Back to Journals » Journal of Asthma and Allergy » Volume 19
Clonal Mast Cell Disorders and Mutation Profile in Adults Initially Presenting with Mast Cell Activation (Anaphylaxis) versus Non-Anaphylactic Presentations: A Joint Allergy–Hematology Cohort
Authors Pornsuthirat P, Owattanapanich W ![]() , Wongsa C, Thongngarm T
, Wongsa C, Thongngarm T ![]() , Sompornrattanaphan M
, Sompornrattanaphan M ![]()
Received 7 January 2026
Accepted for publication 31 March 2026
Published 14 April 2026 Volume 2026:19 594281
DOI https://doi.org/10.2147/JAA.S594281
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Luis Garcia-Marcos
Patchara Pornsuthirat,1 Weerapat Owattanapanich,2 Chamard Wongsa,1,3 Torpong Thongngarm,1,3 Mongkhon Sompornrattanaphan1,3
1Division of Allergy and Clinical Immunology, Department of Medicine, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok, Thailand; 2Division of Hematology, Department of Medicine, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok, Thailand; 3Center of Research Excellence in Allergy and Immunology, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok, Thailand
Correspondence: Mongkhon Sompornrattanaphan, Division of Allergy and Clinical Immunology, Department of Medicine, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok, Thailand, Email [email protected]
Purpose: To compare the diagnostic yield, clinical, and molecular features of clonal mast cell disorders (CMCDs) in adults presenting with mast cell activation (MCA)/anaphylaxis versus non-anaphylactic phenotypes in a joint Allergy-Hematology cohort.
Patients and Methods: In this single-center retrospective study, we reviewed clinician-selected adults (≥ 18 years) evaluated at a Thai tertiary center (2019– 2024) who completed joint Allergy-Hematology evaluation and targeted 66-gene myeloid next-generation sequencing (NGS). Index presentations were classified as MCA/anaphylaxis versus non-MCA. Final diagnoses followed WHO 5th/International Consensus Classification criteria.
Results: Of 24 sequenced adults, 14 (58.3%) had MCA/anaphylaxis and 10 (41.7%) had non-MCA presentations. Among sequenced MCA/anaphylaxis presentations, final diagnoses included idiopathic anaphylaxis (IA, n=11), secondary anaphylaxis (n=2), and systemic mastocytosis (SM, n=1). Conversely, all 10 non-MCA cases had CMCD (SM 6, cutaneous mastocytosis 4). Compared to non-mastocytosis cases, mastocytosis patients were older, predominantly male, with higher basal tryptase (median 22.4 vs 3.2 ng/mL). Pathogenic variants occurred in 12/24 (50%) patients: KIT D816V in 6/24 (25%), with heterogeneous TET2 co-mutations, DNMT3A R882H, and SRSF2 P95 hotspots clustering in SM. IA rarely harbored driver mutations.
Conclusion: CMCD showed a low diagnostic yield (1/14, 7.1%) in the clinician-selected MCA/anaphylaxis subgroup escalated to marrow/NGS, but was universal in the hematologic non-MCA subset (10/10, 100%). These findings suggest that REMA-based, risk-stratified selection may be useful in this setting, but larger prospective studies are needed. While TET2 was the most frequently mutated gene overall, the mutational profile within the SM subgroup—characterized by KIT D816V and frequent TET2, DNMT3A, and SRSF2 co-mutations—parallels established molecular signatures in Western systemic mastocytosis cohorts, suggesting KIT-targeted therapies are biologically applicable in Thai patients. A clinician-selected cohort undergoing NGS includes 24 patients. In the MCA/Anaphylaxis group (n=14), the CMCD yield is 7.1 percent, mainly idiopathic anaphylaxis. In the Non-MCA group (n=10), the CMCD yield is 100 percent, with systemic mastocytosis (SM) in 6 and cutaneous mastocytosis (CM) in 4. The most frequent driver mutation is KIT D816V, dominant in SM at 85.7 percent. Frequent co-mutations in SM (n=7) include TET2 at 71 percent, SRSF2 at 57 percent and DNMT3A at 29 percent. Pathogenic variants are detected in 50 percent of the cohort, while IA driver mutations are rare. The patient profile shows mastocytosis (n=11) with a mean age of 63, tryptase median of 22.4 ng/mL and 72.7 percent male. Non-mastocytosis (n=13) has a mean age of 40, tryptase median of 3.2 ng/mL and 7.7 percent male. Conclusion highlights risk stratification and therapeutic applicability, with REMA/NICAS scores optimizing patient selection for bone marrow and NGS and Thai SM profiles suggesting potential applicability of KIT-targeted therapies.Infographic comparing CMCDs in MCA vs. non-MCA presentations with patient profiles and mutation data.
Keywords: anaphylaxis, KIT D816V, systemic mastocytosis, mast cell activation syndrome, next-generation sequencing, REMA score
Introduction
Clonal mast cell disorders (CMCDs) are clonal myeloid neoplasms characterized by expansion, tissue infiltration, and aberrant immunophenotype of mast cells, usually driven by somatic activating KITsup mutations, most commonly KIT D816V.1–3 CMCDs include systemic mastocytosis (SM)—now classified as a myeloid neoplasm in the 5th edition WHO Haematolymphoid Tumours and the International Consensus Classification—and related clonal mast cell proliferations such as monoclonal mast cell activation syndrome (MMAS), in which KIT-mutated, immunophenotypically aberrant mast cells are present but full SM criteria are not met.1,2 In adults, the most frequent CMCD phenotype is indolent systemic mastocytosis (ISM), which is usually not a high-grade hematologic malignancy, but morbidity is driven by recurrent mast cell mediator–related symptoms (flushing, hypotension or syncope, anaphylaxis) and skeletal involvement (e.g., osteoporosis).1 Accordingly, these patients are seen not only in hematology but also—often first—in the allergy/immunology clinics.1,4
From the allergy/immunology perspective, clinicians are consulted to evaluate “mast cell activation (MCA),” “idiopathic anaphylaxis (IA),” or “possible mast cell activation syndrome (MCAS).”4,5 Severe anaphylaxis in adults—particularly hypotensive anaphylaxis with syncope and absence of cutaneous findings—is now recognized as a clinical red flag for an occult clonal mast cell disorder.5,6 Allergists are therefore expected to identify which adults with IA or MCA may harbor clonal disease and to coordinate early assessment with hematology.1,4,5
However, most data linking severe anaphylaxis to CMCD come from European venom-allergy and mastocytosis centers, particularly in patients with Hymenoptera sting anaphylaxis and hypotension.5,6 To date, no cohort studies from Southeast Asia have reported the diagnostic yield/detection of CMCD among selected adults referred for suspected MCA or IA.3,5,7 Hematology-based series often focus on disease classification, KIT mutation status, and prognosis but rarely describe the initial clinical presentation in a way that informs allergy practice.1,3 Targeted next-generation sequencing (NGS) panels are now routinely used in myeloid neoplasms to detect recurrent somatic mutations across signaling, epigenetic, and splicing genes (eg., KIT, TET2, ASXL1, SRSF2, RUNX1), refine prognostic stratification, and identify high-risk constellations such as the SRSF2/ASXL1/RUNX1 (“S/A/R”) genotype in advanced systemic mastocytosis.8 From a clinical allergy perspective, a key unanswered question is whether routine escalation to bone marrow examination and molecular testing is justified in adults presenting with recurrent MCA or IA in real-world practice, particularly outside specialized mastocytosis referral centers.
