the immune complexes or autoantibody or CIC-stimulated inflammation cause glomerular injury and proteinuria in MN are not fully understood. In addition to all various types of immunoglobulins, the immunofluorescence analyses of the immune complex deposits in renal biopsies show that the MN lesions are positively stained with glomerular expression of C3, C5b-9, C1q, C4d, factor B and mannose-binding lectin (MBL),10,12,13 and in vitro anti-PLA2R1 autoantibodies activate complement-dependent cytotoxicity (CDC),14 suggesting that activation of complements by classical and/or lectin pathways may mediate the podocyte or glomerular injury in the pathogenesis of MN.10 However, the role of complement regulatory proteins (both soluble and local membrane-bound) in the development of MN is barely investigated.
Clusterin (CLU) is a circulating glycoprotein in the physiological fluids (ie, plasma, semen, milk and cerebrospinal fluid), and it is a disulfide-linked heterodimer (approximately 75–80 kDa) encoded by a single gene.15,16 Various biological functions of CLU have been reported, including complement regulation, lipid transport, sperm maturation, apoptosis initiation, endocrine function, membrane protection and cell-to-cell interaction.15 Using CLU knockout (CLU-KO) mice compared to wild type (WT) control mice, several studies have demonstrated that CLU plays an important role in the development of different kidney diseases. CLU-KO aging mice spontaneously develop progressive glomerulopathy,17 and CLU-KO enhances angiotensin II–induced renal fibrosis.18 In addition, our group has demonstrated that CLU deficiency results in an increase in renal tubular epithelial cell apoptosis in the cultures and worsens renal ischemia-reperfusion injury (IRI) and delays repair after IRI in mice.19,20 These studies may imply a cytoprotective role of CLU in the kidney. More interestedly, it has been reported that the levels of serum CLU in MN patients are significantly lower than those in healthy controls,21 and the levels of glomerular CLU in renal biopsies of MN are inversely correlated with C5b-9 and are a single factor being associated with a reduction of proteinuria after a follow-up of 1.5 years.22 However, whether CLU is an important negative regulator of the immune complex-mediated glomerular injury in MN is not investigated yet.
Early experimental studies report that immunization with cationic bovine serum albumin (cBSA) induces the secondary-like MN with mesangial expansion and focal segmental endocapillary proliferation in dogs or rabbits.23,24 One clinical study also confirms that a small group of children with MN have high levels of circulating cBSA and anti-BSA IgG (but not CIC), and the cBSA is co-localized in the subepithelial immune deposits,25 indicating the clinical relevance of cBSA-induced MN in animals. Furthermore, cBSA-induced MN can be found in both BALB/c and ICR strains of mice but not in C57BL/6j (B6) strain.26–28 The objective of this study was to investigate the role of CLU in the resistance to MN in B6 mice using CLU-KO B6 mice as compared to WT controls.
Materials and Methods
Animals and Cells
Both WT B6 and CLU-KO mice in B6 background (female, 7–8 weeks old) were received from the breeding colonies in the animal care facility at Jack Bell Research Centre (Vancouver, British Columbia, Canada).
Conditionally immortalized heat-sensitive mouse podocytes (HSMPs) were a kind gift from Dr. Stuart Shankland (School of Medicine, University of Washington, Seattle, WA, USA).29 These cells were grown at 33°C in RPMI 1640 culture medium (Life Technologies, Inc., Gaithersburg, MD, USA) supplemented with 10% fetal bovine serum (FBS), 2 mmol/L of glutamine (Sigma-Aldrich Canada, Oakville, ON, Canada), 10 mmol/L of HEPES (Sigma-Aldrich Canada), 1 mmol/L of sodium pyruvate (Sigma-Aldrich Canada), and 10 U/mL of recombinant mouse interferon (IFN)-γ (Life Technologies, Inc.). For the experiments, HSMPs were induced into quiescence and the differentiated phenotype at 37°C in the same medium without addition of IFN-γ.
Induction of MN in Mice by cBSA and Experimental Groups
MN in mice (WT or CLU-KO) was induced by immunization with cBSA according to the protocol described previously with minor modification.26 In brief, cBSA solution (2 mg/mL cBSA in PBS, pH 7.4) (Chondrex, Inc., Redmond, WA, USA) was fully emulsified with an equal volume of complete Freund’s adjuvant (Sigma-Aldrich Canada). The resultant emulsion (0.1 mL/mouse, 0.1 mg cBSA) was subcutaneously injected at the base of the tail. After two weeks of the immunization, the mice were intraperitoneally (IP) injected cBSA solution (0.2 mL/mouse, 0.4 mg cBSA) every other day (q.o.d.) for a total of 5 times. Two WT control groups were included: one treated with vehicle PBS (herein PBS-WT) and the other treated with cBSA (herein cBSA-WT). The experimental group was CLU-KO mice treated with cBSA (herein cBSA-KO).
Urine Collection and Protein Determination
Urine samples were collected by using a metabolic cage (3600M021, Tecniplast, Toronto, ON, Canada). In brief, the mice were individually housed in the metabolic cage for 8 h during the dark phase of a 12-h light/dark cycle without adaptive training. The mice were fed ad libitum on the same food and the drinking water as in the “home” cages. Urine specimens (1–1.5 mL per mouse) were collected and centrifuged at 5000 ×g to pellet cellular debris, and the supernatant (urine) was stored in aliquots at −80°C until use. Total protein levels in the urine specimens were measured using the Dimension Vista® 1500 System (Siemens Healthineers Canada, Oakville, ON, Canada) in the Core Chemistry Laboratory at Vancouver General Hospital (Vancouver, BC, Canada).
Measurement of Serum Creatinine (SCr) and Blood Urea Nitrogen (BUN)
At the endpoint of experiments (day 50 after initial cBSA immunization), blood samples were collected via cardiac puncture from euthanized mice, and the serum was stored at −80 °C until use. Both SCr and BUN were measured in the Core Chemistry Laboratory at Vancouver General Hospital (Vancouver, BC, Canada).
Histological Examinations of MN in Kidney Sections
Kidney tissues were preserved by formalin-fixed and paraffin-embedded. For histological scoring of severity of MN, tissue sections (~4 μm thickness) were routinely stained with hematoxylin and eosin (HE). The stained tissue slides were scanned with Leica SCN400 Slide scanner (Leica Microsystens Inc., Concord, ON, Canada). The number of glomerular atrophies (GA) or atrophic glomerulus in each high-powered field (hpf) (400 ×magnification) was counted in each HE-stained section in a blinded fashion (Figure 1), and an average number of at least 20 randomly selected fields in the kidney sections represented the severity of the GA of a mouse.
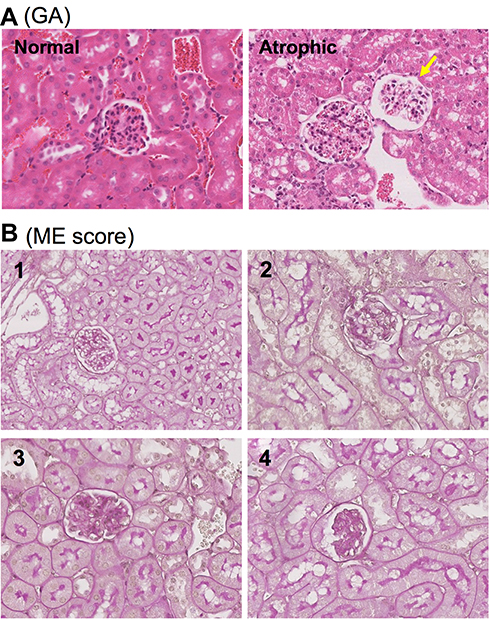 |
Figure 1 Semi-quantitative scoring system for histological evaluation of MN in mice. (A) Typical images of normal and atrophic glomerulus (pointed by yellow arrow). (B) The scoring of mesangial expansion (ME) from 1 to 4. 1: 0–24% of the area affected with densely stain, normal to minimal; 2: 25–49%, mild; 3: 50–74%, moderate; and 4: >75%, severe. |
The mesangial expansion (ME) of the glomeruli as an atypical lesion of the secondary MN was examined by using a semi-quantitative scoring system in periodic acid Schiff (PAS)-stained tissue sections. The scoring system consisted of 1 to 4 scale to determine the severity of the extracellular matrix (ECM) deposition into both the capillary walls (the incrassation of basement membrane) and the mesangium based on the percentage of the area stained strongly with PAS, indicating the ME in an affected glomerulus: 1 (0–24% of the area affected with densely stain, normal to minimal), 2 (25–49%, mild), 3 (50–74%, moderate), and 4 (>75%, severe) (Figure 1). A range of 180 to 250 glomeruli were counted for each mouse and were averaged for the severity of ME.