We therefore conducted a retrospective study in a Thai academic center to (1) estimate the diagnostic yield of CMCD among adults selected for joint Allergy–Hematology work-up and NGS; (2) compare clinical features and final diagnoses across MCA versus non-anaphylactic presentations; and (3) characterize the underlying mutation profile using targeted next-generation sequencing.
Materials and Methods
Study Design and Setting
We conducted a retrospective observational cohort study at Siriraj Hospital, Bangkok, Thailand, from 1 January 2019 to 29 February 2024. During this time, the internal medicine Allergy/Immunology service evaluated several hundred adult encounters for suspected mast cell activation (MCA) presentations, as well as additional cases presenting with non-MCA phenotypes. Rather than employing a systematic screening approach, referral to hematology, bone marrow evaluation, and NGS followed a clinician-directed escalation pathway based on perceived clinical risk. Because no standardized protocol was applied, this process inherently created a highly selected, high-risk subgroup of patients. This analysis includes only adults who completed a full Allergy–Hematology evaluation and underwent targeted NGS. The study was approved by the Siriraj Institutional Review Board under expedited review (SIRB No. 072/2024 [IRB3]), and informed consent was waived due to the retrospective design.
Participants
Eligible patients were aged ≥18 years and had a final multidisciplinary diagnosis consistent with a clonal mast cell disorder (CMCD) or a mast cell activation–type diagnosis (eg., anaphylaxis). Importantly, inclusion was strictly limited to the selected subset of patients who completed a full diagnostic evaluation and underwent targeted 66-gene myeloid NGS from a peripheral blood or bone marrow sample during the study period. Patients were excluded if they had an incomplete evaluation, defined as the absence of both baseline serum tryptase and KIT mutation testing.
Index Presentation Classification
For analysis, each patient was assigned an “index presentation phenotype” based on the initial documented clinical presentation that prompted specialist referral, not on downstream diagnostic findings.
MCA/anaphylaxis presentation
Defined as any of the following at first presentation: Idiopathic or hypotensive anaphylaxis requiring acute care, with physician-documented anaphylaxis; Recurrent stereotyped multisystem episodes clinically assessed as mast cell activation by an allergist/immunologist, typically involving ≥2 organ systems; Syncope or presyncope with generalized flushing or erythema without an alternative explanation. Paired acute and baseline serum tryptase and the “20% + 2 ng/mL” formula were recorded when available, but were not required to assign this category.
Non-MCA presentation
Defined as an initial presentation not dominated by mast cell mediator symptoms or anaphylaxis but instead featuring findings that prompted hematologic evaluation for a mast cell disorder. These included unexplained cytopenias, splenomegaly, abnormal bone marrow morphology identified during evaluation for another hematologic condition, persistent cutaneous lesions suggestive of mastocytosis, or chronic gastrointestinal or constitutional symptoms without collapse.
If a patient exhibited both patterns over time, categorization was based on the earliest clinical presentation that led to specialist consultation.
Diagnostic Assessment and Final Adjudication
All patients underwent coordinated evaluation by the Allergy and Hematology services. The following variables were abstracted from the medical record: age, sex, index presentation (MCA/anaphylaxis vs non-MCA), hemodynamic features (hypotension, syncope), suspected trigger, key examination findings (eg., urticaria pigmentosa, organomegaly), baseline laboratory results (complete blood count, serum albumin, baseline tryptase), bone marrow findings (when available), and persistence of mast cell activation symptoms after diagnosis.
Final diagnoses were determined by a multidisciplinary team (allergy/immunology, hematology, dermatology) using an integrated assessment that included clinical features, acute and baseline tryptase trends, mast cell immunophenotyping (CD25, CD2, CD30 when performed), bone marrow morphology, and molecular findings. Diagnostic adjudication followed the WHO 5th edition and International Consensus Classification frameworks, which recognize mastocytosis as a clonal myeloid neoplasm. Diagnostic categories included systemic mastocytosis (SM), cutaneous mastocytosis (CM), idiopathic anaphylaxis (IA), and secondary anaphylaxis (for non-clonal presentations). Monoclonal mast cell activation syndrome (MMAS) was defined as a clonal KIT-mutated, immunophenotypically aberrant mast cell population that did not fulfill full criteria for SM.1,9
We also abstracted two published clinical risk scores—the Red Española de Mastocitosis (REMA) score10 and the NIH Idiopathic Clonal Anaphylaxis Score (NICAS)11—for descriptive analysis; however, these scores were not used to determine study inclusion.
Next-Generation Sequencing
Genomic testing was performed on DNA extracted from peripheral blood or bone marrow aspirate. NGS was conducted using the QIAseq Targeted DNA Pro Panel (Qiagen, Hilden, Germany), covering 66 genes recurrently mutated in myeloid and mast cell neoplasms; the complete gene list is provided in Supplementary Methods 1. Libraries were prepared from 30 ng of genomic DNA according to the manufacturer’s protocol and sequenced on a NextSeq 1000 platform (Illumina, San Diego, CA, USA) using a P1 XLEAP-SBS reagent kit (300 cycles; paired-end 2×150 bp). Analytical performance for KIT D816V detection was estimated at 60–80% sensitivity and >99% specificity.
Raw FASTQ files were processed through a validated QIAGEN bioinformatics pipeline. Secondary analysis was performed in CLC Genomics Workbench (QIAGEN), including alignment to the human reference genome (hg38) and calling of single-nucleotide variants and small insertions/deletions using the built-in variant detection workflow. Variant interpretation and clinical classification were performed in QIAGEN Clinical Insight Interpret (QCI-I). Pathogenic or likely pathogenic variants with a variant allele frequency (VAF) >3% were reported; selected clinically relevant hotspot variants with VAF >0.5% were also considered. Variant classifications were further manually reviewed against COSMIC and VarSome, and discrepant interpretations were resolved by investigator consensus. Classification followed American College of Medical Genetics and Association for Molecular Pathology recommendations.
Outcomes
Primary outcome was the diagnostic yield of CMCD among patients with an MCA/anaphylaxis index presentation who completed escalated work-up including NGS who were ultimately diagnosed with a clonal mast cell disorder (SM, CM, or other CMCDs) after full Allergy–Hematology work-up. Secondary outcomes included: (1) the distribution of final diagnoses (SM, CM, MMAS, IA, secondary anaphylaxis) across MCA/anaphylaxis versus non-MCA presentations; (2) presenting clinical features (eg., hypotension, suspected trigger, urticaria pigmentosa, cytopenias, platelet count, serum albumin, baseline serum tryptase); and (3) the mutational profile of clonal mast cell disease, including KIT D816V and co-occurring myeloid mutations.