Immunohistochemistry
The protein expression (CLU, Igs and complement C3) in the kidney sections was detected by using a routine method of immunohistochemical staining. In brief, tissue sections were boiled in citrate buffer for antigen retrieval for 30 min. After cooling down at room temperature (RT), the endogenous peroxidase was blocked for 20 min in 0.3% hydrogen peroxide in water. The sections were blocked with normal rabbit serum for 30 min, and then incubated with primary rabbit monoclonal anti-mouse complement C3 (EPR19394, Abcam, Toronto, ON, Canada), goat anti-mouse Igs (IgG + IgM + IgA) (ab102445, Abcam), and goat anti-CLU (C18, Santa Cruz Biotech., Santa Cruz, CA, USA) overnight at 4°C, followed by incubation with an appropriate biotin-labelled secondary antibody in a humidifier at RT for 60 min. All the target proteins bound with the antibodies were detected by using VECTASTAIN® ABC (Avidin-Biotin Complex) kit (Vector Lab., Burlingame, CA, USA) and counterstained with Mayer’s hematoxylin (Sigma-Aldrich Canada).
Enzyme-Linked Immunosorbent Assay (ELISA)
Serum levels of anti-cBSA IgG (subclass IgG1 and IgG2a) were measured by ELISA. Briefly, Nunc-Immuno 96 MicroWell solid plates (Sigma-Aldrich Canada) were coated with 0.5 µg/well of cBSA (Chondrex, Inc.) in carbonate-bicarbonate coating buffer (50 mM Na2CO3, 50 mM NaHCO3, pH 9.6) at 4 °C overnight, and were blocked with 1% BSA (Sigma-Aldrich Canada) in PBS-T (PBS containing 0.05% Tween-20) buffer for 1 h at RT. After washing with PBS-T buffer, serum samples (100 µL/well) in triplicate, serially diluted with ELISA buffer (150 mM NaCl, 1 mM KH2PO4, 10 mm Na2HPO4, 2.6 mm KCl, 0.5% BSA, 0.1% Tween-20, pH 7.4), were incubated at 4 °C overnight. The serum anti-cBSA IgG1 and anti-cBSA IgG2a were detected by using biotinylated goat anti-mouse IgG1 (ab98691, Abcam, 1:1000 diluted) and anti-mouse IgG2 (ab98696, Abcam, diluted from 1:1000), respectively. The color was developed with streptavidin-horseradish peroxidase (HRP) and tetramethylbenzidine (Sigma-Aldrich Canada) and was stopped with 2 M H2SO4 solution. Similarly, serum complement C3 was determined by using mouse complement C3 ELISA kit (ab157711, Abcam) according to the manufacturer’s protocol. The serum titers of each substance were expressed as optical density (OD) at 450 nm (OD450). The “noise” associated with this protocol was confirmed to be low (approximately 5%) by three technical replicates.
Immunoprecipitation
CLU-bound proteins were isolated from the serum of cBSA-WT group by immunoprecipitation. In brief, 25 µL of pooled serum from cBSA-WT mice compared to cBSA-KO mice was diluted with 225 µL PBS and was added 3 µL of goat anti-CLU antibodies (C-18, Santa Cruz Biotech.). After overnight incubation with rotating shaking at 4°C, 10 µL of anti-goat IgG biotin and streptavidin-agarose beads (S1638, Millipore-Sigma Aldrich Canada) were add to the serum solution as secondary antibodies at 4°C overnight. The immunoprecipitate of CLU and its associated proteins was collected by centrifugation at 8000 ×g and was washed with PBS three times (5 min each time) prior to further analyses.
Western Blot Analysis
To confirm the presence of CLU protein in the immunoprecipitate, the immunoprecipitate was mixed with the equal amount of SDS sample buffer (50 mM Tris-HCl, pH6.8, 2% SDS, 10% glycerol, 0.1 M DTT and 0.02% bromophenol blue) and boiled for 5 min. The proteins were fractioned by 10% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and were transferred to nitrocellulose membranes (Bio-Rad Laboratories, Mississauga, ON, Canada). The blot was blocked with 5% milk in TBST (50 mM Tris-HCl pH7.4, 150 mM NaCl, 0.1% Tween 20) for 1 h, followed by incubation with goat anti-CLU (C-18, Santa Cruz Biotech) primary antibodies and then anti-goat IgG conjugated with HRP secondary antibodies. After washing with TBST, the CLU protein in the blot was detected by using enhanced chemiluminescence (ECL) reagents (ThermoFisher Scientific, Rockford, IL, USA).
To examine if serum CLU bound to anti-cBSA IgG in cBSA-WT group, the proteins were eluted out from the immunoprecipitate with 6 M guanidine HCl solution and were used as “primary antibodies” in the detection of cBSA on the blot, which was prepared by electrophoresis of different amounts of cBSA (40 μg or 20 μg in the lane) in 10% SDS-PAGE, followed by transfer to the nitrocellulose membranes. After incubation with the eluted proteins (“primary antibodies”), the cBSA blot was incubated with secondary anti-mouse IgG conjugated with HRP. The presence of anti-cBSA IgG in the immunoprecipitate was indicated by the positive detection of cBSA on the blot using ECL reagents (ThermoFisher Scientific).
Liquid Chromatography-Mass Spectrometry (LC-MS)
Serum CLU-bound proteins in the immunoprecipitate were identified by using LC-MS as described previously with some modification.30 The proteins were extracted from the immunoprecipitate by boiling with the SDS sample buffer for 5 min and were fractioned by 10% SDS-PAGE. The proteins in the gel were stained with Coomassie blue dye. After destaining with 50% methanol in 10% acetic acid, the specific protein bands and the bands with higher staining density in cBSA-WT samples as compared to nonspecific, background protein bands in CLU-KO controls were selected for protein identification using LC-MS technique.
In brief, protein bands of interest were treated with 100 mM ammonium bicarbonate twice for 10 min and dehydrated with 50% acetonitrile twice for 10 min before drying under vacuum. Reduction and alkylation were carried out with 10 mM DTT for 35 min at 65 ºC followed by 55 mM iodoacetamide for 30 min at RT in the dark after removal of excess DTT. Gel pieces were washed twice with 50 mM ammonium bicarbonate for 10 min and dried as above with 50% acetonitrile prior to digestion with trypsin (10 μg/mL). Supernatant peptide solutions were transferred to Eppendorf and gel pieces extracted 15 min twice with 50% acetonitrile in 5% formic acid. Extracts were pooled with initial supernatant and the volume brought to about 15 µL and transferred to sample plates for analysis.
An Orbitrap Fusion Lumos MS platform (Thermo Scientific) was used for analysis. Samples were introduced using an Easy-nLC 1200 system (Thermo) with a 100 µm ID x 20 cm in house-made column (ReproSil-Pur 120 C18-AQ 1.9 µm – Dr. Maisch GMbH HPLC). Prior to each sample injection, the analytical column was equilibrated at 400 bar for a total volume of 8 μL. After injection, sample loading was carried out for a total volume of 8 μL at 500 bar. The injection volumes for all samples was 4 µL and runs using a gradient of mobile phase A (water and 0.1% formic acid) and B (80% acetonitrile with 0.1% formic acid) at 0.3 µL/min, 3–35% B from 2 to 40 min followed by 35–40% B over 5 min, 40–95% B over 3 min and 6 min at 95%.
Data acquisition (control software version 3.1.2412.17) was with a data-dependent method and MS2 in the Orbitrap. The Lumos was operated with a positive ion spray voltage of 2100 and a transfer tube temperature of 325°C. The default charge state was set as 2. Survey scans (MS1) were acquired in the Orbitrap at a resolution of 60K, across a mass range of 375–1500 m/z, with RF lens setting of 60, an AGC target of 4e5, max injection time of 86 ms in profile mode. For MS2 scans, charge state filtering of 2–5, and dynamic exclusion for 60 seconds with 10 ppm tolerances was used with a 1.2 m/z window prior to HCD fragmentation of 37%. MS2 data acquisition carried out in the Orbitrap used a 15K resolution, a fixed first mass of 120m/z, an AGC target of 1e5, and a max injection time of 54 ms in centroid mode.