Statistical Analysis
Continuous variables were summarized as median (interquartile range [IQR]) or mean ± standard deviation (SD), and categorical variables as counts and percentages. Group comparisons—mastocytosis (SM+CM) vs non-mastocytosis, and MCA/anaphylaxis vs non-MCA index presentation—were conducted using the Mann–Whitney U-test for continuous variables and Fisher’s exact or Chi-square test for categorical variables. Proportions were reported with two-sided 95% confidence intervals (Wilson method). A two-sided p-value <0.05 was considered statistically significant. All analyses were performed using R (R Foundation for Statistical Computing).
Results
Cohort and Diagnostic Pathways
Between 1 January 2019 and 29 February 2024, the service evaluated 728 adult encounters with MCA-type presentations and 235 encounters with non-MCA presentations. Escalation to joint Allergy–Hematology assessment, bone marrow evaluation, and NGS was initiated by the clinician. Twenty-four unique adults completed a full evaluation, including NGS, and formed the analytic cohort (Figure 1). Because this escalation was based on clinical judgment, the analytic cohort represents a highly selected subgroup; therefore, the downstream percentages reflect diagnostic yield in this escalated group and do not represent prevalence across all referrals. Of these, 14/24 (58.3%) had an MCA/anaphylaxis index presentation, and 10/24 (41.7%) had a non-MCA presentation, as defined in the Methods. Restricting the molecular findings to this sequenced analytic cohort, 12/24 (50.0%) harbored at least one pathogenic or likely pathogenic variant; 4/14 (28.6%) in the MCA/anaphylaxis group and 8/10 (80.0%) in the non-MCA group.
 |
Figure 1 Flow of study participants. Abbreviations: ASM, aggressive systemic mastocytosis; CM, cutaneous mastocytosis; CMCD, clonal mast cell disorder; IA, idiopathic anaphylaxis; ISM, indolent systemic mastocytosis; MCA, mast cell activation; NGS, next-generation sequencing; SM-AHN, systemic mastocytosis with an associated hematologic neoplasm. Notes: Only patients who underwent NGS (n=24) formed the analytic cohort; other encounters are shown to illustrate overall service volume and were not systematically assessed for clonal disease. |
Clinical Characteristics and Final Diagnoses
Baseline characteristics are summarized in Table 1. Of the 24 sequenced patients, 9 (37.5%) were male. The median age at diagnosis was 49 years (range 18–78), and 7 (29.2%) were ≥60 years. Fourteen patients (58.3%) had mediator-type symptoms at presentation consistent with MCA/anaphylaxis. Urticaria pigmentosa was documented in 5/24 (20.8%) patients; 4/5 (80.0%) of these were ultimately diagnosed with cutaneous mastocytosis. Cytopenias were uncommon: hemoglobin <10 g/dL occurred in 3/24 (12.5%) and platelets <100×109/L in 3/24 (12.5%). Hypoalbuminemia (albumin <3.5 g/dL) was present in 2/20 evaluable patients (10.0%), and elevated alkaline phosphatase in 6/19 (31.6%).
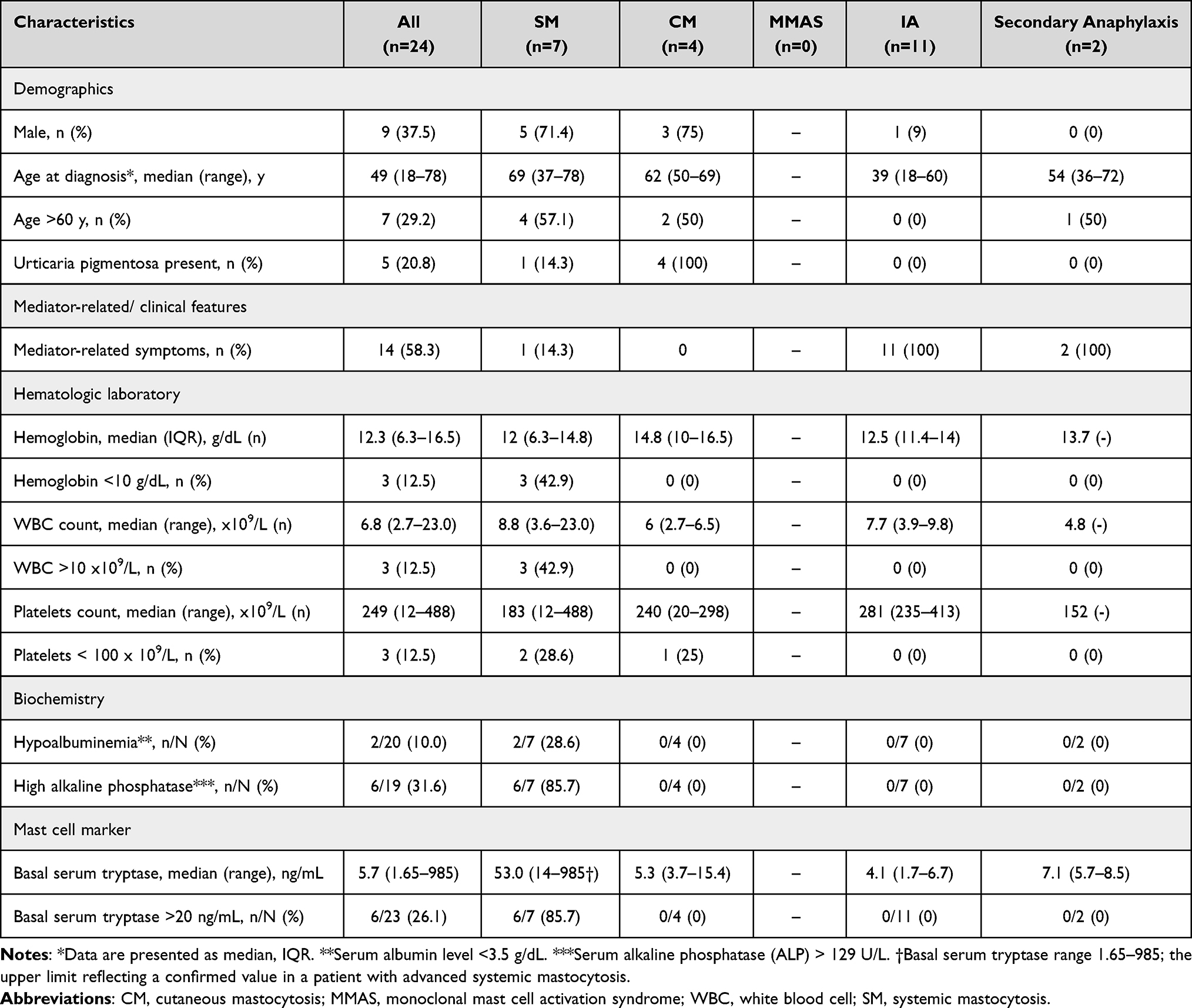 |
Table 1 Baseline Clinical Characteristics of the NGS-Sequenced Cohort (N=24) |
The median baseline serum tryptase for the cohort was 5.7 ng/mL (range 1.65–985†; the upper limit reflecting a confirmed value in a patient with advanced systemic mastocytosis), and 6/23 (26.1%) had a basal tryptase >20 ng/mL. Final multidisciplinary diagnoses were idiopathic anaphylaxis (IA) in 11/24 (45.8%), systemic mastocytosis (SM) in 7/24 (29.2%), cutaneous mastocytosis (CM) in 4/24 (16.7%), and secondary anaphylaxis in 2/24 (8.3%); no patient was adjudicated as MMAS.
Mastocytosis versus Non-Mastocytosis
For descriptive comparison, patients were grouped as mastocytosis (SM+CM; n=11) versus non-mastocytosis (IA or other identified cause of anaphylaxis [secondary causes]; n=13) (Table 2). Patients with mastocytosis were older than those without mastocytosis (mean 63.0 ± 13.5 vs 40.0 ± 16.2 years; p=0.001) and more often ≥60 years (72.7% vs 7.7%; p=0.023). They were predominantly male (72.7% vs 7.7%; p=0.002). Mediator-related symptoms were present in 1/11 (9.1%) in the mastocytosis group compared with 13/13 (100%) in the non-mastocytosis group (p<0.001).
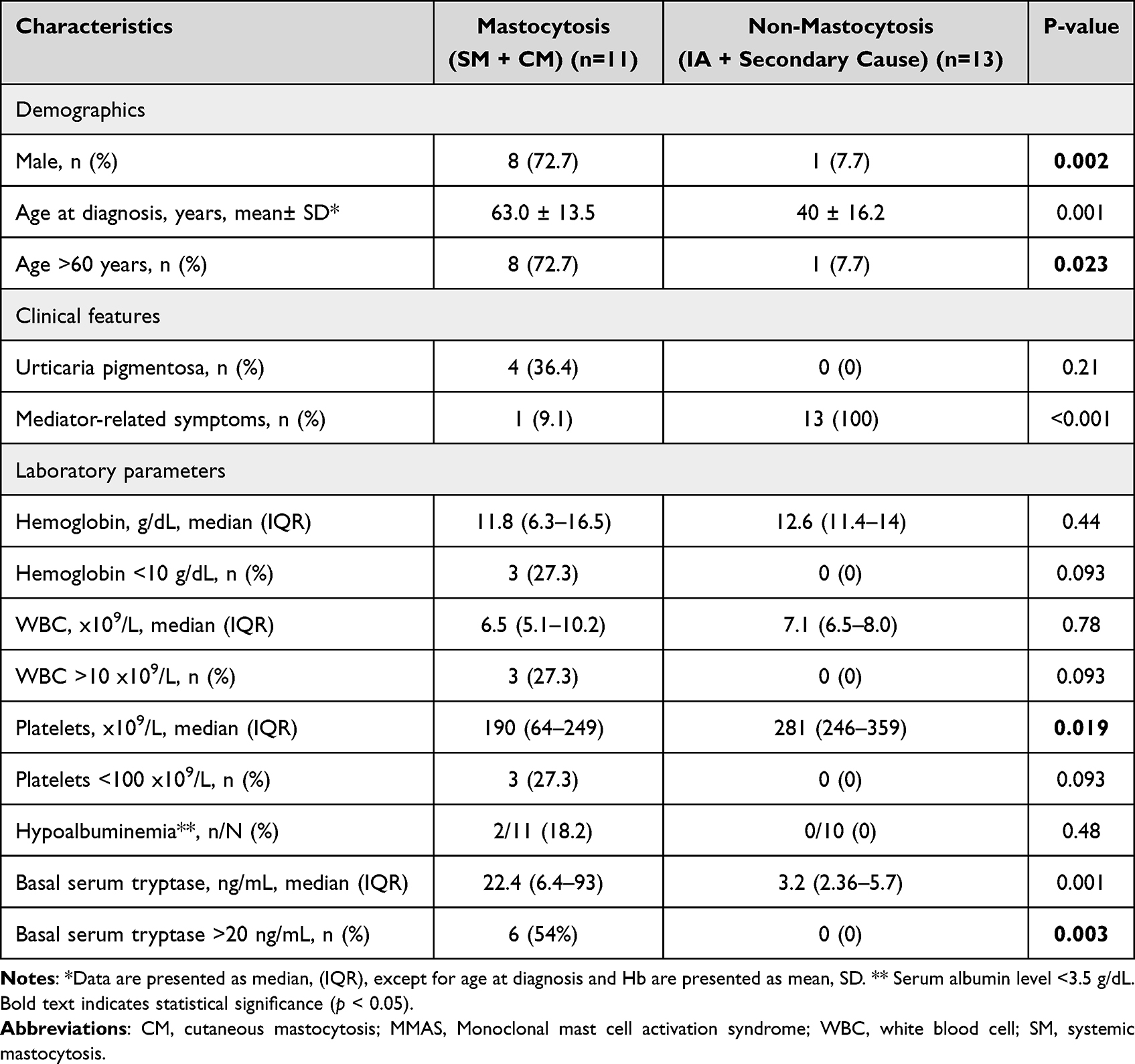 |
Table 2 Clinical and Laboratory Characteristics of Patients with Mastocytosis versus Non-Mastocytosis (N=24) |
Platelet counts were lower in mastocytosis than in non-mastocytosis (median 190×109/L [IQR 64–249] vs 281×109/L [IQR 246–359]; p=0.019). Basal mast cell markers also differed: median baseline serum tryptase was higher in mastocytosis (22.4 ng/mL [IQR 6.4–93]) than in non-mastocytosis (3.2 ng/mL [IQR 2.36–5.7]; p=0.001), and a basal tryptase >20 ng/mL occurred only in the mastocytosis group (6/11 [54%] vs 0/13 [0%]; p=0.003).
CMCD by Presentation Pathway
Across the 24 sequenced adults, 14/24 (58.3%) had an MCA/anaphylaxis index presentation and 10/24 (41.7%) had a non-MCA presentation. The diagnostic yield of CMCD in the selected subgroup was lower in the MCA pathway: 1/14 (7.1%; SM; 95% CI 1.3–31.5%) versus 10/10 (100%; SM 6, CM 4; 95% CI 72.2–100%), respectively among sequenced patients (Figure 1). Thus, CMCDs clustered in the hematology/non-MCA pathway, whereas MCA/anaphylaxis referrals were rarely clonal in this cohort.
Molecular Findings
Overall cytogenetic and molecular results are presented in Table 3, with an oncoplot of SM cases shown in Figure 2. Pathogenic or likely pathogenic variants were identified in 12/24 patients (50.0%). The most frequent mutations were TET2 in 7/24 (29.2%), KIT D816V in 6/24 (25.0%), DNMT3A in 4/24 (16.7%), and SRSF2 in 4/24 (16.7%). Additional recurrent mutations included ASXL1 (2/24, 8.3%), CBL (2/24, 8.3%), MPL (2/24, 8.3%), RUNX1 (1/24, 4.2%), and WT1 (1/24, 4.2%). Beyond KIT D816V, specific hotspot mutations such as DNMT3A R882H and SRSF2 P95 clustered within the SM cohort. In contrast, driver mutations were rarely observed in IA cases.