The accumulated data were analyzed using ProteinLynx Global Server software (PLGS) set with peptide and fragment mass accuracies of 25 ppm and 0.1 Da (Waters, Mississauga, ON, Canada), respectively. The peptide/protein search engine was applied to the SwissProt database. A search with similar parameters was also carried out using Mascot using the peaks generated in PLGS. Proteins with Mascot scores P32 and P2 peptide matches were considered statistically significant (p < 0.05) and included as affirmative identification.
Fluorescence-Activated Cell Sorting (FACS) Analysis
The cell surface binding of serum antibodies to HSMP cells was determined by using FACS analysis. In brief, 5 × 104 of HSMP cells in 100 µL PBS were incubated with 1 µL of serum (1:100 dilution, cBSA-WT versus cBSA-KO) at 4°C for 30 min, followed by staining with anti-mouse IgG conjugated with fluorescein isothiocyanate (FITC) (Sigma-Aldrich Canada) for 15 min in the dark. The antibody binding was detected by using a flow cytometry and further quantified using FlowJo software (Tree Star Inc., Ashland, OR, USA). The cells stained with secondary FITC-anti-mouse IgG only were used as background control.
Complement-Dependent Cytotoxicity (CDC) Assay
In brief, 0.2 × 106 HSMP cells were seeded per well in 24-well plates overnight, followed by incubation with 1:100 diluted serum (1 µL serum in 100 µL PBS) for 3 min at RT. Cell lysis started by addition of 10 µL/well of Low-Tox®-M Rabbit Complement (Cedarlane, Burlington, ON, Canada) and then incubated at 37°C for 2 h. The HSMP cells incubated with 110 µL of PBS containing 0.2% Triton X-100 were used as a positive control (100% of cell lysis). The cellular debris in the resultant supernatant was pelleted by centrifugation at 10,000 ×g for 5 min, and the levels of lactate dehydrogenase (LDH) in the supernatants were determined by using the Cytotoxicity Detection Kit (LDH) (Roche) (Sigma-Aldrich Canada) following manufacturer’s protocol. The CDC was calculated as follows: CDC (%) = sample OD450/control OD450 × 100%.
Statistical Analysis
Data in each group are presented as mean ± standard deviation (SD) and were statistically analyzed by using GraphPad Prism (version 8.0.0) (GraphPad, La Jolla, CA, USA). The difference of the mean between two groups based on a sample of the data was determined using two-tailed t-test. The mean differences between groups split on two independent variables or factors were compared using two-way analysis of variance (two-way ANOVA). A p-value of <0.05 was considered statistically significant.
Results
An Increase in SCr and Urinary Protein (Proteinuria) in CLU-KO B6 Mice After cBSA Immunization
At the endpoint of experiment (50 days after initial cBSA immunization), the levels of SCr were not different between PBS-WT control (2.92 ± 0.38 mg/dL, n = 4) and cBSA-WT mice (2.63 ± 0.45 mg/dL, n = 15) (p = 0.2646). As compared to the WT control groups, the SCr in cBSA-KO mice (3.53 ± 0.49 mg/dL, n = 11) was significantly elevated (cBSA-KO vs cBSA-WT, p < 0.0001; PBS-WT vs cBSA-KO, p = 0.0434) (Figure 2A). However, there was no significant difference of BUN between these three groups, indicated by the BUN levels of PBS-WT mice (42.84 ± 4.87 mg/dL, n = 4), cBSA-WT mice (45.17 ± 7.6 mg/dL, n = 15) and cBSA-KO (45.99 ± 8.75 mg/dL, n = 11) (PBS-WT vs cBSA-WT, p = 0.5769; cBSA-WT vs cBSA-KO, p = 0.8011) (Figure 2B). Similar to the changes of SCr levels in these mice, there was no increased proteinuria in cBSA-WT mice (2.88 ± 1.92 g/L, n = 9) as compared to 2.40 ± 1.05 g/L (n = 4) in PBS-WT mice (p = 0.6495), but an increase in proteinuria was seen in cBSA-KO (6.98 ± 3.96 g/L, n = 8) as compared with that of cBSA-WT controls (cBSA-WT vs cBSA-KO, p = 0.0181) or of PBS-WT mice (PBS-WT vs cBSA-KO, p = 0.0501) (Figure 2C). Taken together, the data suggested that genetic KO of CLU expression increased susceptibility to cBSA-induced renal dysfunction in B6 strain of mice.
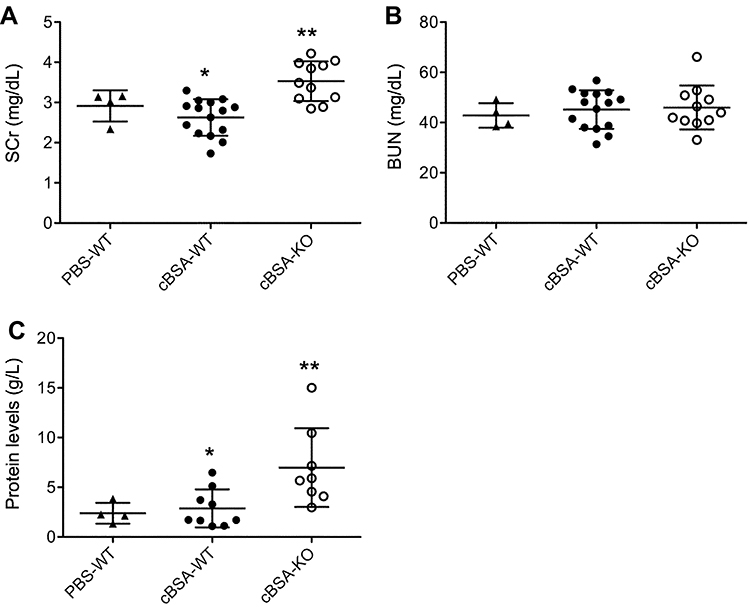 |
Figure 2 The changes of kidney function after cBSA immunization in CLU-KO mice. The parameters of kidney function was determined at day 50 after initial cBSA immunization. (A) Serum creatinine (SCr), *p = 0.2646 (cBSA-WT vs PBS-WT); **p < 0.0001 (cBSA-KO vs cBSA-WT). (B) Blood urea nitrogen (BUN), there was no statistically significant difference between groups. p = 0.5769 (cBSA-WT vs PBS-WT); p = 0.8011 (cBSA-KO vs cBSA-WT). (C) Urinary protein. *p = 0.6495 (cBSA-WT vs PBS-WT); **p = 0.0181 (cBSA-KO vs cBSA-WT). Each value presents the measure of one animal. |
The Association of Renal Dysfunction with Tissue Damage in CLU-KO B6 Mice After cBSA Immunization
To confirm the cBSA-induced MN in CLU-KO B6 mice, the glomerular morphology or damage including both GA and ME (Figure 1) was examined in cBSA-KO mice as compared to WT controls (cBSA-WT and PBS-WT mice). Figure 3A shows a typical microscopic image of renal cortex of the kidney from the mice in cBSA-WT or cBSA-KO group. The tissue sections were stained with PAS. There were not any difference in the glomerular morphology and pathology between PBS-WT and cBSA-WT groups. Two glomeruli with GA and the some with higher scores (3–4) of ME were seen in the tissue sections of the cBSA-KO (Figure 3A). The numbers of GA per hpf in HE-stained kidney sections of cBSA-KO mice were 14.76 ± 4.48 (n = 9), which were significantly higher than 5.99 ± 0.97 (n = 8) in cBSA-WT (p < 0.0001) or 5.3 ± 0.85 (n = 4) in PBS-WT mice (p = 0.0018), whereas the numbers of GA were not different between the cBSA-WT and PBS-WT mice (p = 0.2567) (Figure 3B). Similar data were seen in the scoring of ME of glomeruli in PAS stained sections, indicating the higher scores of the glomerular ME in cBSA-KO mice (1.69 ± 0.18, n = 9) as compared to those in cBSA-WT (1.45 ± 0.08, n = 8) (cBSA-KO vs cBSA-WT, p = 0.0026) or in PBS-WT controls (1.42 ± 0.09, n = 4) (cBSA-KO vs PBS-WT, p = 0.0156). Again, there was no difference in the ME between PBS-WT and cBSA-WT groups (cBSA-WT vs PBS-WT, p = 0.5974) (Figure 3C). Taken together, these data confirmed the presence of pathological changes of glomeruli in cBSA-KO mice but not in WT mice, which positively correlated with cBSA-induced renal dysfunction in CLU-KO B6 mice as shown in Figure 2.