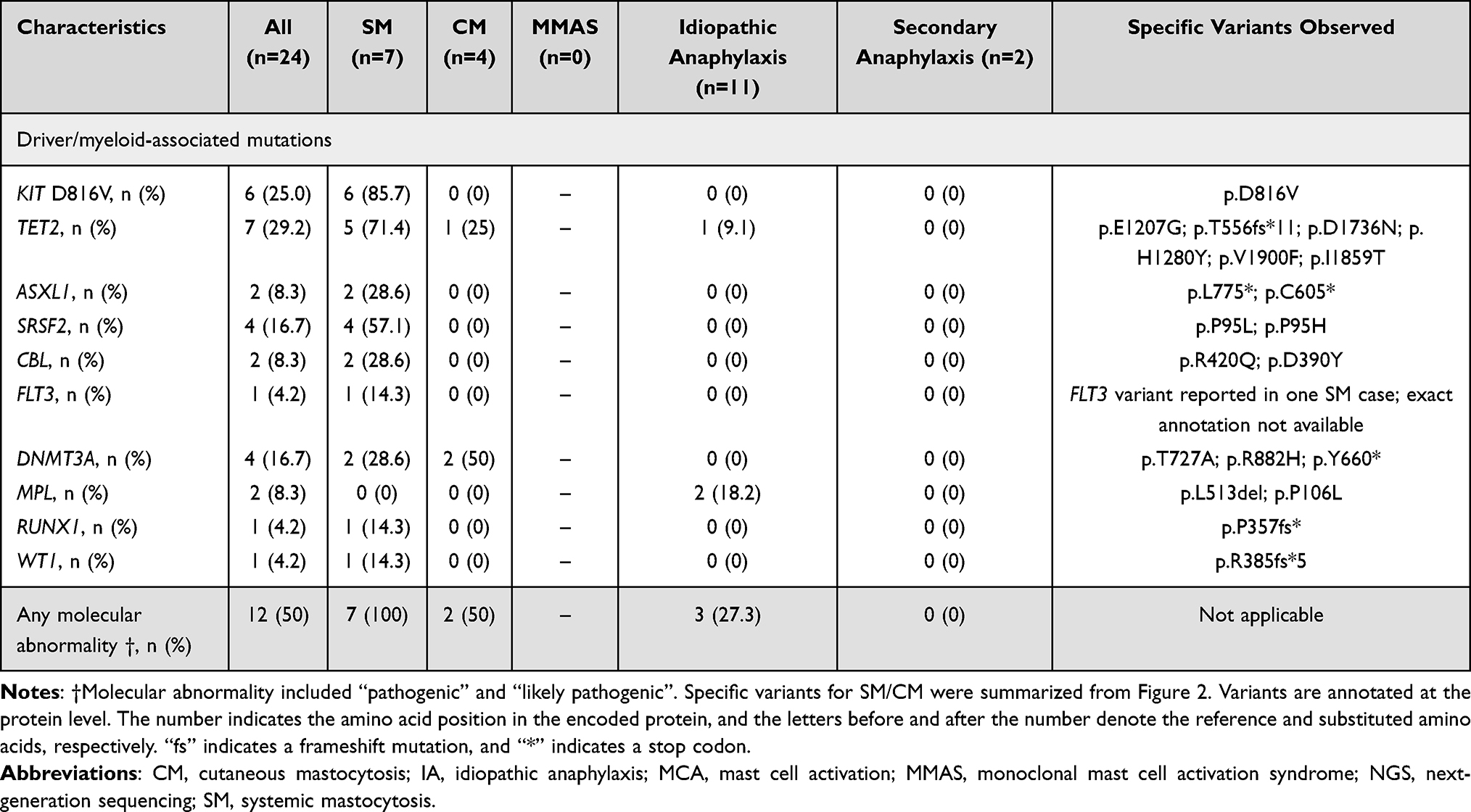 |
Table 3 Cytogenetic and Molecular Findings in the NGS-Sequenced Cohort (N=24) |
 |
Figure 2 Oncoplots, an alignment of gene mutations with MCAD subtype. Each row represents an individual patient. Columns list the 10 genes that were interrogated for mutations. Individual genes are arranged from left to right reflecting those with highest to lowest mutation frequency (N=9). Orange columns represent patients with Advanced Systemic Mastocytosis (AdvSM), and yellow columns represent patients with Non-advanced Systemic Mastocytosis (Non-AdvSM). fs indicates a frameshift mutation, and “*” indicates a stop codon. Abbreviations: AdvSM, advanced systemic mastocytosis; Non-AdvSM, Non-advanced systemic mastocytosis; N/A, Not Available; ASXL1, Additional sex combs like transcriptional regulator 1; CBL, Casitas B-lineage Lymphoma; DNMT3A, DNA (cytosine- 5)-methyltransferase 3A; FLT3, FMS-like tyrosine kinase3; KIT, tyrosine-protein kinase; MPL, MPL proto-oncogene, thrombopoietin receptor; RUNX1, Runt-related transcription factor 1; SRSF2, Serine/arginine-rich splicing factor 2; TET2, Tet Methylcytosine Dioxygenase 2; WT1, Wilms tumor suppressor gene; ASM, AdvSM Includes Aggressive Systemic Mastocytosis; SM-AHN, Systemic Mastocytosis with Associated Hematologic Neoplasm; MCL, Mast Cell Leukemia; ISM, Non-AdvSM Includes Indolent Systemic Mastocytosis; SSM, Smoldering Systemic Mastocytosis; BMM, Bone Marrow Mastocytosis. |
Within MCA presentations, excluding the single SM case, driver mutations were infrequent and non-KIT. Among patients with IA (n=11), MPL variants were detected in 2/11 (18.2%) and TET2 in 1/11 (9.1%); no other recurrent drivers were identified. No pathogenic variants were detected in the two patients with secondary anaphylaxis. Including the single SM case, any pathogenic variant was present in 4/14 (28.6%) MCA/anaphylaxis presentations, and KIT D816V was not detected in IA or in secondary anaphylaxis.
Clonal signals were concentrated in SM. KIT D816V was detected in 6/7 SM patients (85.7%) and in none of the IA or other identified-cause cases. TET2 (5/7, 71.4%), SRSF2 (4/7, 57.1%), and DNMT3A (2/7, 28.6%) were also frequent in SM. By contrast, most IA cases lacked detectable driver mutations: only 1/11 (9.1%) carried TET2, and none carried KIT D816V. CM showed a weaker clonal signal: KIT D816V was not detected, although TET2 (1/4, 25.0%) and DNMT3A (2/4, 50.0%) mutations were observed. Overall, 7/7 SM patients (100%) and 2/4 CM patients (50.0%) harbored at least one pathogenic variant, compared with 3/11 (27.3%) in the IA group and 0/2 in the other-cause group. All 4 CM cases entered via the non-MCA pathway.
Triggers Within MCA Presentations
Within the MCA/anaphylaxis index group (n=14), final diagnoses were IA in 11/14 (78.6%), other identified cause of anaphylaxis (secondary causes) in 2/14 (14.3%), and SM in 1/14 (7.1%). Detailed subclassification of MCA episodes by specific triggers (eg., venom, food, drug) was not available for all patients. Among the 11 patients with mastocytosis (7 SM and 4 CM), the only identified trigger for mast cell–mediated symptoms was a honeybee sting in 1 patient with indolent SM that resulted in severe anaphylaxis. No drug or food allergies were reported in the remaining mastocytosis patients based on nurse-administered screening during chart review.
Additional longitudinal characteristics of the MCA/anaphylaxis subgroup with complete follow-up data (n=12)—including number of anaphylaxis episodes during follow-up, emergency visits and hospitalizations, use of intramuscular adrenaline, and REMA and NICAS risk scores—are summarized in Table 4.
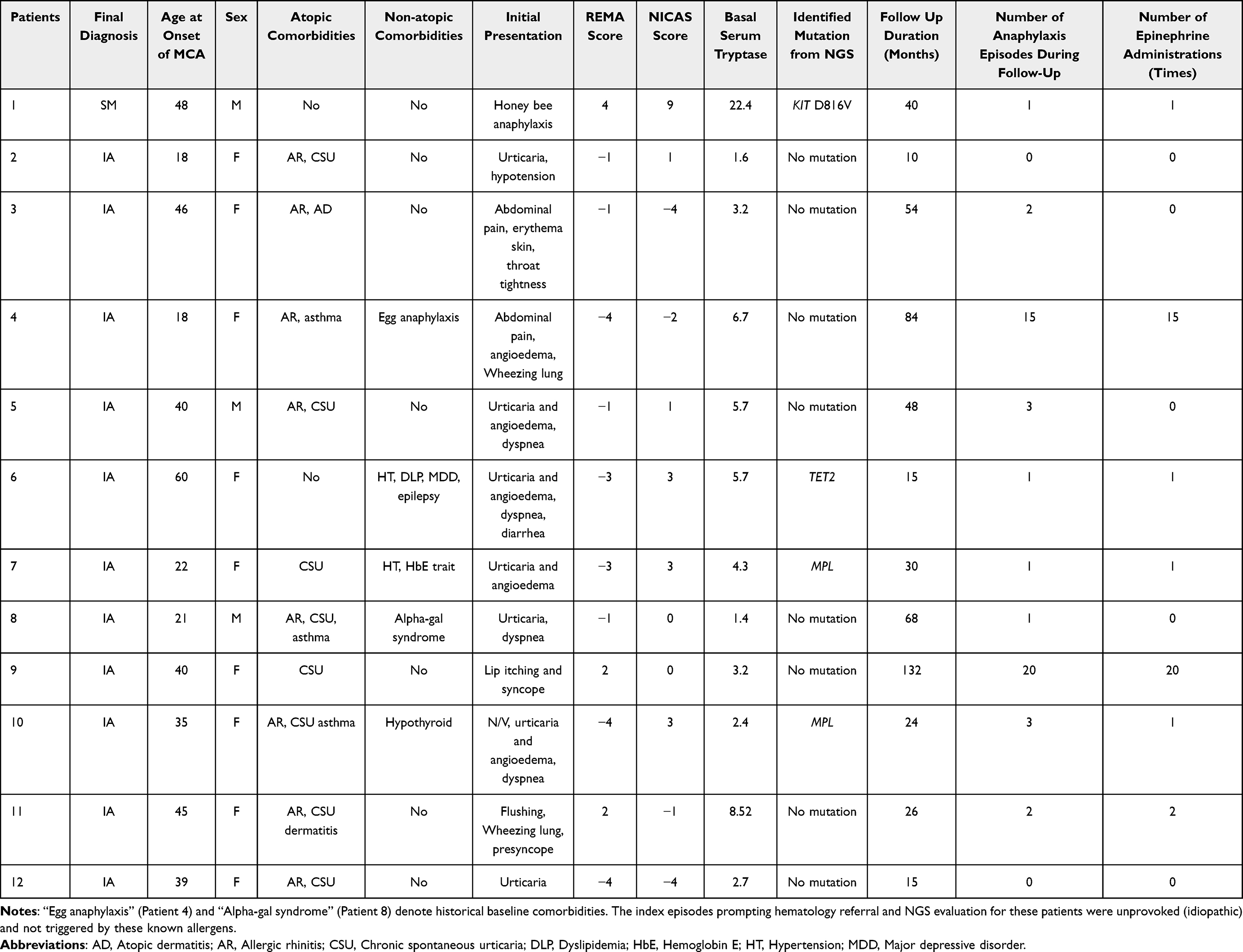 |
Table 4 Clinical Characteristics and Natural Course of Patients with MCA/Anaphylaxis Index Presentations (n=12) |
Advanced Systemic Mastocytosis
Six patients met criteria for advanced systemic mastocytosis (aggressive SM, SM with an associated hematologic neoplasm, or mast cell leukemia). Their mutational profile included KIT D816V in 5/6 (83.3%), TET2 in 5/6 (83.3%), SRSF2 in 3/6 (50.0%), DNMT3A in 2/6 (33.3%), ASXL1 in 2/6 (33.3%), and CBL in 2/6 (33.3%), with FLT3, RUNX1, and WT1 mutations each detected in 1/6 (16.7%) (Figure 2).
Discussion
In this Thai tertiary cohort, the diagnostic yield of CMCD was low within the clinician-selected MCA/anaphylaxis subgroup escalated to marrow/NGS (1/14, 7.1%) but was universal among those entering via non-MCA hematology pathways (10/10, 100%). Within MCA presentations, most patients were ultimately classified as IA or secondary anaphylaxis, and NGS seldom demonstrated KIT D816V. The detected variants, such as TET2 or MPL, in IA are more compatible with age-related clonal hematopoiesis than with CMCD. By contrast, SM showed the expected KIT-centric architecture with frequent co-mutations in TET2, SRSF2, and DNMT3A, while CM included KIT-negative cases carrying myeloid co-mutations. Taken together, in this single-center, clinician-escalated cohort, CMCD was identified more often through non-MCA hematology pathways than among patients escalated from MCA/anaphylaxis presentations.
The low diagnostic yield of CMCD in our clinician-escalated MCA/anaphylaxis subgroup (7.1%) contrasts markedly with the 47% yield reported in Western cohorts of patients with unexplained anaphylaxis lacking cutaneous mastocytosis.12 This discrepancy highlights a fundamental methodological distinction. Western series frequently employ systematic screening in high-risk referral populations—often presenting with severe Hymenoptera venom anaphylaxis and syncope—where routine bone marrow assessment yields a high proportion of occult clonal disease. In contrast, our cohort reflects a real-world, clinician-directed escalation approach across a broader pre-test probability range, where structured venom-allergy referral pathways are less formalized and triggers are often retrospectively heterogeneous. Direct comparison of our 7.1% diagnostic yield with the prevalence-like estimates from European cohorts is therefore structurally constrained. Consequently, these differences in case selection, diagnostic criteria, and regional referral profiles likely account for the divergent diagnostic yields observed.
Clinically, these results support a risk-stratified, rather than routine, clonal work-up in adults with recurrent MCA/anaphylaxis. The REMA score, which incorporates sex, basal tryptase, presence of urticaria pigmentosa and features of hypotensive anaphylaxis, has been validated as a tool to enrich for mast cell clonality and SM in patients with systemic MCA symptoms.10 The NICAS model further refines selection in IA by combining clinical variables, tryptase and molecular markers to predict underlying clonal disease.11 Applied pragmatically, such scores can help select IA or hypotensive MCA patients for KIT D816V testing and marrow, while avoiding routine invasive work-up in all recurrent anaphylaxis. In parallel, our data underscore the need for high vigilance in hematology-first presentations (older age, cytopenias, organomegaly, abnormal marrow), where the diagnostic yield for CMCD was high.