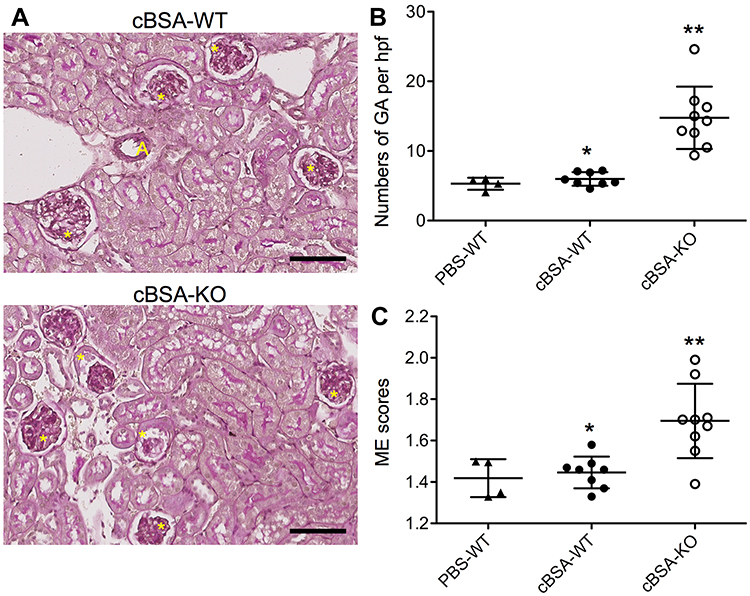 |
Figure 3 The severity of glomerular injury after cBSA immunization in CLU-KO mice. The severity of glomerular injury was determined in kidney sections by using semi-quantitative histological analyses. (A) Typical microscopic images of the renal cortex of mice in cBSA-WT and cBSA-KO groups. The tissue sections were PAS stained. A: artery, yellow star: glomerulus. Scale bar: 80 µm. (B) Glomerular atrophies (GA), the numbers of atrophic glomeruli in a high-powered field (hpf) of HE-stained sections. Each value presents the number of each mouse. *p = 0.2567 (cBSA-WT vs PBS-WT); **p < 0.0001 (cBSA-KO vs cBSA-WT). (C) Mesangial expansion (ME) was scored according to the PAS staining of thickening basement membrane or ECM deposit along the outer surface of the basement membrane. Each value presents the average score of 180 to 250 glomeruli of one animal. *p = 0.5974 (cBSA-WT vs PBS-WT); **p = 0.0026 (cBSA-KO vs cBSA-WT). |
The Association of Complement C3 Glomerular Deposits with Kidney Damage in CLU-KO B6 Mice After cBSA Immunization
One of pathological changes for MN is the deposition of immune complexes on the epithelial side of glomerular capillary loop.31 As shown in Figure 4, a high level of CLU protein was detected in the glomeruli of both PBS-WT and cBSA-WT mice, and it was induced in the renal capsule epithelium in cBSA-WT. Further, this protein was co-localized with Igs but lack of C3 presence in the cBSA-WT group, whereas the immunization with cBSA induced the glomerular deposition of both Igs and C3 in CLU-KO B6 mice. These data suggested the association of C3 glomerular deposits with MN or glomerular injury in CLU-KO B6 mice.
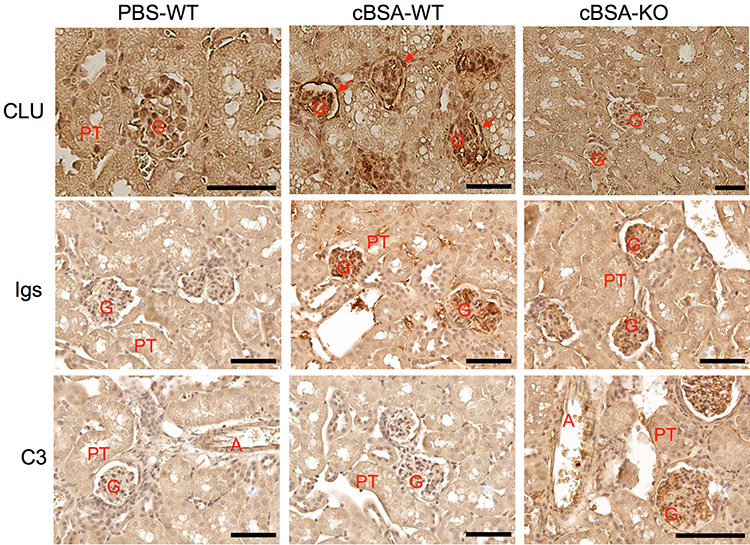 |
Figure 4 The glomerular deposit of Igs and C3 in mice after cBSA immunization in CLU-KO mice. The glomerular expression of CLU, Igs or C3 was detected by using immunohistochemical staining. Upper panel: CLU protein. Red arrow: capsular epithelium. Middle panel: Igs. Bottom panel: C3. G: glomerulus, PT: proximal tubule, A: artery. Data are presented in a typical microscopic image of immunohistochemical staining of each target protein. Brown color: positive staining. Scale bar: 80 µm. |
The levels of anti-cBSA IgG1 and IgG2a, and C3 in the serum in a serial dilution were measured by using ELISA. As shown in Figure 5, there were very little differences between cBSA-WT and cBSA-KO mice in the levels of anti-cBSA IgG1 (p = 0.7822, cBSA-WT vs cBSA-KO) (Figure 5A), anti-cBSA IgG2a (p = 0.1141, cBSA-WT vs cBSA-KO) (Figure 5B), and C3 (p = 0.1783, cBSA-WT vs cBSA-KO) (Figure 5C), indicating that the presence of glomerular injury or C3 in CLU-KO B6 mice was not due to an increase in the C3 expression or anti-cBSA antibodies in the circulation. Furthermore, the OD values of IgG1 in 50,000-fold diluted serum samples were similar to those of IgG2a in 400-fold diluted serum samples, suggesting that IgG1 was a major isotype of antibody in response to cBSA immunization.
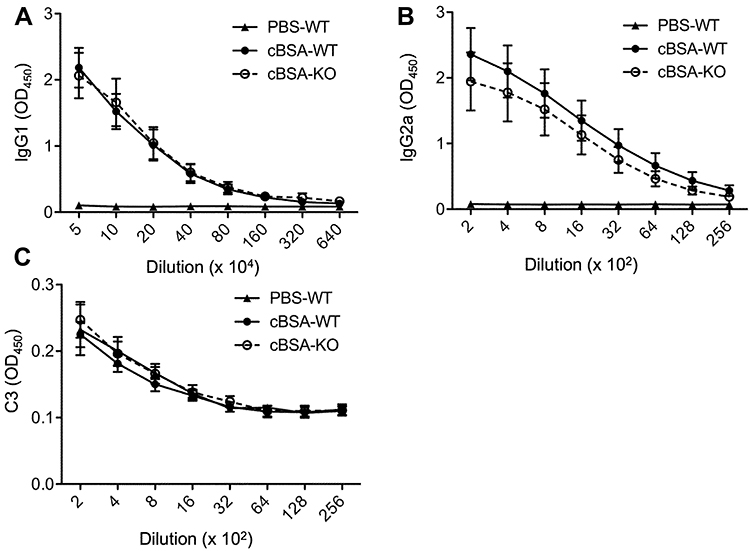 |
Figure 5 Serum levels of anti-cBSA IgG and C3 after cBSA immunization. The serum anti-cBSA IgG subclasses and C3 were measured in PBS-WT (n = 3), cBSA-WT (n = 12) or cBSA-KO (n = 11) by using ELISA. (A) IgG1 was measured in the serum samples serially diluted from 1:50,000. p = 0.7822 (cBSA-KO vs cBSA-WT); p < 0.0001 (PBS-WT vs cBSA-WT). (B) IgG2a was measured in the serum samples serially diluted from 1:200. p = 0.1141 (cBSA-KO vs cBSA-WT); p < 0.0001 (PBS-WT vs cBSA-WT). (C) C3 was measured in the serum samples serially diluted from 1:200. p = 0.1783 (cBSA-KO vs cBSA-WT); p = 0.4792 (PBS-WT vs cBSA-WT). |
Circulating CLU Bound to Anti-cBSA Antibody and Other Proteins in WT B6 Mice After cBSA Immunization
Serum CLU was present in WT but not in KO mice (Figure 6). To further reveal the insight into the mechanisms by which a lack of CLU expression in CLU-KO B6 mice caused glomerular injury after cBSA immunization, CLU-bound proteins in the serum in cBSA-WT mice were immuno-precipitated out from the serum by using anti-CLU antibody. The proteins in the immunoprecipitates from cBSA-WT mice compared to cBSA-KO mice were fractioned in 10% SDS-PAGE and were stained with Coomassie blue dye (Figure 7A). As listed in Table 1, total of 16 proteins were identified around the selected fractions (approximately at 260, 51, 36 and 20 kDa) by LC-MS technique, including CLU, Ig chain, complement C8, proteinase/protease inhibitors (α2 macroglobulin, kininogen 1, and serine protease inhibitor A3K) and anti-oxidative peroxidases (glutathione peroxidase and peroxiredoxin 2).