At the molecular level, our genomic findings demonstrate parallel signatures alongside notable phenotypic differences when compared to published Western cohorts. Consistent with European and North American series, KIT D816V remained the dominant driver mutation, and co-occurring TET2, DNMT3A, and SRSF2 variants clustered predominantly within the SM subgroup.8,13 Importantly, no novel gene variants were observed in our study; all detected mutations have been previously characterized. Although ASXL1, CBL, RUNX1, and WT1 mutations were detected in a minority of our SM cases, their absolute frequencies appeared lower than those heavily enriched in large Western advanced systemic mastocytosis (AdvSM) cohorts. In such advanced Western populations, the presence of additional myeloid aberrations generally confers an adverse prognosis.14 More specifically, high-risk multimutated profiles involving the SRSF2, ASXL1, and/or RUNX1 (S/A/R) genotype are strongly associated with inferior survival.13 While direct frequency comparisons are inherently limited by our small sample size, the detection of these specific high-risk co-mutations among our SM patients—the majority of whom met criteria for AdvSM—confirms that this adverse molecular architecture is biologically conserved in the Southeast Asian population.
Regarding clonal ontogeny, foundational models of myeloid pathogenesis demonstrate that mutations in epigenetic modifiers—particularly TET2 and DNMT3A—often arise early in multipotent hematopoietic progenitors. This establishes an antecedent state of clonal hematopoiesis, which subsequently drives clonal expansion and can evolve into overt malignancies upon the acquisition of cooperating driver mutations such as KIT D816V.15,16 The TET2 co-mutations and DNMT3A R882H hotspot observed in our SM subgroup are compatible with this established model; notably, DNMT3A R882H is a well-characterized variant associated with clonal hematopoiesis of indeterminate potential (CHIP). The SRSF2 P95 variant, an RNA splicing factor mutation distinct from epigenetic modifiers, similarly clusters with clonal myeloid disease in this context. Of note, a single IA case harbored a TET2 variant in the absence of KIT D816V or other mast cell–specific driver mutations; given the known prevalence of age-related CHIP, this isolated finding most likely represents incidental clonal hematopoiesis rather than mast cell clonality, and this patient did not fulfill diagnostic criteria for CMCD.
A distinctive finding in our cohort was the detection of MPL mutations in two IA cases (18.2%), with no MPL variants identified in any SM or CM case. MPL encodes the thrombopoietin receptor. While canonical activating MPL mutations typically involve codon W515 in myeloproliferative neoplasms (MPN),17 the variants observed here (p.L513del and p.P106L) occurred at different loci in these KIT-negative IA patients.
The exclusive occurrence of MPL variants in the IA subgroup raises two clinically relevant possibilities: these may represent coincidental CHIP-associated variants in patients undergoing broad myeloid panel sequencing, or they may signal an occult or concurrent MPN in a subset of patients presenting with anaphylaxis. Although detailed hematologic follow-up data are limited in this retrospective cohort, these findings underscore the potential for myeloid NGS panels to uncover incidental clonopathies beyond the primary diagnostic question, and prospective studies with longitudinal hematologic surveillance will be important to clarify the clinical significance of MPL variants in anaphylaxis populations.
To our knowledge, this represents the first systematic application of a myeloid NGS panel in a Southeast Asian CMCD cohort, demonstrating a broadly conserved KIT-centric mutational architecture across diverse genetic backgrounds, with phenotypic distribution shaped by local referral patterns and disease subtypes.
NGS added important biological granularity. SM cases in this cohort showed a mutational pattern consistent with international AdvSM series, with KIT D816V in most patients and frequent co-mutations in TET2, SRSF2, ASXL1, DNMT3A, RUNX1, and CBL. Additional mutations in SRSF2, ASXL1, and/or RUNX1 (the S/A/R genotype) define a high-risk molecular subgroup associated with inferior survival and are now recognized as a key adverse constellation in AdvSM.13 KIT D816V allele burden itself correlates with WHO subtype and outcome, further supporting the prognostic value of quantitative molecular assessment beyond simple mutation positivity.18 A prognostic model that incorporates additional myeloid mutations has also been shown to improve survival prediction over clinical scores alone in SM.19 Together, these data—mirrored by our finding of frequent KIT-centered co-mutation patterns—support using myeloid-focused NGS in Thai patients with CMCD to refine risk stratification, follow-up intensity, and treatment planning.13,14,18,20
The observation that some KIT-negative CM cases carried myeloid co-mutations suggests overlap between clonal hematopoiesis and mast cell disease in a subset of patients, consistent with broader molecular series in SM and related myeloid neoplasms.14,20 This argues against relying on KIT alone and supports using expanded myeloid panels when evaluating suspected CMCD, particularly in adults with cytopenia or other hematologic abnormalities.
These molecular patterns are directly relevant to targeted therapy. Avapritinib, a highly selective KIT D816V inhibitor, has produced high clinical, hematologic, and molecular response rates in AdvSM in the EXPLORER and PATHFINDER trials, including patients with adverse myeloid co-mutations.21,22 Contemporary reviews emphasize that mutational risk stratification—incorporating KIT D816V allele burden and high-risk co-mutations such as S/A/R—now underpins modern treatment algorithms and prognostic counselling in AdvSM.20 Our finding that Thai SM patients display a broadly similar KIT-centered, co-mutated genomic architecture strongly suggests that these targeted strategies are biologically applicable in this population, provided issues of access, comorbidity, and toxicity monitoring can be addressed.
Several limitations warrant caution. The cohort is small and clinically selected. Importantly, the reported 7.1% should not be interpreted as prevalence among all MCA/anaphylaxis referrals, but as diagnostic yield in a highly selected escalated subgroup. Escalation from index presentation to KIT testing, marrow, and NGS depended on clinician judgment rather than predefined REMA/NICAS thresholds, so the 24 sequenced adults represent a higher-risk subset rather than all MCA/anaphylaxis referrals. A further limitation is the possible misclassification of the index phenotype. Because this retrospective study used a pragmatic referral-based MCA definition rather than formal MCAS criteria, objective documentation of hemodynamics (hypotension), “20%+2” tryptase kinetics, and standardized anaphylaxis criteria was incomplete. This introduces a risk that some cases may represent misdiagnosed food or drug allergies, or severe flares of chronic spontaneous urticaria. In addition, this is a single tertiary academic center with embedded hematology, dermatopathology, and molecular diagnostics; results may not generalize to lower-resource settings or to regions with different venom, food, and drug exposure patterns. These results should be viewed as preliminary pilot data from a single-center Southeast Asian cohort.