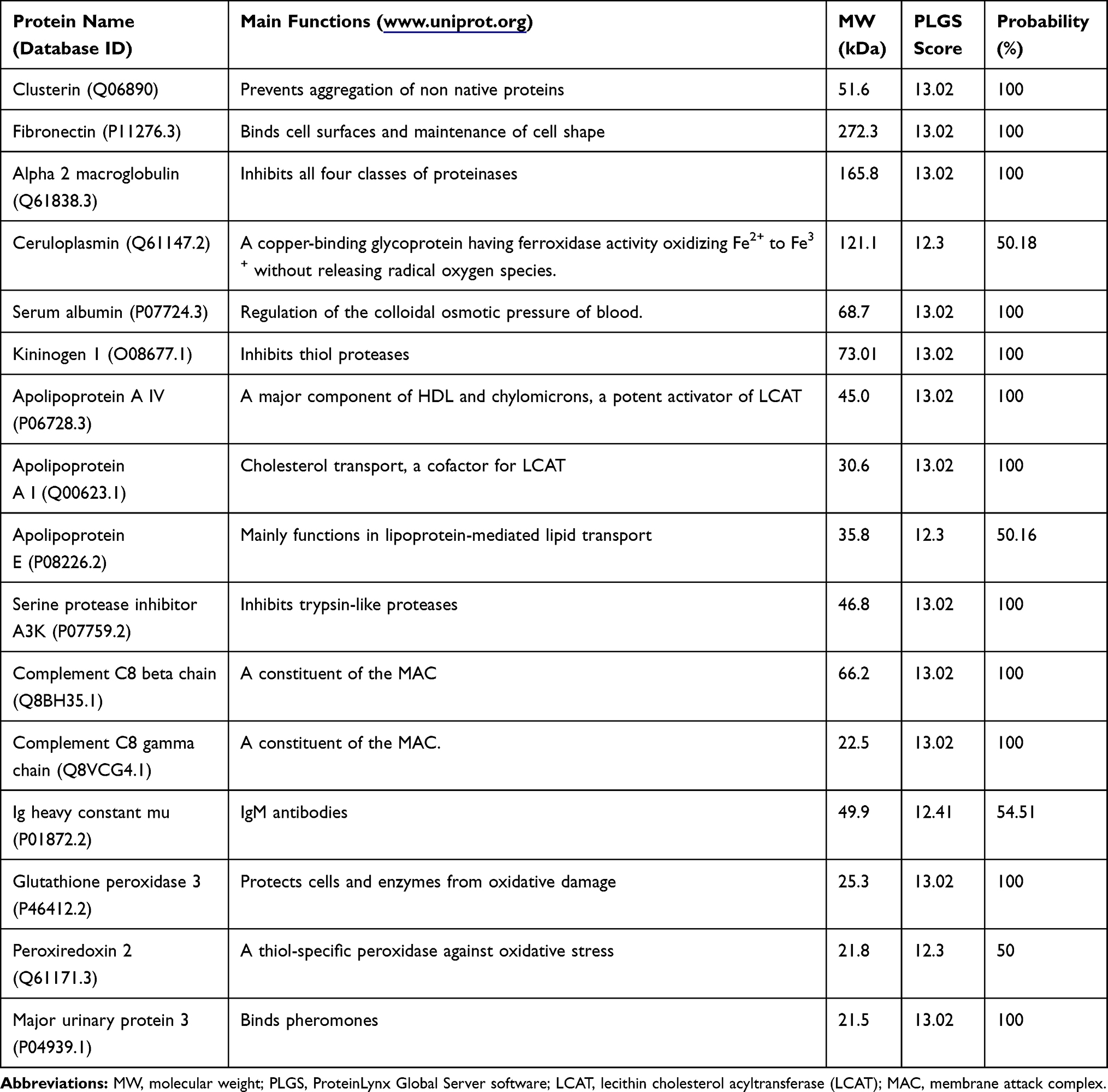 |
Table 1 CLU-Bound Proteins |
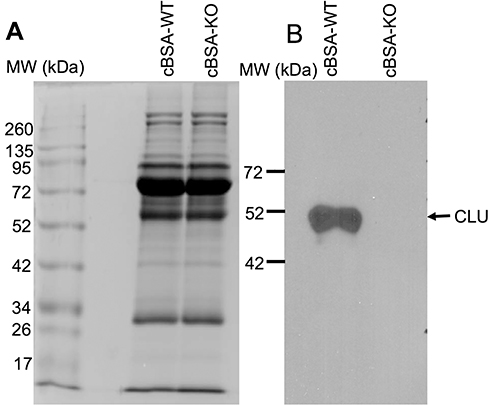 |
Figure 6 Serum CLU in WT mice. The serum proteins (15 µL per lane) were fractioned in 10% SDS-PAGE. (A) Coomassie blue staining of protein fractions. (B) CLU protein was detected by Western blot analysis. |
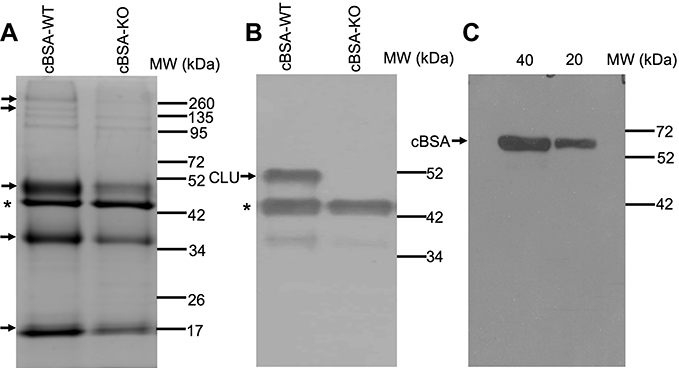 |
Figure 7 CLU-bound proteins in the serum of mice after cBSA immunization. Serum CLU and its bound proteins were immunoprecipitated by using anti-CLU antibodies. (A) A typical Coomassie blue staining of fractions of immunoprecipitate in 10% SDS-PAGE. Arrow: the protein bands of interest for LC-MS analysis. (B) Western blot analysis of CLU protein in the immunoprecipitate. * The heavy chain of anti-CLU antibody. (C) Western blot analysis of anti-cBSA IgG in the immunoprecipitate, in which the immunoprecipitates were used as first antibody in the detection of cBSA in the blot. Data present a typical stained PAGE or Western blot of three separate experiments. |
The presence of CLU in the precipitates from WT mice was confirmed by the Western blotting (Figure 7B). To verify if there was any anti-cBSA IgG specifically bound to the CLU in the serum of cBSA-WT mice, the isolated precipitate was used as a primary “antibody” in the detection of cBSA in Western blot analysis. As shown in Figure 7C, the precipitate contained anti-cBSA antibody that reacted with cBSA in the blot. Taken together, these data indicated that CLU as an extracellular chaperone bound to Igs including anti-cBSA antibody and complement components (ie, C8).
The Association of CLU Deficiency with Increased CDC of the Serum Against Cultured Podocytes
To understand the impact of CLU deficiency on the complement-mediated glomerular injury in CLU-KO mice, the CDC of the sera from CLU-KO mice was determined by the lysis of HSMP cells as compared with that from WT control. As shown in Figure 8, although KO group might have slightly more antibody-binding to the cell surface of HSMP than WT group, the difference was not significant between these two groups (p = 0.3784, n = 7). The CDC of CLU-KO sera was 77.97 ± 3.64% that was significantly higher than 71.18 ± 1.95% of CLU-containing sera from cBSA-WT mice (p = 0.0010, n = 7) in this in vitro experiment (Figure 9), suggesting that a lack of CLU in the serum might result in more potency in the induction of complement-mediated podocyte cell death.
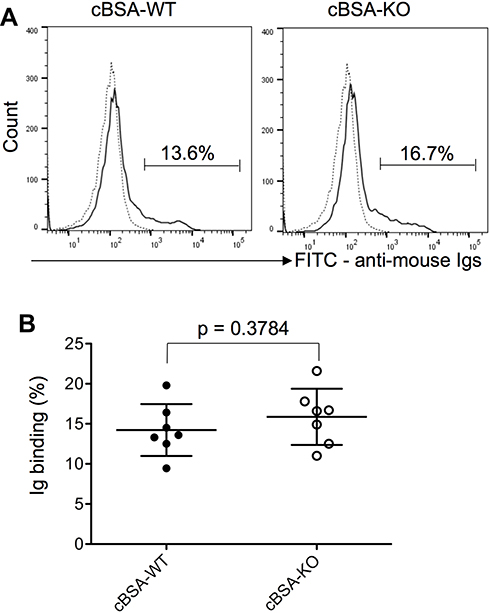 |
Figure 8 Serum antibody-binding on the cell surface of cultured heat-sensitive mouse podocytes (HSMPs). Serum samples were diluted 1:100, and the levels of serum antibody-binding to the cell surface of HSMSs were determined by using FACS analysis. (A) A typical FACS histograph of serum antibody-binding. Dote line: Background staining. Solid line: antibody-binding levels. (B) Each value presents the measure of one animal (n = 7). p = 0.3784 (cBSA-KO vs cBSA-WT). |
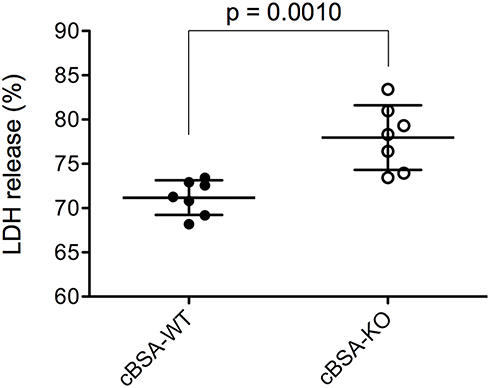 |
Figure 9 Effect of CLU in serum on CDC after cBSA immunization. The CDC against cultured HSMSs in the presence of 1:100 diluted serum was measured by the percentage of LDH release. Each value presents the measure of one animal (n = 7). p = 0.0010 (cBSA-KO vs cBSA-WT). |
Discussion
CLU was initially identified in the testis fluid 37 years ago,32 but its biological or physiological functions are still not fully understood. CLU is a major glycoprotein in the physiological fluids,15 and in human sera, it is found in a range of 35–353 µg/mL in different studies.33–36 In WT B6 mice, CLU protein was detected in the serum by using Western blot and is about 200 µg/mL in these mice based on the measurement with ELISA (data not shown). CLU expression is found in cultured T-HMC (human mesangial cell line)19 and is upregulated locally in the glomerular mesangial cells by the complement stimulation.37 In this study, we demonstrated that a lack of CLU expression in the circulation and glomerular cells transformed MN resistance of B6 mice to MN sensitivity in response to cBSA immunization, which was evidenced by an increase in proteinuria and the presence of glomerular injury and antibodies/C3 deposit in CLU-KO mice. Similarly, a previous study shows that CLU-KO results in the formation of immune complexes (IgG, IgM, IgA, and in some cases C1q, C3, and C9)-associated progressive glomerular injury in aging mice.17 In MN patients, the serum levels of CLU (220.77 ±110.9 µg/mL, n = 19) are markedly lower than that (366.6 ± 62.0 µg/mL, n = 50) in healthy controls (p < 0.001),21 and the levels of glomerular CLU in renal biopsies are associated with a reduction of proteinuria after a follow-up of 1.5 years at multivariate analysis.22 Thus, both clinical and experimental data may suggest a protective role of CLU from the local and/or the circulation against MN development.
In pathology, MN is typically characterized by the formation of immune deposits in the lamina rara externa of the GBM – the space between the podocytes and the GBM that cause a membrane-like thickening of the capillary wall.7,9–11 These immune complexes or deposits induce glomerular injury by damaging and/or activating podocytes or glomerular visceral epithelial cells through complement-dependent processes.10,11 So that the complement-mediated podocyte death plays an important role in the pathogenesis of MN. How CLU prevents inappropriate complement activation against podocytes in the pathogenesis of MN is not completely known. By immunohistochemical staining, we showed similar Igs deposit in the glomeruli of WT as compared with that of CLU-KO mice, and there was little difference in the serum levels of anti-cBSA IgG and C3 between these two groups of mice. However, whether their anti-cBSA IgG deposition in the kidney was the same between these two groups was not specifically examined. Recent evidence suggests that MN is mainly caused by the complement (C3, C4 and C5b-9)-mediated podocyte injury, which is initiated by the formation of immune complexes in the subepithelial area from circulating antibodies (ie, anti-PLA2R1 IgG4) binding to the implanted or autoantigens (ie, PLA2R1) on the podocytes.10,11 CLU is considered to be one of the most important extracellular chaperones in the plasma.38,39 Early studies have demonstrated that CLU is a complement-regulatory protein that directly binds to and inhibits C5b-9 or membrane attack complex (MAC) formation in different experimental systems including anti-Fx1A antibody-induced Heymann nephritis.40–43 In cBSA-immunized WT B6 mice, CLU in the serum bound to complement C8, a constituent of the MAC, and IgM and anti-cBSA IgG, and its expression was associated with the absence of C3 deposit in the glomeruli. These data may indicate that CLU locally and/or systemically prevents early complement pathway in this strain (B6) of mice. The presence of CLU in the serum reduced CDC against cultured podocytes as compared to CLU deficient control. Although the difference was not so large in this in vitro assay, it at least indicated that the presence of CLU could reduce the complement-mediated cytotoxicity, and it could bind to complement and Ig directly. Furthermore, CLU initiates the endocytic uptake and removal of misfolded protein aggregates, a process which is also termed proteostasis,44 suggesting a potential mechanism of CLU-mediated removal of glomerular immune complex deposit via endocytosis of the podocytes. Taken together, all these data suggest that CLU may inhibit or prevent antibody-activated MAC against the podocytes by the direct inhibition of complement activation and/or endocytosis-mediated removal of the activated immune complex in the pathogenesis of MN, at least in B6 mice after cBSA immunization.
In addition to the MAC and antibodies, CLU also binds to apolipoprotein A-I,45 ceruloplasmin, fibrinogen, and albumin.46 By using immunoprecipitation, Western blot and proteomic analysis although the specificity of this approach was concerned, we also found albumin, ceruloplasmin, fibronectin, and apolipoprotein A-E among the list of CLU-associated proteins that were pulled out from the serum of cBSA-immunized B6 mice. Fibronectin is an essential component of ECM, and genetically reducing the plasma fibronectin results in preventing ME and decreasing proteinuria in diabetic mice,47 but whether or not a lack of CLU binding causes more fibronectin deposit into the GBM or results in higher scores of ME in CLU-KO mice in this study remains further investigation. Other proteins incorporated into this “CLU complex” included proteinase/protease inhibitors (α2 macroglobulin, kininogen 1, and serine protease inhibitor A3K) and anti-oxidative peroxidases (glutathione peroxidase and peroxiredoxin 2). The functions of these proteins in the protection of podocytes in MN are unknown, but α2 macroglobulin inhibits mannose-binding protein (MBP)/MASP-derived complement activation (the lectin pathway)48 and serine protease inhibitor A3K prevents cell death by inhibiting benzalkonium chloride-stimulated TNF-α production in the cornea.49 In addition to these two peroxidases, both serine protease inhibitor A3K and ceruloplasmin are also found to have antioxidant activity as well.50–52 Taken together, these findings in literature may imply that the “CLU complex” in WT B6 mice not only inhibits complement activation via the binding of CLU and a2 macroglobulin but also protects the podocytes from oxidative stress-induced cell death by the activities of peroxidases, serine protease inhibitor A3K and ceruloplasmin, which however remains further investigation.