Future work in Southeast Asia should therefore focus on prospective, multicenter cohorts of unselected MCA/anaphylaxis referrals, with systematic recording of triggers, tryptase, REMA/NICAS scores, and predefined criteria for KIT testing and marrow. Such studies are needed to define the true prevalence of CMCD among IA and other MCA presentations in the region, to validate REMA/NICAS performance in Asian populations, and to determine whether specific co-mutation patterns predict clinical course or response to targeted therapy in real-world practice.
Conclusion
These pilot data from a Southeast Asian single-center cohort suggest a lower diagnostic yield of CMCD in clinician-escalated MCA/anaphylaxis presentations than in hematology-first presentations. This suggests that REMA/NICAS-guided, risk-stratified selection of MCA/anaphylaxis patients for KIT testing and bone marrow may be considered, rather than routine clonal work-up of all recurrent anaphylaxis. At the molecular level, NGS revealed that while TET2 was the most frequently mutated gene overall, the systemic mastocytosis (SM) subgroup exhibited a distinct KIT D816V–centered mutational landscape. Notably, we observed specific pathogenic hotspots, including DNMT3A R882H and SRSF2 P95, clustering alongside TET2 mutations in this subgroup. This genomic architecture closely parallels Western cohorts and supports the biological applicability of KIT-targeted therapies in Thai patients, though these findings should be confirmed in larger prospective regional studies before informing broader practice.
Ethics Approval
The study was approved by the Siriraj Institutional Review Board under expedited review (SIRB No. 072/2024 [IRB3]); the requirement for informed consent was waived due to the retrospective design.
Patient data confidentiality was strictly maintained, and the study was conducted in compliance with the Declaration of Helsinki.
Acknowledgments
We sincerely thank Ms. Aree Jameekornrak Taweechue, Ms. Orathai Theankeaw, Ms. Kitnittha Poladao, for their invaluable research assistance.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
There is no funding to report.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Pardanani A. Systemic mastocytosis in adults: 2023 update on diagnosis, risk stratification and management. Am J Hematol. 2023;98(7):1097–13. doi:10.1002/ajh.26962
2. Khoury JD, Solary E, Abla O, et al. The 5th edition of the World Health Organization classification of haematolymphoid tumours: myeloid and histiocytic/dendritic neoplasms. Leukemia. 2022;36(7):1703–1719. doi:10.1038/s41375-022-01613-1
3. Navarro-Navarro P, Alvarez-Twose I, Perez-Pons A, et al. KITD816V mutation in blood for the diagnostic screening of systemic mastocytosis and mast cell activation syndromes. Allergy. 2023;78(5):1347–1359. doi:10.1111/all.15584
4. Castells M, Giannetti MP, Hamilton MJ, et al. Mast cell activation syndrome: current understanding and research needs. J Allergy Clin Immunol. 2024;154(2):255–263. doi:10.1016/j.jaci.2024.05.025
5. Golden DBK, Wang J, Waserman S, et al. Anaphylaxis: a 2023 practice parameter update. Ann Allergy Asthma Immunol. 2024;132(2):124–176. doi:10.1016/j.anai.2023.09.015
6. Zanotti R, Lombardo C, Passalacqua G, et al. Clonal mast cell disorders in patients with severe Hymenoptera venom allergy and normal serum tryptase levels. J Allergy Clin Immunol. 2015;136(1):135–139. doi:10.1016/j.jaci.2014.11.035
7. Valent P, Hoermann G, Bonadonna P, et al. The normal range of baseline tryptase should be 1 to 15 ng/mL and covers healthy individuals with HalphaT. J Allergy Clin Immunol Pract. 2023;11(10):3010–3020. doi:10.1016/j.jaip.2023.08.008
8. Gonzalez-Lopez O, Munoz-Gonzalez JI, Orfao A, Alvarez-Twose I, Garcia-Montero AC. Comprehensive analysis of acquired genetic variants and their prognostic impact in systemic mastocytosis. Cancers. 2022;14(10):2487. doi:10.3390/cancers14102487
9. Leguit RJ, Wang SA, George TI, Tzankov A, Orazi A. The international consensus classification of mastocytosis and related entities. Virchows Arch. 2023;482(1):99–112. doi:10.1007/s00428-022-03423-3
10. Alvarez-Twose I, Gonzalez-de-olano D, Sanchez-Munoz L, et al. Validation of the REMA score for predicting mast cell clonality and systemic mastocytosis in patients with systemic mast cell activation symptoms. Int Arch Allergy Immunol. 2012;157(3):275–280. doi:10.1159/000329856
11. Carter MC, Desai A, Komarow HD, et al. A distinct biomolecular profile identifies monoclonal mast cell disorders in patients with idiopathic anaphylaxis. J Allergy Clin Immunol. 2018;141(1):180–188e3. doi:10.1016/j.jaci.2017.05.036
12. Gulen T, Hagglund H, Sander B, Dahlen B, Nilsson G. The presence of mast cell clonality in patients with unexplained anaphylaxis. Clin Exp Allergy. 2014;44(9):1179–1187. doi:10.1111/cea.12369
13. Jawhar M, Schwaab J, Schnittger S, et al. Additional mutations in SRSF2, ASXL1 and/or RUNX1 identify a high-risk group of patients with KIT D816V(+) advanced systemic mastocytosis. Leukemia. 2016;30(1):136–143. doi:10.1038/leu.2015.284
14. Schwaab J, Schnittger S, Sotlar K, et al. Comprehensive mutational profiling in advanced systemic mastocytosis. Blood. 2013;122(14):2460–2466. doi:10.1182/blood-2013-04-496448
15. An J, Ko M. Epigenetic modification of cytosines in hematopoietic differentiation and malignant transformation. Int J Mol Sci. 2023;24(2):1727. doi:10.3390/ijms24021727
16. Sperling AS, Gibson CJ, Ebert BL. The genetics of myelodysplastic syndrome: from clonal haematopoiesis to secondary leukaemia. Nat Rev Cancer. 2017;17(1):5–19. doi:10.1038/nrc.2016.112
17. Pikman Y, Lee BH, Mercher T, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3(7):e270. doi:10.1371/journal.pmed.0030270
18. Hoermann G, Gleixner KV, Dinu GE, et al. The KIT D 816 V allele burden predicts survival in patients with mastocytosis and correlates with the WHO type of the disease. Allergy. 2014;69(6):810–813. doi:10.1111/all.12409
19. Mannelli F, Gesullo F, Rotunno G, et al. Validation of the Mayo alliance prognostic system for mastocytosis. Blood Cancer J. 2019;9(2):18. doi:10.1038/s41408-019-0179-7
20. Pardanani A. Systemic mastocytosis in adults: 2021 Update on diagnosis, risk stratification and management. Am J Hematol. 2021;96(4):508–525. doi:10.1002/ajh.26118
21. DeAngelo DJ, Radia DH, George TI, et al. Safety and efficacy of avapritinib in advanced systemic mastocytosis: the Phase 1 EXPLORER trial. Nat Med. 2021;27(12):2183–2191. doi:10.1038/s41591-021-01538-9
22. Gotlib J, Reiter A, Radia DH, et al. Efficacy and safety of avapritinib in advanced systemic mastocytosis: interim analysis of the Phase 2 PATHFINDER trial. Nat Med. 2021;27(12):2192–2199. doi:10.1038/s41591-021-01539-8
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.