Administration of cBSA induces MN in ICR and BALB/c but not in B6 mice.26 The reasons why the susceptibility to cBSA-induced MN is different between these strains remain unknown. Similar to ICR or BALB/c mice,26,28 IgG1 was relatively predominate as compared to IgG2a in B6 mice (both WT and CLU-KO) in the response to cBSA immunization, suggesting that there was Th2 immune response that could favor anti-cBSA IgG1 production in B6 mice. However, it is still unknown if there is any difference in the kinetics of antibody responses between these strains of mice, including their production levels, the levels of specific anti-cBSA antibody and the numbers of cBSA-reactive B lymphocytes in the immune system. It is also interesting to note that there were some different clinical and histopathological features of cBSA-induced MN between ICR or BALB/c and CLU-KO mice; the MN induced an increase in SCr and ME and glomerular damage in CLU-KO B6 mice but not in WT ICR or BALB/c mice,26,28 suggesting that CLU in these ICR or BALB/c mice may reduce cBSA/anti-cBSA antibody complex-mediated glomerular injury as discussed above. In addition to CLU, there are many other complement regulatory proteins, such as decay-accelerating factor (DAF, CD55), membrane cofactor proteins (MCP, CD46), complement receptor 1 (CR1), Crry and CD59,53 these molecules may negatively affect the development of anti-cBSA IgG-induced MN in mice. As of today, the differences in the expression of these complement regulatory proteins including CLU between ICR, BALB/c and B6 mice are not investigated.
Conclusion
Clinical studies have reported a negative correlation of serum CLU levels with MN in a group of patients,21 and glomerular CLU is a factor that is associated with reduced proteinuria of MN patients.22 The data from this experimental study showed that CLU bound to antibodies including anti-cBSA IgG, complement components and probably anti-oxidative enzymes, which together may result in inactivation of inappropriate anti-cBSA IgG-activated complement activation or inflammation as well as prevention of oxidative stress-induced cell death within the glomeruli in B6 mice. However, the specific regulatory mechanisms of CLU, including CLU-mediated endocytosis of resident glomerular cells (eg, podocytes and mesangial cells) for removal of the immune complex and its inhibition of complement-mediated glomerular injury in the pathogenesis of primary and secondary MN needs further investigation.
Ethical Statement
The animal care and use for the experiments were performed in accordance with the Canadian Council on Animal Care guideline under the protocols (A16-0123, A18-0035) approved by the Animal Use Subcommittee at the University of British Columbia (Vancouver, BC, Canada). The mice were euthanized using inhalant anesthetic isoflurane, followed by CO2 according to the approved standard operating procedure (SOP: ACC CCM 2012-03 Euthanasia of Adult Rodents) from the University of British Columbia Animal Care Committee.
Acknowledgments
This work was financially supported by the Fourth Hospital of Harbin Medical University (Harbin, China) (to PS), the Institute of Reconstructive Urology at West China Hospital of Sichuan University (Chengdu, China) (to SF), and the Canadian Institutes of Health Research, the Natural Sciences and Engineering Research Council of Canada and Michael Smith Foundation for Health Research of Canada (to CD).
Disclosure
The authors declare that they have no conflicts of interest to disclose in this work.
References
1. Tinawi M. Update on the etiology, classification, and management of glomerular diseases. Avicenna J Med. 2020;10(2):61–67. doi:10.4103/ajm.ajm_136_19
2. Couser WG. Primary membranous nephropathy. Clin J Am Soc Nephrol. 2017;12(6):983–997. doi:10.2215/CJN.11761116
3. Ronco P, Debiec H. Pathophysiological advances in membranous nephropathy: time for a shift in patient’s care. Lancet. 2015;385(9981):1983–1992. doi:10.1016/S0140-6736(15)60731-0
4. Cattran D, Brenchley P. Membranous nephropathy: thinking through the therapeutic options. Nephrol Dial Transplant. 2017;32(suppl_1):i22–i29. doi:10.1093/ndt/gfw404
5. Maisonneuve P, Agodoa L, Gellert R, et al. Distribution of primary renal diseases leading to end-stage renal failure in the United States, Europe, and Australia/New Zealand: results from an international comparative study. Am J Kidney Dis. 2000;35(1):157–165. doi:10.1016/S0272-6386(00)70316-7
6. Simon P, Ramee MP, Autuly V, et al. Epidemiology of primary glomerular diseases in a French region. Variations according to period and age. Kidney Int. 1994;46(4):1192–1198. doi:10.1038/ki.1994.384
7. Fogo AB, Lusco MA, Najafian B, Alpers CE. AJKD atlas of renal pathology: membranous nephropathy. Am J Kidney Dis. 2015;66(3):e15–17. doi:10.1053/j.ajkd.2015.07.006
8. Polanco N, Gutierrez E, Covarsi A, et al. Spontaneous remission of nephrotic syndrome in idiopathic membranous nephropathy. J Am Soc Nephrol. 2010;21(4):697–704. doi:10.1681/ASN.2009080861
9. Trujillo H, Alonso M, Praga M. New ways of understanding membranous nephropathy. Nephron. 2020;144(6):261–271. doi:10.1159/000506948
10. Ma H, Sandor DG, Beck LH
11. Liu W, Gao C, Dai H, et al. Immunological pathogenesis of membranous nephropathy: focus on PLA2R1 and its role. Front Immunol. 2019;10:1809. doi:10.3389/fimmu.2019.01809
12. Hui M, Uppin MS, Prayaga AK, Raju SB, Rajasekhar L. C4d immunohistochemistry in membranous nephropathy. J Lab Physicians. 2014;6(2):76–79. doi:10.4103/0974-2727.141500
13. Hayashi N, Okada K, Matsui Y, et al. Glomerular mannose-binding lectin deposition in intrinsic antigen-related membranous nephropathy. Nephrol Dial Transplant. 2018;33(5):832–840. doi:10.1093/ndt/gfx235
14. Lateb M, Ouahmi H, Payre C, et al. Anti-PLA2R1 antibodies containing sera induce in vitro cytotoxicity mediated by complement activation. J Immunol Res. 2020;2019:1324804.
15. Jones SE, Jomary C. Clusterin. Int J Biochem Cell Biol. 2002;34(5):427–431. doi:10.1016/S1357-2725(01)00155-8
16. Guan Q, Alnasser HA, Nguan CYC, Du C. From humans to experimental models: the cytoprotective role of clusterin in the kidney. Med Surg Urol. 2014;3:134.
17. Rosenberg ME, Girton R, Finkel D, et al. Apolipoprotein J/clusterin prevents a progressive glomerulopathy of aging. Mol Cell Biol. 2002;22(6):1893–1902. doi:10.1128/MCB.22.6.1893-1902.2002
18. Jung GS, Jeon JH, Jung YA, et al. Clusterin/apolipoprotein J attenuates angiotensin II-induced renal fibrosis. PLoS One. 2014;9(8):e105635. doi:10.1371/journal.pone.0105635
19. Zhou W, Guan Q, Kwan CC, et al. Loss of clusterin expression worsens renal ischemia-reperfusion injury. Am J Physiol Renal Physiol. 2010;298(3):F568–578. doi:10.1152/ajprenal.00399.2009
20. Nguan CY, Guan Q, Gleave ME, Du C. Promotion of cell proliferation by clusterin in the renal tissue repair phase after ischemia-reperfusion injury. Am J Physiol Renal Physiol. 2014;306(7):F724–733. doi:10.1152/ajprenal.00410.2013
21. Ghiggeri GM, Bruschi M, Candiano G, et al. Depletion of clusterin in renal diseases causing nephrotic syndrome. Kidney Int. 2002;62(6):2184–2194. doi:10.1046/j.1523-1755.2002.00664.x
22. Rastaldi MP, Candiano G, Musante L, et al. Glomerular clusterin is associated with PKC-alpha/beta regulation and good outcome of membranous glomerulonephritis in humans. Kidney Int. 2006;70(3):477–485. doi:10.1038/sj.ki.5001563
23. Bass PS, Wang Y, Al Nawab M, Evans B, Thomas H, Davies DR. The effect of cyclosporin A on cationized bovine serum albumin-induced nephropathy in NZW rabbits. J Pathol. 1992;167(1):41–47. doi:10.1002/path.1711670108
24. Wright NG, Mohammed NA, Eckersall PD, Nash AS. Experimental immune complex glomerulonephritis in dogs receiving cationized bovine serum albumin. Res Vet Sci. 1985;38(3):322–328. doi:10.1016/S0034-5288(18)31803-4
25. Debiec H, Lefeu F, Kemper MJ, et al. Early-childhood membranous nephropathy due to cationic bovine serum albumin. N Engl J Med. 2011;364(22):2101–2110. doi:10.1056/NEJMoa1013792
26. Chen JS, Chen A, Chang LC, et al. Mouse model of membranous nephropathy induced by cationic bovine serum albumin: antigen dose-response relations and strain differences. Nephrol Dial Transplant. 2004;19(11):2721–2728. doi:10.1093/ndt/gfh419
27. Wu CC, Chen JS, Chen SJ, et al. Kinetics of adaptive immunity to cationic bovine serum albumin-induced membranous nephropathy. Kidney Int. 2007;72(7):831–840. doi:10.1038/sj.ki.5002426
28. Wu CC, Chen JS, Lin SH, Chen A, Sytwu HK, Lin YF. Experimental model of membranous nephropathy in mice: sequence of histological and biochemical events. Lab Anim. 2008;42(3):350–359. doi:10.1258/la.2007.06016e
29. Mundel P, Reiser J, Zuniga Mejia Borja A, et al. Rearrangements of the cytoskeleton and cell contacts induce process formation during differentiation of conditionally immortalized mouse podocyte cell lines. Exp Cell Res. 1997;236(1):248–258. doi:10.1006/excr.1997.3739
30. Elwood CN, Lo J, Chou E, et al. Understanding urinary conditioning film components on ureteral stents: profiling protein components and evaluating their role in bacterial colonization. Biofouling. 2013;29(9):1115–1122. doi:10.1080/08927014.2013.829049
31. Sinico RA, Mezzina N, Trezzi B, Ghiggeri GM, Radice A. Immunology of membranous nephropathy: from animal models to humans. Clin Exp Immunol. 2016;183(2):157–165. doi:10.1111/cei.12729
32. Blaschuk O, Burdzy K, Fritz IB. Purification and characterization of a cell-aggregating factor (clusterin), the major glycoprotein in ram rete testis fluid. J Biol Chem. 1983;258(12):7714–7720.
33. Murphy BF, Kirszbaum L, Walker ID, d’Apice AJ. SP-40,40, a newly identified normal human serum protein found in the SC5b-9 complex of complement and in the immune deposits in glomerulonephritis. J Clin Invest. 1988;81(6):1858–1864. doi:10.1172/JCI113531
34. Choi NH, Tobe T, Hara K, Yoshida H, Tomita M. Sandwich ELISA assay for quantitative measurement of SP-40,40 in seminal plasma and serum. J Immunol Methods. 1990;131(2):159–163. doi:10.1016/0022-1759(90)90186-Y
35. Morrissey C, Lakins J, Moquin A, Hussain M, Tenniswood M. An antigen capture assay for the measurement of serum clusterin concentrations. J Biochem Biophys Methods. 2001;48(1):13–21. doi:10.1016/S0165-022X(00)00137-8
36. Stahl AL, Kristoffersson A, Olin AI, et al. A novel mutation in the complement regulator clusterin in recurrent hemolytic uremic syndrome. Mol Immunol. 2009;46(11–12):2236–2243. doi:10.1016/j.molimm.2009.04.012
37. Yamada K, Hori Y, Hanafusa N, et al. Clusterin is up-regulated in glomerular mesangial cells in complement-mediated injury. Kidney Int. 2001;59(1):137–146. doi:10.1046/j.1523-1755.2001.00474.x
38. Rohne P, Prochnow H, Koch-Brandt C. The CLU-files: disentanglement of a mystery. Biomol Concepts. 2016;7(1):1–15. doi:10.1515/bmc-2015-0026
39. Wilson MR, Easterbrook-Smith SB. Clusterin is a secreted mammalian chaperone. Trends Biochem Sci. 2000;25(3):95–98. doi:10.1016/S0968-0004(99)01534-0
40. Jenne DE, Tschopp J. Molecular structure and functional characterization of a human complement cytolysis inhibitor found in blood and seminal plasma: identity to sulfated glycoprotein 2, a constituent of rat testis fluid. Proc Natl Acad Sci U S A. 1989;86(18):7123–7127. doi:10.1073/pnas.86.18.7123
41. Silkensen JR, Schwochau GB, Rosenberg ME. The role of clusterin in tissue injury. Biochem Cell Biol. 1994;72(11–12):483–488. doi:10.1139/o94-065
42. Murphy BF, Saunders JR, O’Bryan MK, Kirszbaum L, Walker ID, d’Apice AJ. SP-40,40 is an inhibitor of C5b-6-initiated haemolysis. Int Immunol. 1989;1(5):551–554. doi:10.1093/intimm/1.5.551
43. Saunders JR, Aminian A, McRae JL, O’Farrell KA, Adam WR, Murphy BF. Clusterin depletion enhances immune glomerular injury in the isolated perfused kidney. Kidney Int. 1994;45(3):817–827. doi:10.1038/ki.1994.108
44. Wyatt AR, Yerbury JJ, Berghofer P, et al. Clusterin facilitates in vivo clearance of extracellular misfolded proteins. Cell Mol Life Sci. 2011;68(23):3919–3931. doi:10.1007/s00018-011-0684-8
45. Jenne DE, Lowin B, Peitsch MC, Bottcher A, Schmitz G, Tschopp J. Clusterin (complement lysis inhibitor) forms a high density lipoprotein complex with apolipoprotein A-I in human plasma. J Biol Chem. 1991;266(17):11030–11036.
46. Wyatt AR, Wilson MR. Identification of human plasma proteins as major clients for the extracellular chaperone clusterin. J Biol Chem. 2010;285(6):3532–3539. doi:10.1074/jbc.M109.079566
47. Klemis V, Ghura H, Federico G, et al. Circulating fibronectin contributes to mesangial expansion in a murine model of type 1 diabetes. Kidney Int. 2017;91(6):1374–1385. doi:10.1016/j.kint.2016.12.006
48. Terai I, Kobayashi K, Matsushita M, Fujita T, Matsuno K, Okumura K. Alpha 2-macroglobulin binds to and inhibits mannose-binding protein-associated serine protease. Int Immunol. 1995;7(10):1579–1584. doi:10.1093/intimm/7.10.1579
49. Lin Z, Zhou Y, Wang Y, et al. Serine protease inhibitor A3K suppressed the formation of ocular surface squamous metaplasia in a mouse model of experimental dry eye. Invest Ophthalmol Vis Sci. 2014;55(9):5813–5820. doi:10.1167/iovs.13-13546
50. Shiva S, Wang X, Ringwood LA, et al. Ceruloplasmin is a NO oxidase and nitrite synthase that determines endocrine NO homeostasis. Nat Chem Biol. 2006;2(9):486–493. doi:10.1038/nchembio813
51. Wiggins JE, Goyal M, Wharram BL, Wiggins RC. Antioxidant ceruloplasmin is expressed by glomerular parietal epithelial cells and secreted into urine in association with glomerular aging and high-calorie diet. J Am Soc Nephrol. 2006;17(5):1382–1387. doi:10.1681/ASN.2005111239
52. Zhang B, Ma JX. SERPINA3K prevents oxidative stress induced necrotic cell death by inhibiting calcium overload. PLoS One. 2008;3(12):e4077.
53. Miwa T, Song WC. Membrane complement regulatory proteins: insight from animal studies and relevance to human diseases. Int Immunopharmacol. 2001;1(3):445–459. doi:10.1016/S1567-5769(00)00043-6
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.